

Abstract
NagZ is an exo-N-acetyl-β-glucosaminidase, found within Gram-negative bacteria, that acts in the peptidoglycan recycling pathway to cleave N-acetylglucosamine residues off peptidoglycan fragments. This activity is required for resistance to cephalosporins mediated by inducible AmpC β-lactamase. NagZ uses a catalytic mechanism involving a covalent glycosyl enzyme intermediate, unlike that of the human exo-N-acetyl-β-glucosaminidases: O-GlcNAcase and the β-hexosaminidase isoenzymes. These latter enzymes, which remove GlcNAc from glycoconjugates, use a neighboring-group catalytic mechanism that proceeds through an oxazoline intermediate. Exploiting these mechanistic differences we previously developed 2-N-acyl derivatives of O-(2-acetamido-2-deoxy-d-glucopyranosylidene)amino-N-phenylcarbamate (PUGNAc), which selectively inhibits NagZ over the functionally related human enzymes and attenuate antibiotic resistance in Gram-negatives that harbor inducible AmpC. To understand the structural basis for the selectivity of these inhibitors for NagZ, we have determined its crystallographic structure in complex with N-valeryl-PUGNAc, the most selective known inhibitor of NagZ over both the human β-hexosaminidases and O-GlcNAcase. The selectivity stems from the five-carbon acyl chain of N-valeryl-PUGNAc, which we found ordered within the enzyme active site. In contrast, a structure determination of a human O-GlcNAcase homologue bound to a related inhibitor N-butyryl-PUGNAc, which bears a four-carbon chain and is selective for both NagZ and O-GlcNAcase over the human β-hexosamnidases, reveals that this inhibitor induces several conformational changes in the active site of this O-GlcNAcase homologue. A comparison of these complexes, and with the human β-hexosaminidases, reveals how selectivity for NagZ can be engineered by altering the 2-N-acyl substituent of PUGNAc to develop inhibitors that repress AmpC mediated β-lactam resistance.
Keywords: NagZ, β-glucosaminidase, β-hexosaminidase, O-GlcNAcase, β-lactam, X-ray crystal structure, glycoside hydrolase, glycosidase, peptidoglycan recycling
Introduction
exo-N-acetyl-β-glucosaminidases are found in diverse organisms ranging from bacteria to humans. These enzymes catalyze the removal of terminal N-acetylglucosamine (GlcNAc) from a wide range of glycoconjugates and saccharides. Humans express three exo-N-acetyl-β-glucosaminidases and considerable attention has been given to these enzymes as they have been implicated the etiology of various diseases. The isoenzymes β-hexosaminidase A (HexA) and β-hexosaminidase B (HexB) share high sequence similarity and belong to glycoside hydrolase family 20 (GH20, for a discussion of the classification system of glycoside hydrolases see Refs.1 and 2). The importance of these enzymes is highlighted by heritable deficiencies in HexA activity that cause GM2-ganglioside to accumulate in the nervous system, resulting in lethal neurodegenerative disorders known as Tay-Sachs and Sandhoff diseases (for review see Ref.3). The third human exo-N-acetyl-β-glucosaminidase, termed O-GlcNAcase (OGA), belongs to family 84 (GH84) and acts to remove O-linked β-N-acetylglucosamine residues (O-GlcNAc) from the side chain hydroxyls of serine and threonine residues of nuclear and cytoplasmic proteins.4,5 Although the consequences of impaired O-GlcNAcase activity in humans are unknown, it appears the enzyme may work in conjunction with protein phosphorylation to control a number of key cellular processes.
A fourth functionally related exo-N-acetyl-β-glucosaminidase from glycoside hydrolase family 3 (GH3), although of nonhuman origin, has also been implicated in human disease. Known as NagZ, this enzyme is part of the highly conserved Gram-negative peptidoglycan cell wall recycling pathway.6 The enzyme releases GlcNAc from the cytosolic peptidoglycan recycling GlcNAc-1,6-anhydroMurNAcpeptide intermediates to create 1,6-anhydroMurNAcpeptides.7 These catabolites serve as a signal to induce transcription of chromosomal AmpC β-lactamase8 and thereby promote resistance of Gram-negative bacteria to an extended-spectrum of β-lactam antibiotics including cephalosporins, cephamycins, and monobactams.9 This inducible resistance mechanism is found in a number of Gram-negative bacteria, including, for example, the opportunistic human pathogens Pseudomonas aeruginosa and Enterobacter cloacae.10 Given the prevalence of bacterial pathogens that harbor inducible chromosomal AmpC, and the recent spread of inducible plasmid-borne ampC to additional bacterial pathogens,11–15 new strategies to suppress this resistance mechanism are urgently needed. One approach, which we have taken, is to generate inhibitors of NagZ that act to hinder the production of AmpC and therefore render bacteria that harbour AmpC more sensitive to β-lactams.16 One challenge facing this approach however is that inhibitors used for this purpose need to be selective for NagZ and not potently inhibit the functionally related human enzymes.
Although GH3 enzymes, including NagZ, are functionally related to the GH20 human β-hexosaminidase isoenzymes and GH84 O-GlcNAcase, recent kinetic17–19 and structural studies16,20–22 have revealed that GH3 enzymes use a catalytic mechanism that differs from that used by GH20 and GH84 enzymes. This distinction should therefore offer a tractable route to generating selective inhibitors of NagZ, and clear comparisons of these enzymes at a structural level would greatly accelerate these efforts.
Family GH3 exo-N-acetyl-β-glucosaminidases use a two-step double displacement mechanism (Fig. 1, path A), which involves the formation and breakdown of a covalent glycosyl-enzyme intermediate via highly dissociative oxocarbenium ion-like transition states [Fig. 1(A)].17 In the first step of the reaction, the enzymatic nucleophile attacks the anomeric center as the aglycon leaving group departs, with the net result being formation of a covalent glycosyl-enzyme intermediate. A second catalytic residue facilitates departure of the aglycon by providing general acid catalysis. In the second step of the reaction, this same general acid/base residue promotes breakdown of the covalent intermediate by enhancing the nucleophilicity of a water molecule that is positioned to attack the anomeric center of the covalent intermediate. This water molecule displaces the enzymatic nucleophile, resulting in the formation of a hemiacetal product having anomeric stererochemistry matching that of the substrate. This general mechanism has been found to be operative for the vast majority of configuration retaining β-glycosidases, recently reviewed in Ref.23.
Figure 1.
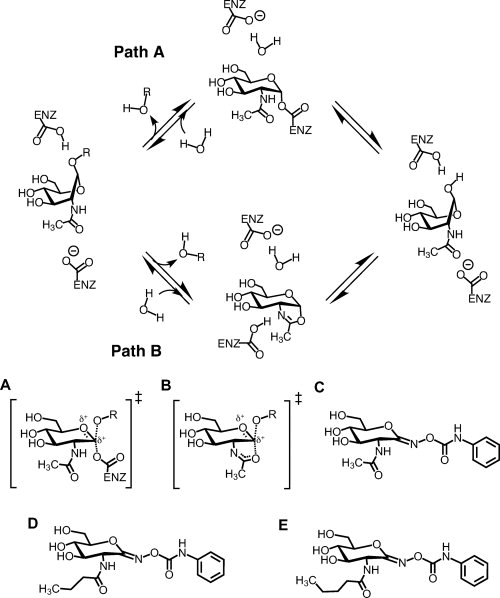
Catalytic mechanisms of family GH3 exo-N-acetyl-β-hexosaminidases (NagZ) (path A) and the family GH20 human β-hexosaminidases and GH84 O-GlcNAcase (path B), and chemical structures of PUGNAc and its 2-N-acyl derivatives. The distinguishing feature between GH3 enzymes verses GH20 and GH84 enzymes is the identity of the nucleophile. GH3 enzymes use an enzymic carboxylate, transiently forming a covalent α-glycosyl-enzyme intermediate; whereas GH20 and GH84 enzymes use a substrate assisted catalytic mechanism involving the 2-acetamido group of the substrate to form a bicyclic oxazoline or oxazolinium ion intermediate. In both cases, a molecule of water attacks the anomeric centre, breaking down the intermediate and leading to the formation of the hemiacetal product with retained stereochemistry. A: Proposed transition state for GH3 enzymes. B: Proposed transition state for GH20 and GH84 enzymes. Also shown are the chemical structures of PUGNAc (C), N-butyryl-PUGNAc (D), and N-valeryl-PUGNAc (E).
The family GH20 human β-hexosaminidases and GH84 O-GlcNAcase use a different catalytic mechanism that requires assistance from the 2-acetamido group of the substrate (Fig. 1, path B).19–21 In the first step of this mechanism, the carbonyl oxygen of the 2-acetamido group takes the place of an enzymic nucleophile, yielding a bicyclic oxazoline or oxazolinium ion intermediate. The nucleophilicity of the 2-acetamido group is enhanced by an essential catalytic residue that accepts a hydrogen-bond from this substituent. In the second step of the reaction the oxazoline ring is opened by attack of a molecule of water at the anomeric center to yield a hemiacetal product with overall retained stereochemistry. In a manner analogous to that seen in the classic double displacement mechanism used by GH3 enzymes (Fig. 1, path A), an enzymatic general acid/base catalyst facilitates both steps of the reaction.
Structural studies of N-acetyl-β-glucosaminidases from families GH3,16 GH20,20,21,24,25 and GH8422,26 have revealed significant differences in the active site structures of enzymes carrying out substrate assisted catalysis versus those proceeding via a glycosyl-enzyme intermediate. For GH3 enzymes, it has been found that the 2-acetamido group of the substrate is not essential for cleavage of the glycosidic bond, consistent with these enzymes using an enzymic nucleophile.19,27 The X-ray structure of NagZ from Vibrio cholerae (VcNagZ) in complex with its GlcNAc product (PDB entry: 1Y65) corroborates this finding, clearly showing that the 2-acetamido group of bound GlcNAc is not positioned to participate directly in C—O bond cleavage. Crystallographic studies of GH20 human HexA25 and HexB,24 and GH84 bacterial homologues of human O-GlcNAcase from Bacteroides thetaiotaomicron (BtGH84)22 and Clostridium perfringens26 have revealed that these families of enzymes bind the 2-acetamido group in a strikingly different manner. To position the carbonyl oxygen of the 2-acetamido group as a nucleophile and favor formation of the bicyclic oxazoline intermediate, these enzymes rotate the amide moiety into a deep hydrophobic pocket that tightly envelops this substituent, positioning it within striking distance of the anomeric center. Close examination of the active sites of these GH20 and GH84 enzymes reveal subtle structural differences including, most notably, the depth of the pocket that cradles the substrate 2-acetamido group. This pocket is slightly deeper in O-GlcNAcase than it is in the GH20 β-hexosaminidases.22 Thus, not only do large structural differences exist between the NagZ active site and the active sites of GH20 β-hexosaminidases and GH84 O-GlcNAcase, there also exist less prevalent yet distinct differences in the active site structures of the latter two enzyme families.
One class of glycoside hydrolase inhibitors that have gained interest are the O-(d-glycopyranosylidene)amino-N-phenylcarbamates.42 These inhibitors are thought to emulate the oxocarbenium ion-like transition state that is proposed for the vast majority of glycoside hydrolases.23 In accord with this expectation, O-(2-acetamido-2-deoxy-d-glucopyranosylidene)amino-N-phenylcarbamate [PUGNAc; Fig. 1(C)] has been shown to be a non-selective yet potent inhibitor of NagZ, human β-hexosaminidases, and O-GlcNAcase (Table I).16,18,28 To generate inhibitors specific for NagZ, we exploited the structural differences between NagZ, GH20 human β-hexosaminidases, and GH84 O-GlcNAcase and generated 2-N-acyl derivatives of PUGNAc.16,28 Moreover, we have demonstrated that AmpC mediated β-lactam resistance can be attenuated by inhibiting NagZ with these selective inhibitors. Attenuation has been achieved in a model system of E. coli harboring a plasmid encoding an ampC-ampR operon,16 and more recently in the opportunistic pathogen P. aeruginosa.29 The most selective inhibitor found to date is ∼100-fold selective for NagZ over the human β-hexosaminidase isoenzymes and O-GlcNAcase. This derivative of PUGNAc, N-valeryl-PUGNAc [Fig. 1(E)], has a five carbon N-acyl group. Interestingly, a minimum of a five carbon acyl group is required to achieve reasonable selectivity for NagZ over both human enzymes; N-butyryl-PUGNAc, which is a 2-acyl derivative bearing a four carbon N-acyl group [Fig. 1(D)], did not discriminate efficiently between NagZ and O-GlcNAcase although it was a poor inhibitor of human β-hexosaminidases (Table I). Here we detail the molecular basis for the selective inhibition of NagZ over O-GlcNAcase through crystallographic studies of NagZ bound to N-valeryl-PUGNAc and to N-butyryl-PUGNAc, and the crystallographic structure of a human GH84 O-GlcNAcase homologue, BtGH84, bound to N-butyryl-PUGNAc. Comparison of these complexes to each other, and to models based on the X-ray structure of the human β-hexosaminidase A, reveals active site features of NagZ that enable generation of 2-acyl derivatives of PUGNAc that are selective for NagZ over the functionally related human enzymes. Given that inhibitors selective for NagZ have potential as an effective strategy to improve the efficacy of a broad spectrum of β-lactam antibiotics, these structures provide a valuable blueprint for the design of inhibitors that are amenable for testing in vivo.
Table I.
Inhibition Constants and Selectivity Ratios of PUGNAc and 2-N-acyl Derivatives for Glycoside Hydrolase Families GH3, GH84, and GH20
| Compound | Family 3 VcNagZ KI (μM)a | Family 84 O-GlcNAcase KI (μM)a | Family 20 ß-hexosaminidase KI(μM)a | Selectivity ratio KI (O-GlcNAcase)/KI (VcNagZ) | Selectivity ratio KI (ß-hexosaminidase)/KI (VcNagZ) |
|---|---|---|---|---|---|
| PUGNAc | 0.048 | 0.046 | 0.036 | 1 | 0.75 |
| N-butyryl-PUGNAc | 0.26 | 2.5 | 26 | 9.6 | 100 |
| N-valeryl-PUGNAc | 0.33 | 40 | 220 | 121 | 667 |
Results and Discussion
To gain insight into the molecular basis of NagZ inhibition and how NagZ can accommodate increased bulk present on selective derivatives of PUGNAc we determined the X-ray structure of VcNagZΔSC in complex with N-valeryl-PUGNAc and N-butyryl-PUGNAc to 2.4 and 2.3 Å resolution, respectively [Fig. 2(A–D)]. VcNagZΔSC is a variant of wild-type VcNagZ that has mutations of three surface residues (E19A, Q22A, and K54A) located on the opposite side of the enzyme relative to the active site. We have determined that this NagZ variant retains full catalytic activity relative to that of the wild type enzyme (data not shown) and a superposition of the X-ray structures of VcNagZΔSC with wild-type VcNagZ reveals the active site remains unchanged. Our motivation for generating this variant stemmed from our previous findings showing that, in the crystalline state, these side chains partially occluded the active site of the enzyme via intermolecular packing with a symmetry related copy of the enzyme, which impaired our ability to obtain structures with ordered ligands bound in the active site. The removal of these side chains by mutation to Ala did not hinder crystallization, yet provided space necessary to free a mobile loop (Met246-Val251) on the enzyme. Although external to the active site pocket, movement of this loop resulted in increased inhibitor occupancy relative to our previous studies. As described below, analysis of the X-ray structures of VcNagZΔSC in complex with N-butyryl-PUGNAc or N-valeryl-PUGNAc superimposed on the structure of wild-type VcNagZ bound to PUGNAc (PDB entry: 2oxn) (RMSD ∼0.48 Å for both complexes) revealed that, while freeing this loop increased inhibitor occupancy, it did not change the position of the derivatives within the active site pocket relative to PUGNAc.
Figure 2.
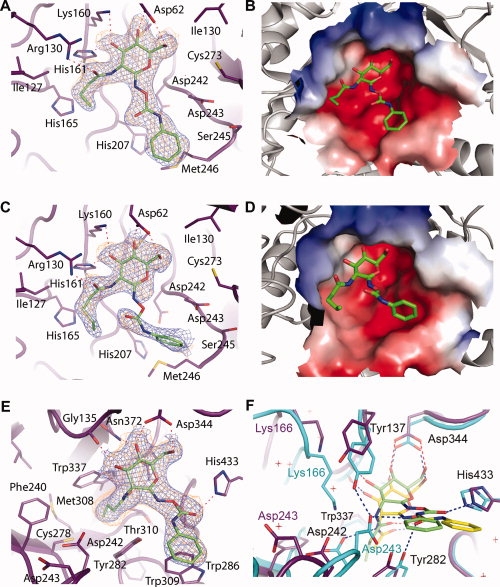
Crystal structures of N-butyryl-PUGNAc and N-valeryl-PUGNAc bound to VcNagZΔSC, and N-butyryl-PUGNAc bound to BtGH84. The active site of VcNagZΔSC binds N-butyryl-PUGNAc (A,B) and N-valeryl-PUGNAc (C,D) primarily through hydrogen bonding (dashed lines) with the pyranose ring of these inhibitors. The N-phenylcarbamate moiety of each inhibitor does not form direct hydrogen bonds with the enzyme and appear mobile. The average B-factor for the pyronose ring of N-butyryl-PUGNAc is 28 Å2, whereas its phenyl ring has an average value of 37 Å2. The pyronase ring of N-valeryl-PUGNAc has an average B-factor of 36.3 Å2; however, the average value for the phenyl ring was 63 Å2. Because of the high average B-factor and lack of continuous electron density, atoms of the N-phenylcarbamate of N-valeryl-PUGNAc are not included in PDB entry 3gsm; however, a refined position is shown here. Electrostatic surface potential maps (negative, red; positive, blue) calculated using APBS30 show the N-butyryl and N-valeryl extensions bound within a large, solvent accessible binding groove within the active site. E: BtGH84 bound to N-butyryl-PUGNAc. The inhibitor is bound via hydrogen-bonds (dashed lines), including one between the carbonyl oxygen of the N-phenylcarbamate extension and His433, which appears to order this moiety relative to the flexible binding observed within VcNagZΔSC. F: Superposition of N-butyryl-PUGNAc and PUGNAc bound31 complexes of BtGH84 reveals large structural distortions and displacement of N-butyryl-PUGNAc within the active site. Blue electron densities are maximum-likelihood weighted 2Fobs − Fcalc syntheses contoured at 0.24 e/Å3. Orange densities are maximum-likelihood weighted Fobs − Fcalc syntheses contoured at 0.11 e/Å3 following refinement with inhibitor models omitted. Superposition was carried out using CCP4.32 Figures were created with PyMOL.33
Electron density maps for crystallographic structures of VcNagZΔSC bound to N-butyryl-PUGNAc [Fig. 2(A)] or N-valeryl-PUGNAc [Fig. 2(C)], reveal the presence of these inhibitors bound within the active site pocket of the enzyme. Both the five carbon N-valeryl and four carbon N-butyryl acyl groups are accommodated within a large binding groove [Fig. 2(B,D)]. Because of the solvent accessible nature of this groove and lack of steric interference, both the N-valeryl and N-butyryl groups were found to adopt unstrained conformations showing favorable van der Waals' packing interactions with the side chain of Ile127 and a hydrogen-bonding interaction between the side chain guanidino group of Arg130 and carbonyl oxygen atoms of both inhibitors. Further comparisons of the N-butyryl-PUGNAc and N-valeryl-PUGNAc complexes to the crystal structures of VcNagZ bound to GlcNAc (PDB entry: 1Y65) or PUGNAc,16 show that the additional bulk of the N-valeryl and N-butyryl groups does not induce any conformational changes of the residues comprising the 2-acetamido binding groove nor does it affect interactions with the pyranose ring [Fig. 2(A–D)]. Both the O4 and O6 hydroxyls of each inhibitor form hydrogen bonds with the side chain carboxyl of Asp62, while the O3 hydroxyls of both inhibitors donate a hydrogen bond to the imidazole ring of His161 and receive a hydrogen bond from the ɛ-amino group of Lys160. These retained interactions and the ordered binding of the acyl chain reflect the ease with which VcNagZΔSC accommodates the bulky 2-acyl substituents of N-valeryl-PUGNAc and N-butyryl-PUGNAc, and is consistent with these inhibitors retaining high potency towards NagZ.
The inhibitory potency of PUGNAc for exo-N-acetyl-β-hexosaminidases has been primarily ascribed to two features. First, the trigonal sp2 center at the pseudo-anomeric position (C1) confers on the molecule a 4H3 conformation, which is believed to mimic the conformation of the proposed oxocarbenium ion-like transition states for the GH3 [Fig. 1(A)], GH20, and GH84 [Fig. 1(B)] catalyzed reactions. Thus, for optimal binding, the pseudoanomeric center of 2-acyl derivatives of PUGNAc likely must retain the ability to bind in the position occupied by the anomeric center in the oxocarbenium ion-like transition states. The structures of both N-valeryl-PUGNAc and N-butyryl-PUGNAc in complex with VcNagZΔSC reveal the pyranose ring of the inhibitors are superimposable with the position of PUGNAc bound to VcNagZ (PDB entry: 2OXN). The second feature conferring potency to PUGNAc is thought to be the pendent N-phenylcarbamate group. Consistent with this view, N-acetylglucosamino-1,5-lactone oxime (LOGNAc), a PUGNAc analogue lacking the N-phenyl group, binds with much lower affinity to VcNagZ (KI = 6400 nM) compared to PUGNAc (KI = 48 nM).16 Like PUGNAc, both N-valeryl-PUGNAc and N-butyryl-PUGNAc also benefit from the presence of the N-phenylcarbamate group in binding to VcNagZ (Table I). The structural basis for this increased affinity is unclear however, since N-phenylcarbamate group of N-valeryl-PUGNAc and N-butyryl-PUGNAc appear mobile, especially N-valeryl-PUGNAc, which makes the electron density for this group less well defined than the pyranose rings and 2-acyl groups. In addition to mobility as a cause for reduced electron density of this group, it is possible that the PUGNAc derivatives could hydrolyze within the enzyme active site, resulting in reduced density for the N-phenylcarbamate group. Indeed, a previous structural study of a GH18 chitinase bound to the PUGNAc analogue N,N′-diacetylchitobionoxime-N-phenylcarbamate found that the inhibitor was broken down, releasing O-((phenylamino)carbonyl)-hydroxylamine, while the lactone product remained bound to the chitinase.34 Most likely, the increased potency associated with the presence of the phenyl carbamate groups stems from desolvation of this substituent upon binding to NagZ, a phenomenon described for other glycoside hydrolases.35 Together, these findings reveal that the NagZ active site accommodates inhibitors having bulky N-acyl moieties while retaining high binding affinity. These structures do not, however, offer insight into the basis of diminished binding of such inhibitors to GH20 and GH84 enzymes.
It has been demonstrated that human GH20 β-hexosaminidases are not inhibited by PUGNAc derivatives bearing bulky 2-N-acyl substituents, yet GH84 O-GlcNAcase activity is inhibited by N-butyryl-PUGNAc.16 Interestingly however, on going from a four carbon acyl group, as found in N-butyryl-PUGNAc, to a five carbon group, found in N-valeryl-PUGNAc, there is an almost 20-fold decrease in binding toward O-GlcNAcase (Table I). Further, compared with PUGNAc, N-butyryl-PUGNAc binds ∼30-fold worse (Table I). To understand the structural basis for this successive and dramatic loss of binding affinity, we determined the crystallographic structure of N-butyryl-PUGNAc in complex with O-GlcNAcase to 2.3 Å resolution [Fig. 2(E)] and compared this complex with that of BtGH84 bound to PUGNAc.31 The electron density maps clearly reveal the presence of N-butyryl-PUGNAc bound within the active site pocket of BtGH84 [Fig. 2(E)]. Unlike the spacious, solvent accessible groove in which the 2-acetamido group binds in the VcNagZ active site, BtGH84, like the human β-hexosaminidases, has a hydrophobic pocket into which the substrate 2-acetamido binds and becomes oriented to participate in catalysis. The four carbon acyl group of N-butyryl-PUGNAc was bound within this pocket; however, its presence appears partly responsible for displacing the inhibitor from being seated deeply within the active site as seen for the BtGH84-PUGNAc complex.31 This alternate binding mode induces a number of gross structural changes throughout the active site region of the enzyme as compared to the structure of the BtGH84-PUGNAc complex.
A superposition of these complexes revealed that the plane of the pyranose ring of N-butyryl-PUGNAc sits ∼1 Å above the corresponding plane of PUGNAc [Fig. 2(F)]. Although the binding of PUGNAc does not distort the active site of BtGH84 from the conformation observed for the unliganded enzyme, the binding of N-butyryl-PUGNAc in this “elevated” position induces a series of dramatic changes in the active site. The complex with PUGNAc reveals this inhibitor is positioned so that the key catalytic residue Asp242 can accept a hydrogen bond from the 2-acetamido group of PUGNAc. The position of N-butyryl-PUGNAc however, places the analogous amide higher up in the active site, bringing the hydrophobic tail of the N-butyryl group adjacent to where the carboxyl group of Asp242 would normally sit. This favors reorientation of the Asp242 side chain to a position 2.5 Å away from where it normally sits within the active site, resulting in a large rearrangement of the loop to which it belongs. Rearrangement of this loop also moves the general acid/base residue, Asp243, a remarkable 7.5 Å away from its normal position, making it unable to interact with the inhibitor. In the X-ray structure of BtGH84 bound to PUGNAc, Asp243 sits in the same position as the unliganded enzyme,22,31 and forms a hydrogen-bond to the carbamate nitrogen of the N-phenylcarbamate moiety of PUGNAc. Although this interaction is lost in the N-butyryl-PUGNAc complex, it is interesting to note that the N-phenylcarbamate moiety of N-butyryl-PUGNAc is flipped 180° so that its carbonyl oxygen can instead accept a hydrogen bond from the imidazole of His443. In addition to the loop rearrangement that results in the loss of interactions with Asp242 and Asp243, the “elevated” placement of N-butyryl-PUGNAc in the active site of BtGH84 relative to that of PUGNAc also causes the additional loss of a hydrogen-bonding interaction between the O3 pyranose hydroxyl and ɛ-amino group of Lys166, a residue that also experiences a dramatic 5.5 Å shift in position. This change most likely stems from the loss of the hydrogen-bond interaction with Asp242 upon binding of the inhibitor.
These active site rearrangements stem from the “elevated” position of N-butyryl-PUGNAc in the active site of BtGH84, but the position of the inhibitor cannot be fully attributed to a steric clash of the N-butyryl side chain with the bottom of the 2-acetamido binding pocket. Previous studies have shown that the transition state analogues NButGT36 [Fig. 3(A,B)] and Thiamet-G [Fig. 3(A,C)],37 which both bear extensions of the same length as the N-butyryl group of N-butyryl-PUGNAc, are properly seated within the active site pocket [Fig. 3(A)]. Indeed, their pyranose rings superpose with the pyranose ring position of PUGNAc. These inhibitors, however, do not posses the N-phenylcarbamate extension of C1, having instead a thiazoline ring that mimics the intermediate or transition state found along Path B (see Fig. 1). Given the distressed state of the enzyme when complexed with N-butyryl-PUGNAc however, it is clear that BtGH84 would not readily accommodate extensions to the carbon chain of N-butyryl-PUGNAc; a suggestion that is entirely consistent with the dramatic 900-fold increase in KI value for N-valeryl-PUGNAc as compared to that of its parent molecule PUGNAc (Table I).
Figure 3.
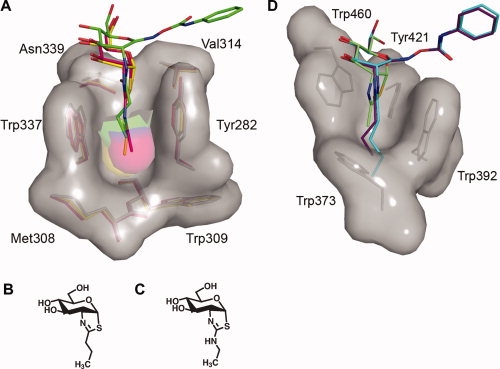
Three-dimensional structures of the 2-acetamido binding pockets of BtGH84 and human β-hexosaminidase A. A: Superposition of the BtGH84 N-butyryl-PUGNAc complex with BtGH84 bound to the thiazoline inhibitors NButGT36 and Thiamet-G37 reveals that these latter inhibitors bind deeper within the BtGH84 active site, indicating that the N-butyryl extension of N-butyryl-PUGNAc is not solely responsible for its “elevated” placement within the BtGH84 active site. N-butyryl-PUGNAc is shown with green carbon atoms, NButGT with yellow carbon atoms, and Thiamet-G with pink carbon atoms. B: Structure of Thiamet-G. C, Structure of NButGT. D: Structure of the 2-acetamido binding pocket of human β-hexosaminidase A in complex with NAG-thiazoline (PDB entry: 2GK1)25 superposed with models of N-butyryl-PUGNAc and N-valeryl-PUGNAc. The superposition clearly indicates the prohibitive steric clashes that would occur between the N-butyryl and N-valeryl groups of these inhibitors and the bottom of the shallow 2-acetamido binding pocket. Residues comprising the binding pockets of both BtGH84 and β-hexosaminidase A are shown as sticks with their corresponding solvent accessible surfaces. Superposition was carried out using CCP4.32 Figures were created with PyMOL.33
A structural comparison of the active sites of both GH20 human β-hexosaminidase A25 and B24 with that of BtGH8422 reveals that the GH20 β-hexosaminidases have a much more shallow 2-acetamido binding pocket than BtGH84 [Fig. 3(D)]. This difference offers a clear rationale as to why these enzymes cannot tolerate any significant bulk at the 2-acyl position of PUGNAc. The bottom of the BtGH84 2-acetamido binding pocket is primarily comprised of the side chain of Cys278, the side chain and backbone carbonyl group of Met308, the main chain of Trp309, and the side chain of Thr310.22 Together, they form a bowl shaped base that is deep enough to accommodate bicyclic intermediate analogues bearing three carbon aliphatic chains that are attached to the thiazoline ring as described earlier36,37 [Fig. 3(A)]. The bottom of the analogous pocket in the β-hexosaminidases, however, is rigidly walled off by the side chain of a Trp residue (HexA Trp337; HexB Trp405), resulting in a shallow pocket that already snugly fits the 2-acetamido group of the substrate [Fig. 3(D)]. Molecular modeling of N-butyryl-PUGNAc and N-valeryl-PUGNAc in the active site of human Hex A (PDB entry: 2GK1) [Fig. 3(D)] highlights the severe steric clashes that would occur between of the 2-acyl extension of either inhibitor with the shallow base of the 2-acetamido binding pocket. These predicted clashes for GH20 human β-hexosaminidases provide a structural rationale for the dramatic increases in the KI values of N-butyryl-PUGNAc (700-fold) and N-valeryl-PUGNAc (6000-fold) relative to PUGNAc (Table I).
Materials and Methods
Construction of a VcNagZ variant with improved crystallization characteristics
VcNagZ (Genbank AAF93857.1) fused to a C-terminal His10-tag, crystallizes with the active site partially occluded by three surface residues (E19, Q22, and K54) of an interacting symmetry mate within the crystal lattice. The residues are located on the opposite side of the enzyme from the active site and were not predicted to participate in catalysis. To provide additional space around the active site of VcNagZ and promote improved inhibitor binding in the crystalline state, three mutations (E19A, Q22A, and K54A) were introduced into the VcNagZ expression plasmid pVcNagZ16 using the Phusion Site Directed Mutagesis® method from New England Biolabs (NEB). The primers (Alpha DNA, Montreal) 5′-P-GAAGATCGCGCAATTCTGGCGCACCCTACAGTGG-3′ and 5′-GGCACTCAGTTCGTAACCGGCCACATCCAACCAAAG-3′ were used to PCR amplify a liner variant of pVcNagZ encoding ΔE19A and ΔQ22A. The amplicon was circularized using T4DNA ligase and used to transform into E. coli BL21 (DE3) GOLD (Stratagene). Plasmid pVcNagZΔ2SC was isolated from a single transformant and verified by DNA sequencing. The plasmid was then used as template to incorporate the third mutation, ΔK54A using the primers 5′-P-CGTCAAGCGGCGGCAAGACCGATTTTG-3′ and 5′-GATCGCTTTATTGAGTGCCAGCAATTGCTG-3′. The amplicon was circularized using T4DNA ligase and transformed into E. coli BL21 (DE3) GOLD. Plasmid containing the three mutations, pVcNagZΔSC, was isolated from a single transformant and verified by DNA sequencing.
Structure determination of VcNagZΔSC in complex with N-butyryl-PUGNAc and N-valeryl-PUGNAc
C-terminally His10-tagged recombinant VcNagZΔSC was expressed and purified as described previously for VcNagZ and exchanged into 150 mM NaCl, 20 mM Bis-Tris, pH 6.5. To confirm that the surface residue mutations (E19A, Q22A, and K54A) had not affected catalytic activity, purified VcNagZΔSC was assayed using pNP-GlcNAc as substrate to confirm that it retained full catalytic activity as compared to wild type VcNagZ. VcNagZΔSC was concentrated to 5–6 mg/mL then mixed with N-butyryl-PUGNAc (dissolved in water) at a 1:35 molar ratio, or N-valeryl-PUGNAc (dissolved in 21% DMSO) at a 1:25 molar ratio and cocrystallized by hanging drop vapor diffusion using a mother liquor comprised of 13% PEG 20,000, 10% glycerol and 100 mM Bis-Tris pH 6.2. Diffraction quality crystals grew overnight and were flash cooled with liquid N2 in a cryosolution consisting of 13% PEG 20,000, 30% glycerol and 100 mM Bis-Tris pH 6.2. Diffraction data were obtained to 2.3 Å for VcNagZΔSC bound to N-butyryl-PUGNAc, and 2.4 Å for VcNagZΔSC bound to N-valeryl-PUGNAc at 100 K using a Rigaku RAXIS VI ++ detector and generator. Data were integrated using MOSFLM38 and scaled and reduced with SCALA from the CCP4 suite of programs.32 Despite the equivalence in length of the crystallographic b and c axes (Table II), crystals of the VcNagZΔSC inhibitor complexes are in orthorhombic space group P212121. The theoretical and experimental centric and acentric cumulative intensity distributions were correlated, and analysis of the diffraction data using the Merohedral Crystal Twinning Server (http://nihserver.mbi.ucla.edu/Twinning/) demonstrated these crystals were not twinned.
Table II.
Crystallographic Statistics
| Crystal Information | VcNagZΔSCN-butyryl-PUGNAc | VcNagZΔSCN-valeryl-PUGNAc | BtGH84 N-butyryl-PUGNAc |
|---|---|---|---|
| Space group | P212121 | P212121 | C2 |
| Solvent content (%) | 48 | 48 | 51 |
| Data Collection (values in parentheses refer to the high-resolution shell) | |||
| Unit cell dimensions (Å) | a = 47.778 b = 86.021 c = 86.217 | a = 47.980 b = 86.439 c = 86.440 | a = 186.880 b = 52.540 c = 82.100 |
| α = β = γ = 90° | α = β = γ = 90° | α = 90 β = 98.34 γ = 90 | |
| Wavelength (Å) | 1.54 | 1.54 | 0.98 |
| Resolution range (Å) | 38.55–2.30 | 32.11–2.40 | 57.07–2.30 |
| High-resolution shell (Å) | 2.42–2.30 | 2.53–2.40 | 2.42–2.30 |
| Total observations | 54168 (5254) | 43120 (6219) | 142592 (21101) |
| Unique reflections | 16304 (2226) | 14516 (2099) | 35434 (5126) |
| (I/σ) | 9.8 (2.6) | 11.6 (2.2) | 13.2 (2.6) |
| Completeness (%) | 99.3 (95.4) | 99.5 (100) | 100 (100) |
| R merge | 0.087 (0.348) | 0.100 (0.49) | 0.093 (0.53) |
| Multiplicity | 3.3 (2.4) | 3.0 (3.0) | 4.0 (4.1) |
| Refinement | |||
| R work | 0.18 | 0.21 | 0.20 |
| R free | 0.24 | 0.27 | 0.24 |
| Number of atoms | |||
| Protein | 2499 | 2512 | 4703 |
| Heterogen | 39 | 30 | 27 |
| Water | 175 | 126 | 194 |
| Average B (Å2) | 25 | 28 | 32 |
| RMSD from ideal geometry | |||
| Bond lengths (Å) | 0.01 | 0.01 | 0.01 |
| Bond angles (deg.) | 1.28 | 1.31 | 1.37 |
| Ramachandran plot | |||
| Most favored (%) | 90.2 | 88.8 | 95.8 |
| Additionally allowed | 9.8 | 11.2 | 4.2 |
Structures of the complexes were determined by molecular replacement using PHASER39 and a heteroatom free structure of VcNagZ (PDB entry: 2oxn) as a search model. The solution was refined by rigid body refinement, followed by rounds of model rebuilding and restrained refinement using COOT,40 and REFMAC,41 respectively. The N-butyryl-PUGNAc and N-valeryl-PUGNAc ligands were modeled and refined using COOT and REFMAC, respectively, using stereochemical target values based upon ideal coordinates generated with QUANTA (Accelerys, San Diego, CA). Solvent molecules were added using COOT and checked manually. Crystallographic statistics and structure quality are shown in Table II.
Structure determination of BtGH84 in complex with N-butyryl-PUGNAc
N-terminal His6-tagged recombinant BtGH84 was expressed and purified as described previously.22 Purified protein in 20 mM HEPES pH 7.5, 300 mM NaCl was concentrated to 11 mg/mL and used for crystallization. Crystals were grown from a solution containing 15% PEG3350, 0.1M MES pH 6.0, 0.3M ammonium acetate and 20% glycerol. To form the N-butyryl-PUGNAc complex, a minute amount of the ligand was added to the crystallization mother liquor in which crystals of apo-BtGH84 were soaked at room temperature before flash cooling in liquid N2. Diffraction data were collected to 2.30 Å resolution on beamline ID23.1 of the European Synchrotron Radiation Facility (ESRF, Grenoble). Data were integrated using MOSFLM38 and scaled and reduced with SCALA from the CCP4 suite of programs.32
The structure of BtGH84 in complex with N-butyryl-PUGNAc was determined using PHASER39 with the PDB entry 2CHO as the search model. Manual corrections to the model were made with COOT40 and refinement cycles were performed with REFMAC.41 Water molecules and ligand were added using COOT with stereochemical target values for the ligand based upon ideal coordinates generated with QUANTA (Accelerys). Crystallographic statistics and structure quality are shown in Table II.
Modeling N-butyryl-PUGNAc and N-valeryl-PUGNAc in the α-subunit active site of human β-hexosaminidase A
Using the crystallographic structure of human β-hexosaminidase A in complex with the intermediate analogue NAG-thiazoline (PDB entry: 2GK1),25 the refined molecular model of N-butyryl-PUGNAc from the BtGH84 complex was placed into the HexA active site using COOT40 so that the pyranose ring of the inhibitor superposed onto the pyranose ring of NAG-thiazoline. Given the similar active site architectures of BtGH84 and the human β-hexosaminidase isoenzymes, this placement yielded a model where the only significant steric clash with the enzyme occurred between the N-butyryl-group and the base of the 2-acetamido binding pocket. A model of N-valeryl-PUGNAc was superposed onto the placed N-butyryl-PUGNAc model, also revealing that the only significant steric clashes with the enzyme occur between the N-valeryl group and the base of the 2-acetamido binding pocket.
Conclusions
The crystallographic structures of VcNagZΔSC bound to N-valeryl-PUGNAc and N-butyryl-PUGNAc demonstrate that the enzyme can easily accommodate substantial bulk at the 2-acyl position without affecting the binding mode of the inhibitor within the active site. Conversely, the crystallographic structure of N-butyryl-PUGNAc bound to BtGH84 provides a glimpse into the distressed conformational state the enzyme must adopt to accommodate this inhibitor, suggesting that even modestly bulky 2-acyl groups, as in N-valeryl-PUGNAc, would be sufficient to confer distinct selectivity for NagZ over this enzyme. Modeling N-valeryl-PUGNAc and N-butyryl-PUGNAc into the active site of human Hex A also reveals why the GH20 human β-hexosaminidases cannot readily accommodate any additional bulk beyond the 2-acetamido group found on natural substrates; the shallow pocket tightly packs against the methyl group of the 2-acetamido and any elaborations would clash with the walls of this pocket.
Most likely, due to the fundamental role of NagZ in Gram-negative peptidoglycan recycling, the enzyme is highly conserved in Gram-negative bacteria.17 Given the high sequence similarity of these NagZ homologues, and knowledge that the sequence related glycoside hydrolases share a conserved structure and catalytic mechanism,23 we expect that NagZ selective 2-acyl derivatives of PUGNAc will be potent inhibitors of NagZ from many Gram-negative pathogens. Thus, the approach of suppressing AmpC mediated resistance through inhibition of NagZ may provide a strategy that could be effective against a broad spectrum of Gram-negative bacteria harbouring this resistance mechanism. Having defined the molecular basis for how 2-acyl derivatives of PUGNAc can selectively inhibit NagZ over the functionally related human enzymes, we envision considerable structure-guided improvements of these inhibitors that should enhance their selectivity and potency for NagZ. We believe these improvements will lead to candidates with properties that are amenable for testing in vivo.
Accession codes
Coordinates and structure factors have been deposited with the Protein Data Bank under the following accession codes: 3gs6; VcNagZΔSC in complex with N-butyryl-PUGNAc, 3gsm; VcNagZΔSC in complex with N-valeryl-PUGNAc, 2wca; BtGH84 in complex with N-butyryl-PUGNAc.
Acknowledgments
M.D.B. and T.W.J. hold studentships from the Manitoba Health Research Council. D.J.V. is a Canada Research Chair in Chemical Glycobiology and a Scholar of the Michael Smith Foundation for Health Research. G.J.D. is a Royal-Society/Wolfson Research Merit award recipient.
References
- 1.Henrissat B. A classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem J. 1991;280(Part 2):309–316. doi: 10.1042/bj2800309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 2009;37(Database issue):D233–D238. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gravel RA, Clarke JTR, Kaback MM, Mahuran D, Sandhoff K, Suzuki K. The GM2 gangliosidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. Vol. 2. New York: McGraw-Hill; 1995. pp. 2839–2879. The metabolic and molecular bases of inherited disease. [Google Scholar]
- 4.Dong DL, Hart GW. Purification and characterization of an O-GlcNAc selective N-acetyl-beta-d-glucosaminidase from rat spleen cytosol. J Biol Chem. 1994;269:19321–19330. [PubMed] [Google Scholar]
- 5.Gao Y, Wells L, Comer FI, Parker GJ, Hart GW. Dynamic O-glycosylation of nuclear and cytosolic proteins: cloning and characterization of a neutral, cytosolic beta-N-acetylglucosaminidase from human brain. J Biol Chem. 2001;276:9838–9845. doi: 10.1074/jbc.M010420200. [DOI] [PubMed] [Google Scholar]
- 6.Park JT, Uehara T. How bacteria consume their own exoskeletons (turnover and recycling of cell wall peptidoglycan) Microbiol Mol Biol Rev. 2008;72:211–227. doi: 10.1128/MMBR.00027-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng Q, Li H, Merdek K, Park JT. Molecular characterization of the beta-N-acetylglucosaminidase of Escherichia coli and its role in cell wall recycling. J Bacteriol. 2000;182:4836–4840. doi: 10.1128/jb.182.17.4836-4840.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jacobs C, Frere JM, Normark S. Cytosolic intermediates for cell wall biosynthesis and degradation control inducible beta-lactam resistance in gram-negative bacteria. Cell. 1997;88:823–832. doi: 10.1016/s0092-8674(00)81928-5. [DOI] [PubMed] [Google Scholar]
- 9.Votsch W, Templin MF. Characterization of a beta-N-acetylglucosaminidase of Escherichia coli and elucidation of its role in muropeptide recycling and beta-lactamase induction. J Biol Chem. 2000;275:39032–39038. doi: 10.1074/jbc.M004797200. [DOI] [PubMed] [Google Scholar]
- 10.Livermore DM. beta-Lactamases in laboratory and clinical resistance. Clin Microbiol Rev. 1995;8:557–584. doi: 10.1128/cmr.8.4.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bradford PA, Urban C, Mariano N, Projan SJ, Rahal JJ, Bush K. Imipenem resistance in Klebsiella pneumoniae is associated with the combination of ACT-1, a plasmid-mediated AmpC beta-lactamase, and the foss of an outer membrane protein. Antimicrob Agents Chemother. 1997;41:563–569. doi: 10.1128/aac.41.3.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barnaud G, Arlet G, Verdet C, Gaillot O, Lagrange PH, Philippon A. Salmonella enteritidis: AmpC plasmid-mediated inducible beta-lactamase (DHA-1) with an ampR gene from Morganella morganii. Antimicrob Agents Chemother. 1998;42:2352–2358. doi: 10.1128/aac.42.9.2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fortineau N, Poirel L, Nordmann P. Plasmid-mediated and inducible cephalosporinase DHA-2 from Klebsiella pneumoniae. J Antimicrob Chemother. 2001;47:207–210. doi: 10.1093/jac/47.2.207. [DOI] [PubMed] [Google Scholar]
- 14.Miriagou V, Tzouvelekis LS, Villa L, Lebessi E, Vatopoulos AC, Carattoli A, Tzelepi E. CMY-13, a novel inducible cephalosporinase encoded by an Escherichia coli plasmid. Antimicrob Agents Chemother. 2004;48:3172–3174. doi: 10.1128/AAC.48.8.3172-3174.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakano R, Okamoto R, Nakano Y, Kaneko K, Okitsu N, Hosaka Y, Inoue M. CFE-1, a novel plasmid-encoded AmpC beta-lactamase with an ampR gene originating from Citrobacter freundii. Antimicrob Agents Chemother. 2004;48:1151–1158. doi: 10.1128/AAC.48.4.1151-1158.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stubbs KA, Balcewich M, Mark BL, Vocadlo DJ. Small molecule inhibitors of a glycoside hydrolase attenuate inducible AmpC-mediated beta-lactam resistance. J Biol Chem. 2007;282:21382–21391. doi: 10.1074/jbc.M700084200. [DOI] [PubMed] [Google Scholar]
- 17.Vocadlo DJ, Mayer C, He S, Withers SG. Mechanism of action and identification of Asp242 as the catalytic nucleophile of Vibrio furnisii N-acetyl-beta-d-glucosaminidase using 2-acetamido-2-deoxy-5-fluoro-alpha-L-idopyranosyl fluoride. Biochemistry. 2000;39:117–126. doi: 10.1021/bi991958d. [DOI] [PubMed] [Google Scholar]
- 18.Macauley MS, Whitworth GE, Debowski AW, Chin D, Vocadlo DJ. O-GlcNAcase uses substrate-assisted catalysis: kinetic analysis and development of highly selective mechanism-inspired inhibitors. J Biol Chem. 2005;280:25313–25322. doi: 10.1074/jbc.M413819200. [DOI] [PubMed] [Google Scholar]
- 19.Vocadlo DJ, Withers SG. Detailed comparative analysis of the catalytic mechanisms of beta-N-acetylglucosaminidases from families 3 and 20 of glycoside hydrolases. Biochemistry. 2005;44:12809–12818. doi: 10.1021/bi051121k. [DOI] [PubMed] [Google Scholar]
- 20.Tews I, Perrakis A, Oppenheim A, Dauter Z, Wilson KS, Vorgias CE. Bacterial chitobiase structure provides insight into catalytic mechanism and the basis of Tay-Sachs disease. Nat Struct Biol. 1996;3:638–648. doi: 10.1038/nsb0796-638. [DOI] [PubMed] [Google Scholar]
- 21.Mark BL, Vocadlo DJ, Knapp S, Triggs-Raine BL, Withers SG, James MN. Crystallographic evidence for substrate-assisted catalysis in a bacterial beta-hexosaminidase. J Biol Chem. 2001;276:10330–10337. doi: 10.1074/jbc.M011067200. [DOI] [PubMed] [Google Scholar]
- 22.Dennis RJ, Taylor EJ, Macauley MS, Stubbs KA, Turkenburg JP, Hart SJ, Black GN, Vocadlo DJ, Davies GJ. Structure and mechanism of a bacterial beta-glucosaminidase having O-GlcNAcase activity. Nat Struct Mol Biol. 2006;13:365–371. doi: 10.1038/nsmb1079. [DOI] [PubMed] [Google Scholar]
- 23.Vocadlo DJ, Davies GJ. Mechanistic insights into glycosidase chemistry. Curr Opin Chem Biol. 2008;12:539–555. doi: 10.1016/j.cbpa.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 24.Mark BL, Mahuran DJ, Cherney MM, Zhao D, Knapp S, James MN. Crystal structure of human beta-hexosaminidase B: understanding the molecular basis of Sandhoff and Tay-Sachs disease. J Mol Biol. 2003;327:1093–1109. doi: 10.1016/s0022-2836(03)00216-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lemieux MJ, Mark BL, Cherney MM, Withers SG, Mahuran DJ, James MN. Crystallographic structure of human beta-hexosaminidase A: interpretation of Tay-Sachs mutations and loss of G(M2) ganglioside hydrolysis. J Mol Biol. 2006;359:913–929. doi: 10.1016/j.jmb.2006.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rao FV, Dorfmueller HC, Villa F, Allwood M, Eggleston IM, van Aalten DM. Structural insights into the mechanism and inhibition of eukaryotic O-GlcNAc hydrolysis. EMBO J. 2006;25:1569–1578. doi: 10.1038/sj.emboj.7601026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mayer C, Vocadlo DJ, Mah M, Rupitz K, Stoll D, Warren RA, Withers SG. Characterization of a beta-N-acetylhexosaminidase and a beta-N-acetylglucosaminidase/beta-glucosidase from Cellulomonas fimi. FEBS J. 2006;273:2929–2941. doi: 10.1111/j.1742-4658.2006.05308.x. [DOI] [PubMed] [Google Scholar]
- 28.Stubbs KA, Zhang N, Vocadlo DJ. A divergent synthesis of 2-acyl derivatives of PUGNAc yields selective inhibitors of O-GlcNAcase. Org Biomol Chem. 2006;4:839–845. doi: 10.1039/b516273d. [DOI] [PubMed] [Google Scholar]
- 29.Asgarali A, Stubbs KA, Oliver A, Vocadlo DJ, Mark BL. Inactivation of the glycoside hydrolase NagZ attenuates antipseudomonal beta-lactam resistance in Pseudomonas aeruginosa. Antimicrob Agents Chemother. 53:2274–2282. doi: 10.1128/AAC.01617-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Macauley MS, Bubb AK, Martinez-Fleites C, Davies GJ, Vocadlo DJ. Elevation of global O-GlcNAc levels in 3T3-L1 adipocytes by selective inhibition of O-GlcNAcase does not induce insulin resistance. J Biol Chem. 2008;283:34687–34695. doi: 10.1074/jbc.M804525200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Collaborative computational project number 4. The CCP4 Suite: programs for protein crystallography. Acta Crystallogr D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 33.DeLano WL. Palo Alto, CA, USA: DeLano Scientific; 2002. The PyMOL Molecular Graphics System. [Google Scholar]
- 34.Vaaje-Kolstad G, Vasella A, Peter MG, Netter C, Houston DR, Westereng B, Synstad B, Eijsink VG, van Aalten DM. Interactions of a family 18 chitinase with the designed inhibitor HM508 and its degradation product, chitobiono-delta-lactone. J Biol Chem. 2004;279:3612–3619. doi: 10.1074/jbc.M310057200. [DOI] [PubMed] [Google Scholar]
- 35.Gloster TM, Roberts S, Perugino G, Rossi M, Moracci M, Panday N, Terinek M, Vasella A, Davies GJ. Structural, kinetic, and thermodynamic analysis of glucoimidazole-derived glycosidase inhibitors. Biochemistry. 2006;45:11879–11884. doi: 10.1021/bi060973x. [DOI] [PubMed] [Google Scholar]
- 36.Whitworth GE, Macauley MS, Stubbs KA, Dennis RJ, Taylor EJ, Davies GJ, Greig IR, Vocadlo DJ. Analysis of PUGNAc and NAG-thiazoline as transition state analogues for human O-GlcNAcase: mechanistic and structural insights into inhibitor selectivity and transition state poise. J Am Chem Soc. 2007;129:635–644. doi: 10.1021/ja065697o. [DOI] [PubMed] [Google Scholar]
- 37.Yuzwa SA, Macauley MS, Heinonen JE, Shan X, Dennis RJ, He Y, Whitworth GE, Stubbs KA, McEachern EJ, Davies GJ, Vocadlo DJ. A potent mechanism-inspired O-GlcNAcase inhibitor that blocks phosphorylation of tau in vivo. Nat Chem Biol. 2008;4:483–490. doi: 10.1038/nchembio.96. [DOI] [PubMed] [Google Scholar]
- 38.Leslie AGW. In: Crystallographic Computing 5: From Chemistry to Biology. Mores D, Podjarny AD, Thiery JC, editors. Oxford: Oxford University Press; 1991. pp. 27–38. [Google Scholar]
- 39.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Crystallogr D Biol Crystallogr. 2005;61(Patt 4):458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 40.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Part 12 Part 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 41.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53(Part 3):240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 42.Beer D, Maloisel JL, Rast DM, Vasella A. Synthesis of 2-acetamido-2-deoxy-d-gluconhydroximolactone-derived and chitobionhydroximolactone-derived N-phenylcarbamates, potential inhibitors of beta-N-acetylgluco-saminidase. Helv Chim Acta. 1990;73:1918–1922. [Google Scholar]