

Abstract
Empirical or knowledge-based potentials have many applications in structural biology such as the prediction of protein structure, protein–protein, and protein–ligand interactions and in the evaluation of stability for mutant proteins, the assessment of errors in experimentally solved structures, and the design of new proteins. Here, we describe a simple procedure to derive and use pairwise distance-dependent potentials that rely on the definition of effective atomic interactions, which attempt to capture interactions that are more likely to be physically relevant. Based on a difficult benchmark test composed of proteins with different secondary structure composition and representing many different folds, we show that the use of effective atomic interactions significantly improves the performance of potentials at discriminating between native and near-native conformations. We also found that, in agreement with previous reports, the potentials derived from the observed effective atomic interactions in native protein structures contain a larger amount of mutual information. A detailed analysis of the effective energy functions shows that atom connectivity effects, which mostly arise when deriving the potential by the incorporation of those indirect atomic interactions occurring beyond the first atomic shell, are clearly filtered out. The shape of the energy functions for direct atomic interactions representing hydrogen bonding and disulfide and salt bridges formation is almost unaffected when effective interactions are taken into account. On the contrary, the shape of the energy functions for indirect atom interactions (i.e., those describing the interaction between two atoms bound to a direct interacting pair) is clearly different when effective interactions are considered. Effective energy functions for indirect interacting atom pairs are not influenced by the shape or the energy minimum observed for the corresponding direct interacting atom pair. Our results suggest that the dependency between the signals in different energy functions is a key aspect that need to be addressed when empirical energy functions are derived and used, and also highlight the importance of additivity assumptions in the use of potential energy functions.
Keywords: protein structure assessment, knowledge-based potentials, statistical potentials, comparative modeling, protein structure prediction
Introduction
Different approaches to derive empirical energy functions emerged as a consequence of the increasing amount of three-dimensional protein structures solved by experiment and deposited during the last decades in the Protein Data Bank.1 Empirical energy functions consist on the incorporation of Boltzmann statistics to analyze propensities of interaction between atoms from known protein structures.2 These energy functions are commonly known as scoring functions, empirical potentials, knowledge-based potentials, or statistical potentials.3
In contrast to classical force fields, empirical potentials do not classify forces, but instead, based on geometrical descriptors, they extract information about the interactions between two or more bodies from experimental data of known protein structures.4 Using principles borrowed from statistical physics, these knowledge-based potentials describe microstates of interactions within protein structures as probabilities of discrete events normalized in reference to the whole system (i.e., all possible microstates).
Most of the research on empirical potentials has been focused on the setting and optimization of parameters, which include completeness of the sample space,5,6 geometric descriptors such as distance or angles between atoms,7 reduced amino acid alphabets and atom-type definitions,8,9 bodies of interaction and the structure of the potential,4,10,11 and reference systems12–15 among others.
Given that empirical potentials deal with information about specific atomic interactions in proteins, their performance will be directly related to their parameters; in other words, related to the set of variables that are involved in the compression and decompression of the structural information during the derivation and evaluation processes, respectively.
Although the close relationship between information theory and classic statistical mechanics has been recognized a long time ago,16 only recently this connection was extrapolated to understand the empirical potentials from an information theoretic point of view.17 Based on the similarity between the formulation of these potentials and the classical information theory,18 pseudo energies derived from database statistics can be considered informatic functions.17
Empirical potentials make use of the information encoded in protein structure databases in a two-step process. First, derivation consists on the extraction of information from a database of representative protein structures. This information is compressed in terms of probability distributions and translated into a series of energy functions that constitute the potential. In a second step, the potential is used to evaluate a given protein structure. Although the purpose of the derivation step is to extract structural information to finally build a representative potential energy function, the evaluation step seeks to optimize the usage of that information. Both procedures depend on a series of parameters that determine the efficiency of the information extraction and usage.
Commonly, empirical potentials are composed of energy functions describing all atom–atom interactions observed in native proteins. Each energy function has specific information about a particular atom pair interaction. However, proteins are systems of many interacting particles. Most importantly, covalent bonds between atoms generate structural constraints that introduce different levels of dependencies among the obtained distributions that are used to derive the energy functions.
A common assumption in the evaluation procedure with potential energy functions is the additivity principle.5,19 The Fourth Law of Thermodynamics, as this principle has been called, states that the free energy contribution of two or more phenomena are additive if and only if independency applies.20 Unfortunately, this is clearly not the case in empirical potentials derived from known protein structures.
Dependency among physical phenomena has its statistical counterpart in the concept of correlation. Multiple atomic interactions, as those observed in protein structures, may give rise to complex correlation patterns that are the origin of deviations from additivity. The purpose of energy functions is to capture these complex patterns.
Different approaches have been developed in this direction, which include studies focused on multiple body interactions,21 cooperativity estimated from the comparison of energy functions,11 and geometric filtering of pairwise atomic contacts.22,23
Major improvements in energy function performance are related to the problem of additivity. Short-distance range and nonlocal energy functions reduce the dependency among interactions by considering only direct interacting particles that are not constraint to be close in three-dimensional space (i.e., by defining the nonlocal component in the potential, the two interacting atoms belong to amino acids that are far away in the protein chain; by defining a short maximum distance range in the potential, the effect of atom connectivity is also reduced, because only the closest interacting atomic shells will be considered). This observation settled the basis to derive empirical energy functions based on a reduced number of atom types and consisting only of nonlocal interactions at short distances. The statistical potential obtained, called ANOLEA,24 is based on a reduced definition of 40 atom types8 and incorporates only nonlocal information (sequence separation or topological factor k larger than nine residues) at a short distance range (maximum of 7.0 Å), therefore excluding some of those shielded atomic interactions that mostly arise from atom connectivity constraints when a large maximum distance range is used to derive the potential.
In an effort to further improve the performance of empirical potentials, energy functions combining explicit physical and statistical components have also been developed. These include physical energy functions replacing noncovalent interactions terms with a nonlocally derived statistical energy function.25 The calculation of 1–4 and above nonbonded terms of classical force fields from an empirical potential allows to obtain a more precise description of local interactions, further improving the discrimination between native and near-native protein structures.26
Here, we propose that energy function performance can be managed by controlling the processes of information extraction and usage at the derivation and evaluation steps, respectively. The derivation process should attempt to maximize the extraction of information from nonlocal interactions and to minimize spurious dependencies among energy functions by excluding noninformative interactions; however, the evaluation step should maximize the total number of atom–atom interactions considered.
To this end, we describe a simple geometric procedure to identify those atoms that are interacting directly in three-dimensional space (e.g., those atom pairs whose interactions are not shielded by other atoms). This method is able to capture the extent at which covalent or noncovalently linked atoms determine the effectiveness of the atomic interactions. While using this procedure, only a subset of all possible interactions per atom (i.e., those that are not shielded by any other atom) are considered informative, and thus called effective atomic interactions. Even though these selected atomic interactions are usually scattered through the three-dimensional interacting sphere, they represent a reliable first shell of atomic contacts. Here, we show that this methodology, when used for the calculation of empirical potentials from a database of protein structures, has a clear effect upon the shape of the resulting energy functions, improving their performance at discriminating between native and near-native protein structures.
Our findings suggest that dependency among atomic interactions is a key aspect that needs to be considered when empirical energy functions are derived and used, and emphasize the importance of information and additivity assumptions in the use of potential energy functions.
Results
Effective atomic interactions
We first present a simple geometric method to define the effectiveness of pairwise atomic interactions. The method consists on estimating the exposure between two atoms taking into account the relative position of all other atoms inside an interacting sphere, which is centered in the atom under analysis. The physical exposure between two atoms is evaluated by calculating the angles among all possible constrained three-body combinations inside the contacting sphere of a given atom [Fig. 1(A)]. The combinations are constrained because atoms X and Y must be the flanking points while calculating the angle. Briefly, the effectiveness of the interaction between two hypothetic atoms X and Y is evaluated by measuring all the X-Wi-Y angles, where Wi is every non-X and -Y atom found inside the X interacting sphere. A given interaction is effective if, and only if, all calculated angles are equal to, or smaller than, a fixed shielding angle Ω; otherwise the interaction is defined as shielded by other atoms and thus it is not considered in the calculations (see Methods section).
Figure 1.
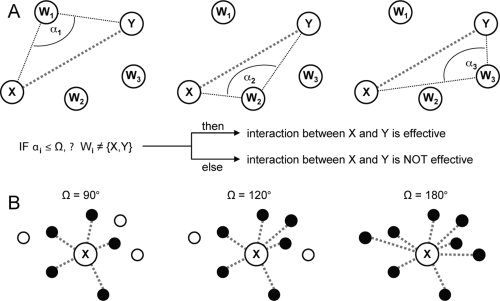
Definition of effective atomic interactions. (A) To determine the effectiveness of the interaction between atoms X and Y, all other atoms inside the X interacting sphere (Wi atoms) are evaluated by comparing each αi angle (i.e., the angle of X-Wi-Y atoms) with a defined shielding angle value Ω. If all the αi angles observed are smaller than Ω, then the interaction between X and Y is defined as effective (for details see Methods section). (B) Two-dimensional view of three interacting spheres of X, which differ in the value of Ω. Filled circles represent those atoms interacting effectively with X. Open circles represent atoms not interacting effectively with X, as they are shielded by other atoms inside the interacting sphere of X, according to the Ω value defined and used. A definition of Ω = 90° commonly captures the first interacting atom shell. By using Ω = 180°, as it is the case of traditional pairwise potentials, all the interactions observed inside the contacting sphere are considered as effective.
The goal of this simple procedure is to attempt the recognition of the first atomic interacting shell for each atom in a three-dimensional protein structure, the extension of which can be fine-tuned by controlling the value of the shielding angle [Fig. 1(B)]. This parameter value determines how strictly the effective interactions are defined. The most permissive scenario, when Ω = 180°, defines all the interactions occurring within the maximum accepted distance range as effective [Fig. 1(B)].
Variants of empirical potential derivation and utilization
In previous work, we have introduced the concept that an empirical potential can be derived with a fixed set of parameters, and then used to calculate the energy of a protein structure with a different set of parameters.27 In that work, a potential that was derived only for the nonlocal interactions was then used to calculate the energy of local and nonlocal interactions (i.e., the 1–4 and above nonbonded interactions). The energy of the local interactions was obtained by direct extrapolation from a potential that did not contain those terms explicitly. For example, this potential was derived by considering only those atom pairs that belonged to amino acids separated by more than nine residues in the protein chain. This nonlocal restriction at the derivation step assures that the atom pair is not restraint to be close in three-dimensional space because of chain connectivity effects. However, when this potential was used to calculate the total energy of the protein, local interactions were also assessed, although they were not considered at the derivation step for the reasons explained earlier. In that work, we evaluated this particular strategy of assessing local interacting terms (i.e., 1–4 nonbonded interactions and above), by using the information contained in the potential that was obtained from nonlocal interactions only in native protein structures. We suggested that this approach allows the maximization of the information quality and quantity at the potential's derivation and utilization steps, respectively.27
Here, we apply the same concept, but in a different context. We differentially derive and use nonlocal potentials with distinct definition schemes of the atom–atom interactions. Effectiveness at the derivation (D) is defined if the empirical potential is calculated considering any Ω value that is smaller than 180°. Accordingly, effectiveness at the utilization (U) is defined if the interactions are estimated at any Ω value that is smaller than 180°, and the other parameters are the same as those used to derive the potential. The traditional approach is also defined at derivation or utilization, setting the Ω value to 180°. Thus, a Dπ-U90 combination means that the empirical potential was derived with Ω = 180° and then used to calculate only the energy of the effective atomic interactions defined by setting Ω = 90°. This strategy allows us to decouple or to dissociate the processes of information extraction and utilization when calculating and using statistical potentials. The advantages of this approach have been already demonstrated for the calculation of close nonbonded interactions.27 The nonbonded interactions cannot be directly calculated from native proteins because of connectivity constraints (i.e., the observed distances between atom pairs in native proteins are almost the same as those obtained in the reference system, thus leading to the obtention of flat energy curves with energy values near to zero). However, if the energy functions are derived only for the nonlocal interactions between atom pairs, then these functions can be used to infer the energy of close nonbonded terms (assuming that the energy curve derived from interactions free of constraints will better represent the true energy curve of a given atom pair, irrespectively that the observed interaction is constrained or not by connectivity effects).
According to this methodology, four combinations for derivation and utilization of potentials are possible, which consist of: (i) Combination Dπ-Uπ: the potential is derived for all interactions within the defined distance range by using a shielding angle of 180° and then used to calculate the energy of the same type of interactions. This corresponds to the traditional approach described in the literature. (ii) Combination DΩ-UΩ: the potential is derived for the effective interactions only (as defined by Ω) and then used to calculate the energy of the effective interactions (as defined by the same Ω). In this potential, the total number of interactions observed will depend on the value of Ω. (iii) Combination Dπ-UΩ: the potential is derived for all interactions within the defined distance range by using a shielding angle of 180° and then used to calculate the energy of the effective interactions only (as defined by Ω). (iv) Combination DΩ-Uπ: the potential is derived for the effective interactions only (as defined by Ω) and then used to calculate the energy of all interactions (by using a shielding angle of 180°).
The four combinations for derivation and utilization of the potentials described earlier, together with the definition of distinct parameters such as the maximum distance range and shielding angles, led to 49 different schemes of derivation and utilization of the potentials tested in this work (Table I).
Table I.
Combinations of Derivation/Utilization of Potentials Tested in This Work
| Derivation (D) |
Utilization (U) |
|||||
|---|---|---|---|---|---|---|
| Name | Distance range (Å) | Type of interactions | Shielding angle (°) | Distance range (Å) | Type of interactions | Shielding angle (°) |
| Dπ-Uπ | 7.0 | Noneffective | 180 | 7.0 | Noneffective | 180 |
| Dπ-U60 | 7.0 | Noneffective | 180 | 7.0 | Effective | 60 |
| Dπ-U70 | 7.0 | Noneffective | 180 | 7.0 | Effective | 70 |
| Dπ-U80 | 7.0 | Noneffective | 180 | 7.0 | Effective | 80 |
| Dπ-U90 | 7.0 | Noneffective | 180 | 7.0 | Effective | 90 |
| Dπ-U100 | 7.0 | Noneffective | 180 | 7.0 | Effective | 100 |
| Dπ-U110 | 7.0 | Noneffective | 180 | 7.0 | Effective | 110 |
| Dπ-U120 | 7.0 | Noneffective | 180 | 7.0 | Effective | 120 |
| Dπ-U130 | 7.0 | Noneffective | 180 | 7.0 | Effective | 130 |
| Dπ-U140 | 7.0 | Noneffective | 180 | 7.0 | Effective | 140 |
| Dπ-U150 | 7.0 | Noneffective | 180 | 7.0 | Effective | 150 |
| Dπ-U160 | 7.0 | Noneffective | 180 | 7.0 | Effective | 160 |
| Dπ-U170 | 7.0 | Noneffective | 180 | 7.0 | Effective | 170 |
| D60-Uπ | 7.0 | Effective | 60 | 7.0 | Noneffective | 180 |
| D70-Uπ | 7.0 | Effective | 70 | 7.0 | Noneffective | 180 |
| D80-Uπ | 7.0 | Effective | 80 | 7.0 | Noneffective | 180 |
| D90-Uπ | 7.0 | Effective | 90 | 7.0 | Noneffective | 180 |
| D100-Uπ | 7.0 | Effective | 100 | 7.0 | Noneffective | 180 |
| D110-Uπ | 7.0 | Effective | 110 | 7.0 | Noneffective | 180 |
| D120-Uπ | 7.0 | Effective | 120 | 7.0 | Noneffective | 180 |
| D130-Uπ | 7.0 | Effective | 130 | 7.0 | Noneffective | 180 |
| D140-Uπ | 7.0 | Effective | 140 | 7.0 | Noneffective | 180 |
| D150-Uπ | 7.0 | Effective | 150 | 7.0 | Noneffective | 180 |
| D160-Uπ | 7.0 | Effective | 160 | 7.0 | Noneffective | 180 |
| D170-Uπ | 7.0 | Effective | 170 | 7.0 | Noneffective | 180 |
| D60-U60 | 7.0 | Effective | 60 | 7.0 | Effective | 60 |
| D70-U70 | 7.0 | Effective | 70 | 7.0 | Effective | 70 |
| D80-U80 | 7.0 | Effective | 80 | 7.0 | Effective | 80 |
| D90-U90 | 7.0 | Effective | 90 | 7.0 | Effective | 90 |
| D100-U100 | 7.0 | Effective | 100 | 7.0 | Effective | 100 |
| D110-U110 | 7.0 | Effective | 110 | 7.0 | Effective | 110 |
| D120-U120 | 7.0 | Effective | 120 | 7.0 | Effective | 120 |
| D130-U130 | 7.0 | Effective | 130 | 7.0 | Effective | 130 |
| D140-U140 | 7.0 | Effective | 140 | 7.0 | Effective | 140 |
| D150-U150 | 7.0 | Effective | 150 | 7.0 | Effective | 150 |
| D160-U160 | 7.0 | Effective | 160 | 7.0 | Effective | 160 |
| D170-U170 | 7.0 | Effective | 170 | 7.0 | Effective | 170 |
| Dπ-Uπ-R5 | 5.0 | Noneffective | 180 | 5.0 | Noneffective | 180 |
| Dπ-Uπ-R12 | 12.0 | Noneffective | 180 | 12.0 | Noneffective | 180 |
| Dπ-Uπ-R15 | 15.0 | Noneffective | 180 | 15.0 | Noneffective | 180 |
| Dπ-U90-R5 | 5.0 | Noneffective | 180 | 5.0 | Effective | 90 |
| Dπ-U90-R12 | 12.0 | Noneffective | 180 | 12.0 | Effective | 90 |
| Dπ-U90-R15 | 15.0 | Noneffective | 180 | 15.0 | Effective | 90 |
| D90-Uπ-R5 | 5.0 | Effective | 90 | 5.0 | Noneffective | 180 |
| D90-Uπ-R12 | 12.0 | Effective | 90 | 12.0 | Noneffective | 180 |
| D90-Uπ-R15 | 15.0 | Effective | 90 | 15.0 | Noneffective | 180 |
| D90-U90-R5 | 5.0 | Effective | 90 | 5.0 | Effective | 90 |
| D90-U90-R12 | 12.0 | Effective | 90 | 12.0 | Effective | 90 |
| D90-U90-R15 | 15.0 | Effective | 90 | 15.0 | Effective | 90 |
Benchmark test set
To assess the effect of changing the parameters in the performance of empirical potentials, we used a benchmark set of near-native comparative protein structures models and their corresponding experimental native structures. Using the empirical potentials, we calculated the total normalized energy for each protein structure and evaluated the performance of the potentials at discriminating between the two data populations: near-native protein structure models and their native protein structure counterparts. The evaluation of the performance of each potential as a binary classifier (i.e., classification of native and near-native protein structures) was carried out by receiver operating characteristic (ROC) curve analysis (see Methods section). More specifically, the area under the ROC curves (AUC), which is a robust indicator of classifier performance, was used to assess the performance of the potentials in this task.
Effect of the shielding angle
A critical parameter of the methodology presented here is the shielding angle Ω, which determines those atomic interactions that will be defined as effective. To study the influence of this parameter on the performance of an energy function, we derived and used several empirical potentials with different Ω values in the range between 60° and 180° (Table I) and tested them as binary classifiers on our benchmark set of models (Fig. 2). Three main regions of varying performance are clearly observed for the different shielding angles defined. First, a set of Ω angles lying between 60° and 90°, where the performance of DΩ-UΩ is clearly better than that obtained for Dπ-UΩ or DΩ-Uπ. Then, a second region of transition with Ω angles lying between 100° and 140°, where the performance of DΩ-Uπ rapidly improves as the Ω angle increases and the performances of DΩ-UΩ and Dπ-UΩ decrease. Finally, a third region, with Ω lying between 150° and 170°, where the three types of potentials present a similar performance and converge to Dπ-Uπ when Ω = 180°.
Figure 2.
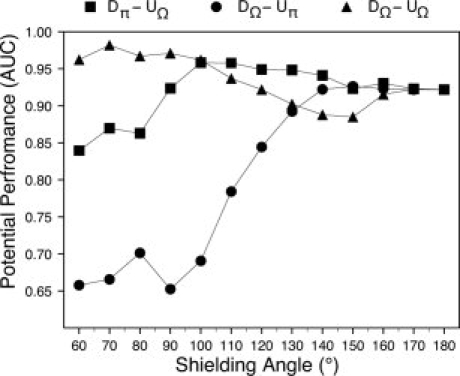
The shielding angle influences the discrimination between native and near-native protein structures by energy functions defining effective atomic interactions. Thirteen empirical potentials were derived using different Ω values ranging from 60° to 180°; a radial distance range of 7.0 Å and a distance bin of 0.2 Å define 35 distance classes (see Methods section). These potentials were used to evaluate the discrimination of native structures from their near-native counterparts using different combinations of the parameters at derivation or evaluation steps. Squares (Dπ-UΩ) indicate the performances using the potential derived at 180° and evaluating considering effective interactions at the different Ω values. Circles (DΩ-Uπ) indicate the performances using the corresponding potentials derived considering effective interactions at variables Ω values and evaluating considering all the interactions (Ω = 180°). Triangles (DΩ-UΩ) indicate the performances considering effective interactions at both derivation and evaluation. Each point in this figure corresponds to a particular classifier. The combination Dπ-Uπ is represented by only one point (Ω = 180°) at which all the three curves converge.
The statistical significance of the differences in performance observed between the potentials was assessed by a nonparametric test (see Methods section and supporting information). According to the AUC values obtained by ROC analysis, the best classifier using effective interactions is the empirical potential D70-U70 (see Fig. 2). However, the differences in performance between this potential and the D60-U60, D80-U80, D90-U90, and D100-U100 potentials are not statistically significant at a confidence level of 95%. The observed differences in the performance of these four D-U potentials and all other potentials are statistically significant at the same confidence level (Supp. Info. Table 1). In the following and because of geometric, statistical, and performance criteria, we decided to use the effective empirical potentials defined by a shielding angle of 90°.
Effect of the maximum distance range
The maximum distance range defines the extent at which an energy function operates. Given that the proposed geometric method captures the first atomic interacting shell, it seems appropriate to test whether different distance ranges have some impact on the performance of empirical potentials using effective interactions.
We tested the four types of potentials mentioned earlier (i.e., DΩ-UΩ, DΩ-Uπ, Dπ-UΩ, and the canonical Dπ-Uπ potential), but with a varying maximum distance range of 5, 7, 12, and 15 Å. The same maximum distance range established in each case was adopted both to derive and to use the potential. In the case of effective interactions, as mentioned earlier, the Ω parameter was set to 90°. We evaluated the performance of these potentials at discriminating between the two sets of native and near-native protein structures (Fig. 3).
Figure 3.
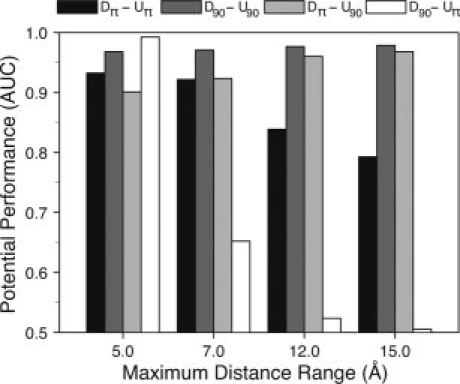
Effect of different radial distance ranges upon the discrimination between native and near-native protein structures by energy functions defining effective atomic interactions. Eight empirical potentials were derived for four maximum distance range values (5.0, 7.0, 12.0, 15.0 Å) and two Ω values (90° and 180°). Combinations at both derivation and evaluation parameters were used to evaluate the discrimination of native structures from their near-native counterparts. Black bars indicate the performance of the potential derived and used as a canonical scoring function (Dπ-Uπ). Dark gray bars indicate the effective potential at derivation and evaluation (D90-U90). Light gray bars show the performance of the potentials derived cannonically but used effectively (Dπ-U90). Finally, the results of using the combination D90-Uπ are shown in white bars.
Both the canonical Dπ-Uπ and the D90-Uπ potentials decrease significantly their performances, in terms of AUCs, as the maximum distance range increases from 5 to 15 Å. While the former does it parsimoniously, the latter falls abruptly. Interestingly, the performance of the Dπ-U90 potential significantly improves as the maximum distance range increases. As expected, the performance of D90-U90 potential remains constant, independently of the maximum distance range defined. The detailed ROC curve analysis not only confirm these results but also gives some additional insights about the trade-off between sensitivity and specificity of these potentials when used as binary classifiers of structural modeling accuracy in proteins (Fig. 4).
Figure 4.
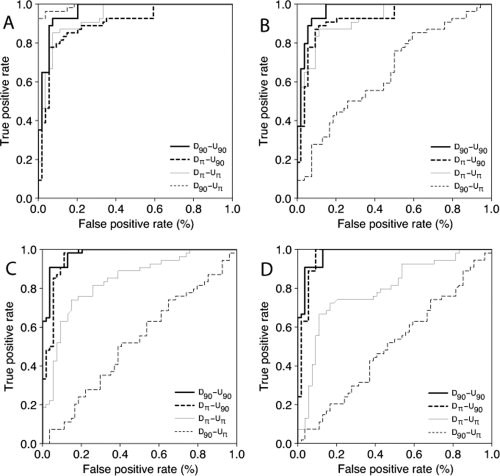
ROC curve analysis of empirical potentials using effective interactions. A detailed comparison of potentials when used as binary classifiers is carried out by means of ROC curve analysis. The ROC plots for the four derivation/utilization combinations of potentials with maximum distance ranges of (A) 5.0, (B) 7.0, (C) 12.0, and (D) 15.0 Å from Figure 3 are shown.
The limited performance observed for other currently used potentials in protein structure assessment, DFIRE,13 RAPDF,12 and PROSA,28 demonstrates that the benchmark used in this work constitutes a difficult test (Fig. 5). However, it must be mentioned that PROSA potential only includes Cα and Cβ atoms, and thus the comparison of this potential against full atom potentials in this particular benchmark is not totally fair. The statistical significance analysis of the observed differences in performance of these potentials is provided as Supporting Information (Supp. Info. Tables 2 and 3).
Figure 5.
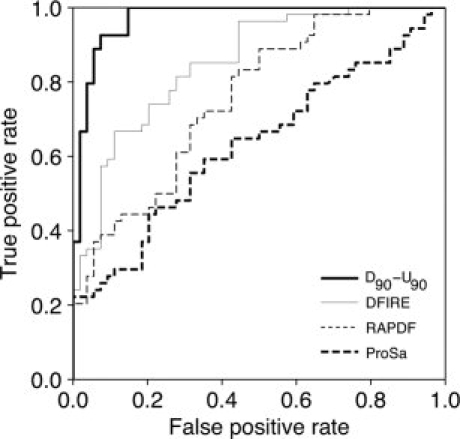
Comparison between effective potentials and other empirical potentials. The ROC curves for DFIRE, ProSa, and RAPDF are compared with that obtained by the D90-U90 potential from Figure 4(B) in the same benchmark.
Atom–atom energy functions
A pairwise distance-dependent potential contains a complete collection of possible combinations of atom–atom interactions observed in proteins. Therefore, the fundamental basis to understand its performance should be found at the detailed description of the atom–atom energy functions. Thus, as an attempt to find an explanation of the differences in performance of the potentials developed and tested in this work, we explored and compared some representative atom–atom energy functions between the potentials derived effectively at shielding angles of 90° and 120° and the canonical potential derived at Ω = 180°.
The total number of energy functions depends on the atom-type definition used. Empirical potentials derived in this work adopt the atom-type definition previously described8 and used in ANOLEA potential.24 This classification groups all nonhydrogen atoms (i.e., heavy atoms) observed for the 20 standard amino acids into 40 atom types. The atom-type definition is mainly based on three criteria: chemical nature, bond connectivity, and location level (side chain or backbone). Some atom types group more than one heavy atom, whereas others are unique.
First, we focused on the typical energy function of hydrogen bonds occurring between main-chain N and O atoms, which is important in the formation of regular secondary structure in proteins. When this specific energy function from the potentials derived effectively (Ω < 180°) and canonically (Ω = 180°) is compared, minor differences are observed [Fig. 6(A)]. The impact of using shielding angles of 120° and 90° to describe the effective interactions translates into a decreasing maximum value of the energy functions and also causes a slight modification of the shape of the energy function for larger distances after the global minimum, which occurs at 3.0 Å. The reduced maximum value of the energy functions that describe effective interactions is explained by the smaller value of the weighting factor Mij (which simply consists of the total number of observations for a particular atom pair) because in the case of effective potentials fewer observations are recorded after masking all those interactions that are shielded by other atoms. This effect is obviously larger for smaller values of the shielding angle, where more atoms are masked and then fewer atom–atom interactions recorded [Fig. 6(A)].
Figure 6.
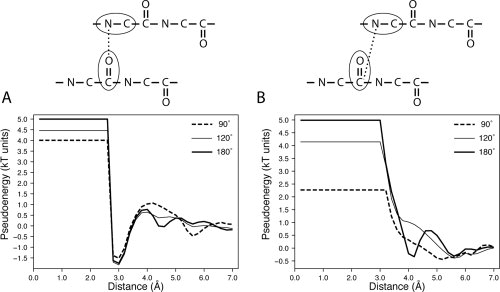
Potential energy function for a hydrogen bond derived at Ω values of 90°, 120°, and 180°. (A) Energy functions of interacting atom types 3 and 5. Atom type 3 groups all backbone nitrogen atoms except Pro-N. Atom type 5 groups all backbone oxygen atoms. (B) Energy functions of interacting atom types 3 and 4. Atom type 4 groups all backbone carbonyl carbon atoms.
However, the situation abruptly changes when the effective and canonical energy functions corresponding to the interaction between main-chain N atom and the main-chain carbonyl atom (which is covalently bonded to the main-chain O atom) are analyzed [Fig. 6(B)]. In this case, it can be clearly observed that the canonical energy function inherits in a large extent both the energy minimum and the corresponding “locking elbow” after the minimum that is characteristic of hydrogen bond energy functions.29 As expected, the energy minimum of this function occurs at a larger distance (at about 4.0 Å). On the other hand, the effective energy functions that describe the interaction of these atoms do not inherit the shape of the energy function for N and O main-chain atoms and consist mostly of repulsive terms [Fig. 6(B)]. The effect of the weighting factor in the amplitude of the effective energy functions is much larger in this case than that observed for the interaction of N and O main-chain atoms. As expected, this observation is consistent with the fact that the shielding effect should be higher for those interactions occurring at a larger distance range.
The differences between effective and canonical energy functions discussed earlier for hydrogen bonds and their covalently linked atoms are even clearer when the energy functions for disulfide and salt bridges are analyzed. In the case of disulfide bridges, the effective energy functions for the interaction between Cys-Sγ and Cys-Sγ are very similar to the canonical one [Fig. 7(A)]. However, very distinct functions are obtained for the interaction between Cys-Cβ and Cys-Cβ [Fig. 7(B)], where only the shielding angle of 90° removes the effect of observing an energy minimum at a larger distance (at about 4.0 Å). When salt bridges were analyzed, the same effect was observed (data not shown).
Figure 7.
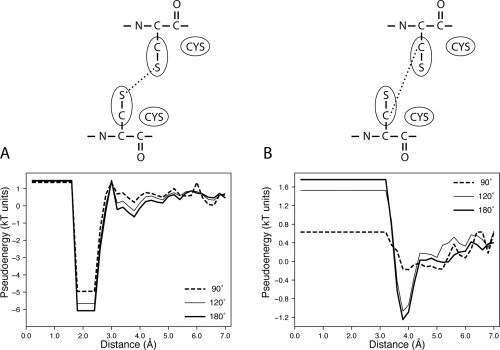
Potential energy function for the disulfide bridge derived at Ω values of 90°, 120°, and 180°. (A) Energy functions of interacting atom types 19 and 19. This atom type corresponds to Cys-Sγ. (B) Energy functions of interacting atom types 29 and 29. This atom type groups Cys-Cβ and Met-Cγ.
Similarly to that found for the pairwise energy functions of directly interacting functional atoms [i.e., hydrogen bonding in Fig. 6(A), disulfide bridges in Fig. 7(A) and salt bridges, data not shown], the effective and canonical energy functions for hydrophobic interactions are also quite similar (Fig. 8). Different Ω values for the shielding angle do not produce major changes in the canonical (Ω = 180°) atom–atom energy functions, as illustrated in the interaction between two aliphatic atoms [Fig. 8(A)] and between two aromatic atoms [Fig. 8(B)]. However, in these cases, the shapes of the energy functions are slightly stylized, with a narrower and better-defined energy minimum, and also with repulsive terms arising at a shorter distance range.
Figure 8.
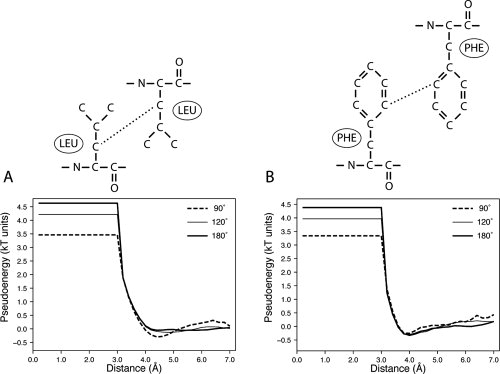
Example potential energy functions for hydrophobic interactions derived at Ω values of 90°, 120°, and 180°. (A) Energy functions of interacting atom types 8 and 8. Atom type 8 groups Arg-Cβ, Arg-Cγ, Asn-Cβ, Asp-Cβ, Gln-Cβ, Gln-Cγ, Glu-Cβ, Glu-Cγ, His-Cβ, Ile-Cγ1, Leu-Cβ, Lys-Cβ, Lys-Cγ, Lys-Cδ, Met-Cβ, Phe-Cβ, Pro-Cβ, Pro-Cγ, Trp-Cβ, and Tyr-Cβ. (B) Energy functions of interacting atom types 12 and 12. Atom type 12 groups Phe-Cδ1, Phe-Cδ2, Phe-Cɛ1, Phe-Cɛ2, Phe-Cζ, Trp-Cɛ3, Trp-Cζ, Trp-Cζ3, Trp-Cη2, Tyr-Cδ1, Tyr-Cδ2, Tyr-Cɛ1, and Tyr-Cɛ2.
Information content of potentials
Recently published work has formally established a direct connection between the pseudo energies obtained from statistical potentials and some basic information-theoretic quantities.17 More specifically, it was shown that the total divergence calculated from a nonlocal residue contact potential allows to predict the fold discrimination success that is achieved by the same potential in a threading exercise.30 This finding reconciles some contradictory results from previous work where unoptimized contact potentials were found to bear a modest amount of information31,32 and indicates that the amount of information encoded in contact potentials is clearly increased when the potentials are previously optimized for a particular task.30
Inspired on this, we decided to explore the association between the statistical potentials derived in this work and their information content, expressed as the information product. The information product relies both in the average score per interaction in the set of native protein structures used to derive a potential and in the mean number of score events observed when the potential is used in the same set of native proteins (see Methods section). The average score per interaction constitutes the best estimate of mutual information for the distance-dependent potentials derived in this work (see Methods section). Therefore, the information product is an indirect measure of the amount of mutual information of a potential that naturally incorporates a correction for sparse data.30
We calculated the information product for each of the potentials derived at different Ω angles [Fig. 9(A)]. The results clearly show the trend that as the Ω angle decreases, the amount of information product of a potential increases (R2 = 0.98). This statement is valid for almost all shielding angles used, with the only exception of Ω = 60, where the amount of information product is reduced when compared with that of Ω = 70. When the relationship between the information product and performance of the potentials was assessed [Fig. 9(B)], the overall trend of increasing performance for increasing information product is clearly observed and, more importantly, the potential with the largest information product is the one with the best performance in our benchmark (Ω = 70).
Figure 9.
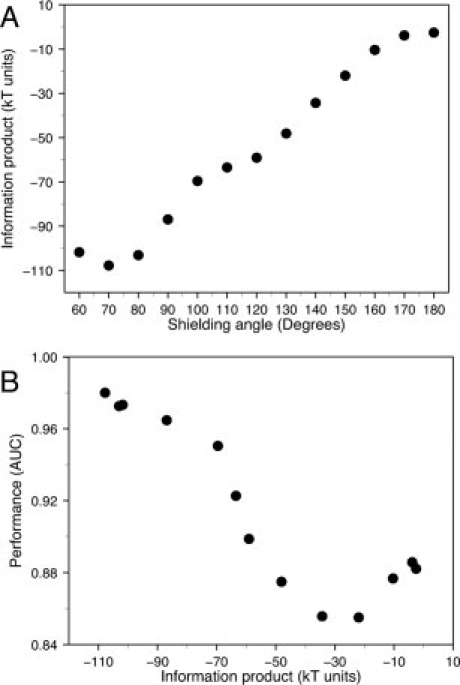
Information product of effective potentials. (A) The information product of a potential is plotted as a function of the shielding angle used to derive it. (B) The observed performance of the potential is plotted as a function of the information product of the potential. All these potentials were derived with a maximum distance range of 7 Å.
Discussion
Benchmark test
Although energy functions present a wide range of applications, in this work, we tested their ability to discriminate between two sets of protein structures: near-native and native protein conformations. We would like to emphasize that achieving a good discrimination in the particular benchmark test used here is difficult for two reasons. First, the near-native structures are quite accurate. Second, the specific discrimination test is performed not individually for each native and non-native protein pair, but simultaneously includes a mix of proteins having different folds, secondary structure composition, and sizes. The difficulty of the benchmark test was demonstrated by the poor performance observed for other empirical potentials that are commonly used in protein structure assessment (see Fig. 5). The benchmark test used here allows a more detailed comparison of the discrimination capability of energy functions that have been derived with similar parameters (e.g., small variation of the shielding angle).
Methodology for the estimation of effective atomic interactions
We presented a new procedure to derive and use empirical energy functions, which consists in the estimation of effective atomic interactions (see Fig. 1). We showed that a significant improvement of potential's performance is achieved by filtering out those atomic interactions that are shielded by other atoms. The procedure to detect the effective atomic interactions consists in estimating the physical exposure between atoms by taking into account the relative position of all other atoms inside an interacting sphere, which is centered in the atom under analysis. The set of atomic interactions selected by this procedure approximate the first interacting atomic shell.
The procedure described here is not the unique method to detect the first atomic interacting shell, other previously described methods could also be used to this end. Geometric exact and approximate methods mainly based on Voronoi diagrams,33 accessible surface area, and visible volume,34 and combinations of them have been described to optimize specific tasks such as threading22 and decoy discrimination.11 However, the impact of using these methodologies on the specific shape of the resulting energy functions was not reported. Additionally, the methodology presented here is the only one that is capable of being fine-tuned by changing a single parameter (i.e., the shielding angle). Based on these results, our approach has shown to be computationally efficient and, spite of its simplicity, accurate and flexible enough to explore the influence of the structural constraints among interacting atoms.
The shielding effect is not expected to be the same for different triplets of XWiY atoms. Distinct behaviors are expected based on the chemical nature and electronegativity of the three atoms used to define an effective interaction. For example, strong induced dipoles can take place in the case of some Wi atoms, thus bridging instead of masking interactions. In this work, we have assumed that all intermediate Wi atoms that are occluding or perturbing a given X-Y interaction cause that the interaction is neglected (i.e., not considered as effective). Although we initially incorporated electronegativity and atom size as additional restraints in our algorithm (data not shown), we did not observe significant improvement over the more simple version presented here. However, we do not discard the impact of these variables on other applications since the benchmark set used in this study is a very exigent task. Understanding the influence of these factors in different application benchmarks constitutes an interesting subject of further investigation.
Effective atomic interactions and performance of the potentials
We observed that the performance obtained while using different combinations of derivation and utilization of empirical energy functions is highly influenced by the Ω angle adopted. The performance of the canonical energy functions, derived and used by considering the complete interacting sphere as effective (i.e., Ω = 180°, Dπ-Uπ), is significantly improved if effective atomic interactions are defined in the derivation and evaluation steps (i.e., DΩ-UΩ). The rate of variation for the observed performance of the potentials is particularly sensitive to Ω angles ranging between 90° and 120° (see Fig. 2). We suggest that a possible explanation for these critical Ω values arises from the inherent molecular geometry imposed by the common hybridization states of the atoms present in the 20 standard amino acids. In fact, the most abundant atom in protein structures is the carbon atom, which in proteins can commonly be found in two of its three possible hybridization states: sp2 and sp3. The sp2 hybridization state arranges three coplanar substitutions with an ideal angle of 120° between them (e.g., carbonyl carbon atoms in backbone; carboxyl and amide carbon atoms in Glu, Asp, Gln, and Asn; aromatic carbons in Phe, Tyr, Trp, and His; etc). The sp3 hybridization state arranges four substitutions in a tetrahedron with an angle between the substitutions that, depending on the electronegativities of the substituent atoms, ranges between 105° and 110°.35 Since the atomic interactions evaluated by an empirical energy function correspond to nonbonded interactions, we would expect to observe a direct influence of the hybridization geometry in cases such as hydrogen bonds, where the contacting atoms and their directly bonded atoms are collinear. In spite of that, our results suggest that at least partially, the functional form of canonical empirical energy functions is due to restraints imposed by the inherent geometry of bonded protein atoms.
Since the procedure used here to define effective interactions should mainly capture the first interacting atomic shell, we observed a significant influence of the maximum distance range adopted over the performance of canonical potentials when compared with that obtained for effective potentials (see Fig. 3). Potentials that do not use an effective atomic definition at the evaluation step (i.e., Dπ-Uπ and D90-Uπ) are particularly sensitive to the maximum distance range defined and perform better when a short maximum distance range is defined; in other words, when the maximum distance range defined approximates the first atomic interacting shell.
In contrast, empirical energy functions that use an effective atomic definition at the evaluation step (i.e., Dπ-U90 and D90-U90) generally perform better, whether or not a definition of effective interactions is used at the derivation step. This last feature is evident for the D90-U90 potential, which has a constant high performance for different maximum distance ranges (see Fig. 3). In this case, the independence on the maximum distance range adopted clearly arises from the use of effective interactions at the evaluation step. However, when a short maximum distance range is defined (i.e., 5.0 and 7.0 Å), the D90-U90 potential performs better than the Dπ-U90 potential. This observation suggests that a critical trade-off between information content and number of observations exists (see later) at the derivation and utilization steps of the potentials, which has a significant impact on their performance at discriminating between native and near-native protein conformations. In other words, less amount of information at the derivation step can be somehow counterbalanced only if a larger amount of interactions is used at the evaluation step. This is illustrated by the fact that the good performance observed for the Dπ-U90 potential is only achieved for large distance ranges (12 and 15 Å), but decreases when the maximum distance range is smaller (5 and 7 Å). A possible explanation for this unexpected result would be a distinct abundance in native and near-native proteins of some specific effective atomic interactions occurring at distances larger than 5 Å such as (1) surface–surface polar atomic interactions, (2) buried salt bridges, and (3) stacking of aromatic groups, both occurring effectively at distances larger than 7.0 Å.
In summary, and irrespectively of the particular potential used, our results clearly highlight the importance of an accurate definition for the first interacting atomic shell when attempting to discriminate the “true” interacting microenvironment for each atom in the structure. Regarding the overall performance, the determination of effective interactions seems to be more relevant at the utilization step rather than at the derivation step, when the maximum distance range of the potential is large enough to account for two or more atomic shells. This observation implies that the performance of currently existing potentials that were derived by considering all interactions should simply improve if they are only used to calculate the pseudo energies of the effective interactions.
Effective atomic interactions and functional shape of energy functions
We would expect that the main features responsible for a good performance of a potential be ultimately found at its specific atom–atom energy functions. We selected four different energy functions (i.e., hydrogen bonding, disulphide bonding, salt bridges, and hydrophobic interactions) that represent most of the atomic interactions observed in protein structures. To analyze the impact of the definition of effective atomic interactions on the shape of the energy functions, we compared the representative energy function ij with the energy function of the atoms directly bonded to i or j (Figs. 6–8). The results clearly showed that effective energy functions derived with a shielding angle of 90° do not contain secondary energy minima (Figs. 5 and 6), which constitute in most cases an artifact that arises from connectivity effects. Moreover, effective energy functions are smoother and apparently have a better energy scaling in terms of magnitude. Therefore, the calculation of effective interactions when deriving a potential does not change the shape of those energy functions that describe a direct interacting atom pair (e.g., disulfide bridges, hydrogen bonds, salt bridges, van der Waals interactions of nonpolar atoms), but it has a large impact on the functional form of those energy functions that describe the interaction of atom pairs that are bonded to the interacting atoms. In these cases, the canonical energy functions inherit most of their shape from the energy function that describe the direct interacting pair. This behavior was clearly observed for hydrogen bonds (see Fig. 6), disulfide bridge formation (see Fig. 7) and salt bridges (data not shown). Effective energy functions corresponding to aliphatic and aromatic interactions did not show large differences when compared with their homolog canonical energy functions (see Fig. 8). This can be explained by the frequent stacking of aromatic residues, by the highly packed hydrophobic core of proteins, or by the reference system used to derive the potentials. The uniform density model4 constitutes a robust reference system for describing long distance range pairwise interactions in proteins because is less sensitive than the quasi-chemical approximation36 to the incorporation of indirect atomic contacts (because it is averaged over all atom pairs and not only over a particular one).
Effective atomic interactions and information content
As an alternative approach to assess the amount of information contained in effective and canonical potentials, we calculated the information product for all the distance-dependent energy functions derived at different Ω angles. The results obtained showed the clear trend that lower Ω angles increase the amount of information in the potential.
Our results also confirm previous observations indicating that the performance of a potential is subjected to a trade-off between the amount of information that it contains and the number of observations taken into account during the evaluation process.30 This trade-off is due to the fact that the amount of information increases at shorter distance ranges though the number of contacts is reduced considerably.
Moreover, our findings show that the performance of truly effective potentials (i.e., derived and used effectively) is insensitive to the maximum distance range (see Fig. 3). This suggests that the real factor influencing the performance of a distance-dependent potential is not the maximum distance range adopted, but rather, it indicates that shorter distances represent a good approximation to capture the first contacting shell of a given atom. Since effective potentials capture the first contacting shell of interacting atoms independently of the maximum distance range adopted, the total number of observations upon reduction of the maximal distance range is not as affected as in the case of canonical potentials. This implies that the information product could be used as a measure to optimize the performance of potentials without the need of a specific benchmark, as previously proposed.30
Although statistical potentials have been criticized for their lack of theoretical foundations,19,37 our results are in agreement with most of previous works in this field and suggest that propensities expressed as probability distributions of events are closely connected to the physical properties found in protein structures. We observed that direct physical interactions rather than distance seem to be the main source increasing the information content of empirical potentials. Although in the study of protein structure, both physics and statistics can be exploited as totally different phenomena, they are somehow reconciled in statistical energy functions and thus can be seen as two sides of the same coin.
It has been recently shown that statistical potentials can be seen as informatic functions and that higher amounts of information are in agreement with the performance of an specific potential.17 It is also known that mutual information is a nonlinear measure of correlation.38 From these observations, we conclude that the goal of an energy function is to infer the correlation patterns of atomic interactions observed in protein structures. The higher the correlation between functions, the higher is their nonadditivity. Other sources of studies interpret these observations as cooperativity or anticooperativity depending on the sign of the correlation.11
Nonadditivity between energy functions (i.e., cooperativity) has been shown to be fundamental in explaining the topology dependence of the folding rates observed in protein domains.39 In fact, thermodynamic cooperativity accelerates folding by smoothing the energy landscape.40 Nonadditivity seems to be a crucial component of energy functions that carefully captured could improve the performance of potentials and ultimately foster our understanding of structural biology. The findings reported here represent an effort in that direction.
Methods
Experimental protein structures for calculating the potentials
A set of 518 nonredundant and well-refined protein structures solved by X-ray crystallography was used. This set does not contain proteins with duplicated or missing atoms, structural gaps, or proteins with less than 100 residues. All the protein chains share less than 25% sequence identity, have a resolution below 3.0 Å and contain full atomic coordinates for all amino acids. The list of protein structures is available as Supporting Information at http://protein.bio.puc.cl/sup-mat.html.
Definition of effective atomic interactions
A given atom X in a protein structure can have many neighbor atoms in the three-dimensional space, which are typically defined by setting up a fixed maximum distance threshold. In the absence of additional definitions, all these atoms found in the neighborhood of atom X are considered to be interacting with it. However, by using this simple approach, many indirect interactions that in fact are shielded by other atoms and thus could not be relevant from a physical point of view will still be included in the analysis. To avoid this problem, we have developed a simple method that relies on the definition of additional restraints to select only the direct interactions between two atoms.
Direct or effective interactions are defined as those atom–atom interactions that are not shielded or masked by any other atom in the three-dimensional space. We propose here a simple geometric algorithm to assess the shielding effect that any atom has on the interaction of two other atoms (see Fig. 1). Based on this new methodology, we are able to classify the interactions as being either effective or not.
Before formalizing the algorithm, we define the following: (a) Let X be the atom under evaluation. Then, its spatial coordinates constitute the center of its interacting sphere. (b) The radius of the interacting sphere is defined by the maximum distance range adopted. (c) Let N be the total number of atoms, different from X, that are found inside the interacting sphere of X. (d) Let Z be the spatially closest atom to X inside the interacting sphere of X. By definition, Z is interacting effectively with X, since no other atoms can mask this interaction. (e) Let M be the total number of Y atoms, which are different from X and Z, and are found inside the interacting sphere of X (i.e., M = N − 1). (f) Let Ω be an angle ranging between 60 and 180°.
The following algorithm evaluates if the interaction between atoms X and Y is effective or not:
A = array of N atoms sorted according to their distance to X, in ascending order.
for j = 2 to N do
Yj ← A[j]
for i = 1 to (j − 1) do
Wi ← A[i]
αi ← angle(XWiYj)
if αi ≤ Ω then
the interaction between X and Yj is not shielded by the atom Wi
else
the interaction between X and Yj is shielded by the atom Wi
end if
end for
the interaction between X and Yj is effective ⇔ ∀ i, 1 ≤ i < j, αi ≤ Ω.
end for
The goal of this procedure is to detect only the direct pairwise atomic interactions that are not being shielded or masked by any other atom [Fig. 1(A)]. The masking effect can be easily fine-tuned by varying a single parameter: the Ω shielding angle. After applying this methodology, only a subset of all possible interactions per atom (i.e., those that are not shielded by any other atom) are further considered. Altogether, these atomic interactions should represent a reliable approximation to the first contacting shell of any atom in the structure [Fig. 1(B)].
Additionally, other restraints can also be incorporated to define those effective atomic interactions of interest. In this study, we have focused on the effective nonlocal interactions between atoms. This means that we have only calculated the effective interactions between atoms X and Y when these two atoms belong to amino acids that are separated along the protein chain by nine or more residues or when they belong to amino acids found in different protein chains.24
Calculation of potentials
A total of 19 different types of distance-dependent potentials were calculated (Table I). They differ only in the maximum distance range and the shielding angle Ω adopted to define the effective interactions when deriving the potential. Typical statistical potentials use a shielding angle of 180°, that is, the shielding effect of other atoms is not considered or, in other words, all the atomic interactions found below the maximum distance range are considered as effective [Fig. 1(B)]. In addition to the canonical potential with Ω = 180°, statistical energy functions with Ω values of 60°, 70°, 80°, 90°, 100°, 110°, 120°, 130°, 140°, 150°, 160°, and 170° were calculated. All these statistical energy functions were derived by taking into account nonlocal interactions only. We define nonlocal interacting atoms as those interactions occurring between any two atoms that belong to amino acids found in the same chain with a separation along the sequence equal or larger than nine residues, or atoms that belong to amino acids from different chains. A total of 40 atom types were defined for all nonhydrogen atoms observed in the 20 standard amino acids.8 The distance-dependent energy functions were calculated as previously described.8,24,28 The following equation was used:
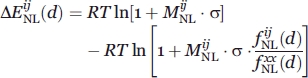 |
where 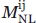 is the total number of nonlocal interactions observed between atom types i and j below the maximum distance range defined and was calculated as follows:
is the total number of nonlocal interactions observed between atom types i and j below the maximum distance range defined and was calculated as follows:
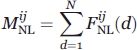 |
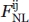 (d) is the absolute frequency of nonlocal observations between atom types i and j at the distance class d, and N is the total number of classes of distance. The potentials were calculated using maximum distance ranges of 5.0, 7.0, 12.0, and 15.0 Å (Table I). In all cases, homogeneous distance bins of 0.2 Å were defined. The constant weight factor σ given to each pairwise energy function was set to 0.02, as previously described.4
(d) is the absolute frequency of nonlocal observations between atom types i and j at the distance class d, and N is the total number of classes of distance. The potentials were calculated using maximum distance ranges of 5.0, 7.0, 12.0, and 15.0 Å (Table I). In all cases, homogeneous distance bins of 0.2 Å were defined. The constant weight factor σ given to each pairwise energy function was set to 0.02, as previously described.4
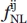 (d) is the relative frequency of nonlocal observations between atom types i and j at the distance class d and is defined as follows:
(d) is the relative frequency of nonlocal observations between atom types i and j at the distance class d and is defined as follows:
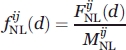 |
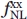 (d) is the reference system and corresponds to the relative frequency of nonlocal observations between any two atom types in the distance class d. This quantity was calculated using the following equation:
(d) is the reference system and corresponds to the relative frequency of nonlocal observations between any two atom types in the distance class d. This quantity was calculated using the following equation:
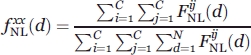 |
where C is the number of different atom types and N is the number of distance classes. The temperature T was set to 293 K, so that RT is equivalent to 0.582 kcal/mol.
Utilization of potentials
The potentials were used to calculate the energy of protein structure models with the same definition of nonlocal interactions and maximum distance range used to derive them. However, different combinations of derivation and utilization procedures are possible depending on the definition of effective atomic interactions at any of both steps. Effectiveness at the derivation (DΩ) is defined if the empirical potential was calculated from the database of native protein structures considering any Ω value smaller than 180°. Accordingly, effectiveness at the utilization (UΩ) of the potential is defined if the interactions are estimated at any Ω value smaller than 180° and all other parameters are the same as those used to derive the potential. On the other hand, the typical or canonical approach that considers all interactions found within the maximum distance range as being effective is defined at derivation (Dπ) or utilization (Uπ) by setting the Ω value at 180°. A total of 49 different combinations of derivation and utilization of potentials were tested in this work (Table I).
The energies were calculated as follows: (a) for each atom in the molecule, all its nonlocal effective atomic interactions are determined at a given Ω value (see Definition of Effective Atomic Interactions section); (b) for each nonlocal effective pairwise interaction, the energy value is taken from the distance-dependent energy function; (c) the total energy per atom is calculated by summing up all its energy terms; (d) the total energy of the structure is the sum of the energies of all its atoms. When expressing the normalized energy of a protein, the total energy is divided by the total number of nonlocal effective atomic interactions observed. The final energy value is expressed in RT units.
External potentials
In addition to the potentials described earlier, we also tested the performance in our benchmark of other potentials typically used in the assessment of protein structure models. The potentials tested were DFIRE,13 ProSa,28 and RAPDF.12 ProSa was initially developed in 1993 but here we used the most recent version of this software, which was released in 2003. The software was downloaded from http://www.came.sbg.ac.at.
Benchmark set of native protein structures and near-native protein structure models
To assess the performance of knowledge-based potentials at discriminating between native and near-native conformations, a subset of a previous set of comparative protein structure models was used.26 Briefly, the original set contains 152 native protein structures and a single near-native protein structure model for each of them (i.e., 152 near native models). These models have a length equal or larger than 100 amino acids, have at least 90% equivalent α-carbons with their corresponding native structures, a target chain coverage equal or larger than 90%, and a total or global root mean square deviation (RMSD) of less than 3.0 Å for all α-carbons. All models were built for target monomeric proteins. To avoid any bias when testing the performance of the potentials, we have removed all models from the original set that shared more than 70% sequence identity with any structure in the X-ray set of 518 proteins used to derive the potentials and also with any other model in the set. After filtering the initial set, we ended up with 54 near-native protein models and their corresponding native structures, which were used to test the performance of the potentials. According to SCOP classification of protein structures,41 the 54 protein chains in this set contain a total of 62 SCOP folds, of which a total of 54 are unique (i.e., 54 different SCOP folds are represented in this set of proteins). According to CATH classification of protein structures,42 28% of the models contain only alpha helix secondary structure elements, 20% have only beta sheets, 49% contain alpha and beta, and only two proteins (3%) have few secondary structures. The details about the construction of the original set of 152 models can be found in Ref.26. The list of 54 models selected for this work along with the 3D coordinates of the native protein structures and their models are available in PDB format as Supporting Information at http://protein.bio.puc.cl/sup-mat.html.
Assessment of the performance of potentials
The performance of potentials as binary classifiers was assessed by ROC analysis as previously described.27 The measure used was the area under the ROC curve (AUC). Briefly, each potential was used to obtain a normalized total energy for each protein model in the set and for each native protein structure. Upon a given normalized energy score threshold, a binary classifier was built for each potential, where each protein was predicted or classified as native or near-native, depending whether its normalized energy score value fell below or above the fixed threshold, respectively. In the “real classification,” a positive instance was defined as a near-native protein. A negative instance was defined as a native protein. The predictions generated by each classifier at each possible normalized energy score threshold for all proteins, named “hypothetical classifications,” were then compared with those previously defined by the real classification of proteins and ROC analysis performed. The statistical significance of the observed differences between any two potentials used as binary classifiers was evaluated with the StaR web server.43 This server relies on a nonparametric test for the difference of the AUCs that accounts for the correlation of the ROC curves.
Calculation of the information product of potentials
The information product (P) of a potential was calculated as previously described,17,30 by using the following equation:
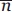 is the mean number of interactions that will be observed in a typical protein when using the potential and corresponds to:
is the mean number of interactions that will be observed in a typical protein when using the potential and corresponds to:
where ni is the number of score events (i.e., those interactions that will be considered by a potential according to its utilization parameters) in native protein i and N is the total number of native proteins used to derive the potential. 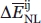 is the average score or energy value per interaction observed in those native proteins used to derive the potential:
is the average score or energy value per interaction observed in those native proteins used to derive the potential:
where x corresponds to any valid score event or interaction observed in the native proteins when the potential is used to calculate their total score. Therefore, X corresponds to:
In the case of the distance-dependent potentials calculated here, constitutes the best estimate of mutual information because it naturally takes into account the sensible issue of sparse data in the calculation of informatic quantities and adjusts the estimate of energy accordingly.30
Acknowledgments
The authors thank Dr. Andrej Sali, Dr. Manfred Sippl, and Dr. Armando Solis for critical reading of this manuscript and valuable suggestions that, in our opinion, significantly contributed to improve its quality. They are also grateful to Alex W. Slater, doctoral student, for the contributions to the analysis of SCOP and CATH folds in our model dataset.
References
- 1.Berman H, Henrick K, Nakamura H, Markley JL. The worldwide Protein Data Bank (wwPDB): ensuring a single, uniform archive of PDB data. Nucleic Acids Res. 2007;35:D301–D303. doi: 10.1093/nar/gkl971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tanaka S, Scheraga HA. Statistical mechanical treatment of protein conformation. I. Conformational properties of amino acids in proteins. Macromolecules. 1976;9:142–159. doi: 10.1021/ma60049a026. [DOI] [PubMed] [Google Scholar]
- 3.Melo F, Feytmans E. Scoring functions for protein structure prediction. In: Schwede T, Peitsch M, editors. Computational structural biology. World Scientific Publishing Co. Pte. Ltd.: Singapore; 2008. pp. 61–88. [Google Scholar]
- 4.Sippl MJ. Calculation of conformational ensembles from potentials of mean force. An approach to the knowledge-based prediction of local structures in globular proteins. J Mol Biol. 1990;213:859–883. doi: 10.1016/s0022-2836(05)80269-4. [DOI] [PubMed] [Google Scholar]
- 5.Furuichi E, Koehl P. Influence of protein structure databases on the predictive power of statistical pair potentials. Proteins. 1998;31:139–149. doi: 10.1002/(sici)1097-0134(19980501)31:2<139::aid-prot4>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 6.Zhang C, Liu S, Zhou H, Zhou Y. The dependence of all-atom statistical potentials on structural training database. Biophys J. 2004;86:3349–3358. doi: 10.1529/biophysj.103.035998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Betancourt MR, Skolnick J. Local propensities and statistical potentials of backbone dihedral angles in proteins. J Mol Biol. 2004;342:635–649. doi: 10.1016/j.jmb.2004.06.091. [DOI] [PubMed] [Google Scholar]
- 8.Melo F, Feytmans E. Novel knowledge-based mean force potential at atomic level. J Mol Biol. 1997;267:207–222. doi: 10.1006/jmbi.1996.0868. [DOI] [PubMed] [Google Scholar]
- 9.Melo F, Marti-Renom MA. Accuracy of sequence alignment and fold assessment using reduced amino acid alphabets. Proteins. 2006;63:986–995. doi: 10.1002/prot.20881. [DOI] [PubMed] [Google Scholar]
- 10.Melo F, Sanchez R, Sali A. Statistical potentials for fold assessment. Protein Sci. 2002;11:430–448. doi: 10.1002/pro.110430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li X, Liang J. Geometric cooperativity and anticooperativity of three-body interactions in native proteins. Proteins. 2005;60:46–65. doi: 10.1002/prot.20438. [DOI] [PubMed] [Google Scholar]
- 12.Samudrala R, Moult J. An all-atom distance-dependent conditional probability discriminatory function for protein structure prediction. J Mol Biol. 1998;275:895–916. doi: 10.1006/jmbi.1997.1479. [DOI] [PubMed] [Google Scholar]
- 13.Zhou H, Zhou Y. Distance-scaled, finite ideal-gas reference state improves structure-derived potentials of mean force for structure selection and stability prediction. Protein Sci. 2002;11:2714–2726. doi: 10.1110/ps.0217002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu H, Lu L, Skolnick J. Development of unified statistical potentials describing protein–protein interactions. Biophys J. 2003;84:1895–1901. doi: 10.1016/S0006-3495(03)74997-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Min-Yi S, Sali A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006;15:2507–2524. doi: 10.1110/ps.062416606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jaynes ET. Information theory and statistical mechanics. Phys Rev. 1957;106:620–630. [Google Scholar]
- 17.Solis AD, Rackovsky S. Improvement of statistical potentials and threading score functions using information maximization. Proteins. 2006;62:892–908. doi: 10.1002/prot.20501. [DOI] [PubMed] [Google Scholar]
- 18.Shannon CE. A mathematical theory of communication. Bell Syst Tech J. 1948;27:379. [Google Scholar]
- 19.Ben-Naim A. Statistical potentials extracted from protein structures: are these meaningful potentials? J Chem Phys. 1997;107:3698–3706. [Google Scholar]
- 20.Dill KA. Additivity principles in biochemistry. J Biol Chem. 1997;272:701–704. doi: 10.1074/jbc.272.2.701. [DOI] [PubMed] [Google Scholar]
- 21.Carter CW, LeFebvre BC, Cammer SA, Tropsha A, Edgell MH. Four-body potential reveal protein-specific correlations to stability changes caused by hydrophobic core mutations. J Mol Biol. 2001;311:625–638. doi: 10.1006/jmbi.2001.4906. [DOI] [PubMed] [Google Scholar]
- 22.Bienkowska JR, Rogers RG, Jr, Smith TF. Filtered neighbors threading. Proteins. 1999;37:346–359. [PubMed] [Google Scholar]
- 23.Zomorodian A, Guibas L, Koehl P. Geometric filtering of pairwise atomic interactions applied to the design of efficient statistical potentials. Comput-Aided Geom Des. 2006;23:531–544. [Google Scholar]
- 24.Melo F, Feytmans E. Assessing protein structures with a non-local atomic interaction energy. J Mol Biol. 1998;277:1141–1152. doi: 10.1006/jmbi.1998.1665. [DOI] [PubMed] [Google Scholar]
- 25.Fiser A, Do RK, Sali A. Modeling of loops in protein structures. Protein Sci. 2000;9:1753–1773. doi: 10.1110/ps.9.9.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ferrada E, Vergara IA, Melo F. A knowledge-based potential with an accurate description of local interactions improves discrimination between native and near-native protein conformations. Cell Biochem Biophys. 2007;49:111–124. doi: 10.1007/s12013-007-0050-5. [DOI] [PubMed] [Google Scholar]
- 27.Ferrada E, Melo F. Nonbonded terms extrapolated from nonlocal knowledge-based energy functions improve error detection in near-native protein structure models. Protein Sci. 2007;16:1410–1421. doi: 10.1110/ps.062735907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sippl MJ. Recognition of errors in three-dimensional structures of proteins. Proteins. 1993;17:355–362. doi: 10.1002/prot.340170404. [DOI] [PubMed] [Google Scholar]
- 29.Sippl MJ. Helmholtz free energy of peptide hydrogen bonds in proteins. J Mol Biol. 1996;260:644–648. doi: 10.1006/jmbi.1996.0427. [DOI] [PubMed] [Google Scholar]
- 30.Solis AD, Rackovsky S. Information and discrimination in pairwise contact potentials. Proteins. 2008;71:1071–1087. doi: 10.1002/prot.21733. [DOI] [PubMed] [Google Scholar]
- 31.Cline MS, Karplus K, Lathrop RH, Smith TF, Rogers RG, Jr, Haussler D. Information-theoretic dissection of pairwise contact potentials. Proteins. 2002;49:7–14. doi: 10.1002/prot.10198. [DOI] [PubMed] [Google Scholar]
- 32.Crooks GE, Wolfe J, Brenner SE. Measurements of protein sequence-structure correlations. Proteins. 2004;57:804–810. doi: 10.1002/prot.20262. [DOI] [PubMed] [Google Scholar]
- 33.Dupuis F, Sadoc JF, Jullien R, Angelov B, Mornon JP. Voro3D: 3D Voronoi tessellations applied to protein structures. Bioinformatics. 2005;21:1715–1716. doi: 10.1093/bioinformatics/bth365. [DOI] [PubMed] [Google Scholar]
- 34.Lo Conte L, Smith TF. Visible volume: a robust measure for protein structure characterization. J Mol Biol. 1997;273:338–348. doi: 10.1006/jmbi.1997.1298. [DOI] [PubMed] [Google Scholar]
- 35.Vollhardt KPC. Organic chemistry. New York: Freeman Corp; 1987. [Google Scholar]
- 36.Miyazawa S, Jernigan RL. Estimation of effective interresidue contact energies from protein crystal structures: quasi-chemical approximation. Macromolecules. 1985;18:534–552. [Google Scholar]
- 37.Thomas PD, Dill KA. Statistical potentials extracted from protein structures: how accurate are they? J Mol Biol. 1996;257:457–469. doi: 10.1006/jmbi.1996.0175. [DOI] [PubMed] [Google Scholar]
- 38.Cover TM, Thomas JA. Elements of information theory. New York: Wiley; 1991. [Google Scholar]
- 39.Jewett AI, Pande VS, Plaxco KW. Cooperativity, smooth energy landscapes and the origins of topology-dependent protein folding rates. J Mol Biol. 2003;326:247–253. doi: 10.1016/s0022-2836(02)01356-6. [DOI] [PubMed] [Google Scholar]
- 40.Faisca PFN, Plaxco KW. Cooperativity and the origins of rapid, single-exponential kinetics in protein folding. Protein Sci. 2006;15:1608–1618. doi: 10.1110/ps.062180806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Murzin AG, Brenner SE, Hubbard TJ, Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 42.Orengo CA, Jones DT, Thornton JM. Protein superfamilies and domain superfolds. Nature. 1994;372:631–634. doi: 10.1038/372631a0. [DOI] [PubMed] [Google Scholar]
- 43.Vergara IA, Norambuena T, Ferrada E, Slater AW, Melo F. StAR: a simple tool for the statistical comparison of ROC curves. BMC Bioinformatics. 2008;9:1–11. doi: 10.1186/1471-2105-9-265. [DOI] [PMC free article] [PubMed] [Google Scholar]