

Abstract
Previously published 3-D structures of a prototypic ATP-binding cassette (ABC) transporter, MsbA, have been recently corrected revealing large rigid-body motions possibly linked to its catalytic cycle. Here, a closely related multidrug bacterial ABC transporter, BmrA, was studied using site-directed spin labeling by focusing on a region connecting the transmembrane domain and the nucleotide-binding domain (NBD). Electron paramagnetic resonance (EPR) spectra of single spin-labeled cysteine mutants suggests that, in the resting state, this sub-domain essentially adopts a partially extended conformation, which is consistent with the crystal structures of MsbA and Sav1866. Interestingly, one of the single point mutants (Q333C) yielded an immobilized EPR spectrum that could arise from a direct interaction with a vicinal tyrosine residue. Inspection of different BmrA models pointed to Y408, within the NBD, as the putative interacting partner, and its mutation to a Phe residue indeed dramatically modified the EPR spectra of the spin labeled Q333C. Moreover, unlike the Y408F mutation, the Y408A mutation abolished both ATPase activity and drug transport of BmrA, suggesting that a nonpolar bulky residue is required at this position. The spatial proximity of Q333 and Y408 was also confirmed by formation of a disulfide bond when both Q333 and T407 (or S409) were replaced jointly by a cysteine residue. Overall, these results indicate that the two regions surrounding Q333 and Y408 are close together in the 3-D structure of BmrA and that residues within these two sub-domains are essential for proper functioning of this transporter.
Keywords: ABC transporter, electron paramagnetic resonance, site-directed spin labeling, resting state, multidrug transporter
Introduction
ABC (“ATP-Binding Cassette”) transporters are one of the most abundant families of membrane proteins and are widely distributed in all living kingdoms.1 They are involved in the transport, either import or export, of a large variety of substrates including sugars, ions, amino acids, peptides, large cytotoxic molecules and even proteins.2,3 In humans, the dysfunction of several ABC transporters is associated with severe pathologies such as cystic fibrosis, adrenoleukodystrophy or hyperinsulinemia.4 Additionally, human transporters such as MDR1 or MRP1 are overexpressed in cancer cells thereby conferring multidrug resistance (MDR) phenotypes. Related MDR transporters are also found in prokaryotes, yeasts or parasites where they mediate resistance to xenobiotics, including antimicrobial compounds.5
ABC transporters are composed of four protein domains: two transmembrane domains (TMDs) are involved in substrate recognition and translocation, and two cytosolic nucleotide-binding domains (NBDs) bind and hydrolyze ATP to energize the transporter.6 These four domains may be fused together to form a full-length transporter (such as MDR1 or CFTR), or found on separate subunits from two (half-transporter) to four. Many nucleotide-binding domains from either importers or exporters have now been crystallized and all share a common topology,3 suggesting that a similar mechanism of energy transduction is conserved throughout the ABC family. Regarding complete ABC transporters, 3-D structures have been determined at high resolution for some importers with various specificities and membrane topologies.7–11 In the case of exporters, the 3-D structure of a putative bacterial multidrug half-transporter from Staphylococcus aureus, Sav1866,12 led to the retraction of three previously published erroneous structures of a related transporter, MsbA,13 which were recently reinterpreted and completed by a fourth structure.14 This allowed the description of a putative catalytic cycle for MsbA where a large rigid-body motion was proposed to occur.
MsbA and Sav1866 are homologous to other bacterial ABC exporters shown to be involved in multidrug efflux such as LmrA from Lactococcus lactis15 and the more recently characterized BmrA from Bacillus subtilis.16,17 We previously showed that, in the resting state of BmrA, an intramolecular disulfide bond could be formed between two engineered cysteine residues, one in the NBD and a second one in the first intracellular loop (ICL1).18 This disulfide bond was modulated by nucleotide binding suggesting that some motion of the NBD relative to ICL1 may be permitted during the resting state of BmrA. This assumption was however challenged by the model of rigid-body motion proposed for MsbA where the NBD moves in concert with both ICL1 from the same subunit and ICL2 from the other subunit of the dimer.14 Some flexibility was nevertheless put forward to explain the recent crosslinking experiments performed either on CFTR or MDR1 between their ICL4 (equivalent to ICL2 in the second half of full-length transporters) and their NBD1, using crosslinkers of variable length.19,20 Also, solid-state NMR performed on the resting state of LmrA led to the conclusion that the NBD is highly mobile as compared to the transmembrane domain including ICL1 and ICL2.21 Therefore, if BmrA is indeed able to sample multiple conformations in the resting state, switching between different conformations would require structural rearrangements of some part of the transporter, and ICL3 that forms a large loop which wraps around the NBD is a possible candidate.
In the present work, the environment of residues located in ICL3 of BmrA (residues 329–336) was probed using a site-directed spin labeling approach, a particularly sensitive technique allowing the study of local environment and/or conformation of protein sub-domains.22,23 The results suggest that, in the resting state of BmrA, this sequence preferentially adopts a partially extended conformation. Interestingly, the electron paramagnetic resonance (EPR) spectrum of the spin-labeled Q333C mutant was unique, suggesting a possible direct interaction with a hydroxyl group of a nearby tyrosine residue. Examination of the different BmrA models based on either Sav1866 or MsbA 3-D structures revealed that a single tyrosine residue, Y408, might be close enough to the spin probe attached to Q333C. Mutation of this tyrosine to a phenylalanine residue resulted in a much more mobile residue at position 333 as revealed by the EPR spectrum. While the Y408F mutation had little functional effect on BmrA, by contrast the Y408A mutant showed undetectable levels of both Hoechst transport and ATPase activity. The spatial proximity of Y408 with Q333 was further confirmed by introducing two cysteine residues simultaneously at positions 333 and 407 (or 409) thereby allowing the formation of a disulfide bond. Overall, the present results suggest that, in the resting state of BmrA, residues 329–336 of ICL3 adopt preferentially an extended conformation in agreement with that found in the different 3-D structures of either Sav1866 or MsbA. Moreover, the 329–336 sequence appears spatially close to, and likely interacting with, residues surrounding Y408 within the NBD, and this interaction and/or proximity seems to be required for proper functioning of BmrA.
Results
To probe the local structural organization of BmrA in the resting state, the strategy used was to introduce a nitroxide side chain at specific sites after cysteine substitution by site-directed mutagenesis, followed by reaction with a sulfhydryl specific nitroxide reagent (see Scheme 1). Eight single successive cysteine mutants, targeting residues V329 to N336 in ICL3 of BmrA, were first generated [Fig. 1(A)]. The rationale for focusing on this limited region of ICL3 from BmrA was initially based on the comparison of two previous structures of MsbA indicating a sharp transition from a helix-loop-helix structure in the “open” conformation of EcMsbA to an extended ‘unfolded’ structure in the “closed” conformation of VcMsbA at the level of the ICL3 region. Although after retraction of the two previous structures,13 the newly corrected MsbA structures showed apparently no major structural differences in ICL3 between the different conformations of MsbA14 or Sav186612 [see Fig. 1(B,C) for a comparison of the structure of ICL3 of BmrA modeled on either Sav1866 or EcMsbA], our results reported here revealed nevertheless an important role played by residues of ICL3, which are likely interacting with a region of the NBD located between the Walker-A motif and the Q-loop.
Scheme 1.
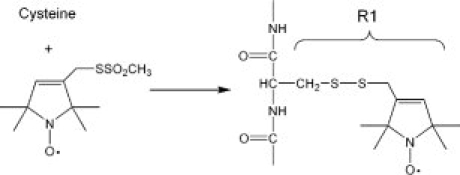
Scheme of the spin labeling reaction. Reaction of the methanethiosulfonate with cysteine generated a disulfide side chain R1.
Figure 1.
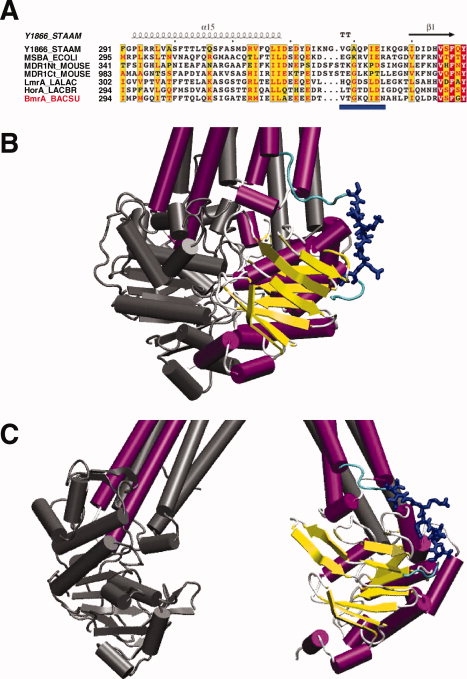
Sequence alignment of the region connecting the TMD and the NBD in ABC exporters and 3-D models of BmrA. (A) sequences were obtained on the ExPASY server (http://us.expasy.org/) and the alignment was generated by ClustalW (1.8) on the NPA@ server (http://npsa-pbil.ibcp.fr). The figure was made with ESPript 2.0 (http://prodes.toulouse.inra.fr/ESPript/). The abbreviations used are: Y1866_STAAM, Sav1866 from Staphylococcus aureus; MSBA_ECOLI, MsbA from E. coli; MDR1Nt_MOUSE and MDR1Ct_MOUSE, P-glycoprotein from mouse, N-terminus and C-terminus halves, respectively; LALAC, Lactococcus lactis; LACBR, Lactobacillus brevis; and BACSU, Bacillus subtilis. Blue frames were drawn when at least 50% of the residues were identical (white characters in red boxes for fully identical residues, or red characters otherwise). The Sav1866 secondary structure is shown above the sequence. BmrA residues mutated to cysteine residues are underlined in blue. (B) and (C) Models of the 3-D structure of BmrA based either on Sav1866 or E. coli MsbA, respectively. One monomer is shown in color with the underlined residues in A shown in blue (the backbone of other residues from ICL3 are colored in cyan) while the second monomer is shown in grey. Panels B and C were drawn with VMD.24
Importance of G331 for BmrA function
Cysteine mutants were then introduced into a cysteine-less background mutant previously generated and where the unique cysteine of BmrA, Cys436, was replaced by a serine residue without significantly altering the properties of the transporter.25 Each single mutant was overexpressed at the same level as the wild-type BmrA in the membrane fraction, and was purified with similar yield and level of purity as wild-type BmrA (not shown). First, the effect of mutations was studied by measuring transport activities on the membrane fractions. As shown in Figure 2, a cysteine residue can be introduced in all mutants without a major impact on Hoechst 33342 transport, except for two mutants: the K332C mutant, which showed a strongly reduced rate of transport, and the G331C mutant where the transport rate was virtually absent. Unlike K332, G331 is conserved in two other bacterial MDR transporters, LmrA and HorA, and also in both halves of mouse P-glycoprotein (MDR1). In EcMsbA and Sav1866, a glycine is also found one residue ahead in the sequence, and it is involved in a turn motif in Sav1866 3-D structure [see Fig. 1(A)]. G331 was also mutated to an alanine or a serine residue, two other small residues. These two BmrA mutants retained a very low transport activity [Fig. 2(C)], similar to that seen with the K332C mutant, suggesting that a glycine residue is required at this position for proper functioning of BmrA.
Figure 2.
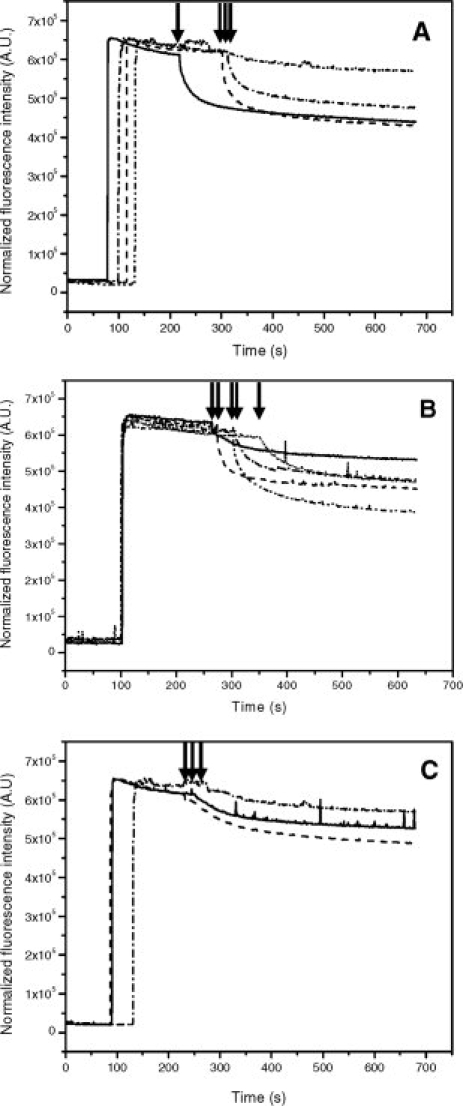
Transport of Hoechst 33342 by inside-out E. coli membrane vesicles containing over-expressed BmrA. (A) inverted-membrane vesicles (200 μg) containing Cys-less (solid line), V329C (dashed line), T330C (dash-dotted line) or G331C (dash-dot-dotted line) were added to the cuvette and after ∼2 min incubation at 37°C, 2 μM Hoechst 33342 were added. After ∼1–2 min, ATP was added to initiate Hoechst transport (black arrow). (B) Same as (A) except that mutants K332C (solid line), Q333C (dashed line), I334C (dash-dotted line), E335C (dash-dot-dotted line), N336C (dotted line) were studied. (C) Effect of different mutations at position 331. The glycine at position 331 was mutated to either cysteine (dash-dotted line), alanine (dashed line) or serine (solid line) and Hoechst transport was monitored as in (A).
Val329 to Asn336 of BmrA adopts preferentially a partially extended conformation in the resting state
Methane thiosulfonate was then used to covalently attach a spin label moiety to each mutant, giving rise to an R1 side chain (Scheme 1). First, to ascertain that introduction of the nitroxide probe did not disrupt BmrA structure, ATPase activities were measured for all mutants after reconstitution into proteoliposomes. All spin-labeled mutants conserved a high level of ATPase activity except for the G331C mutant which was virtually inactive (not shown), consistent with the lack of Hoechst transport ability seen with the unlabeled mutant. The cysteine and Cys-less mutants were treated in the same way with MTSL, and their EPR spectra were recorded. As shown in Figure 3(A), some nonspecific background labeling occurred with the Cys-less mutant and was subtracted from the spectrum of the V329R1 mutant to obtain the net EPR signal produced by labeling at this specific site of BmrA. The same correction was systematically applied to the other R1 mutants, assuming a similar level of background labeling [Fig. 3(B)].
Figure 3.
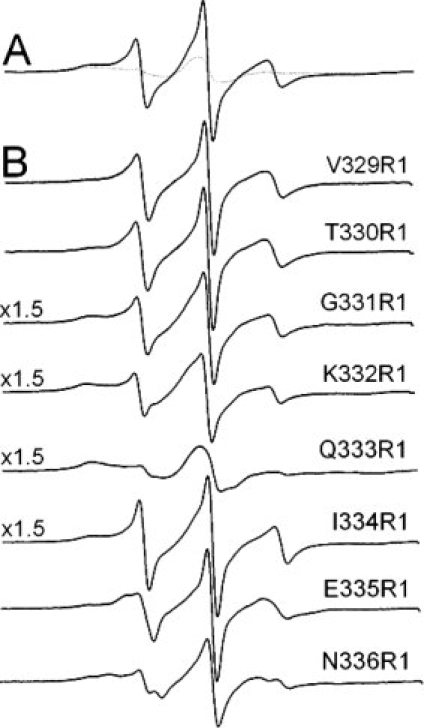
X band spectra of spin labeled BmrA mutants reconstituted into proteoliposomes. All spectra were recorded at room temperature, with a 100 G scan width. (A) overlay of the non-normalized spectra of the Cys-less (dotted line) and the V329R1 mutant (plain line). (B) spectra of the mutants were corrected from the background labeling (the Cys-less spectra in (A)) in a one-to-one ratio and using the non normalized data, and the subtracted 0 spectra are shown.
The EPR spectrum of the R1 label provides information about the dynamics of the nitroxide on the nanosecond timescale. This is due to rotation around the bonds linking the nitroxide side-chain to the backbone, as well as local backbone flexibility.26 As shown previously, this motion frequently takes place as a result of rotation about the last two bonds attaching the nitroxide side ring to the backbone. Spectra from labels at positions 329, 330, 331, 332, and 334 are similar and clearly contain two motional components: one exhibiting a relatively fast correlation time and a second that represents a nitroxide having more restricted motion. The more mobile component dominates at positions 329 and 330, but its fraction decreases, relative to the immobile component, as one progresses to positions 331 and 332. Values for the scaled mobility, Ms, which were obtained from the central linewidth of the EPR spectrum, as well as the correlation times and fractions of the components obtained by simulation of these spectra with the MOMD model (see experimental procedures) are shown in Table I. The Ms values indicate that these spectra, with the exception of the spectrum of Q333R1, arise from relatively mobile R1 side chains. The more mobile components in the spectra from R1 at positions 329, 330, 331, 332, and 334 can be approximated by isotropic motion on the 1 to 2 ns timescale, and indicate that the backbone segment of this region of the protein may be highly dynamic and possibly unstructured. Spectra from R1 at sites 335 and 336 also arise from a component undergoing fast motion; however, a restoring potential was necessary to simulate these spectra, and indicates that the protein segment is more structured in this region and probably assumes some regular secondary structure. The presence of two quite different motional components in many of these spectra suggests that the protein backbone may undergo a conformational exchange, which is slow on the EPR timescale. This might result from both folded and unfolded conformations. There is no obvious periodicity in the EPR spectra as one progress along this sequence, and the EPR spectra indicate that the segment is overall quite dynamic. This result is consistent with the backbone of this BmrA region being predominantly in a loop structure.
Table I.
Scaled Mobilities and Motional Parameters from the EPR Spectra Derived from Figure 3
| Residue | Ms | τc | Fraction |
|---|---|---|---|
| V329R1 | 0.77 | 1.6 ns | 0.7 |
| 7.8 ns | 0.3 | ||
| T330R1 | 0.69 | 1.4 ns | 0.8 |
| 7.8 ns | 0.2 | ||
| G331R1 | 0.63 | 1.5 ns | 0.6 |
| 6.6 ns | 0.4 | ||
| K332R1 | 0.58 | 1.5 ns | 0.47 |
| 8.3 ns | 0.53 | ||
| Q333R1 | 0.24 | 8.3 ns | 1.0 |
| I334R1 | 0.72 | 0.9 ns | 0.43 |
| 7.4 ns | 0.57 | ||
| E335R1 | 0.72 | 1.4 nsa | 0.82 |
| 6.3 ns | 0.18 | ||
| N336R1 | 0.58 | 1.3 nsa | 0.5 |
| 10 ns | 0.5 |
Spectra were simulated using the program NLSL (see methods).
For these components, an ordering potentials of C20 = 1.0 and 3.8 was used for E335R1 and N336R1, respectively. The subtraction of the background labeling introduced a possible variation in the calculation of the two parameters that we estimated to be lower than 20%.
The EPR spectrum of Q333R1 is strongly influenced by the spatially close Y408 residue
Unlike the other spectra from nitroxides along this segment, the spectra from Q333R1 can be fitted by a single correlation time of about 8 ns. This would be characteristic of a nitroxide either undergoing strong tertiary contact or buried within the protein. Previously, it was reported that the R1 side chain in T4 lysozyme could be constrained by the formation of a hydrogen bond to a vicinal tyrosine residue.27 If a similar scenario occurred here, examination of BmrA model based on the Sav1866 structure might reveal which tyrosine residue of BmrA is possibly involved in a hydrogen bond with the R1 moiety. BmrA contains a total of 15 tyrosine residues in its sequence. As shown in Figure 4(A), a single tyrosine residue, Y408, is predicted to lie close enough to Q333 (less than 6 Å between the two Cα) so as to perturb the R1 moiety tethered to the Q333C mutant. Similar results were obtained when models of BmrA based on each MsbA structures were used instead (not shown), which is not really surprising given the fact that, apart from a rigid-body motion, these structures were similar to that obtained with Sav1866. In the BmrA sequence, Y408 is located between the Walker-A motif and the Q-loop and is often replaced by a hydrophobic residue (I, L, V or F) in other ABC exporters [Fig. 4(B)].
Figure 4.
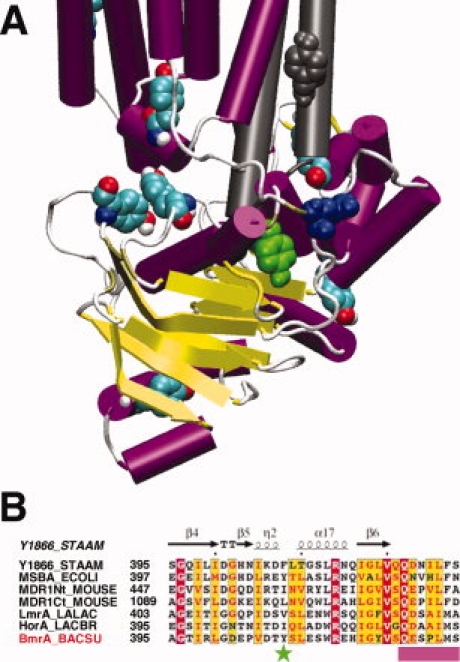
Position of the tyrosine residues in the model of the 3-D structure of BmrA based on Sav1866 structure and partial sequence alignment of the NBD of some ABC exporters. (A) one monomer is shown in color and, for sake of clarity, only ICL2 from the second monomer is shown in grey. Tyrosine residues are shown as spheres colored by element (with C in cyan, N in blue, O in red and H in white) except for two tyrosines residues, one from ICL2 of the second monomer colored grey and Y408 colored green. The Q333 residue is shown as spheres colored blue. The figure was drawn with VMD. (B) for the legend of the sequence alignment, see Figure 1(A). Y408 of BmrA is indicated by a green star and the position of the Q-loop is shown as a pink box. The figure was made with ESPript 2.0.
To check whether Y408 in BmrA was involved in a hydrogen bond with the R1 moiety tethered to the Q333C mutant, it was replaced by a phenylalanine residue in a Q333C mutant background. After purification and labeling of the Y408F/Q333C mutant by MTSL, Y408F/Q333R1 yielded a dramatically different EPR spectrum consistent with rapid ns motion of the R1 side chain (see Fig. 5). This spectrum is similar to that seen with the other spin-labeled mutants of BmrA and indicates that, in the presence of Y408F, this segment of the backbone becomes again highly dynamic. A similar result was also reported when the tyrosine residue involved in a hydrogen bond with the R1 moiety in the lysozyme mutant was replaced by a phenylalanine residue.27 Therefore, this suggests that some motion has been restored for the R1-moiety tethered to the Y408F/Q333C mutant, and this supports the contention that, in the original Q333C mutant, the R1 moiety was involved in a hydrogen bond with the vicinal Y408 residue. An alternative explanation is that replacement of Y408 by a phenylalanine residue might have sufficiently altered the structure of BmrA to permit a greater flexibility of the R1-moiety. If this were the case, one would then expect that the mutation Y408F would greatly perturb the drug transport activity of BmrA. Therefore, the transport properties of the Y408F mutant, and additionally of the Y408A mutant, were further studied.
Figure 5.
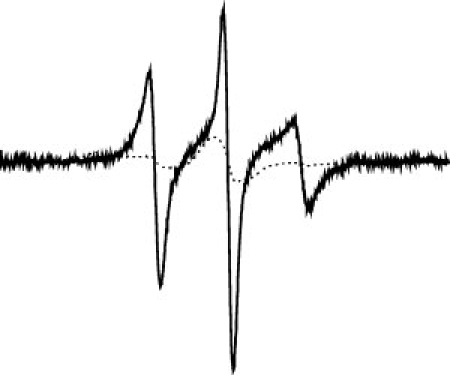
Spectral overlay of Q333R1 (dotted line) and Y408F/Q333R1 (solid line) after correction from the background labeling (the cys-less spectra was subtracted in a one-to-one ratio and using the non normalized spectra). The normalized spectra are shown.
A nonpolar bulky residue is likely required at position 408 in BmrA
As shown in Figure 6, mutation of Y408 to phenylalanine still maintained a significant level of Hoechst 33342 transport, and a similar result was obtained when this mutation was introduced into the Q333C background mutant (not shown). This indicates that the mutation Y408F preserves the overall structure of BmrA, suggesting that the increased flexibility recovered in the Y408F/Q333R1 is likely due to the loss of a hydrogen bond between the R1 moiety and F408. By contrast, mutation of Y408 to alanine fully abrogated the Hoechst transport. After purification of the two mutant enzymes and reconstitution into proteoliposomes, their ATPase activity was measured. Table II shows that while the Y408F mutant conserved a significant level of ATPase activity, albeit reduced as compared to the Cys-less BmrA, no detectable ATPase activity could be measured for the Y408A mutant, consistent with its inability to transport the Hoechst compound.
Figure 6.
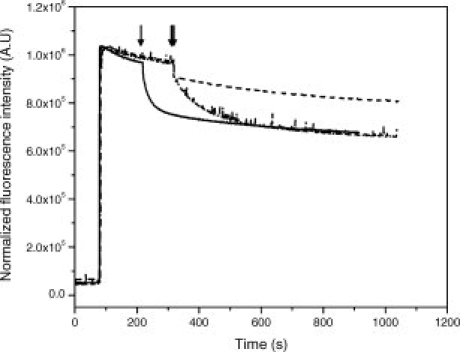
Effect of mutations of tyrosine 408 on the transport activity of BmrA. The tyrosine 408 residue was mutated to either phenylalanine (dash-dotted line) or alanine (dashed line), and the transport of Hoechst was monitored at 37°C as described in Figure 2. The transport rate of the cys-less mutant (solid line) is shown for comparison.
Table II.
ATPase activities of the Cys-less, Q333C and Y408F/Q333C mutants, either before or after labeling by the R1 moiety
| BmrA proteins | Cys-less | Q333C | Q333C-R1 | Y408A | Y408F | Y408F/Q333C | Y408F/Q333C-R1 |
|---|---|---|---|---|---|---|---|
| ATPase activitya | 7.3 ± 0.72 | 5.01 ± 0.28 | 7.58 ± 0.26 | N.D. | 3.43 ± 0.21 | 4.15 ± 0.42 | 3.66 ± 0.44 |
ATPase activity is expressed in μmoles ATP hydrolyzed/min/mg proteins. The values given are the means of four different samples and the standard deviation is indicated. N.D., not detectable. For comparison, the value of ATPase activity of wild-type protein was previously reported as being 6.5 μmoles ATP hydrolyzed/min/mg proteins.17,28
The proximity of Q333 and Y408 is confirmed by formation of an intramolecular disulfide bond between ICL3 and the NBD
To further assess the spatial proximity of Q333 and Y408 in the 3-D structure of BmrA, we attempted to create a disulfide bond between ICL3 and the NBD. Because of the requirement for an aromatic residue at position 408, we decided to mutate the two adjacent residues, T407 and S409, to introduce a new cysteine residue. Either the T407C or S409C mutation was introduced into a Q333C mutant background, and membranes from these double mutants, T407C/Q333C or S409C/Q333C, were prepared and analyzed by SDS-PAGE. As shown here for the T407C/Q333C double mutant, even without any treatment, a disulfide bond occurred spontaneously in some mutants (Fig. 7, lane 3, grey arrow). A prior treatment with copper, which favored the oxidation, increased the population of double mutant having a disulfide bond between C407 and C333 (Lane 6), and this disulfide bond was cleaved by a subsequent treatment with DTT (Lanes 4 and 7, black arrow). Similar results were obtained when these experiments were performed with membranes from the S409C/Q333C double mutant (not shown here). The nature of the two forms of BmrA in the T407C/Q333C double mutant was further analyzed by in-gel trypsin digestion followed by mass spectra analysis of the digested peptides. In contrast to the slowest migrating band on SDS-PAGE (which likely represents the reduced form of BmrA), a new fragment at m/z 3287.50 Da was detected in the fastest migrating band (which presumably corresponds to the oxidized form of BmrA; see Fig. 8). This new ion corresponds to the predicted disulfide-linked peptides [333–345] and [400–414] (theoretical monoisotopic mass: 3287.55 Da) of the oxidized form (internally disulfide bonded peptide between 333C and 407C). Similar results were also obtained when the S409C/Q333C double mutant was analyzed by mass spectra experiments (not shown here).
Figure 7.
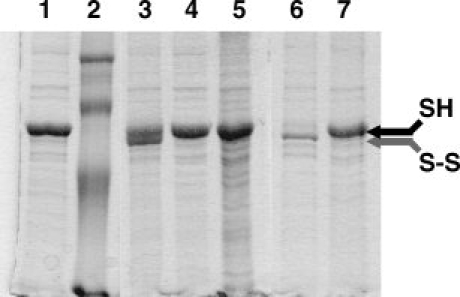
Formation of an intramolecular disulfide bond between T407C and Q333C monitored on a nonreducing 12% SDS-PAGE. Ten micrograms of membrane proteins were loaded into each lane. Lane 1, membranes containing the overexpressed BmrA double mutant were incubated with 10 mM N-ethyl-maleimide during 90 min prior to electophoresis. Lane 2, molecular weight markers (97 kDa, 66 kDa, 45 kDa, and 30 kDa from top to bottom, respectively). Lane 3, membranes containing overexpressed BmrA double mutant were resuspended in Laemli buffer, without any reducing agent. Lane 4, same as lane 3 but membranes were further treated with DTT during 10 min at room temperature prior to electrophoresis. Lane 5, membrane containing the overexpressed Cys-less mutant. Lane 6, membranes containing the overexpressed BmrA double mutant were treated with 200 μM CuSO4 for 90 min, then the reaction was stopped with NEM for 30 min prior to electrophoresis. Lane 7, same as lane 6 but after stopping the reaction with 10 mM N-ethyl-maleimide during 30 min, membranes were further treated with 10 mM DTT prior to electophoresis.
Figure 8.
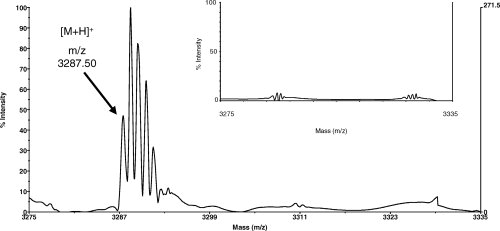
MALDI-TOF mass spectra of the BmrA T407C/Q333C double mutant. The tryptic peptides were analyzed without a reduction step. A new signal at m/z 3287.50 Da was observed for the BmrA band which migrated faster on the SDS-PAGE and which corresponded to the oxidized form of BmrA. This signal can be attributed to the peptides [333–345] and [400–414] bonded by a disulfide bridge between 333C and 407C (theoretical monoisotopic mass 3287.55 Da). Please note that due to the high resolution of this technique, the different isotopes of the disulfide-bonded peptide were resolved here. The same mass range is shown in the insert for the BmrA band which migrated slower on the SDS-PAGE. In this case, no signal was observed since it corresponded to the reduced form of BmrA, without a disulfide-bonded peptide.
Discussion
ATP-Mg binding is believed to cause a significant reorganization of the domains of ABC transporters during the catalytic cycle,29 but how large is the conformational modification associated with is still a matter of debate. The 3-D structure of Sav1866 revealed a tightly packed homodimer with a swapping domain between the two monomers. Because, this structure was solved in an ADP-bound state that is very similar to the ATP sandwich conformation of the LolD dimer of NBDs,30 the authors suggested that limited conformational modifications must occur throughout the cycle of ABC transporters.12 In contrast, we recently showed that the very peculiar, highly stable, ring structures formed by BmrA in the resting state, which supported an open conformation of the transporter,31 were quite sensitive to the addition of ATP-Mg that produced a conformational change large enough to destabilize these rings.32 On the other hand, fluorescence resonance energy transfer led to the conclusion that, in the resting state, most BmrA dimers adopt a rather closed conformation.25 To investigate this apparent conundrum and get further insights into the resting state conformation of BmrA, we used an EPR approach which is a powerful technique to probe the dynamic of proteins and is becoming widely used in the field of membrane proteins in general, and of ABC transporters in particular.33–38 The results obtained strongly support that the ICL3 region targeted here, which is part of the physical link between the TMD and the NBD, mainly adopts a partially extended conformation during the resting state of BmrA. This conclusion is based on the lack of periodicity of the EPR spectra and on the presence of a fast mobile component in some of the spectra. Nevertheless, we cannot exclude that among the BmrA population, a small proportion of dimers may adopt a different conformation thus resulting in a more structured region. This latter possibility would be consistent with the presence of a slow mobile component in some of the spectra, although not all the residues targeted here showed such a characteristic, i.e. Q333R1.
An important question directly related to our study and which warrants some comments here is whether or not the resting state conformation of ABC transporters is physiologically relevant? It has been proposed that, given on the one hand the apparent affinity of some ABC transporters for ATP (e.g. ∼100 μM in proteoliposomes for the maltose transporter,39 or 33 μM for the ABCR retinal transporter40) and on the other hand the concentration of ATP present in the cells (in the mM range for most living cells41,42), the NBDs are probably always saturated with ATP in vivo.10 Although this is possibly true when the food supply is plentiful, starving or stressful conditions have been shown to dramatically reduce the ATP level in plant,43 yeast,44 bacteria (down to ∼0.1 pM,45) or even mammal cells.46 In Bacillus species, dreadful conditions will trigger the formation of spores which contains up to 250-fold less ATP than the vegetative cells.47 Therefore, at least in ‘extreme’ conditions that are a commonplace in the natural habitat of most microorganisms, the ABC transporters can return to their resting state, i.e. with their NBDs free of nucleotides. Thus, the characterization of this conformation in vitro is a mandatory step if one wants to understand the catalytic cycle of ABC transporters.
An interesting feature emerging from this study is the peculiar behavior of Q333R1. It showed a ‘constrained’ spectrum which was completely relieved in the Y408F/Q333R1 protein. This strongly argues for the presence of a hydrogen bond between Y408 and the nitroxide tethered to Q333. However, in the native enzyme, one cannot conclude whether or not a hydrogen bond also occurs between Y408 and Q333. The close proximity between Y408 in the NBD and Q333 in ICL3 was further shown by the disulfide bond formation when T407C (or S409C) and Q333C were mutated jointly to cysteine residues. Overall, these two independent lines of evidence support, at least locally, the 3-D conformation of a monomer of Sav1866 within the dimer structure, because our BmrA model used to pinpoint the tyrosine residue close to Q333 was initially based on the Sav1866 structure. As compared to MsbA, given the fact that the global conformation of one monomer of MsbA is similar to that of Sav1866 regardless of the dimeric conformation of MsbA considered, either open or closed,14 our results are also in agreement with the 3-D conformation of MsbA monomers within either the open or closed dimers.
Another important result that stems from this study is the crucial role played by some residues that belong either to the region connecting the TMD and the NBD, namely G331 and to a lesser extent K332, or to the NBD, that is, Y408. A global perturbation of the structure of BmrA due to different mutations of either residue is very unlikely because some of the mutations (i) maintained a significant level of transport and ATPase activities (see for instance the Y408F mutant), (ii) exhibited a typical EPR spectrum after R1 modification (i. e. the G331C mutant), and (iii) allowed a disulfide crosslink between ICL3 and the NBD. Therefore, the results observed with the mutants are presumably due to a local perturbation around the mutated residue. The mutation of either G331 or Y408 to an alanine residue completely abrogated the ATPase activity of the transporter, thus preventing its drug transport ability. Because these two residues are located far from the ATP-binding site, it is unlikely that their mutation may directly affect ATP binding. A more likely hypothesis would be that these residues are involved in some motions of the transporter which are normally required to permit ATP hydrolysis, for instance to allow the tight dimerization of the two NBDs. Regarding G331, it is conserved in the MDR related ABC transporter (see Fig. 1) and in Sav1866 the equivalent glycine residue forms a β-turn in the 3-D structure. An equivalent turn might be required in BmrA which would explain the lack of functionality of the various G331 mutants. The functional and/or structural importance of having glycine residues in the intracellular loops linking two transmembrane helices was shown for human MDR1,48 but the equivalent glycine residue as the one reported here was not targeted in this previous study. For Y408, although it is not strictly conserved in ABC exporters, a nonpolar bulky residue is often found at the same place. Also, Y408 is located close to the Q-loop motif, and in BtuD, the α-helix equivalent to helix 17 of Sav1866 [see Fig 4(B)] has been shown to be involved in interaction with the BtuC subunit.7
Because of the spatial proximity of ICL3 with the Y408 residue of the NBD, it is tempting to propose that an interaction, and/or a loss of interaction, between these two regions must occur during the catalytic cycle of BmrA, and that some mutations in either region will affect similarly the ATPase activity of the transporter. An alternative explanation is related to the fact that both ICL3 and Y408 in the NBD are located close to the two intracellular loops of the transporter, ICL1 and ICL2. Given the importance of these two loops in the coupling mechanism between ATP hydrolysis and drug transport,20,49–51 it is possible that the role of ICL3 and Y408 is to secure a proper interaction between the NBD and ICL1/ICL2, thereby allowing an efficient cross-talk between the TMD and the NBD. ICL3, or its equivalent linker in the second half of full-length ABC transporters, is missing in the importer where the TMD and the NBD are located on separate polypeptides. Based on NMR spectroscopy experiments, it has been recently proposed that in the resting state of LmrA, the NBD displays a high mobility as opposed to the TMD (including ICL2 and ICL3).21 For the NBD to move independently from the TMD, a physical disconnection must occur between the two domains and the relative motion of the NBD would require some flexibility in ICL3. Whether a high flexibility of the NBD is required in full-length ABC transporters is an important issue. Conformational changes have been shown to take place during the catalytic cycle of P-glycoprotein,6,29,52–54 but how large are these movements is still unknown. Because of the presence of a linker between both halves of full-length ABC transporters, this likely imposes a physical constraint that might limit the motion of, at least, NBD1.
Methods
Unless stated otherwise, products were purchased from Sigma. Lipids were purchased from Avanti Polar lipids, the nitroxide spin label MTSL (2,2,5,5-tetramethylpyrrolin-3-methanethiosulfonate spin label) was purchased from Toronto Research Chemicals (Ontario, Canada), and n-dodecyl-β-D-maltoside (DDM) was obtained from Alexis Biochemicals (San Diego, CA). Restriction enzymes were from Fermentas.
Site-directed mutagenesis
pET23b(+)/C436S was used as a template for site-directed mutagenesis to introduce a single cysteine using Kunkel's method as previously described.28 The cysteine-less protein has been shown to have native-like transport properties in inverted membrane vesicles, and unaltered ATPase activity upon reconstitution into proteoliposomes.25 To screen for positive clones, oligonucleotides with the following sequences were designed to introduce the desired mutations, and in most cases to simultaneously introduce a restriction site without modifying the protein sequence.
V329C, 5′-CAATTTGTTTTCCTGTGCACGTATCTTCCTCTTC-3′ (+ Alw 441)
T330C, 5′-CAATTTGTTTTCCACACACTGTATCTTCCTCTTC-3′ (no site added)
G331C, 5′-CATTTTCAATTTGTTTACACGTGACTGTATCTTCC-3′ (+Eco72 I)
K332C, 5′-GCATTTTCAATTTGACAACCGGTCACTGTATCTTC-3′ (+Cfr10 I)
Q333C, 5′-GTGCATTTTCAATGCATTTACCGGTCACTGTATC-3′ (+MpH 1103 I)
I334C, 5′-GATGTGCATTTTCGCATTGTTTACCGGTCACTGTATCTTC-3′ (+Cfr10 I)
E335C, 5′-GGCAGATGTGCATTGCAAATTTGTTTACCGGTCACTGTATCTTC-3′ (+Cfr10 I)
N336C, 5′-GGCAGATGTGCGCATTCAATTTGTTTTCC-3′ (+ NsbI)
G331S, 5′-GCATTTTCAATTTGTTTGGACGTCACTGTATCTTC-3′ (no site added)
The G331A mutation was performed using a QuickChange Site-Directed mutagenesis kit (Stratagene). Oligonucleotides were designed as followed: G331A-A 5′-GAGGAAGATACAGTGACAGCAAAACAAATTGAAAATGCACATCTG-3′ and G331A-B 5′- AGATGTGCATTTTCAATTTGTTTTGCTGTCACTGTATCTTCCTC-3′.
For Y408A/Q333C, Y408F/Q333C, T407C/Q333C and S409C/Q333C, the plasmid Q333C was used as template and the mutations were introduced using the Quick Change site-directed mutagenesis kit (Stratagene).
Y408A - A: 5′- GAGCCGGTCGATACTGCCTCGCTTGAATCGTGGAGG-3′
Y408A - B: 5′- CCTCCACGATTCAAGCGAGGCAGTATCGACCGGCTC-3′
Y408F - A: 5′-GAGCCGGTCGATACTTTCTCGCTTGAATCGTGGAGG-3′
Y408F - B : 5′-CCTCCACGATTCAAGCGAGAAAGTATCGACCGGCTC-3′
T407C - A : 5′-GACGAGCCGGTCGATTGTTACTCGCTTGAATCGTGG-3′
T407C - B : 5′-CCACGATTCAAGCGAGTAACAATCGACCGGCTCGTC-3′
S409C - A : 5′-CCGGTCGATACTTACTGCCTTGAATCGTGGAGGGAG-3′
S409C - B : 5′- CTCCCTCCACGATTCAAGGCAGTAAGTATCGACCGG-3′
All mutations were checked by DNA sequencing.
Preparation of BmrA enriched membranes
E. coli inverted membrane vesicles containing overexpressed BmrA were prepared as described,16 and kept frozen in liquid nitrogen. Protein concentration, usually ∼50–70 mg/mL, was estimated by a Lowry Protein Assay (Pierce), after precipitation by trichloroacetic acid.
Transport activity
Variation in fluorescence intensity was monitored with a Photon Technology International Quanta Master I fluorimeter, using excitation and emission wavelengths of 355 and 457 nm, and slit widths of 2 and 4 nm, respectively, as previously described.17 Membrane vesicles (200 μg protein) were added into a 3-mL cuvette, containing 2 mL of 50 mM Hepes/KOH, pH 8, 2 mM MgCl2, 8.5 mM NaCl, 120 μg pyruvate kinase (EC 2.7.1.40), and 4 mM phosphoenolpyruvate. After 1-min incubation at 37°C, 2 μM Hoechst was added, and its fluorescence was recorded for 1–2 min. ATP was then added to a final concentration of 2 mM, and the fluorescence intensity was monitored for several minutes.
ATPase activity
ATPase hydrolytic activity was measured in the presence of 50 mM Hepes/KOH pH 8, 10 mM ATP, 10 mM MgCl2 and a regenerating ATP system (5 mM phosphoenolpyruvate and 50 μg/mL pyruvate kinase) in a final volume of 2.5 mL. The reaction was started by adding 10 μg of protein reconstituted into liposomes, and samples were collected at different times for Pi determination by a colorimetric assay at 740 nm,55 using Na2HPO4 as a standard. Briefly, 500 μL of solution were withdrawn at four different times and the reaction was stopped by denaturing the protein with 500 μL of 10% SDS. 500 μL of Taussky and Schorr reagent were then added, and the absorbance was read at 740 nm after 15 min of color development at room temperature.
Protein labeling
After solubilization of membrane vesicles with buffer containing 50 mM Hepes/KOH pH 8, 100 mM NaCl, 10% glycerol, 10 mM imidazole, 1 mM PMSF, 1% DDM (w/v) at 4°C for 1 h, ultracentrifugation was performed at 100,000 g for 1 h. The supernatant was used for spin labeling: typically, 250 μL of 30 mM MTSL solution was added to 25 mL of supernatant and incubated at 37°C for 1 h in the dark, allowing to tether a nitroxide moiety to the desired position (Scheme 1). The samples were then incubated for 1 h in the presence of Ni-NTA (Qiagen) previously equilibrated with buffer A (50 mM Hepes/KOH pH 8, 100 mM NaCl, 10% glycerol, 10 mM imidazole). The slurry was loaded into a column and was washed with buffer containing 50 mM Hepes/KOH pH 8, 100 mM NaCl, 10% glycerol, 20 mM imidazole, 0.05% DDM. Protein was eluted with buffer containing 50 mM Hepes/KOH pH 8, 100 mM NaCl, 10% glycerol, 250 mM imidazole and 0.05% DDM. The fractions were pooled and subjected to extensive dialysis in 50 mM Hepes/KOH pH 8, 50 mM NaCl, 10% glycerol and 0.05% DDM. The purity of the samples was determined by SDS-PAGE and the protein concentration was determined by using a “Coomassie Plus protein assay reagent” from Pierce.
Protein reconstitution and sample preparation
Eighty microliter of E. coli phospholipids total extract (Avanti Polar lipids) at 25 mg/mL (water) were incubated under stirring at room temperature with 20 μL of 10% DDM. After 1 h, 100 μg of protein were added to a final volume of 500 μL (50 mM Hepes/KOH, pH 8). After 45 min incubation, three successive additions of 40 mg Bio-beads SM2 (Bio-rad) were performed every hour, and the proteoliposomes formed were kept at 4°C. Two hundred microliter of proteoliposomes were concentrated using a Beckman Airfuge. The pellet was resuspended with 2 μL of 50 mM Hepes/KOH pH 8, and used for EPR measurements.
Electron paramagnetic resonance spectroscopy
For EPR analysis, 5 μL of sample were loaded into glass capillary tubes with a 0.6-mm internal diameter (VitoCom, Mountain Lakes, NJ). Continuous wave (CW) EPR spectroscopy was performed on a Varian E-line 102 series X-band spectrometer fitted with a loop-gap resonator (Medical Advances, Milwaukee, WI). Data collection and analysis were carried out by using Labview software which was generously provided by Drs. Christian Altenbach and Wayne Hubbell (UCLA). EPR spectra were obtained at room temperature using a modulation amplitude of 1G and an incident microwave power of 2 mW, with a field modulation of 100 kHz. The scan range for each spectrum was 100 G. Spectra represent the average of 32 scans with a scan time of 30 s. EPR spectra are normalized to the same number of spins and are shown after normalization and background subtraction: the non specific labeling observed for the cysteine-less mutant was subtracted from the spectra of the different mutants. Quantitation of mobile and immobile components were determined using the Jack Freed simulation.
Analysis of EPR spectra
The scaled mobility (Ms) provides an approximate measure of nitroxide motion relative to the least and most mobile R1 side chains seen in proteins. Values of Ms were calculated from the peak-to-peak linewidth of the EPR central resonance, δ, according to: 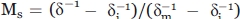 , where δm and δi represent the linewidths of the most and least mobile R1 side chains previously observed in proteins.26 (Columbus and Hubbell 2004).
, where δm and δi represent the linewidths of the most and least mobile R1 side chains previously observed in proteins.26 (Columbus and Hubbell 2004).
EPR spectra were simulated and fitted using the microscopic order macroscopic disorder (MOMD) model described previously by Freed and coworkers.56 In this model, the nitroxide motion is described by an effective correlation time or diffusion rate that is under the restraint of a restoring potential, U. In the simulations used here we use an axially symmetric restoring potential which is defined by the scaling factor C20, where U (θ) = 1/2kBTC20(3 cos2θ−1), and θ represents the instantaneous angle between the z axis of the rotational diffusion tenor and the director defining the symmetry axis of the restoring potential. The values used for Axx, Ayy, and Azz were 6.2, 5.9, and 37, respectively, and for gxx, gyy, and gzz were 2.0076, 2.0050, and 2.0023, respectively. The diffusion tilt angles defining the relationship between the coordinate frame of the diffusion tensor and the magnetic tensor were βD = 36.2 and αD = 4.0 degrees. In each case, we employed an axially symmetric diffusion tensor, where the geometric mean of the diffusion tensor was described by 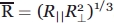 . In most cases, an asymmetry parameter N = R||/R⊥ (of approximately 1.0 was found to adequately fit the data. This choice of magnetic parameters and the model used for motion are described in detail elsewhere.57 It should be noted that these parameters have been primarily applied to exposed surface sites on proteins, but in most cases worked well to approximate the EPR spectra observed here, consistent with the location of ICL3 at the surface of BmrA.
. In most cases, an asymmetry parameter N = R||/R⊥ (of approximately 1.0 was found to adequately fit the data. This choice of magnetic parameters and the model used for motion are described in detail elsewhere.57 It should be noted that these parameters have been primarily applied to exposed surface sites on proteins, but in most cases worked well to approximate the EPR spectra observed here, consistent with the location of ICL3 at the surface of BmrA.
Cross-linking experiment
E. coli membrane vesicles containing over-expressed BmrA were diluted at 1 mg/mL (final volume of 50 μL) in 50 mM Hepes/KOH pH 8, 50 mM NaCl, 5 mM MgCl2. Cysteine oxidation was promoted by addition of 200 μM CuSO4, and samples were incubated during 90 min at 30°C. The reaction was stopped by addition of a final concentration of 10 mM N-ethyl maleimide for 30 min at room temperature. When indicated, the disulfide bridge was reduced by 10 mM DTT. Ten microgram of protein were resolved by SDS-PAGE (12%) using a Laemmli buffer in the absence of reducing agent, and stained with SimplyBlue SafeStain (Invitrogen).
MALDI-TOF
One microliter of fresh membrane were resuspended with 15 μL of Laemmli buffer without reducing agent, and incubated at room temperature for 10 min. The reaction was stopped with 10 mM NEM. Ten micrograms of protein were loaded onto a 12% SDS PAGE, and the protein bands were detected by Coomassie Blue Staining. The two bands corresponding to BmrA with or without a putative intramolecular disulfide bonds were excised and cut into 1 mm pieces to perform in gel trypsin digestion. In gel digestion was performed as described by Shevchenko et al.,58 without reduction and alkylation, using 30 μL of a trypsin (Promega, Charbonnières, France) solution (20 ng/μL in 50 mM NH4HCO3). MALDI-TOF mass spectra were recorded on a Voyager DE-PRO (Applied Biosystems, Courtaboeuf, France) mass spectrometer in positive ion mode. A 0.5 mg/100 μL solution of α-cyano-4-hydroxycinnamic acid (Laser-BioLab, Sophia-Antipolis, France) with 50% CH3CN, 0.1% TFA in water was used as the matrix. Delayed extraction source and reflector equipments allowed sufficient resolution to consider [M+H]+ of monoisotopic masses. Internal calibration was done with trypsin autolysis fragments at m/z 842.5100 and 2211.1046 Da.
Molecular modeling
Homology models of BmrA were built from the structure of Sav1866 and from the four structures of MsbA recently published, using the MODELLER program (version 9v1).59 The program was able to generate plausible all atom models even when alpha carbons only were present. Particular attention was brought to introduce in BmrA models both the symmetry of the 2 monomers and the correct secondary structures (alpha helices and beta sheets). This was done through the introduction of special additional restraints in MODELLER, namely 48 α-helix and 18 β-strand restraints. These secondary structure restraints for BmrA were deduced from the alignment with Sav1866 and secondary structure information found in the PDB file 2HYD. In each case, 10 models were generated with MODELLER and the model with the lowest objective function was retained. The unreliable N- and C-terminal residues, which were not aligned in the alignment files, were removed from the models. Finally, the models of complete BmrA dimers, including residues Leu-12 to Lys-579 were relaxed by energy minimization with the Molecular Dynamics program CHARMM.60 Strong harmonic restraints (force constant 5 kcal/mol/≈2) were applied on the backbone atoms of the models during the minimization to prevent unfolding or large atomic displacements which might be caused by the absence of solvent and lipids in this simple molecular modeling. The most stable model was the one built from the structure of Sav1866 with lowest total energy (−25526 kcal/mol). The interaction energies between loop ICL3 (residues 329 to 336) and residue Y408 was −21 (kcal/mol) in the model based on Sav1866 but they were comparable for all generated models.
Acknowledgments
The authors thank Dr. Wayne Hubbell for providing us the atomic coordinates of the Cys residue tethered with the R1 moiety in the T4 lysozyme, Dr Geoffrey Chang for sharing with us the atomic coordinates of MsbA prior to publication, and Dr. Anne Durand for her critical reading of the manuscript.
Glossary
Abbreviations:
- ABC
ATP-Binding Cassette
- Cys-less
cysteine-less
- DDM
n-dodecyl-β-D-maltoside
- EcMsbA
VcMsbA and StMsbA, MsbA from E. coli, V. cholerae, and S. typhimurium, respectively
- EPR
Electron Paramagnetic Resonance
- ICL
IntraCellular Loop
- NBD
Nucleotide-Binding Domain
- TMD
TransMembrane Domain.
References
- 1.Ren Q, Kang KH, Paulsen IT. TransportDB: a relational database of cellular membrane transport systems. Nucl Acids Res. 2004;32:D284–D288. doi: 10.1093/nar/gkh016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dassa E, Bouige P. The ABC of ABCS: a phylogenetic and functional classification of ABC systems in living organisms. Res Microbiol. 2001;152:211–229. doi: 10.1016/s0923-2508(01)01194-9. [DOI] [PubMed] [Google Scholar]
- 3.Davidson AL, Chen J. ATP-binding cassette transporters in bacteria. Annu Rev Biochem. 2004;73:241–268. doi: 10.1146/annurev.biochem.73.011303.073626. [DOI] [PubMed] [Google Scholar]
- 4.Borst P, Elferink RO. Mammalian abc transporters in health and disease. Annu Rev Biochem. 2002;71:537–592. doi: 10.1146/annurev.biochem.71.102301.093055. [DOI] [PubMed] [Google Scholar]
- 5.Borges-Walmsley MI, McKeegan KS, Walmsley AR. Structure and function of efflux pumps that confer resistance to drugs. Biochem J. 2003;376:313–338. doi: 10.1042/BJ20020957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Higgins CF, Linton KJ. The ATP switch model for ABC transporters. Nat Struct Mol Biol. 2004;11:918–926. doi: 10.1038/nsmb836. [DOI] [PubMed] [Google Scholar]
- 7.Locher KP, Lee AT, Rees DC. The E. coli BtuCD structure: a framework for ABC transporter architecture and mechanism. Science. 2002;296:1091–1098. doi: 10.1126/science.1071142. [DOI] [PubMed] [Google Scholar]
- 8.Hollenstein K, Frei DC, Locher KP. Structure of an ABC transporter in complex with its binding protein. Nature. 2007;446:213–216. doi: 10.1038/nature05626. [DOI] [PubMed] [Google Scholar]
- 9.Hvorup RN, Goetz BA, Niederer M, Hollenstein K, Perozo E, Locher KP. Asymmetry in the structure of the ABC transporter-binding protein complex BtuCD-BtuF. Science. 2007;317:1387–1390. doi: 10.1126/science.1145950. [DOI] [PubMed] [Google Scholar]
- 10.Oldham ML, Khare D, Quiocho FA, Davidson AL, Chen J. Crystal structure of a catalytic intermediate of the maltose transporter. Nature. 2007;450:515–521. doi: 10.1038/nature06264. [DOI] [PubMed] [Google Scholar]
- 11.Pinkett HW, Lee AT, Lum P, Locher KP, Rees DC. An inward-facing conformation of a putative metal-chelate-type ABC transporter. Science. 2007;315:373–377. doi: 10.1126/science.1133488. [DOI] [PubMed] [Google Scholar]
- 12.Dawson RJ, Locher KP. Structure of a bacterial multidrug ABC transporter. Nature. 2006;443:180–185. doi: 10.1038/nature05155. [DOI] [PubMed] [Google Scholar]
- 13.Chang G, Roth CB, Reyes CL, Pornillos O, Chen YJ, Chen AP. Retraction Sci. 2006;314:1875. doi: 10.1126/science.314.5807.1875b. [DOI] [PubMed] [Google Scholar]
- 14.Ward A, Reyes CL, Yu J, Roth CB, Chang G. Flexibility in the ABC transporter MsbA: alternating access with a twist. Proc Natl Acad Sci USA. 2007;104:19005–19010. doi: 10.1073/pnas.0709388104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Veen HW, Venema K, Bolhuis H, Oussenko I, Kok J, Poolman B, Driessen AJ, Konings WN. Multidrug resistance mediated by a bacterial homolog of the human multidrug transporter MDR1. Proc Natl Acad Sci USA. 1996;93:10668–10672. doi: 10.1073/pnas.93.20.10668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Steinfels E, Orelle C, Dalmas O, Penin F, Miroux B, Di Pietro A, Jault JM. Highly efficient over-production in E. coli of YvcC, a multidrug-like ATP-binding cassette transporter from Bacillus subtilis. Biochim Biophys Acta. 2002;1565:1–5. doi: 10.1016/s0005-2736(02)00515-1. [DOI] [PubMed] [Google Scholar]
- 17.Steinfels E, Orelle C, Fantino JR, Dalmas O, Rigaud JL, Denizot F, Di Pietro A, Jault JM. Characterization of YvcC (BmrA), a multidrug ABC transporter constitutively expressed in Bacillus subtilis. Biochemistry. 2004;43:7491–7502. doi: 10.1021/bi0362018. [DOI] [PubMed] [Google Scholar]
- 18.Dalmas O, Orelle C, Foucher AE, Geourjon C, Crouzy S, Di Pietro A, Jault JM. The Q-loop disengages from the first intracellular loop during the catalytic cycle of the multidrug ABC transporter BmrA. J Biol Chem. 2005b;280:36857–36864. doi: 10.1074/jbc.M503266200. [DOI] [PubMed] [Google Scholar]
- 19.Zolnerciks JK, Wooding C, Linton KJ. Evidence for a Sav1866-like architecture for the human multidrug transporter P-glycoprotein. FASEB J. 2007;21:3937–3948. doi: 10.1096/fj.07-8610com. [DOI] [PubMed] [Google Scholar]
- 20.Serohijos AW, Hegedus T, Aleksandrov AA, He L, Cui L, Dokholyan NV, Riordan JR. Phenylalanine-508 mediates a cytoplasmic-membrane domain contact in the CFTR 3D structure crucial to assembly and channel function. Proc Natl Acad Sci USA. 2008;105:3256–3261. doi: 10.1073/pnas.0800254105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Siarheyeva A, Lopez JJ, Lehner I, Hellmich UA, van Veen HW, Glaubitz C. Probing the molecular dynamics of the ABC multidrug transporter LmrA by deuterium solid-state nuclear magnetic resonance. Biochemistry. 2007;46:3075–3083. doi: 10.1021/bi062109a. [DOI] [PubMed] [Google Scholar]
- 22.Hubbell WL, Cafiso DS, Altenbach C. Identifying conformational changes with site-directed spin labeling. Nat Struct Biol. 2000;7:735–739. doi: 10.1038/78956. [DOI] [PubMed] [Google Scholar]
- 23.Columbus L, Hubbell WL. A new spin on protein dynamics. Trends Biochem Sci. 2002;27:288–295. doi: 10.1016/s0968-0004(02)02095-9. [DOI] [PubMed] [Google Scholar]
- 24.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38. 27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 25.Dalmas O, Do Cao MA, Lugo MR, Sharom FJ, Di Pietro A, Jault JM. Time-resolved fluorescence resonance energy transfer shows that the Bacterial multidrug ABC half-transporter BmrA functions as a homodimer. Biochemistry. 2005a;44:4312–4321. doi: 10.1021/bi0482809. [DOI] [PubMed] [Google Scholar]
- 26.Columbus L, Hubbell WL. Mapping backbone dynamics in solution with site-directed spin labeling: GCN4–58 bZip free and bound to DNA. Biochemistry. 2004;43:7273–7287. doi: 10.1021/bi0497906. [DOI] [PubMed] [Google Scholar]
- 27.Langen R, Oh KJ, Cascio D, Hubbell WL. Crystal structures of spin labeled T4 lysozyme mutants: implications for the interpretation of EPR spectra in terms of structure. Biochemistry. 2000;39:8396–8405. doi: 10.1021/bi000604f. [DOI] [PubMed] [Google Scholar]
- 28.Orelle C, Dalmas O, Gros P, Di Pietro A, Jault JM. The conserved glutamate residue adjacent to the walker-B motif is the catalytic base for ATP Hydrolysis in the ATP-binding cassette transporter BmrA. J Biol Chem. 2003;278:47002–47008. doi: 10.1074/jbc.M308268200. [DOI] [PubMed] [Google Scholar]
- 29.Callaghan R, Ford RC, Kerr ID. The translocation mechanism of P-glycoprotein. FEBS Lett. 2006;580:1056–1063. doi: 10.1016/j.febslet.2005.11.083. [DOI] [PubMed] [Google Scholar]
- 30.Smith PC, Karpowich N, Millen L, Moody JE, Rosen J, Thomas PJ, Hunt JF. ATP binding to the motor domain from an ABC transporter drives formation of a nucleotide sandwich dimer. Mol Cell. 2002;10:139–149. doi: 10.1016/s1097-2765(02)00576-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chami M, Steinfels E, Orelle C, Jault JM, Di Pietro A, Rigaud JL, Marco S. Three-dimensional structure by cryo-electron microscopy of YvcC, an homodimeric ATP-binding cassette transporter from Bacillus subtilis. J Mol Biol. 2002;315:1075–1085. doi: 10.1006/jmbi.2001.5309. [DOI] [PubMed] [Google Scholar]
- 32.Orelle C, Gubellini F, Durand A, Marco S, Levy D, Gros P, Di Pietro A, Jault JM. Conformational change induced by ATP binding in the multidrug ATP-binding cassette transporter BmrA. Biochemistry. 2008;47:2404–2412. doi: 10.1021/bi702303s. [DOI] [PubMed] [Google Scholar]
- 33.Omote H, Al-Shawi MK. A novel electron paramagnetic resonance approach to determine the mechanism of drug transport by P-glycoprotein. J Biol Chem. 2002;277:45688–45694. doi: 10.1074/jbc.M206479200. [DOI] [PubMed] [Google Scholar]
- 34.Austermuhle MI, Hall JA, Klug CS, Davidson AL. Maltose-binding protein is open in the catalytic transition state for ATP hydrolysis during maltose transport. J Biol Chem. 2004;279:28243–28250. doi: 10.1074/jbc.M403508200. [DOI] [PubMed] [Google Scholar]
- 35.Buchaklian AH, Funk AL, Klug CS. Resting state conformation of the MsbA homodimer as studied by site-directed spin labeling. Biochemistry. 2004;43:8600–8606. doi: 10.1021/bi0497751. [DOI] [PubMed] [Google Scholar]
- 36.Buchaklian AH, Klug CS. Characterization of the Walker A motif of MsbA using site-directed spin labeling electron paramagnetic resonance spectroscopy. Biochemistry. 2005;44:5503–5509. doi: 10.1021/bi047568v. [DOI] [PubMed] [Google Scholar]
- 37.Dong J, Yang G, McHaourab HS. Structural basis of energy transduction in the transport cycle of MsbA. Science. 2005;308:1023–1028. doi: 10.1126/science.1106592. [DOI] [PubMed] [Google Scholar]
- 38.Borbat PP, Surendhran K, Bortolus M, Zou P, Freed JH, McHaourab HS. Conformational motion of the ABC transporter MsbA induced by ATP hydrolysis. PLoS biology. 2007;5:e271. doi: 10.1371/journal.pbio.0050271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davidson AL, Laghaeian SS, Mannering DE. The maltose transport system of Escherichia coli displays positive cooperativity in ATP hydrolysis. J Biol Chem. 1996;271:4858–4863. [PubMed] [Google Scholar]
- 40.Sun H, Nathans J. Mechanistic studies of ABCR, the ABC transporter in photoreceptor outer segments responsible for autosomal recessive Stargardt disease. J Bioenerg Biomembr. 2001;33:523–530. doi: 10.1023/a:1012883306823. [DOI] [PubMed] [Google Scholar]
- 41.Bochner BR, Ames BN. Complete analysis of cellular nucleotides by two-dimensional thin layer chromatography. J Biol Chem. 1982;257:9759–9769. [PubMed] [Google Scholar]
- 42.Matsuno K, Blais T, Serio AW, Conway T, Henkin TM, Sonenshein AL. Metabolic imbalance and sporulation in an isocitrate dehydrogenase mutant of Bacillus subtilis. J Bacteriol. 1999;181:3382–3391. doi: 10.1128/jb.181.11.3382-3391.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shimano F, Ashihara H. Effect of long-term phosphate starvation on the levels and metabolism of purine nucleotides in suspension-cultured Catharanthus roseus cells. Phytochemistry. 2006;67:132–141. doi: 10.1016/j.phytochem.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 44.Thomsson E, Svensson M, Larsson C. Rapamycin pre-treatment preserves viability. ATP level and catabolic capacity during carbon starvation of Saccharomyces cerevisiae. Yeast (Chichester England) 2005;22:615–623. doi: 10.1002/yea.1219. [DOI] [PubMed] [Google Scholar]
- 45.Ganesan B, Stuart MR, Weimer BC. Carbohydrate starvation causes a metabolically active but nonculturable state in Lactococcus lactis. Appl Environ Microbiol. 2007;73:2498–2512. doi: 10.1128/AEM.01832-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kriat M, Fantini J, Vion-Dury J, Confort-Gouny S, Galons JP, Cozzone PJ. Energetic metabolism of glucose, mannose and galactose in glucose-starved rat insulinoma cells anchored on microcarrier beads. A phosphorus-31 NMR study. Biochimie. 1992;74:949–955. doi: 10.1016/0300-9084(92)90079-t. [DOI] [PubMed] [Google Scholar]
- 47.Setlow P, Kornberg A. Biochemical studies of bacterial sporulation and germination. XXII. Energy metabolism in early stages of germination of Bacillus megaterium spores. J Biol Chem. 1970;245:3637–3644. [PubMed] [Google Scholar]
- 48.Loo TW, Clarke DM. Functional consequences of glycine mutations in the predicted cytoplasmic loops of P-glycoprotein. J Biol Chem. 1994;269:7243–7248. [PubMed] [Google Scholar]
- 49.Kwan T, Gros P. Mutational analysis of the P-glycoprotein first intracellular loop and flanking transmembrane domains. Biochemistry. 1998;37:3337–3350. doi: 10.1021/bi972680x. [DOI] [PubMed] [Google Scholar]
- 50.Ambudkar SV, Dey S, Hrycyna CA, Ramachandra M, Pastan I, Gottesman MM. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu Rev Pharmacol Toxicol. 1999;39:361–398. doi: 10.1146/annurev.pharmtox.39.1.361. [DOI] [PubMed] [Google Scholar]
- 51.Omote H, Figler RA, Polar MK, Al-Shawi MK. Improved energy coupling of human P-glycoprotein by the glycine 185 to valine mutation. Biochemistry. 2004;43:3917–3928. doi: 10.1021/bi035365l. [DOI] [PubMed] [Google Scholar]
- 52.Loo TW, Bartlett MC, Clarke DM. Drug binding in human P-glycoprotein causes conformational changes in both nucleotide-binding domains. J Biol Chem. 2003;278:1575–1578. doi: 10.1074/jbc.M211307200. [DOI] [PubMed] [Google Scholar]
- 53.Sharom FJ. Probing of conformational changes, catalytic cycle and ABC transporter function. In: Holland IB, Cole SP, Kuchler K, Higgins C, editors. ABC proteins: from bacteria to man. Elsevier Science Ltd; 2003. pp. 107–133. [Google Scholar]
- 54.Sauna ZE, Ambudkar SV. About a switch: how P-glycoprotein (ABCB1) harnesses the energy of ATP binding and hydrolysis to do mechanical work. Mol Cancer Ther. 2007;6:13–23. doi: 10.1158/1535-7163.MCT-06-0155. [DOI] [PubMed] [Google Scholar]
- 55.Taussky HH, Shorr EA. Microcolorimetric method for the determination of inorganic phosphorus. J Biol Chem. 1953;202:675–685. [PubMed] [Google Scholar]
- 56.Budil DE, Lee S, Saxena S, Freed JH. Nonlinear-least-squares analysis of slow-motion EPR spectra in one and two dimensions using a modified Levenberg-Marquardt algorithm. J Magn Reson Ser A. 1996;120:155–189. [Google Scholar]
- 57.Columbus L, Kalai T, Jeko J, Hideg K, Hubbell WL. Molecular motion of spin labeled side chains in alpha-helices: analysis by variation of side chain structure. Biochemistry. 2001;40:3828–3846. doi: 10.1021/bi002645h. [DOI] [PubMed] [Google Scholar]
- 58.Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 59.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 60.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J Comp Chem. 1983;4:187–217. [Google Scholar]