

Abstract
Islet Amyloid Polypeptide (IAPP or amylin) is a peptide hormone produced and stored in the β-islet cells of the pancreas along with insulin. IAPP readily forms amyloid fibrils in vitro, and the deposition of fibrillar IAPP has been correlated with the pathology of type II diabetes. The mechanism of the conversion that IAPP undergoes from soluble to fibrillar forms has been unclear. By chaperoning IAPP through fusion to maltose binding protein, we find that IAPP can adopt a α-helical structure at residues 8–18 and 22–27 and that molecules of IAPP dimerize. Mutational analysis suggests that this dimerization is on the pathway to fibrillation. The structure suggests how IAPP may heterodimerize with insulin, which we confirmed by protein crosslinking. Taken together, these experiments suggest the helical dimerization of IAPP accelerates fibril formation and that insulin impedes fibrillation by blocking the IAPP dimerization interface.
Keywords: IAPP, amylin, amyloid, aggregation, type II diabetes
Introduction
Islet amyloid polypeptide (IAPP or amylin) has been implicated in the pathology of type II diabetes, a disease that affects an estimated 20 million people in the United States.1 The disease is characterized initially by a condition of insulin resistance and progresses towards insulin dependence.2 Nearly all type II diabetics exhibit amyloid plaques in the pancreas composed primarily of the mature, 37-residue form of IAPP.3 This hormone was discovered in 1987 and is expressed, processed, stored, and secreted from the β-islet cells of the pancreas along with insulin.4–7
The importance of amyloid formation in the etiology of type II diabetes has been established but the details of its involvement are unclear.8 Post-mortem approximately 90% of type II diabetics stain positive for amyloid plaques in their pancreas.9 The severity of the disease appears to correlate with the degree of plaque deposition.10 IAPP has been shown to be highly cytotoxic to cultured islet cells.11–13 This toxicity is believed to be connected to the islet cell loss observed in the diseased state. Since the islet cells also produce insulin, it is believed this is the cause of the progression to insulin dependence.14 Mouse IAPP differs from human in only six of 37 residues but the mouse peptide does not fibrillize.15 Transgenic mice expressing human IAPP can develop the disease, supporting the relationship between amyloid formation and type II diabetes.16
There is evidence of a molecular interaction between IAPP and insulin. IAPP is stored along with insulin in the secretory granules of the β-islet cells at ∼1–4 mM; 1000-fold higher than is required to form amyloid fibrils in vitro.17,18 It has been established that insulin can inhibit the fibril formation of IAPP in a concentration-dependent manner.19,20 The direct association between insulin and IAPP has been characterized and the interacting regions have been determined via molecular mapping.21,22 Insulin exists in molar excess to IAPP in the secretory granules and it has been proposed that the binding of insulin to IAPP prevents the fibrillation of IAPP in the absence of diabetes.19–21
Establishing the structure of human IAPP in the dissolved state has proved difficult because it forms amyloid fibrils in vitro in minutes at modest concentrations under physiological conditions.22,23 Previously published reports suggested IAPP is natively disordered based on circular dichroism.24,25 However, Miranker and coworkers have shown by NMR that mouse IAPP is capable of adopting a transient helical structure in solution.26 They suggest this helical form of IAPP is an intermediate on the pathway to fibrillation. Circular dichroism (CD) as well as infrared reflection absorption spectroscopy (IRRAS), two-dimensional infrared spectroscopy (2D IR), and other NMR studies show an increase in the helical content of IAPP prior to its conversion to the β-sheet rich fibrillar form.24,27–30 Lipids and aqueous solutions of hexafluoroisopropanol (HFIP) promote the helical formation of IAPP and have been shown to accelerate the rate of IAPP fibrillation.31–35 The structure of this N-terminal, helical region has recently been determined by NMR in the presence of lipids.36
Crystallographic studies of this helical form of IAPP are hindered by its aggressive propensity to aggregate. We have overcome this problem by chaperoning IAPP by fusion with a larger, more soluble protein. To acquire X-ray diffraction data from what is essentially an isolated IAPP molecule, we set out to protect IAPP from the inevitable fibrillation that occurs when it is unchaperoned. Our intention is to understand the structural propensity of IAPP at the atomic level and how this propensity is involved in the conversion from the soluble to the fibrillar form.
Results
The IAPP fusion reveals an IAPP dimer
To capture IAPP in its nonfibrillar state, we fused IAPP to maltose binding protein (MBP), following a previous approach facilitating the crystallization of small, recalcitrant proteins.37 To provide a rigid C-terminal helix within MBP and to accommodate crystal packing, our fusion includes three residue replacements at the C-terminus of MBP followed by an Ala-Ala-Ala linker leading immediately into the sequence of IAPP. Crystallization was successful, and we were able to determine the structure to 1.9 Å resolution (Supporting Information Table 1).
The crystal structure of the MBP-IAPP fusion reveals that IAPP adopts an ordered, helical conformation, consistent with the previously reported NMR measurements.26 Our work shows that segments 8–18 and 22–27 are helical (see Fig. 1). This helical structure within the 8–18 segment is in excellent agreement with the NMR structure determined in lipids,36 having a RMSD of 1.1 Å (Supporting Information Fig. S1). The structure also suggests that nonfibrillar state of IAPP can form dimers with the two IAPP molecules interacting at a helical interface centered at the aromatic stack of two Phe15 residues [Fig. 1(B)]. That is, each MBP-IAPP fusion molecule in the crystal binds to a second fusion molecule of the asymmetric unit at the three-turn α-helix from residues 8 to 18. These identical helices meet at a two-fold axis at an angle of 55° between the helices [Fig 1(C)]. The surface complementarity of this interface is large (Sc = 0.86) with a small, but perhaps significant, area buried of 302 Å2. The asymmetric unit contains four fusion molecules, which can be superimposed in pairs with a backbone RMSD of 1.1 Å. Representative electron density for the helical interface derived from the omit map used to model the structure is shown in the Supporting Information Figure S2.
Figure 1.
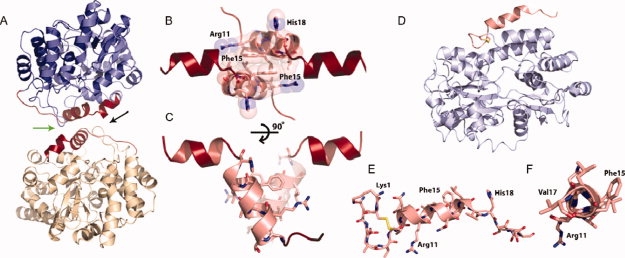
The crystal structure of the maltose binding protein (MBP)-IAPP fusions protein reveals the helical propensity of IAPP. (A) A helix-helix homodimerization interface (green arrow between IAPP molecules) is observed between fusion molecules. The two MBP molecules are shown in blue and yellow, respectively. Residues of the fusion attributable to IAPP are shown in red, each attached to MBP molecules. (B) Space filling representation showing the tight interface between parallel IAPP helices, centered at Phe 15. The view is in the direction of the black arrow in A. (C) By rotating the IAPP homodimer, it can be seen that the 8–18 helices interact at a 55° angle. (D and E) A MBP-IAPP1-22 fusion reveals the cyclic disulfide bond between Cys 2 and Cys 7 and the helical propensity within the N-terminus. MBP is shown in light blue and IAPP is shown in red. (F) View looking down the N-terminal helix of IAPP shows the 310 helix between residues Gln10 and Val17.
Thinking that the C-terminal amyloidogenic domain of IAPP would complicate crystallization, we also created a truncated MBP-IAPP 1–22 fusion. This construct contains only the N-terminal 22 residues of IAPP shown by the structure of Figure 1(C) to contain the first helical domain. The crystal structure was determined to 1.7 Å resolution [Fig. 1(D–F)]. This structure contains the cyclic disulfide bond between Cys2 and Cys7 and is again helical between residues 8 and 17. The crystal contacts of this helix are against an entirely different region of MBP than the full-length MBP-IAPP structure. This suggests that the helicity of this segment is not a crystallization artifact. In contrast to the helix in the full-length structure, this helix appears to be a 310 helix between residues 10 and 17. Thus although the MBP-IAPP 1–22 strengthens the case for a propensity of IAPP to adopt a helical conformation at its N-terminus, it also shows that the precise environment of the N-terminus affects its structure.
Insulin may bind to IAPP as IAPP binds to itself
Our MBP-IAPP fusion crystal structure suggests a molecular explanation for insulin's ability to inhibit IAPP fibrillation: that insulin binds to IAPP in the same way that an IAPP monomer binds to the second monomer of the dimer. We used the following information to build a model of an IAPP:insulin heterodimer: (1) Insulin binds to IAPP and inhibits IAPP fibrillation.19–22 (2) When bound to insulin, IAPP adopts a helical conformation as shown by CD.21 (3) A recent molecular mapping study determined the associating segments from IAPP and insulin molecules and that the 9–20 segment from the insulin B-chain binds to IAPP and prevents IAPP fibrillation.22 (4) The interacting regions from insulin and IAPP are both helical as shown in the structure of insulin and our IAPP fusion structures (Fig. 1). (5) A sequence alignment between the IAPP and insulin segments reveals sequence similarity.21,22 We have used this alignment to determine the orientation of the helices [Fig. 2(A)].
Figure 2.
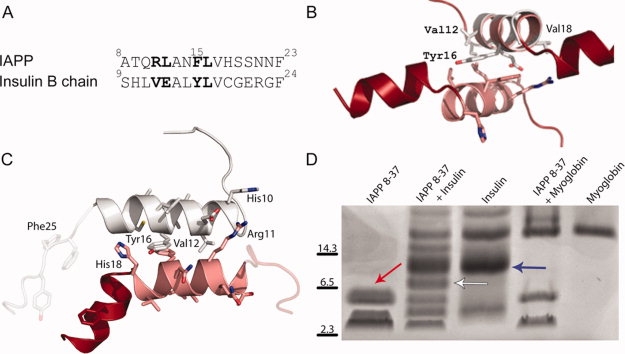
The homodimerization of IAPP suggests an analogous structure for IAPP-insulin heterodimerization. (A) Sequence alignment of residues 8–23 from IAPP with residues 9–24 of the insulin B chain. (B) The putative IAPP-interacting segment from insulin (helical segment composed of residues 9–20 shown in gray) is shown overlaid on the IAPP 8–19 helix. This interaction is centered on the stacked aromatic sidechains (Phe 15 from IAPP and Tyr 16 from insulin) between the two helices. (C) Computationally energy-minimized packing between insulin 9–19 and IAPP 8–18 helices. Notice that the energy-minimized helices are more parallel than the helices in B. (D) Crosslinking of IAPP and Insulin reveals an IAPP dimer (red arrow), an insulin dimer (blue arrow) as well as an IAPP-insulin heterodimer (white arrow). IAPP also crosslinks to higher-order insulin multimers. Myoglobin is included as a negative control for nonspecific crosslinking with IAPP.
The helical segment from the insulin B chain was superimposed onto the N-terminal IAPP helix, suggesting how insulin might interact with IAPP, competing with a second IAPP molecule [Fig. 2(B)]. This putative interface conserves the aromatic stacking seen in the IAPP structure, between two Phe15 residues in IAPP and between Phe15 of IAPP and Tyr16 of insulin. We used the RosettaDock algorithm to explore the helical packing between IAPP and the insulin B chain.38 One of the top scoring insulin-IAPP helical interfaces is shown in Figure 2(C). These molecules rotate to bury more surface area than in the IAPP dimer structure, suggesting the possibility of a tighter interaction between IAPP and insulin than between two IAPP molecules. This model also explains the observation that a truncated mutant of insulin that cannot oligomerize was found to have greater than a 5-fold increase in IAPP binding than wild-type insulin.21 In its hexameric state, the B helix of insulin is not available for binding to IAPP in the way proposed in Figure 2. The physical association of insulin and IAPP has been characterized previously,21,22 but to confirm a 1:1 heterodimeric complex is possible we performed glutaraldehyde crosslinking of IAPP to insulin in solution [Fig. 2(D)]. The positive result supports a 1:1 complex between monomeric insulin and the helical form of IAPP.
The crosslinking experiment in Figure 2D also supports the presence of dimeric IAPP in solution. To investigate whether crosslinked dimers are helical, as in our crystal structures in Figure 1, we performed a CD experiment on crosslinked IAPP in solution (Supporting Information Fig. S3). We found that untreated IAPP in solution has a CD spectrum characteristic of random coil.39 Upon treatment with 1% glutaraldehyde, there is an increase in the helical content of IAPP and a corresponding decrease in the random coil signal. This suggests that the dimeric, helical form of IAPP is in equilibrium with the monomeric, disordered state. In the absence of crosslinking reagent this monomeric, disordered state appears to be greatly favored.
Dimerization promotes, and insulin binding inhibits, IAPP fibril formation
Human IAPP readily forms amyloid fibrils in vitro under physiological conditions at nanomolar concentrations.28 The C-terminal segment of IAPP, composed of residues 23–37, has approximately fourfold slower aggregation kinetics than a construct composed of residues 8–37, suggesting that the N-terminal region of IAPP enhances the rate of IAPP fibril formation (Supporting Information Fig. S4). To explore the effect of the helical interactions we observe in the crystal structure and the putative IAPP-insulin model, fibril formation assays were performed on IAPP with residue substitutions at position 15. Predictions of residue substitutions were made based on their assumed influence on the helical interface guided by computational docking studies of the IAPP homodimers and heterodimers with insulin. Representative models from computational docking and our interpretation of their effects on dimerization are shown in Figure 3.
Figure 3.
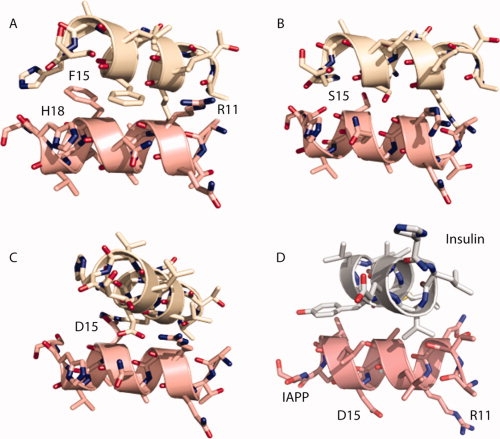
Computational docking experiments yielded an ensemble of solutions that suggest how IAPP might dimerize and the effect on the interaction of residue replacements at Phe15. In (A), a docked dimer between wild-type IAPP segments 8–19 reveals a relative rotation of the helices and tighter association than observed in the crystal structure of IAPP. This association disrupts the aromatic stacking between Phe15 residues in exchange for more contact area between the molecules. This association led us to believe that small residue substitutions for Phe15, such as alanine or serine may allow the helices to pack even closer and bury more surface area, as shown in (B) for F15S. The tighter association places Phe15 in closer proximity to His18 and Arg11 of the adjacent molecule, suggesting that acidic residues could potentially stabilize the interaction while large, positively charged residues, such as lysine or arginine could disrupt helical packing. In (C), the F15D dimer reveals how the negative charge may help stabilize the dimer by interacting with Arg11 in the neighboring molecule. In (D), a top scoring interaction model of the IAPP F15D mutant and insulin suggests an alternative interaction mode between IAPP and insulin as compared to that in Figure 2. The rotated IAPP helix exposes Asp15 to solvent and buries several hydrophobic residues from both IAPP and insulin. This packing was not observed in the MBP-fusion structure presumably because the Phe at position 15 preferred to be buried at the helical interface.
The importance of the helical association centered at Phe15 to the rate of fibril formation was confirmed by fibril formation assays. IAPP 8–37 was synthesized with residue replacements at position 15. Residues 1–7 of IAPP were not included in these peptides because these residues participate in the disulfide bond between Cys 2 and Cys 7; a region shown not to substantially affect the rate of de novo fibril formation of IAPP.23 Thioflavin T binding fluorescence was used to monitor fibril formation. All constructs were first resuspended in HFIP then diluted 100-fold to a final peptide concentration of 10 μM with 1% HFIP. This approach was taken to ensure a uniform starting material since some constructs were more soluble than others. HFIP promotes helical formation of IAPP as shown by CD (Supporting Information Fig. S5). The addition of 1% HFIP to IAPP dissolved in buffer can stimulate an immediate increase in helical content followed by conversion to the fibrillar form (Supporting Information Fig. S6). The enhancement in fibril formation kinetics caused by the helical promoting agent HFIP was observed as previously characterized for IAPP.23,32
Residue replacements that were predicted by molecular modeling to favor IAPP-IAPP helical association (F15S, F15A, and F15D) showed more rapid fibril formation kinetics than wild-type human IAPP [Figs. 3(B,C) and 4(A)]. To disrupt this helical interface, we selected the F15K substitution because it carries a similar charge as residues within this helix. It was therefore believed that both charge repulsion and steric bulk from this substitution would disrupt the helical interface and thereby cause slower kinetics of fibril formation. This prediction was confirmed by the greater lag phase observed for this F15K substitution. Residue replacements at position 17, a residue in the N-terminal helical region but not at the dimerization interface observed in the crystal structure, does not have the same modulating effect as residue replacements at position 15 (Supporting Information Fig. S7).
Figure 4.
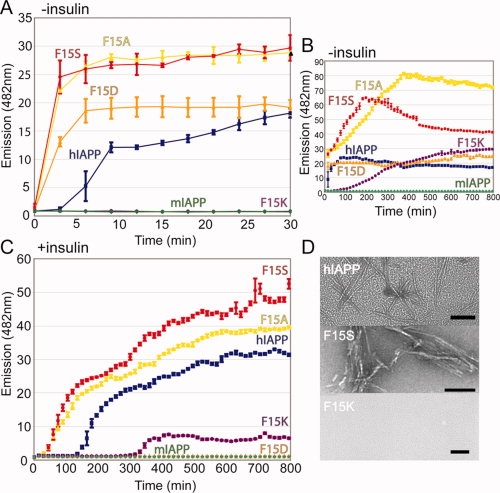
Mutations at position 15 of human IAPP affect the rate of fibril formation and the ability of insulin to inhibit this fibrillation. Residue substitutions computationally predicted to affect IAPP homodimerization and Insulin-IAPP heterodimerization were tested by a fibril formation assay. (A and B) Substitutions predicted to enhance helical association (F15A, F15S, F15D) of IAPP formed amyloid fibrils more rapidly than wild-type IAPP, as determined by Thioflavin T binding. The F15K mutation was predicted to disrupt the helical interface and a marked delay in fibril formation is observed. The 8–37 construct was used for both wild-type and Phe15 mutants. Mouse IAPP serves as a negative control for aggregation and never achieves a fluorescence value higher than baseline. (C) Including equimolar insulin in the assay delays the fibril formation of each mutant tested but has comparatively less of an inhibitory effect on the F15K substitution. (D) Representative EM images taken at 30 min of WT, F15S, and F15K to confirm the presence or absence of fibrils. Images of additional time points are included in the supporting Information. Scale bar represents 100 nm.
The ability of insulin to inhibit the fibril formation of IAPP in vitro increases proportionally with the concentration of insulin, suggesting insulin is a competitive inhibitor of IAPP aggregation.20 To test our model of insulin binding with human IAPP [Fig. 2(C)], the Phe 15 replacements were investigated for their ability to enhance or disrupt this inhibition [Fig. 4(B)]. We found that insulin has less of an inhibitory effect on the F15S and F15A substitutions than on wild-type IAPP. The F15D replacement was effectively inhibited by insulin for the duration of the experiment. In fact, an aliquot of F15D IAPP was examined by electron microscopy one week after incubation with insulin, and no fibrils were seen; only small, spherical aggregates were present (data not shown). Insulin did not have a noticeable inhibitory effect on the F15K mutant as compared with kinetics of fibril formation in the absence of insulin. Taken together, these data support the model in Figure 2 about how insulin binds to and inhibits the fibrillation of IAPP.
Discussion
MBP-IAPP
To determine the prefibrillar form of IAPP we suppressed its problematic proclivity to fibrillize by chaperoning this 37-residue peptide as a fusion protein with the 370-residue maltose binding protein. The structure of the fusion protein reveals that IAPP contains two α-helical segments at residues 8–18 and 22–27. The structure also reveals that IAPP dimerizes, with the three-turn α-helix between residues 8–18 forming a helix-helix interaction. Gazit and coworkers identified a self association domain within the 8–18 region and this is in agreement with the dimer in our MBP-IAPP structure.40 The crystal structure of MBP-IAPP 1–22 is also in agreement with the helical propensity of the N-terminal region of IAPP. The CD spectrum of a 310 helix in solution is remarkably similar to that of soluble IAPP with a peak at 207 nm and a small shoulder at 222 nm.41 Although it may be possible that IAPP forms a stable 310 helix in solution, our own NMR studies performed on the 8–20 segment did not reveal NOEs suggestive of a stable secondary structure in the 8–20 segment (data not shown). The observed 310 structure is more likely to have been a consequence of a particular crystal packing which distorts the helix in this region.
These structures are consistent with previous studies which have indicated that mouse IAPP forms α-helical structure in its N-terminal segment.26,36 A recent NMR study of human IAPP in SDS micelles is in excellent agreement with the crystal structure reported here, including the kink in the 18–20 segment between the two helices.42 At lower concentrations, IAPP may form only transient helical structure.24,26,43 Also supporting helical structure in IAPP is the calcitonin gene-related peptide (CGRP) which is 46% identical to IAPP and has been shown to be helical by NMR.44,45 Sequence alignments of IAPPs from numerous species have revealed that the N-terminus of IAPP is strongly conserved.15 We have shown by fibril formation assay that substitutions at position 15 can influence the kinetics of aggregation, suggesting this dimerization is an important, and perhaps short-lived, intermediate on the path to fibril formation. Taken together, these observations suggest that IAPP can adopt a helical structure and that dimerization facilitated through N-terminal helical domains may be important for the fibrillation of IAPP.
Our structure also suggests that IAPP may bind to insulin using the same helical motif of residues 8–18. This heterodimer uses segments for its interaction that have been previously mapped to the regions of IAPP-insulin interaction.21,22 A recent NMR study suggests a related model for the interaction of mouse IAPP and insulin in agreement with a parallel orientation of associating helices.46 We speculate that IAPP may bind to, and thereby solubilize, multimeric insulin from its crystalline-like state prior to secretion. It has been suggested that insulin deters IAPP from aggregation in the secretory granules in the absence of type II diabetes.19 Understanding the mechanism of inhibition of IAPP aggregation by insulin may lead to a new approach for the clearance of IAPP deposits in the treatment of type II diabetes.
Conversion of IAPP from the native to the fibril state
Our structures for helical IAPP, our model of insulin binding, as well as our fibrillation studies, hint at initial steps for the structural conversion of IAPP from its native to its fibrillar form (Fig. 5). IAPP likely exists in equilibrium between the unstructured and helical forms and the environment may influence this equilibrium [Fig. 5(A,B)]. The dimerization of IAPP we observed in our crystal structure coupled with our fibril formation studies suggest this dimerization is an on-pathway intermediate in fibrillation. This nearly parallel prealignment [Fig. 5(C)] lowers the entropic cost of the parallel β-strand association by bringing together two of the C-terminal amyloidogenic regions (the pigtails of panel D). In the transition from panel D to panel E, the two C-terminal regions stack in a two layer β-sheet, and two such sheets interdigitate their sidechains to form a two-layered nucleus of the fibril.47 The turn of the linker region nucleates the second layer of β-sheets [Fig. 5(F)]. In panel G, the binding of insulin to the IAPP dimer is shown competing with the formation of the pre-aligned dimer, illustrating how insulin may inhibit fibrillation of IAPP.
Figure 5.
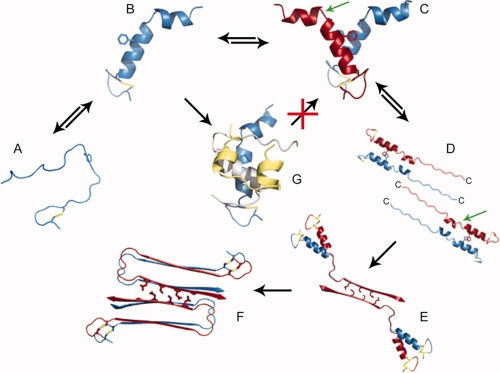
Model of nucleus formation of IAPP fibrils and the inhibition of fibrillation by insulin. (A) IAPP monomers are assumed to be in equilibrium between disordered state and helical, shown in (B) as it appears in the crystal. Phe15 is shown in stick view for reference. (C and D) Two IAPP dimers align their C-terminal amyloid domains in an antiparallel orientation. The C-terminus of each IAPP molecule is labeled. The position of residue S20 is indicated by the green arrow. When this residue is mutated to Glycine, fibrils form more rapidly. (E) This region, residues 23–37, forms the steric zipper spine of the fibril, by interdigitating sidechains with a second IAPP dimer. (F) Formation of the N-terminal strand occurs subsequent to spine formation. (G) Insulin prevents homodimeric helical association by binding to monomeric IAPP and blocking IAPP homodimerization. The insulin A-chain is shown in gold and the B chain is shown in gray.
The speculative pathway for conversion from native to fibril structure depicted in Figure 5 receives support from CD, IRRAS, and 2D IR studies that show an increase in helical content just prior to conversion to the β-sheet conformation.24,27,28,30 Condensation of helical IAPP molecules has been suggested previously as a mechanism for the nucleation of IAPP fibrillation in the presence of lipids.28,34,48 Helical promoting agents, such HFIP and TFE, accelerate the kinetics of IAPP aggregation suggesting that a helical intermediate is involved in conversion to amyloid fibrils.32 A recent publication from the Langen group suggests the parallel condensation of IAPP molecules at the lipid surface as being an important step on the path to fibril formation.35 There also exists a rare allele of human IAPP (S20G) that causes IAPP to be more amyloidogenic and cytotoxic in vitro and has been associated with early-onset type II diabetes.49 This mutation is located within the loop region separating the 8–18 and 22–27 helices. The S20G mutant protein could accelerate formation of the tetrameric nucleus [Fig. 5(E)] by the greater flexibility of the chain at Gly 20 faciliting formation of the interdigitated dimer of dimers. This behavior may be attributed to the S20G mutation rendering this loop more flexible and making the transition from C to D more favorable.
Methods
Cloning, purification, and crystallization of MBP-IAPP
The IAPP nucleotide sequence was derived by primer annealing of overlapping oligonucleotides. This sequence was cloned into the pMal-a1 vector that was a gift from Prof. Cynthia Wolberger of Johns Hopkins University. The purification was performed by column chromatography first by an amylose column and followed by gel filtration on a Superdex 200. The MBP-IAPP fusion protein was crystallized using a mosquito crystallization robot that screened over 3,000 conditions. Crystals were formed at 20 mg/mL in 2.0M ammonium sulfate, 0.1M sodium acetate pH 4.6. The crystals grew in approximately one week and diffraction data were collected at the Advanced Light Source beamline 8.2.2. The data were scaled and indexed using Denzo and Scalepack from the HKL suite of programs.50 The structure was determined to a resolution of 1.86 Å and the phases were determined by molecular replacement with MBP using Phaser.51 Refinement and model building were performed using Refmac5 and Coot.52,53 Four fusion IAPP-MBP molecules per asymmetric unit were found in space group P21. An omit map was used to determine electron density attributable to IAPP (Supporting Information Fig. S2). The coordinates have been deposited in the Protein Databank with accession codes 3G7W for the 1-22 fusion and 3G7V for the full-length fusion.
IAPP and insulin crosslinking
IAPP 8–37 peptide (CSbio) was solubilized into 20 mM NaAc pH 6.5 to a final concentration of 100 μM. Glutaraldehyde was immediately added to a final concentration of 0.8% (w/v) along with either 100 μM insulin (Sigma Aldrich), 100 μM myoglobin or buffer alone. Samples were allowed to incubate for 15 min on ice and reactions were TCA precipitated and washed with acetone. Equal volumes of nonreducing SDS-PAGE loading buffer and water were used to resuspend the protein pellets. Samples were run on a high-density Phast gel and stained with coomassie.
Fibril formation assays
All peptides (CSbio) were first solubilized in 100% Hexafluoroisopropanol (HFIP) to a stock concentration of 1 mM. Some peptides were more soluble than others and this initial solublization in HFIP was used to assure a uniform starting material for each sample. Reaction conditions were 20 mM sodium acetate pH 6.5 and 10 μM Thioflavin T (ThT). Peptides were diluted 1/100 into this mixture then spin filtered to remove any potential preformed aggregated that may have been created during synthesis. The total reaction volume was 200 μL with final concentrations of 10 μM peptide and 1% HFIP. The addition of 150 mM sodium chloride did not have an effect on the kinetics of fibril formation (as determined by electron microscopy) but caused strong fluctuations in ThT fluorescence. A sample was taken from each reaction immediately after filtering to confirm the absence of any fibrils or preformed aggregates prior to the start of the assay. Samples were performed in triplicate and fluorescence measurements were taken every three minutes. The average of five time points is shown on the representative graph for clarity. The standard deviation of the fluorescence measurement at each time point is shown as error bars. ThT fluorescence was treated as a binary indicator of fibrillation and samples were taken at pertinent time points for analysis by electron microscopy to confirm the presence or absence of fibrils as seen by ThT binding.
Molecular modeling
Computational docking
The RosettaDock algorithm was used to perform rigid body docking to optimize local packing between IAPP homodimers and IAPP-insulin heterodimers.38 The starting structure was the crystal structure of IAPP. The dimers were repacked after local perturbation with rigid body docking and side chain rotamer sampling. Docked structures were ranked by the Rosetta energy function.
Conclusions
By chaperoning IAPP fused to a larger protein, we determined a helical form of IAPP, rarely encountered in vitro because of this molecule's tendency to fibrillize. IAPP molecules are capable of adopting a dimeric structure, with the α-helices at residues 8–18 binding to each other around a stack of the two Phe15 sidechains and orienting the two α-helices in parallel. Mutations at this helical interface affect the rate of fibrillation, suggesting that the dimer is an intermediate on the pathway to fibrillation. We speculate that the parallel orientation of the two helices orients the C-terminal residues of both IAPP molecules also in parallel, permitting them to stack in a two-stranded β-sheet, and that the condensation of two such dimers forms a four-molecule fibril nucleus. The homodimeric structure of IAPP suggests a structure for the heterodimeric structure of insulin bound to IAPP. Taken together, these data suggest a plausible model for the conversion of IAPP from the soluble to the fibril form and how insulin functions to deter this conversion by blocking the N-terminal dimerization domain within IAPP.
Acknowledgments
We thank C. Wolberger for the MBP fusion construct, E. Gazit, R. Langen, A. Miranker, J. Bowie, G. Chanfreau, D. Cascio, and S. Clark for discussion, J. Navarro for crystallization aid, the staff of ALS beamline 8.2.2, and NSF, NTH, and HHMI for support.
References
- 1.Hossain P, Kawar B, El Nahas M. Obesity and diabetes in the developing world—a growing challenge. N Engl J Med. 2007;356:213–215. doi: 10.1056/NEJMp068177. [DOI] [PubMed] [Google Scholar]
- 2.Saltiel AR. New perspectives into the molecular pathogenesis and treatment of type 2 diabetes. Cell. 2001;104:517–529. doi: 10.1016/s0092-8674(01)00239-2. [DOI] [PubMed] [Google Scholar]
- 3.Cooper GJ. Amylin compared with calcitonin gene-related peptide: structure, biology, and relevance to metabolic disease. Endocr Rev. 1994;15:163–201. doi: 10.1210/edrv-15-2-163. [DOI] [PubMed] [Google Scholar]
- 4.Cooper GJS, Willis AC, Clark A, Turner RC, Sim RB, Reid KBM. Purification and characterization of a peptide from amyloid-rich pancreases of type 2 diabetic patients. PNAS. 1987;84:8628–8632. doi: 10.1073/pnas.84.23.8628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Westermark P, Wernstedt C, Wilander E, Hayden DW, O'Brien TD, Johnson KH. Amyloid fibrils in human insulinoma and islets of langerhans of the diabetic cat are derived from a neuropeptide-like protein also present in normal islet cells. PNAS. 1987;84:3881–3885. doi: 10.1073/pnas.84.11.3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schmitz O, Brock B, Rungby J. Amylin agonists: a novel approach in the treatment of diabetes. Diabetes. 2004;53:S233–S238. doi: 10.2337/diabetes.53.suppl_3.s233. [DOI] [PubMed] [Google Scholar]
- 7.Yan L-M, Tatarek-Nossol M, Velkova A, Kazantzis A, Kapurniotu A. Design of a mimic of nonamyloidogenic and bioactive human islet amyloid polypeptide (IAPP) as nanomolar affinity inhibitor of IAPP cytotoxic fibrillogenesis. PNAS. 2006;103:2046–2051. doi: 10.1073/pnas.0507471103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoppener JWM, Nieuwenhuis MG, Vroom TM, Ahren B, Lips CJM. Role of islet amyloid in type 2 diabetes mellitus: consequence or cause? Mol Cell Endocrinol. 2002;197:205–212. doi: 10.1016/s0303-7207(02)00266-6. [DOI] [PubMed] [Google Scholar]
- 9.Hoppener JWM, Ahren B, Lips CJM. Islet Amyloid and Type 2 Diabetes Mellitus. N Engl J Med. 2000;343:411–419. doi: 10.1056/NEJM200008103430607. [DOI] [PubMed] [Google Scholar]
- 10.Esapa C, Moffitt JH, Novials A, McNamara CM, Levy JC, Laakso M, Gomis R, Clark A. Islet amyloid polypeptide gene promoter polymorphisms are not associated with Type 2 diabetes or with the severity of islet amyloidosis. Biochim Biophys Acta. 2005;1740:74–78. doi: 10.1016/j.bbadis.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 11.de Koning EJP, Morris ER, Hofhuis FMA, Posthuma G, Hoppener JWM, Morris JF, Capel PJA, Clark A, Verbeek JS. Intra- and extracellular amyloid fibrils are formed in cultured pancreatic islets of transgenic mice expressing human islet amyloid polypeptide. PNAS. 1994;91:8467–8471. doi: 10.1073/pnas.91.18.8467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kapurniotu A. Amyloidogenicity and cytotoxicity of islet amyloid polypeptide. Pept Sci. 2001;60:438–459. doi: 10.1002/1097-0282(2001)60:6<438::AID-BIP10182>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 13.Konarkowska B, Aitken JF, Kistler J, Zhang S, Cooper GJS. The aggregation potential of human amylin determines its cytotoxicity towards islet βeta-cells. FEBS J. 2006;273:3614–3624. doi: 10.1111/j.1742-4658.2006.05367.x. [DOI] [PubMed] [Google Scholar]
- 14.Ritzel RA, Meier JJ, Lin C-Y, Veldhuis JD, Butler PC. Human islet amyloid polypeptide oligomers disrupt cell coupling, induce apoptosis, and impair insulin secretion in isolated human islets. Diabetes. 2007;56:65–71. doi: 10.2337/db06-0734. [DOI] [PubMed] [Google Scholar]
- 15.Nishi M, Chan SJ, Nagamatsu S, Bell GI, Steiner DF. Conservation of the sequence of islet amyloid polypeptide in five mammals is consistent with its putative role as an islet hormone. PNAS. 1989;86:5738–5742. doi: 10.1073/pnas.86.15.5738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fox N, Schrementi J, Nishi M, Ohagi S, Chan SJ, Heisserman JA, Westermark GT, Leckstrom A, Westermark P, Steiner DF. Human islet amyloid polypeptide transgenic mice as a model of non-insulin-dependent diabetes mellitus (NIDDM) FEBS Lett. 1993;323:40–44. doi: 10.1016/0014-5793(93)81444-5. [DOI] [PubMed] [Google Scholar]
- 17.Hutton JC. The insulin secretory granule. Diabetologia. 1989;32:271–289. doi: 10.1007/BF00265542. [DOI] [PubMed] [Google Scholar]
- 18.Nishi M, Sanke T, Nagamatsu S, Bell GI, Steiner DF. Islet amyloid polypeptide. A new beta cell secretory product related to islet amyloid deposits. J Biol Chem. 1990;265:4173–4176. [PubMed] [Google Scholar]
- 19.Westermark P, Li Z-C, Westermark GT, Leckstrom A, Steiner DF. Effects of beta cell granule components on human islet amyloid polypeptide fibril formation. FEBS Lett. 1996;379:203–206. doi: 10.1016/0014-5793(95)01512-4. [DOI] [PubMed] [Google Scholar]
- 20.Larson JL, Miranker AD. The mechanism of insulin action on islet amyloid polypeptide fiber formation. J Mol Biol. 2004;335:221–231. doi: 10.1016/j.jmb.2003.10.045. [DOI] [PubMed] [Google Scholar]
- 21.Jaikaran E, Nilsson MR, Clark A. Pancreatic beta-cell granule peptides form heteromolecular complexes which inhibit islet amyloid polypeptide fibril formation. Biochem J. 2004a;377:709–716. doi: 10.1042/BJ20030852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gilead S, Wolfenson H, Gazit E. Molecular mapping of the recognition interface between the islet amyloid polypeptide and insulin. Angew Chem Int Ed. 2006;45:6476–6480. doi: 10.1002/anie.200602034. [DOI] [PubMed] [Google Scholar]
- 23.Koo BW, Miranker AD. Contribution of the intrinsic disulfide to the assembly mechanism of islet amyloid. Protein Sci. 2005;14:231–239. doi: 10.1110/ps.041051205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kayed R, Bernhagen J, Greenfield N, Sweimeh K, Brunner H, Voelter W, Kapurniotu A. Conformational transitions of islet amyloid polypeptide (IAPP) in amyloid formation in Vitro. J Mol Biol. 1999;287:781–796. doi: 10.1006/jmbi.1999.2646. [DOI] [PubMed] [Google Scholar]
- 25.Jaikaran ETAS, Clark A. Islet amyloid and type 2 diabetes: from molecular misfolding to islet pathophysiology. Biochim Biophys Acta. 2001;1537:179–203. doi: 10.1016/s0925-4439(01)00078-3. [DOI] [PubMed] [Google Scholar]
- 26.Williamson JA, Miranker AD. Direct detection of transient α-helical states in islet amyloid polypeptide. Protein Sci. 2007;16:110–117. doi: 10.1110/ps.062486907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goldsbury C, Goldie K, Pellaud J, Seelig J, Frey P, Muller SA, Kistler J, Cooper GJS, Aebi U. Amyloid fibril formation from full-length and fragments of amylin. J Struct Biol. 2000;130:352–362. doi: 10.1006/jsbi.2000.4268. [DOI] [PubMed] [Google Scholar]
- 28.Lopes DHJ, Meister A, Gohlke A, Hauser A, Blume A, Winter R. Mechanism of islet amyloid polypeptide fibrillation at lipid interfaces studied by infrared reflection absorption spectroscopy. Biophys J. 2007;93:3132–3141. doi: 10.1529/biophysj.107.110635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yonemoto IT, Kroon GJ, Dyson HJ, Balch WE, Kelly JW. Amylin proprotein processing generates progressively more amyloidogenic peptides that initially sample the helical state. Biochemistry. 2008;47:9900–9910. doi: 10.1021/bi800828u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ling YL, Strasfeld DB, Shim S-H, Raleigh DP, Zanni MT. Two-dimensional infrared spectroscopy provides evidence of an intermediate in the membrane-catalyzed assembly of diabetic amyloid. J Phys Chem B. 2009;113:2498–2505. doi: 10.1021/jp810261x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cort J, Liu ZH, Lee G, Harris SM, Prickett KS, Gaeta LSL, Andersen NH. β-structure in human amylin and 2 designer β-peptides: CD and NMR spectroscopic comparisons suggest soluble β-oligomers and the absence of significant populations of β-strand dimers. Biochem Biophys Res Commun. 1994;204:1088–1095. doi: 10.1006/bbrc.1994.2574. [DOI] [PubMed] [Google Scholar]
- 32.Padrick SB, Miranker AD. Islet amyloid: phase partitioning and secondary nucleation are central to the mechanism of fibrillogenesis. Biochemistry. 2002;41:4694–4703. doi: 10.1021/bi0160462. [DOI] [PubMed] [Google Scholar]
- 33.Knight JD, Miranker AD. Phospholipid catalysis of diabetic amyloid assembly. J Mol Biol. 2004;341:1175–1187. doi: 10.1016/j.jmb.2004.06.086. [DOI] [PubMed] [Google Scholar]
- 34.Jayasinghe SA, Langen R. Lipid membranes modulate the structure of islet amyloid polypeptide. Biochemistry. 2005;44:12113–12119. doi: 10.1021/bi050840w. [DOI] [PubMed] [Google Scholar]
- 35.Apostolidou M, Jayasinghe SA, Langen R. Structure of α-helical membrane-bound human islet amyloid polypeptide and its implications for membrane-mediated misfolding. J Biol Chem. 2008;283:17205–17210. doi: 10.1074/jbc.M801383200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nanga RPR, Brender JR, Xu J, Veglia G, Ramamoorthy A. Structures of rat and human islet amyloid polypeptide IAPP1-19 in micelles by NMR spectroscopy. Biochemistry. 2008;47:12689–12697. doi: 10.1021/bi8014357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ke A, Wolberger C. Insights into binding cooperativity of MATa1/MATα2 from the crystal structure of a MATa1 homeodomain-maltose binding protein chimera. Protein Sci. 2003;12:306–312. doi: 10.1110/ps.0219103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gray JJ, Moughon S, Wang C, Schueler-Furman O, Kuhlman B, Rohl CA, Baker D. Protein-protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. J Mol Biol. 2003;331:281–299. doi: 10.1016/s0022-2836(03)00670-3. [DOI] [PubMed] [Google Scholar]
- 39.Andrade MA, Chacon P, Merelo JJ, Moran F. Evaluation of secondary structure of proteins from UV circular dichroism spectra using an unsupervised learning neural network. Protein Eng. 1993;6:383–390. doi: 10.1093/protein/6.4.383. [DOI] [PubMed] [Google Scholar]
- 40.Mazor Y, Gilead S, Benhar I, Gazit E. Identification and characterization of a novel molecular-recognition and self-assembly domain within the islet amyloid polypeptide. J Mol Biol. 2002;322:1013–1024. doi: 10.1016/s0022-2836(02)00887-2. [DOI] [PubMed] [Google Scholar]
- 41.Toniolo C, Polese A, Formaggio F, Crisma M, Kamphuis J. Circular dichroism spectrum of a peptide 310-Helix. J Am Chem Soc. 1996;118:2744–2745. [Google Scholar]
- 42.Patil SM, Xu S, Sheftic SR, Alexandrescu AT. The dynamic α-helix structure of micelle-bound human amylin. J Biol Chem. 2009;284:11982–11991. doi: 10.1074/jbc.M809085200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jaikaran ETAS, Higham CE, Serpell LC, Zurdo J, Gross M, Clark A, Fraser PE. Identification of a novel human islet amyloid polypeptide β-sheet domain and factors influencing fibrillogenesis. J Mol Biol. 2001;308:515–525. doi: 10.1006/jmbi.2001.4593. [DOI] [PubMed] [Google Scholar]
- 44.Breeze AL, Harvey TS, Bazzo R, Campbell ID. Solution structure of human calcitonin gene-related peptide by proton NMR and distance geometry with restrained molecular dynamics. Biochemistry. 1991;30:575–582. doi: 10.1021/bi00216a036. [DOI] [PubMed] [Google Scholar]
- 45.Hakala JML, Vihinen M. Modelling the structure of the calcitonin gene-related peptide. Protein Eng. 1994;7:1069–1075. doi: 10.1093/protein/7.9.1069. [DOI] [PubMed] [Google Scholar]
- 46.Wei L, Pervushin K, Mu Y. Residual structure in islet amyloid polypeptide mediates its interactions with soluble insulin. Biochemistry. 2009;48:2368–2376. doi: 10.1021/bi802097b. [DOI] [PubMed] [Google Scholar]
- 47.Wiltzius JJW, Sievers SA, Sawaya MR, Cascio D, Popov D, Riekel C, Eisenberg D. Atomic structure of the cross-β spine of islet amyloid polypeptide (amylin) Protein Sci. 2008;17:1467–1474. doi: 10.1110/ps.036509.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Knight JD, Hebda JA, Miranker AD. Conserved and cooperative assembly of membrane-bound α-helical states of islet amyloid polypeptide. Biochemistry. 2006;45:9496–9508. doi: 10.1021/bi060579z. [DOI] [PubMed] [Google Scholar]
- 49.Sakagashira S, Hiddinga HJ, Tateishi K, Sanke T, Hanabusa T, Nanjo K, Eberhardt NL. S20G Mutant amylin exhibits increased in vitro amyloidogenicity and increased intracellular cytotoxicity compared to wild-type amylin. Am J Pathol. 2000;157:2101–2109. doi: 10.1016/S0002-9440(10)64848-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Otwinowski Z, Minor W, Carter CW., Jr . Processing of X-ray diffraction data collected in oscillation mode. In: Carter JW, Sweet RM, editors. Methods in Enzymology: Macromolecular Crystallography, Part A. Vol. 276. Academic Press; 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 51.Storoni LC, McCoy AJ, Read RJ. Likelihood-enhanced fast rotation functions. Acta Crystallogr D Biol Crystallogr. 2004;60:432–438. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- 52.Collaborative Computational Project. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 53.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]