

Abstract
The active sites of caspases are composed of four mobile loops. A loop (L2) from one half of the dimer interacts with a loop (L2′) from the other half of the dimer to bind substrate. In an inactive form, the two L2′ loops form a cross-dimer hydrogen-bond network over the dimer interface. Although the L2′ loop has been implicated as playing a central role in the formation of the active-site loop bundle, its precise role in catalysis has not been shown. A detailed understanding of the active and inactive conformations is essential to control the caspase function. We have interrogated the contributions of the residues in the L2′ loop to catalytic function and enzyme stability. In wild-type and all mutants, active-site binding results in substantial stabilization of the complex. One mutation, P214A, is significantly destabilized in the ligand-free conformation, but is as stable as wild type when bound to substrate, indicating that caspase-7 rests in different conformations in the absence and presence of substrate. Residues K212 and I213 in the L2′ loop are shown to be essential for substrate-binding and thus proper catalytic function of the caspase. In the crystal structure of I213A, the void created by side-chain deletion is compensated for by rearrangement of tyrosine 211 to fill the void, suggesting that the requirements of substrate-binding are sufficiently strong to induce the active conformation. Thus, although the L2′ loop makes no direct contacts with substrate, it is essential for buttressing the substrate-binding groove and is central to native catalytic efficiency.
Keywords: caspase, apoptosis, tyrosine, compensatory structural rearrangements, substrate-binding stabilization, kinetics, loop bundle
Introduction
Apoptosis, or the programmed cellular suicide pathway, has been studied with great interest because of its role in many diseases, including cancer and autoimmune disorders, as well as for its role in development. It is estimated that over half of all diseases result from irregularities in apoptosis.1 During apoptosis, the cell executes a series of irreversible proteolytic events that ultimately cause cell death. The machinery of this cellular destruction is a family of proteins called caspases. Caspases (cysteine aspartate proteases) are part of the CD clan, the cysteine nucleophile endopeptidases.2 Caspases-3, -6, -7, -8, -9, and -10 play roles in apoptosis.
The family of apoptotic caspases function both in the signal propagation of cell death (initiator caspases) and actual cellular disassembly (executioner caspases). Initiator caspases (-8, -9, and -10) work upstream to activate the executioner caspases (-3, -6, and -7). Activation of the initiators is complex, involving formation of large multiprotein assemblies, such as the apoptosome, containing caspase-9, and the DISC complex, containing caspase-8. The canonical view of executioner caspase zymogen activation is more straightforward, relying on cleavage as the critical activating event.3–5
Initiator caspases cleave executioner caspases in two locations to liberate the N-terminal propeptide and cleave the intersubunit linker. Executioner procaspases are dimeric so that cleavage of the intersubunit linker converts each half of the homodimer into one large and one small subunit. Mature caspases then consist of four chains, with two large subunits flanking two small subunits in the mature state. Cleavage of the intersubunit linker results in the formation of two new termini that function as loops involved in substrate binding. The C-terminal end of the large subunit becomes the L2 loop, whereas the N-terminal end of the small subunit becomes the L2′ loop. The catalytic cysteine is contained on the L2 loops, thus each half of the dimer contains one catalytic site.6 The catalytic sites are surrounded by extended substrate-binding grooves. Each substrate-binding groove is composed of three loops (L2, L3, and L4) from one monomer. A fourth loop (L2′) from the opposite monomer interacts with L2 to stabilize the substrate-binding loop bundle in the active state [Fig. 1(A,B)]. Large conformational changes in L2 and L2′ are associated with the transformation between the caspase-7 conformational states, all of which have been structurally characterized. When the inactive procaspase-7 zymogen8,9 is cleaved to generate mature caspase-7,8 it remains in a semiactive conformation until substrate binds, inducing the active caspase-7 conformation.10–12 Caspase-7 can also be allosterically inhibited13 by binding of effectors to a cavity at the dimer interface. In the inactive procaspase-7 zymogen L2 and L2′ are still covalently connected yet they are observed to be pointing away from one another, separated by a stretch of disordered residues. This conformation is very similar to that observed in the allosterically inhibited state, where the L2′ loops interact above the allosteric site [Fig. 1(C,D)]. In the absence of substrate, the mature caspase-7 is in a partially active conformation [Fig. 1(E,F)]. L2 is in the conformation that allows substrate binding, but L2′ is in the down position that is more similar to the zymogen and allosterically inhibited conformations. Thus, the L2′ position correlates with caspase-7 activity.
Figure 1.
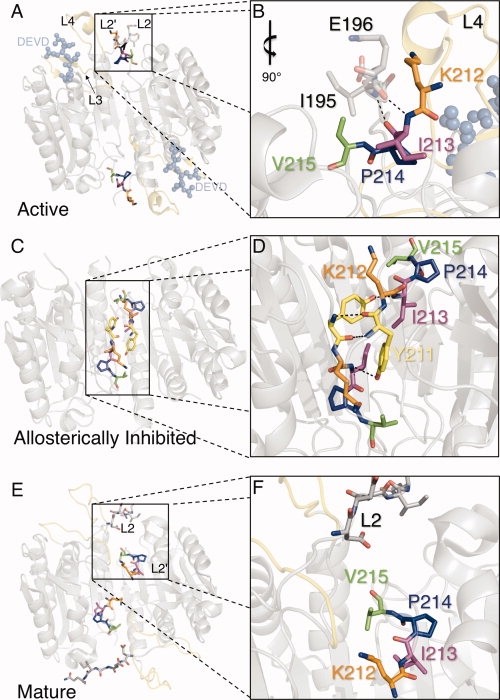
Role of the L2′ loop in the active, allosteric/zymogen and mature conformations. (A) The L2′ loop (colored sticks) is in the “up” position only when substrate binds to the active conformation of caspase-7 (PDB ID 1F1J). Residues selected for mutation (K212, orange; I213, magenta; P214, blue; V215, green) make interactions with the L2 loop from the opposite half of the dimer. The two substrate-binding grooves are bound by inhibitor DEVD (light blue ball-and-sticks), which is a substrate-mimicking compound. Loops L3 and L4 (yellow) contribute to the substrate-binding loop bundle with loops L2 and L2′. (B) Interactions between L2′ (colored sticks) and L2 (gray sticks) stabilize the active form of caspase-7. (C) The L2′ loop (colored sticks) is in the “down” position both in the zymogen and allosterically inhibited (allosteric model shown, PDB ID 1SHJ) conformations. The magnitude of the L2′ loop rearrangement is clearly visible compared to (A). Mutated residue Y211 is visible as yellow in this conformation while resides 212–215 are colored is as in (A). (D) The hydrogen-bonding network (dashed lines) at the dimer interface comprises interactions between L2′ from one monomer and L2′ from the opposite side of the dimer (dashed lines). (E, F) The half-active conformation observed in the mature (cleaved but substrate-free) form of caspase-7 (PDB ID 1K86) is colored as (A). Although the L2 loop is in a similar conformation to the active-site-bound structure, the L2′ loops point down toward the allosteric cavity in a conformation most similar to the allosteric/zymogen conformation. For additional clarity not possible in paper form, see Supporting Information Movie S1. Figures were generated and rendered in PyMol.7
This canonical caspase-7 activation model suggests L2 and L2′ can only interact properly after cleavage by initiator caspases to generate the free loops, meaning that both of the caspase monomers must be cleaved to achieve a fully active caspase. Thus, both the L2 and L2′ loops are central in caspase activation. Recently, however, hybrid dimers of one uncleavable and one cleavable caspase monomer suggest that cleavage of both monomers in not necessary for formation of one catalytically active site.14,15 This implies that the uncleaved intersubunit linker can also adopt the conformation of either L2 or L2′ but not both. The L2 loop has been implicated in an allosteric network in caspase-1 in which the two catalytic sites are coupled through an extensive hydrogen-bond network.16 It remains to be seen if this allosteric coupling is present in all of the caspases and what role the L2′ might play in this mechanism.
The fact that the L2′ loop is not in the correct orientation in the mature and zymogen conformations has been suggested to be the basis for the lack of activity in the zymogen.8 Some mutagenesis inquiries have probed the role of L2 in caspase activity, and report that mutations in this region are deleterious.15,17,18 On the basis of structural observations, we anticipate that the L2′ loop likewise plays an important role in caspase function. To our knowledge, we report the first systematic study of the L2′ loop. Given the difficulties that have been encountered in developing active-site-directed caspase therapeutics, this region is particularly interesting because it is distal from the catalytic site, but appears to be influential for both substrate binding and catalysis.
Results
Analysis of the existing crystallographic structures of caspase-7 indicates that residues in the L2′ loop buttress the L2 loop into the proper configuration of the active-site loop bundle. We investigated the role of the L2′ loop by serial alanine substitution of residues 211–215, which form the core of the buttress. The resulting mutants were tested to determine the severity of these mutations on kinetics and apparent thermal stability. To assess the structural changes in the most severely affected mutant, we solved the crystal structure and compared the results with the existing caspase-7 structures.
Role of the L2′ loop in catalytic efficiency in caspase-7
The placement of L2′ in the active conformation at the base of the active-site loop bundle suggests that it may be essential for substrate binding. Alanine mutants were designed to probe the regions of greatest interaction between L2 and L2′ (211–215) in previous crystal structures of caspase-7 in the active conformation [Fig. 1(A)].10–12 In caspase-3, the residues in the region of the L2′ loop are identical to caspase-7 (see Fig. 2) with the exception of position 211 (caspase-3 position 185). Additionally, in the crystal structures of active caspase-3, the L2′ loop is in an identical conformation10,19–23 suggesting that the buttressing role is conserved within the executioner caspases. Given the buttressing role of the L2′ loop, alanine substitution should be a useful probe for the role of each residue in the formation of the active-site loop bundle as mutations of critical residues would have a strong impact on Km.
Figure 2.

Structurally biased sequence alignment of the L2′ loop in related caspases. Caspase-7 residue 213 (Ile) is conserved in size or type in not only apoptotic executioner caspases (caspase-6, -3, and -7) but also apoptotic initiators (-8, -9, and -10), C. elegans CED3, the first discovered caspase, the inflammatory caspases (-1 and -4) and caspase-14, the epidermal-specific caspase. This residue is not conserved in the initiator caspase-2 or the inflammatory caspase-5.
Kinetics of wild-type and mutant caspase-7 variants were tested as a function of substrate concentration to evaluate Km and kcat (Table I). As anticipated, mutation of two of the residues, K212 and I213, had a significant effect on substrate binding as assessed by Km, while the Km for K212A was ∼10-fold weaker than wild-type caspase-7. The Km for I213A at ∼20-fold weaker than wild type is the most significant effect observed. I213A is the only mutation that has a significant impact, an ∼5-fold decrease, in kcat. Mutation at Y211, P214, and Y215 had minimal impact on the kinetic parameters. Catalytic efficiency (kcat/Km) of only K212A and I213A was significantly different from wild type, at 4- and 100-fold decreases, respectively. These results identify K212 and I213 as the most critical residues in the L2′ buttress.
Table I.
Kinetic Parameters for Caspase-7 Alanine-Substitution Mutants
| Km (μM) | kcat (s−1) | 106 × [kcat/Km] (M−1s−1) | Relative activity (relationship factor of mutant kcat/Km versus WT) | |
|---|---|---|---|---|
| WT | 23 ± 2 | 0.35 ± 3×10−3 | 0.015 | 1.0 |
| Y211A | 18 ± 10 | 0.39 ± 3×10−3 | 0.021 | 1.4 |
| K212A | 210 ± 20 | 0.93 ± 4×10−2 | 0.004 | 0.26 |
| I213A | 480 ± 50 | 0.07 ± 5×10−3 | 1.5 × 10−4 | 0.01 |
| P214A | 8 ± 1 | 0.10 ± 4×10−3 | 0.012 | 0.8 |
| Y215A | 26 ± 8 | 0.70 ± 2×10−2 | 0.026 | 1.7 |
Alignment of the L2′ region of the caspases (see Fig. 2) indicates hydrophobic and size conservation of the 213 position, but not a strict conservation of amino acid identity. Caspase-1, -3, -7, and -10 use isoleucine at this position, whereas other caspases substitute only valine and leucine, which are nearly isosteric with isoleucine. Position 212, which exhibited the second most significant Km effect when mutated to alanine, is not conserved across the caspase family. Position 212 is always hydrophilic; however, sizes vary from small (serine) to long (lysine) or bulky (histidine and tyrosine). In wild-type caspase-7, the exposed aliphatic portion of the lysine side chain is packed against the aliphatic portion of E196 [Fig. 1(B)]. Interestingly, these residues are not strictly conserved in the third executioner caspase, caspase-6. Conservation of this region is weak compared with the region flanking the catalytic cysteine C186 (residues 178–188).
L2′ mutations affect protein stability
Alanine mutations have been reported to generate cavities in protein cores.24 Because alanine mutants have the possibility of introducing a destabilizing cavity, we probed the stability of the protein in the mature (substrate unbound) form using thermal denaturation followed by circular dichroism [Fig. 3(A)]. Inspection of caspase-7 structures suggests that the only important interactions that form in the substrate-bound conformation but not in the mature or zymogen form are between L2 from one monomer and L2′ from the opposite monomer. Thus, interrogation of the stability of wild-type and mutant caspases in the active conformation [with substrate-like inhibitor, the caspase-binding peptide aspartate–glutamate–valine–aspartate (DEVD) bound] was also relevant.
Figure 3.
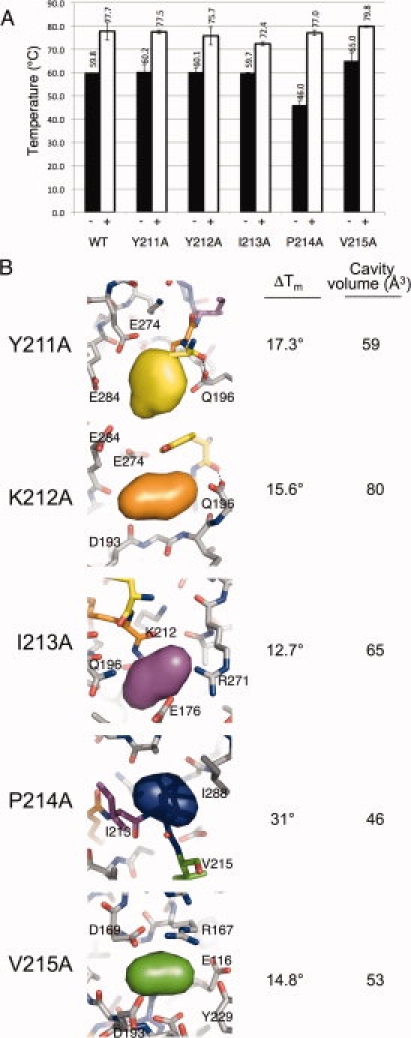
Melting temperatures and cavities generated in caspase-7 variants. (A) Melting temperatures (Tm) were measured by thermal denaturation monitoring the change in CD signal at 222 nm in the presence (white bars) and absence (black bars) of the active-site inhibitor peptide DEVD-CHO. Measurements were made at least in duplicate with independently prepared samples on separate days. Error bars represent SD between replicates. (B) Volumes of cavities in models of mutant proteins generated computationally in PyMol were assessed with Pocketfinder.25 The deleted volumes are shown as colored pillows to illustrate the chemical environment and residues forming the generated cavity. Change in the Tm of caspase-7 dimers in the presence of active-site-binding peptides (DEVD-CHO) relative to ligand free is also listed. Figures were generated and rendered in PyMol.7
The serial alanine mutations appear to have no discernable effect on the overall fold of the protein in either the inhibitor-bound versus unbound states, as the measured spectra at 25°C were unaffected by mutation (data not shown). The melting temperature (Tm) of all mutants in the unbound form was similar to wild type at ∼60°C, except for the P214A mutation which had a Tm of 46°C [Fig. 3(A)]. We had hoped to also measure thermodynamic stabilities of the caspase-7 mutants; unfortunately, unfolding of wild type and all of the mutant caspase-7 variants was irreversible and precluded accurate assessment of ΔGunfolding. We calculated the volume changes each mutation contributed to the overall structure [Fig. 3(B)], and from these figures, we were able to assess the volume of each deletion and what role it might play in the stability of each protein. The Matthews group has systematically assessed the effect of cavity-forming mutations in the core of T4 lysozyme. In one study of 44 large amino acid to alanine substitutions, cavities between 17 and 123 Å3 were observed crystallographically.24 These cavities correlated with decreases in stability (ΔΔG) of 0.9–5.0 kcal/mol. Several cavities were observed in the range of 25 Å3 that had ΔΔG ranging from −3.2 to −2.7 kcal/mol. In T4 lysozyme, a 1.9 kcal/mol change in ΔΔG corresponds to approximately a 5° decrease in Tm.26 Thus, the 14° decrease in Tm of unbound P214A could reflect a ΔΔG of greater than 4 kcal/mol.
Interestingly, the Tm difference between mature and active-site-bound forms was very pronounced, with an average 18.7°C increase in Tm of the active-site-bound form over the unbound form. The single observed structural difference between these two forms is that the inactive, unbound form is able to sample both the up conformation of L2′ and the inactive down conformation of L2′. When substrate is bound, L2′ is locked in the active up conformation. Thus, when the protein adopts the up, bound form, it appears to be significantly stabilized by the interactions between L2′ and L2 from opposite monomers and by interactions with substrate.
I213A has a smaller than average ΔTm of 7°C between the forms (see Fig. 3), indicating that an energetic penalty must be paid to adopt this conformation because the active form is less stable to thermal denaturation. The dramatically destabilized P214A mutant, on the other hand, showed a ΔTm of 31°C between bound and unbound forms. Although the identity of 214 seems to be more influential in the inactive form, in the active form, the stability is strikingly unchanged relative to the wild-type enzyme. This can be explained by structural examination of the active form where P214 seems to have a more limited role in buttressing than I213 (see Fig. 1). In the active conformation, L2–L2′ physical interactions tend to dominate over any geometric constraints imposed by P214. The substitution of P214A has negligible effect on active (bound) enzyme stability; however, in the inactive form, the hydrogen-bonding network present between L2′ and L2′ from the opposite monomer would likely be disrupted as the geometric restraints imposed by the proline at position 214 are removed by the alanine substitution. This substitution, however, does not inhibit dimerization of the protein, as measured by size exclusion chromatography (data not shown). Thus, we can conclude that substrate binding and the concomitant stabilization of interactions observed between the L2 and L2′ loops are the most critical factor in significant stabilization of the active-bound conformation. Indeed, substrate binding appears to be so influential in locking L2′ securely into the up conformation that it can overcome significant thermodynamic destabilization in the unbound state. Because P214A had negligible effect on both the stability of the active form and the kinetics of the enzyme, we focused our investigation on the structural differences caused by the I213A mutation to further understand the structural role of the L2′ loop in substrate binding and catalysis.
Crystallographic studies of I213A in the presence of active-site inhibitor
Crystals of caspase-7 I213A with DEVD bound in the active site to lock the protein in to the active conformation were grown in a variation on known conditions,12 and the structure was solved at 2.6 Å (Table II). To avoid model bias during structure determination, we omitted all of the active-site loops from the molecular replacement search model and thus calculated an omit map that is unbiased in the loop region as our initial map [Fig. 4(A,B)]. Compared to an active caspase-7 structure that crystallized in the same form (1F1J10), the RMSD of all atoms was found to be 0.191 Å, further supporting the observation that the I213A mutation had no effect on the overall fold of the protein. The substrate-binding grooves and active-site inhibitor conformations in our structure were virtually unchanged from wild-type caspase-7.
Table II.
Statistics for the X-Ray Crystal Structure of Caspase-7 I213A
| Diffraction data | |
| Wavelength (Å) | 1.54 |
| Resolution range (Å) | 50–2.61 |
| Measured reflections (n) | 26499 |
| Unique reflections | 10599 |
| Completeness (%) | 97.8 (92.0) |
| Redundancy | 2.5 (2.5) |
| <I/σI> | 22.5 (1.99) |
| Space group | P3221 |
| a = b (Å) | 89.4 |
| c (Å) | 186.1 |
| α = β (°) | 90 |
| γ (°) | 120 |
| Refinement statistics | |
| Atoms (n) | 3919 |
| Water molecules (n) | 119 |
| Rwork (%) | 19.9 |
| Rfree (%) | 25.1 |
| RMSD bond length (Å) | 0.008 |
| RMSD bond angle (°) | 0.035 |
| Average B-factor (Å2) | 51.82 |
Figure 4.
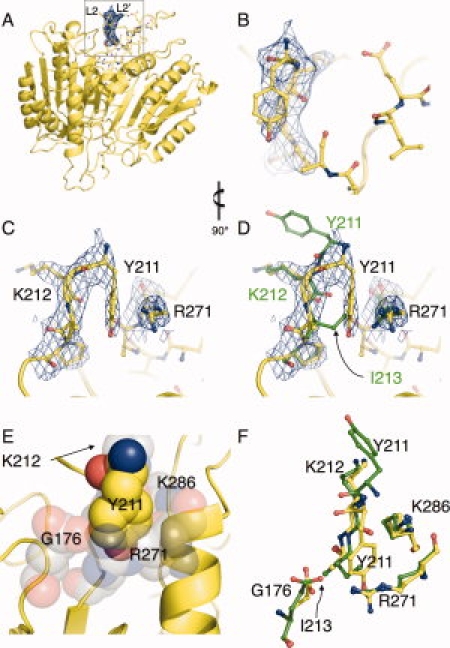
Crystallographic evidence of compensatory mechanism that facilitates active-site-binding in caspase-7 I213A. (A) Caspase-7 Y213A structure with buttress region highlighted in inset. (B) 2Fo-Fc density (blue mesh) from the initial omit map (residues 211–215 omitted from the phase calculation) and final refined model of I213A (yellow sticks) shown in same orientation as (A). (C) is as (B) but rotated 90° for clarity. The unbiased omit map clearly indicates the down position for Y211. (D) Wild-type caspase-7 (green sticks) is superimposed with the final I213A model, demonstrating that the conformation of Y211 fills a structural void when the I213 side-chain is deleted. (E) The environment (gray spheres) into which Y211 (yellow spheres) inserts is hydrophilic in nature to accommodate the hydoxyl moiety. (F) Residues forming the cavity into which Y211 inserts are in a nearly identical conformation in wild-type caspase-7 (green sticks) and I213A (yellow sticks). Figures were generated in and rendered in PyMol.7
The greatest difference between the wild-type and the I213A structure was in the Y211 region [Fig. 4(A–C)]. Y211 is unresolved in most caspase-7 structures of the active form, and only recently has been observed in crystals (PDB ID 2QLF and 2QLJ) of active caspase-7 bound to peptide inhibitors with aldehyde warheads.12 In both these structures, Y211 is solvent exposed [Fig. 4(D,F)]. In our structure, Y211 exhibits a radically different conformation, burying itself into a newly formed surface cavity. This burial appears to be a compensatory mechanism for the I213A cavity-forming mutation [Fig. 4(D,E)]. This position of the tyrosine fills a void in the region, where I213 is found in the wild-type structure. In the wild-type conformation, Y211 burial is sterically occluded by I213 [Fig. 4(E)]. This structural observation helps to explain the diminished kcat, in the context of residual catalytic activity in the I213A mutant. Given that the apparent thermal stability of I213A in the active-site-bound form is lower than any of the other mutants, and the lack of any other structural changes in this mutant crystal structure, we can conclude that the cavity formed by deletion of the I213 side-chain is so deleterious as to prevent proper formation of the loop bundle without some compensatory reorganization. The compensatory conformation we observe allows the formation of a catalytically competent enzyme but not without an energetic penalty.
Discussion
Based on the work presented here, two main points emerge. First, binding of substrate stabilizes caspase-7 via a structurally validated mechanism. Second, the core of the L2′ loop is residue I213 and this buttressing region is an essential component of active caspase-7. If the core of the L2′ loop is perturbed, then the L2′ loop does not properly hold L2 in place, the substrate-binding loop bundle is therefore destabilized and Km of the resulting mutants are weakened.
Caspase-7 is a constitutive dimer and is not believed to exist in the monomeric state to a significant degree.27 In contrast, the initiator, caspase-9, is predominantly an inactive monomer before activation by the apoptosome. Binding of active-site inhibitors drives caspase-9 to the dimeric state.28 Thus, it is plausible that other caspases would likewise be stabilized by binding of active-site inhibitors. To our knowledge, no other group has shown and quantified the dramatic stabilization of the active (active-site bound) form of caspase-7 compared with the unliganded state. Prior computational analysis of the stability of the active versus unbound conformation mirrors our experimental results, indicating that the L2′ up or active conformation of caspase-7 was significantly more stable than the L2′ down conformation.29
The roles of individual residues in the L2′ loop obviously change depending on the state (active-site-bound or unbound) of the protein. The structures of the mature unbound form of caspase-7 [Fig. 1(E,F)] when compared with the active [Fig. 1(A,B)] and allosterically inactive [Fig. 1(C,D)] forms suggest an ensemble of states, which are in dynamic equilibrium with one another. Two of our mutants, I213A and P214A, support the idea that the L2′ loop is critical for stabilizing the active-site-bound conformation.
I213A caspase-7 has the same apparent thermal stability in the unbound form as does wild type. This indicates that the equilibrium constant for unfolding of the unbound form is likewise unchanged from wild type. On the other hand, the Tm of the active-site-bound form is lowered, suggesting that an energetic penalty must be paid to achieve this conformation. The crystal structure of I213A indicates that the decrease in stability is due to the cost of hydrophilic burial when Y211 intercalates into the I213A cavity to properly buttress the loop bundle (Supporting Information Movie S2). We anticipate that in the unbound form of I213A, L2′ is free to sample the down conformation, whereas in our structure, binding at the active site locks L2′ into the active, up, conformation. If caspase-7 natively sampled the active, up, conformation to a significant degree when the active site was empty, this up conformation would lower the stability of the unbound form of the enzyme. Because the I213A unbound apparent thermal stability is unchanged, it seems that the up conformation of the L2′ loop does not contribute significantly to the conformational equilibrium ensemble of the unbound form. Thus, proper ordering of the L2′ loop as a buttress for L2 is so essential for active-site-binding that the enzyme will pay an energetic penalty to maintain this state.
The P214A mutant displays the opposite effect on stability. P214A is destabilized in the unbound state by a significant margin (14°C relative to wild type). Interestingly, when it is locked into the active conformation by active-site binding, the complex is as stable as the wild-type enzyme. This indicates the absence of energetic penalties like those observed for I213A and further suggests that the conformation of this mutant when the active site is bound is identical to wild type. The interactions of L2 with L2′ are the only additional interactions within the caspase protein itself that are observed on going from mature unbound caspase-7 to active-site-bound caspase-7. The fact that a dramatic destabilization of the unbound form of P214A can be overcome by active-site binding indicates that these interactions are fully responsible for the jump in apparent thermal stability observed in all versions of caspase-7. Structural analysis suggests that the L2′-loop up conformation should be more stable as it has better burial of exposed hydrophobic residues. Likely, an entropic penalty for ordering of mobile loops prevents this conformation in the absence of active-site binding. Moreover, the conformational equilibrium in the bound state has essentially no component of the unbound state, which would lead to destabilization of both the unliganded and liganded states.
One of the looming controversies in understanding caspase structure and activity is the structural state of the mature (cleaved) protein in the absence of active-site ligand. There is but one existing crystal structure of unliganded, mature caspase-7.8 In this structure, the active-site loops do not exist in the substrate-bound conformation (L2 and L2′ up), but are in a half-active conformation with L2 up but L2′ down. In contrast, mature caspase-3 has been reported to exist in the liganded conformation (L2 and L2′ up) with an empty active site, crystallized in the presence of what the authors term a noncatalytic site inhibitor, which is not observed in the crystal structure.21 The fact that a mutation in L2′ exists, namely P214A, that dramatically affects the stability of the unliganded state but not the liganded state suggests that without ligand, the L2′ loop is in a conformation that differs from the active state. If the unliganded enzyme “rested” in the active (L2′ up) conformation, as suggested by the mature caspase-3 structure,21 then this mutation should have the same energetic effect on the active-site liganded form as on the unbound form. As this is not the case, our analysis supports the notion that structural rearrangements that are observed in the existing crystal structures of caspase-78,10,12,13,30 are the biologically relevant conformations.
In the context of the other mutagenesis data, the Y211A mutation indicates that interactions between the backbones of the L2′ loops covering the allosteric pocket are the major energetic contributors to the allosterically inhibited/zymogen complex. In the active conformation, the interactions between the L2 loop from one half of the dimer and the L2′ loop from the other half of the dimer have a tremendous stabilizing effect on caspase-7, as the addition of active-site inhibitor increases the apparent thermal stability of the complex by 12–19°C. Thus, the L2′ loop appears to play important roles in both substrate binding and maintaining caspase-7 in an inactive yet still dimeric conformation.
We report a large perturbation (10- to 20-fold) in the Km of caspase-7 variants with alanine substitutions in the L2′ loop. Although the L2′ loop is further away in both amino acid sequence and in space than the L2 loop is from the catalytic residues, our reported mutations had as a great an effect on substrate binding and catalytic efficiency as did mutations in the L2 loop. Clark and coworkers18 mutated caspases-3 residues 167, 169, 173, and 203 (homologous residues in caspase-7 are 190, 192, 196, and 229) to alanine. The residue with the most significantly impaired substrate-binding properties was D169A, in which Km was weakened 35-fold relative to wild type. It is significant that our mutants had the same magnitude effect on substrate binding as mutations in the L2 loop because structurally the two loops also appear to act in concert.
Another region of the caspase substrate-binding loop bundle that has been widely discussed as being critical for caspase activity is the DDD “safety catch” in the L2′ loop in caspase-3. This “safety catch” comprises residues 179–181 (caspase-7 residues 205–207) and was found to be critical for activation of procaspase-3.17 The DDD “safety catch” region is further from the active site, has not been observed in crystal structures, and is unlikely to make physical buttressing interactions like those we observe in the L2′ buttressing region. Although the DDD region is essential for proper cleavage and activation of caspase-3, the L2′ loop may be a more tractable target for new methods of caspase regulation.
Before this work, the L2′ loop was implicated in both the active and zymogen/allosterically inhibited conformations. In the active state, L2′ is part of the substrate-binding loop bundle. In the zymogen/allosterically inhibited state, L2′ forms a lid occluding the allosteric cavity. Interconversion between the active and inactive conformations had been observed, but the energetic contributors of L2′ to these two conformations were unclear. Our work demonstrates that residues 212 and 213 are critical for proper functioning of the L2′ loop, particularly in binding substrate. We have shown that I213, in particular, serves as a critical buttress for holding the L2 loop (which is connected to the catalytic cysteine) in place and thus stabilizing the active form of the enzyme. If L2′ is not in the up conformation, the entire loop bundle is destabilized. This buttressing region is present in the homologous conformation in structures of the other active caspases indicating that the results we report may be relevant to the entire caspase family.
This understanding of the role of the L2′ loop as critical for formation of the active-site loop bundle also provides clues toward new mechanisms for pharmacological control of caspases. Molecules that bind overlapping with the buttressing region occupied by I213 and neighbors should work as allosteric inhibitors of caspase function. Similarly, molecules that bind behind the L2/L2′ interacting region [which is on the back side of caspase-7 in Fig. 1(A,C,E)] should stabilize the active conformation and serve as heterologous caspase activators, which are a long-sought entity. Given recent evidence that the uncleaved form of caspase-7 can support catalysis in a hemicleaved heterodimer,14,15 this mechanism of activation might even prove more relevant for activation of procaspases.
Materials and Methods
Caspase-7 mutant generation, expression, and purification
Wild-type and mutant versions of caspase-7 were expressed from a two plasmid expression system.8 The constructs encoding residues 50–198 comprise the large subunits and were contained in the pRSET(AmpR) plasmid. The constructs encoding residues 199–303 and one codon for Q plus a six-histidine tag are contained in the plasmid pBB75 (KanR) plasmid.31 Mutants in the small subunit were generated using QuikChange (Stratagene) site-directed mutagenesis. The recombinant large and small subunits were coexpressed in E. coli in 2× YT media grown for 18 h after induction with 1 mM Isopropyl β-d-1-thiogalactopyranoside at an OD600 of 0.6. Wild-type and mutant caspase-7 variants were purified using Ni-affinity liquid chromatography (HiTrap Chelating HP, GE). After binding of protein to the affinity column, the protein was eluted with a step gradient from 50 to 250 mM imidazole. Protein was diluted to 50 mM NaCl and then purified using a Macro-Prep High Q ion exchange column (Bio-Scale Mini 5 mL, Bio-Rad) with a linear gradient from 50 to 500 mM NaCl in 20 mM Tris buffer pH 8.0, with 2 mM DTT. Protein eluted in 120 mM NaCl and 20 mM Tris pH 8.0 was assessed for purity by SDS-PAGE to be 98%+ pure and stored in elution buffer at −80°C.
Caspase-7 I213A crystallization and X-ray data collection
To prepare I213A crystals, 29 μM I213A protein in a buffer containing 120 mM NaCl and 20 mM Tris pH 8.0 was incubated at room temperature with a 3:1 molar ratio of DEVD-CHO (asp–glu–val–glu-aldehyde, BioMol) to protein for 3 h. Optimal protein labeling occurred with the addition of enzyme to the small volume of DMSO-solvated inhibitor. The extent of enzyme labeling was confirmed by activity determination. Protein was then concentrated using Millipore Ultrafree 5K NMWL membrane concentrators (Millipore) to 11 mg/mL as assessed by absorbance at 280 nm. Crystal tray setup was conducted on ice with cooled buffers. Crystals were grown in 2 μL hanging drops with mother liquor consisting of 14% polyethylene glycol 3350, 300 mM diammonium hydrogen citrate at pH 5.6, 10 mM guanidine hydrochloride, and 10 mM DTT in a 1:1 ratio of protein mother liquor. Crystals grew to a maximum of 200 μm in 48 h at 4°C. Crystals were cryoprotected in 14% polyethylene glycol 3350, 300 mM diammonium hydrogen citrate at pH 5.6, 21% glycerol, and 10 mM DTT with a 60-s incubation, then frozen by rapid immersion in liquid N2. Data were collected on an R-axis IV++ detector using a Rigaku X-ray generator and showed diffraction data to 2.4 Å. Indexing, integration, and scaling were carried out on HKL2000.32 Low I/σ in the high-resolution shells forced the dataset to be restricted to 2.61Å. Only 47° of data were collected, resulting in low overall data redundancy, because of a power-outage leading to the loss of the crystal.
Structural determination
Phase information was obtained by molecular replacement using 1F1J as the search model with residues 194–196/494–496 and 212–213/512–513 omitted during searches with CCP4 amore.33 Tyrosine at position 211/511 was not present in the parent model. Removed residues were rebuilt into unambiguous density using O,34 then refined with three rounds of Refmac restrained refinement using medium strength NCS averaging constraints. Waters were inserted with multiple rounds of ARP/warp then checked for stereochemical viability manually. The final refined model contains residues 58–196, 211–303 for chain A and 57–196, 211–304 for chain B. The inhibitor is modeled as chains C and D, with residue numbers 701–705 and 801–805, respectively, and 119 ordered waters as chain W.
Caspase activity assays
Enzyme concentrations were determined by active site titration with suicide substrate DEVD-FMK (BioMol) in caspase activity buffer containing 100 mM Hepes pH 7.0, 10% polyethylene glycol 400, 0.1% CHAPS, 5 mM β-mercaptoethanol, and 5 mM CaCl2 against flourogenic substrate, DEVD-AMC, Ex365/Em495 (BioMol). Active site titration setups were incubated over a period of 4 h in 120 mM NaCl, 20 mM Tris pH 8.0 at nanomolar concentrations. Optimal labeling was observed when protein was added to DEVD-FMK solvated in DMSO in 96-well V-bottom plates, sealed with tape, and incubated at room temperature. Ninety microliters of aliquots were transferred to black-well plates in duplicate and assayed with 50-fold molar excess of substrate. For kinetic measurements, 50 nM protein (500 nM in the case of K212A, I213A) was assayed over 0–180 μM DEVD-AMC (0–500 μM for K212A, I213A) over a course of 7 min. Assays were performed at 37°C in 100 μL volumes in microplate format using a Molecular Devices Spectramax M5 spectrophotometer. Initial velocities versus substrate concentration were fit to a rectangular hyperbola using GraFit software (Erithacus Software) to determine kinetic parameters Km and kcat.
Thermal stability determination
Thermal denaturation of wild-type or caspase-7 variants was monitored by loss of circular dichroism signal at 222 nm on a J-715 circular dichroism spectrometer (Jasco) with a PTC-348WI Peltier controller. Inhibited protein was prepared by 4-h incubation with 3M equivalents of DEVD-CHO (asp–glu–val–glu-aldehyde, BioMol) at a concentration of 6–12 μM in 120 mM NaCl, 20 mM Tris pH 8.0. Both inhibited and apo proteins were then buffer exchanged into 10 mM phosphate buffer, pH 7.4 using Millipore Ultrafree 5K NMWL membrane concentrators (Millipore) for repeated concentration and dilution until NaCl concentration was below 1 nM and protein concentration of ∼12 μM as assessed by absorbance at 280 nm. Data were collected in duplicate on separate days and fit using Origin Software (OriginLab) using sigmoid fit.
Assessment generated cavity volumes
Models of the alanine-mutant caspase-7 variants were generated in PyMol7 and analyzed with Pocketfinder25 to calculate the volume of pockets created by alanine mutations.
Coordinates
Coordinates and structure factors have been deposited in the Protein Data Bank with the accession code 3H1P.
Acknowledgments
The authors thank Johnson Agniswamy and Irene Weber for advice on crystallization of caspase-7 with active-site inhibitors and Lila Gierasch for use of her CD spectrometer.
Glossary
Abbreviations:
- DEVD
the caspase-binding peptide aspartate–glutamate–valine–aspartate
- CHO
aldehyde
- FMK
fluoromethyl ketone
- Tm
melting temperature.
References
- 1.Reed JC, Tomaselli KJ. Drug discovery opportunities from apoptosis research. Curr Opin Biotechnol. 2000;11:586–592. doi: 10.1016/s0958-1669(00)00148-8. [DOI] [PubMed] [Google Scholar]
- 2.Rawlings ND, Barrett AJ. Evolutionary families of peptidases. Biochem J. 1993;290:205–218. doi: 10.1042/bj2900205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Srinivasula SM, Ahmad M, MacFarlane M, Luo Z, Huang Z, Fernandes-Alnemri T, Alnemri ES. Generation of constitutively active recombinant caspases-3 and -6 by rearrangement of their subunits. J Biol Chem. 1998;273:10107–10111. doi: 10.1074/jbc.273.17.10107. [DOI] [PubMed] [Google Scholar]
- 4.Denault JB, Salvesen GS. Human caspase-7 activity and regulation by its N-terminal peptide. J Biol Chem. 2003;278:34042–34050. doi: 10.1074/jbc.M305110200. [DOI] [PubMed] [Google Scholar]
- 5.Denault JB, Salvesen GS. Apoptotic caspase activation and activity. Methods Mol Biol. 2008;414:191–220. doi: 10.1007/978-1-59745-339-4_15. [DOI] [PubMed] [Google Scholar]
- 6.Wilson KP, Black JA, Thomson JA, Kim EE, Griffith JP, Navia MA, Murcko MA, Chambers SP, Aldape RA, Raybuck SA, Livingston DJ. Structure and mechanism of interleukin-1 β converting enzyme. Nature. 1994;370:270–275. doi: 10.1038/370270a0. [DOI] [PubMed] [Google Scholar]
- 7.DeLano WL. The PyMOL molecular graphics system. San Carlos, CA: DeLano Scientific; 2002. Available at http://www.pymol.org. [Google Scholar]
- 8.Chai J, Wu Q, Shiozaki E, Srinivasula SM, Alnemri ES, Shi Y. Crystal structure of a procaspase-7 zymogen: mechanisms of activation and substrate binding. Cell. 2001;107:399–407. doi: 10.1016/s0092-8674(01)00544-x. [DOI] [PubMed] [Google Scholar]
- 9.Ridel SJ, Fuentes-Prior P, Renatus M, Kairies N, Krapp S, Huber R, Salveson GS, Bode W. Structural basis for the activation of human procaspase-7. Proc Natl Acad Sci USA. 2001;98:14790–14795. doi: 10.1073/pnas.221580098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wei Y, Fox T, Chambers SP, Sintchak J, Coll JT, Golec JM, Swenson L, Wilson KP, Charifson PS. The structures of caspases-1, -3, -7, and -8 reveal the basis for substrate and inhibitor selectivity. Chem Biol. 2000;7:423–432. doi: 10.1016/s1074-5521(00)00123-x. [DOI] [PubMed] [Google Scholar]
- 11.Chai J, Shiozaki E, Srinivasula SM, Wu Q, Datta P, Alnemri ES, Shi Y. Structural basis of caspase-7 inhibition by XIAP. Cell. 2001;104:769–780. doi: 10.1016/s0092-8674(01)00272-0. [DOI] [PubMed] [Google Scholar]
- 12.Agniswamy J, Fang B, Weber IT. Plasticity of S2-S4 specificity pockets of executioner caspase-7 revealed by structural and kinetic analysis. FEBS J. 2007;274:4752–4765. doi: 10.1111/j.1742-4658.2007.05994.x. [DOI] [PubMed] [Google Scholar]
- 13.Hardy JA, Lam J, Nguyen JT, O'Brien T, Wells JA. Discovery of an allosteric site in the caspases. Proc Natl Acad Sci USA. 2004;101:12461–12466. doi: 10.1073/pnas.0404781101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berger AB, Witte MD, Denault JB, Sadaghiani AM, Sexton KM, Salvesen GS, Bogyo M. Identification of early intermediates of caspase activation using selective inhibitors and activity-based probes. Mol Cell. 2006;23:509–521. doi: 10.1016/j.molcel.2006.06.021. [DOI] [PubMed] [Google Scholar]
- 15.Denault JB, Bekes M, Scott FL, Sexton KM, Bogyo M, Salvesen GS. Engineered hybrid dimers: tracking the activation pathway of caspase-7. Mol Cell. 2006;23:523–533. doi: 10.1016/j.molcel.2006.06.020. [DOI] [PubMed] [Google Scholar]
- 16.Datta D, Scheer JM, Romanowski MJ, Wells JA. An allosteric circuit in caspase-1. J Mol Biol. 2008;381:1157–1167. doi: 10.1016/j.jmb.2008.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roy S, Bayly CI, Gareau Y, Houtzager VM, Kargman S, Keen SL, Rowland K, Seiden IM, Thornberry NA, Nicholson DW. Maintenance of caspase-3 proenzyme dormancy by an intrinsic “safety catch” regulatory tripeptide. Proc Natl Acad Sci USA. 2001;98:6132–6137. doi: 10.1073/pnas.111085198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feeney B, Pop C, Swartz P, Mattos C, Clark AC. Role of loop bundle hydrogen bonds in the maturation and activity of (Pro)caspase-3. Biochemistry. 2006;45:13249–13263. doi: 10.1021/bi0611964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mittl PR, Di Marco S, Krebs JF, Bai X, Karanewsky DS, Priestle JP, Tomaselli KJ, Grutter MG. Structure of recombinant human CPP32 in complex with the tetrapeptide acetyl-Asp-Val-Ala-Asp fluoromethyl ketone. J Biol Chem. 1997;272:6539–6547. doi: 10.1074/jbc.272.10.6539. [DOI] [PubMed] [Google Scholar]
- 20.Choong IC, Lew W, Lee D, Pham P, Burdett MT, Lam JW, Wiesmann C, Luong TN, Fahr B, DeLano WL, McDowell RS, Allen DA, Erlanson DA, Gordon EM, O'Brien T. Identification of potent and selective small-molecule inhibitors of caspase-3 through the use of extended tethering and structure-based drug design. J Med Chem. 2002;45:5005–5022. doi: 10.1021/jm020230j. [DOI] [PubMed] [Google Scholar]
- 21.Ni C, Li C, Wu J, Spada A, Ely K. Conformational restrictions in the active site of unliganded human caspase-3. J Mol Recognit. 2003;16:121–124. doi: 10.1002/jmr.615. [DOI] [PubMed] [Google Scholar]
- 22.Ganesan R, Mittl PR, Jelakovic S, Grutter MG. Extended substrate recognition in caspase-3 revealed by high resolution X-ray structure analysis. J Mol Biol. 2006;359:1378–1388. doi: 10.1016/j.jmb.2006.04.051. [DOI] [PubMed] [Google Scholar]
- 23.Fu G, Chumanevich AA, Agniswamy J, Fang B, Harrison RW, Weber IT. Structural basis for executioner caspase recognition of P5 position in substrates. Apoptosis. 2008;13:1291–1302. doi: 10.1007/s10495-008-0259-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu J, Baase WA, Baldwin E, Matthews BW. The response of T4 lysozyme to large-to-small substitutions within the core and its relation to the hydrophobic effect. Protein Sci. 1998;7:158–177. doi: 10.1002/pro.5560070117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jackson MR, Laurie TRA. 2004. http://www.modelling.leeds.ac.uk/pocketfinder/ University of Leeds Pocket-Finder Pocket Detection.
- 26.Gassner NC, Baase WA, Matthews BW. A test of the “jigsaw puzzle” model for protein folding by multiple methionine substitutions within the core of T4 lysozyme. Proc Natl Acad Sci USA. 1996;93:12155–12158. doi: 10.1073/pnas.93.22.12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shiozaki EN, Chai J, Rigotti DJ, Riedl SJ, Li P, Srinivasula SM, Alnemri ES, Fairman R, Shi Y. Mechanism of XIAP-mediated inhibition of caspase-9. Mol Cell. 2003;11:519–527. doi: 10.1016/s1097-2765(03)00054-6. [DOI] [PubMed] [Google Scholar]
- 28.Renatus M, Stennicke HR, Scott FL, Liddington RC, Salvesen GS. Dimer formation drives the activation of the cell death protease caspase 9. Proc Natl Acad Sci USA. 2001;98:14250–14255. doi: 10.1073/pnas.231465798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Piana S, Sulpizi M, Rothlisberger U. Structure-based thermodynamic analysis of caspases reveals key residues for dimerization and activity. Biochemistry. 2003;42:8720–8728. doi: 10.1021/bi034032l. [DOI] [PubMed] [Google Scholar]
- 30.Riedl SJ, Fuentes-Prior P, Renatus M, Kairies N, Krapp S, Huber R, Salvesen GS, Bode W. Structural basis for the activation of human procaspase-7. Proc Natl Acad Sci USA. 2001;98:14790–14795. doi: 10.1073/pnas.221580098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Batchelor AH, Piper DE, de la Brousse FC, McKnight SL, Wolberger C. The structure of GABPα/β: an ETS domain-ankyrin repeat heterodimer bound to DNA. Science. 1998;279:1037–1041. doi: 10.1126/science.279.5353.1037. [DOI] [PubMed] [Google Scholar]
- 32.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 33.Dodson E, Winn M, Ralph A. Collaborative computational Project, number 4: providing programs for protein crystallography. Methods Enzymol. 1997;277:620–633. doi: 10.1016/s0076-6879(97)77034-4. [DOI] [PubMed] [Google Scholar]
- 34.Jones T, Zou J, Cowan S, Kjeldgaard M. Improved methods for the building of protein models in electron density maps and the locations of errors in these models. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]