

Abstract
11β-Hydroxysteroid dehydrogenase type 1 (11β-HSD1) is a key enzyme in the conversion of cortisone to the functional glucocorticoid hormone cortisol. This activation has been implicated in several human disorders, notably the metabolic syndrome where 11β-HSD1 has been identified as a novel target for potential therapeutic drugs. Recent crystal structures have revealed the presence of a pronounced hydrophobic surface patch lying on two helices at the C-terminus. The physiological significance of this region has been attributed to facilitating substrate access by allowing interactions with the endoplasmic reticulum membrane. Here, we report that single mutations that alter the hydrophobicity of this patch (I275E, L266E, F278E, and L279E in the human enzyme and I275E, Y266E, F278E, and L279E in the guinea pig enzyme) result in greatly increased yields of soluble protein on expression in E. coli. Kinetic analyses of both reductase and dehydrogenase reactions indicate that the F278E mutant has unaltered Km values for steroids and an unaltered or increased kcat. Analytical ultracentrifugation shows that this mutation also decreases aggregation of both the human and guinea pig enzymes, resulting in greater monodispersity. One of the mutants (guinea pig F278E) has proven easy to crystallize and has been shown to have a virtually identical structure to that previously reported for the wild-type enzyme. The human F278E enzyme is shown to be a suitable background for analyzing the effects of naturally occurring mutations (R137C, K187N) on enzyme activity and stability. Hence, the F278E mutants should be useful for many future biochemical and biophysical studies of the enzyme.
Keywords: 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1), recombinant protein expression, hydrophobicity, mutation
Introduction
The interconversion of cortisol and its inactive 11-keto form, cortisone, is performed by the two isoforms of the enzyme 11β-hydroxysteroid dehydrogenase. The type 1 enzyme (11β-HSD1) is expressed in the liver, brain, and adipose tissue with a predominant NADPH-dependent reductase activity (reviewed in1), whereas the type 2 enzyme (11β-HSD2), which acts predominantly as a NAD+-dependent dehydrogenase, is expressed mainly in the kidney.2 The 11β-HSD isoforms constitute an important “prereceptor” mechanism to regulate the physiological effects of active glucocorticoid.3 It has been hypothesized that a dysregulation of cortisol at a tissue level, which would indicate abnormal 11β-HSD1 activity, may be the underlying pathology behind a range of conditions such as type 2 diabetes and the metabolic syndrome (a condition comprising insulin resistance, abnormal lipid and glucose metabolism, hypertension, and central obesity).4 Recent animal studies have shown that 11β-HSD1 is an important regulator of hepatic glucose output and visceral adiposity. Transgenic mice overexpressing 11β-HSD1 in liver and adipose tissue recapitulate features of the metabolic syndrome.5–7 In contrast, recombinant mice lacking 11β-HSD1 show improved glucose tolerance, enhanced insulin sensitivity, and reduced weight gain when given a high-fat diet.8,9 11β-HSD1 has therefore emerged as a novel therapeutic target to treat patients with obesity and insulin resistance, selective 11β-HSD1 inhibitors being tested in rodent10 and mammalian models.11
11β-HSD1 is a microsomal12 34 kDa integral membrane glycoprotein, which is postulated to exist as a homodimer,13 although some studies have suggested it might also exist naturally as a homotetramer.14 11β-HSD1 is a member of the short-chain dehydrogenase reductase (SDR) superfamily of enzymes. Unusually for an SDR enzyme, 11β-HSD1 has an N-terminal membrane anchor,15,16 with a small portion of the N terminal segment of the enzyme present in the cytosol. The C-terminus, which includes the catalytic domain, exists in the lumen of the endoplasmic reticulum.16,17 Although glycosylation was postulated to be essential for activity based on a putative role in maintaining the SDR scaffold,18 it has been shown that 11β-HSD1 is active as a nonglycosylated form when expressed in bacteria.17,19 Ten high-resolution structures of recombinant mouse (PDB entries: 1Y5M, 1Y5R), guinea pig (1XSE), and human (2ILT, 2IRW, 2BEL, 3BYZ, 2RBE, 1XU7, and 1XU9) 11β-HSD1 are currently available. The structures of these proteins are all extremely similar and possess a Rossman fold typical of SDR family members.20,21
Interestingly, comparing the 11β-HSD1 structures from the different species shows that the last two helices (α2 and α3) at the C-terminus in all structures protrude and form a long helical structure near to the steroid-binding site. The residues on the surface of these C-terminal helices are in the form of a nonpolar plateau encircled by positively charged residues (see Fig. 1).24 The existence of this region, which is conserved across all species, probably explains previous difficulties with expression and purification of this protein in a bacterial system, because it presumably promotes aggregation and precipitation in the absence of the membrane. If one of the suggested orientations of 11β-HSD1 in vivo24 is to be believed, this plateau exists in the nonpolar center of the ER membrane, with the charged residues forming salt bridges with the displaced phospholipid and sulfolipid head groups. Because of the hydrophobic nature of the substrates of 11β-HSD1, this could constitute a potential “membrane dipping” mechanism to funnel hydrophobic substrates from the membrane into the 11β-HSD1 active site.24 A similar “membrane dipping” mechanism has been proposed for the mammalian microsomal cytochrome P450 monooxygenase.25,26 However, it has alternatively been suggested that this nonpolar plateau in 11β-HSD1 could be involved in dimer–dimer interactions, forming a “closed” tetrameric form of the enzyme.14
Figure 1.
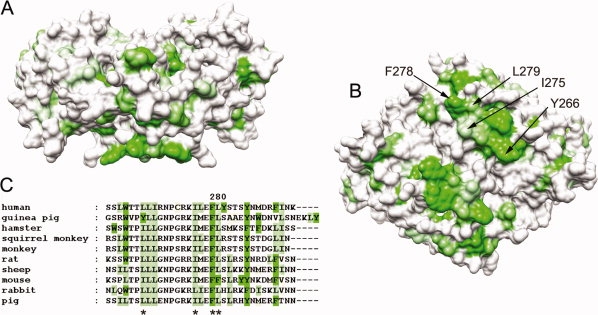
Putative membrane-dipping region of the guinea pig 11β-HSD1 structure (PDB code:1XSE). Structures and sequences are colored by the experimentally determined hydrophobicity scale for proteins at membrane surfaces,22 with hydrophobicity shown as increasing green. (A) Surface representation of a “:side” view of 11β-HSD1 with the putative membrane-associating region at the bottom of the structure, showing protrusion of the hydrophobic patch. (B) “Bottom” of the protein showing the hydrophobic surface region that might come into contact with the membrane. Hydrophobic residues contributing to this region are labeled for one of the two chains. (C) Sequence alignment of the C-termini of mammalian 11β-HSD1 proteins, showing the conserved hydrophobicity. Residues mutated in this study are asterisked. Structural images produced using UCSF Chimera from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH P41 RR-01081).23
To initiate investigations into the importance of this hydrophobic region, we have replaced a series of key residues within human and guinea pig 11β-HSD1 with the hydrophilic residue glutamate. Here, we report the effects of these mutations on the solubility, monodispersity, and activity of the proteins expressed in E. coli, and on the crystal structure of one of the guinea pig mutants, F278E.
Results
Effect of mutations on the expression and purification of recombinant 11β-HSD1
A series of single-point mutations were made to hydrophobic residues at the C-terminus of both guinea pig and human 11β-HSD1 proteins. Target residues for mutation were chosen by analysis of the crystal structures for the human and guinea pig 11β-HSD1 (pdb 2BEL and 1XSE, respectively). The positions of these hydrophobic residues are shown in Figure 1.
The human mutants L266E, I275E, F278E, and L279E and the guinea pig mutants Y266E, I275E, F278E, and L279E were analyzed for their effect on protein expression. Analysis of bacterial lysates using SDS-PAGE showed that the solubility of recombinant 11β-HSD1 was dramatically increased by the mutations I275E, F278E, and L279E in the guinea pig sequence and F278E in the human sequence (see Fig. 2). This was not simply because of an increase in total protein expression, as there was not only an increase of protein in the soluble fraction but also a concomitant decrease in the amount of protein in the insoluble fraction (data not shown). The effect of the various mutations on the final yield of soluble protein when the protein was purified from larger cultures by IMAC and gel-filtration is shown in Figure 3. All the mutations increased the final yield of soluble protein, with the human F278E and guinea pig I275E mutants showing the most dramatic increases (∼8- and 23-fold increases compared with respective wild-type constructs).
Figure 2.
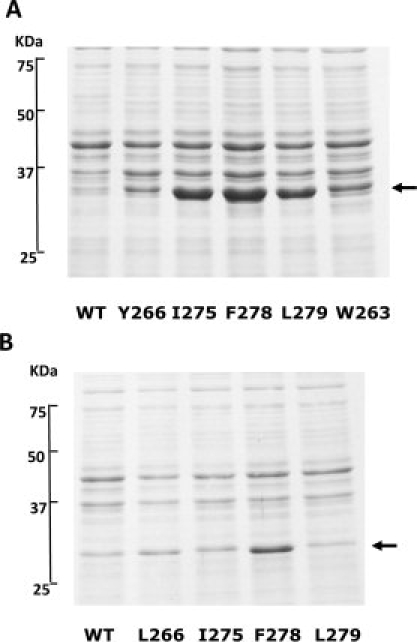
SDS-Page analysis of wild-type (WT) and mutant 11β-HSD1 recombinant proteins. Supernatant fractions from lysates of bacterial cultures expressing (A) guinea pig 11β-HSD1 proteins and (B) human 11β-HSD1 proteins indicate that the I275E, F278E, and L point mutation dramatically increased the production of soluble 11β-HSD1. Arrows indicate positions of respective guinea pig and human recombinant proteins.
Figure 3.
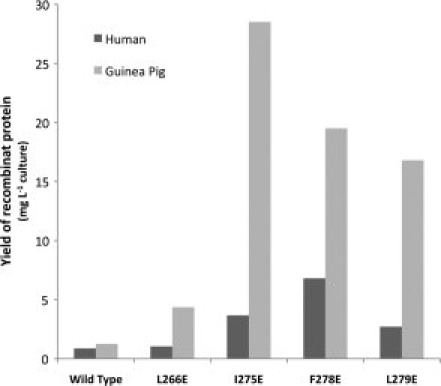
Relative yields of wild-type and mutant human and guinea pig 11β-HSD1 proteins in the bacterial expression system. Proteins were produced and purified as per Materials and Methods. Levels of purified protein produced are shown in mg protein per liter LB broth and are representative of more than one experiment. Introduction of glutamate at position 275, 278, and 279 in the guinea pig sequences, and 278 in the human sequence, dramatically increased the yield of recombinant 11β-HSD1 protein.
Effect of mutations on enzyme kinetics
The effect of the mutations on the kcat and Km for the steroid substrates of 11β-HSD1 was analyzed in both dehydrogenase and reductase directions (Tables I and II). For the guinea pig enzyme, the F278E mutant had virtually unchanged Km and kcat compared with wild type, in both dehydrogenase and reductase directions (Table II). The Y266E and L279E mutants showed a slight decrease in turnover rate but with little change in Km. The only pronounced variation from wild type was seen with the I275E mutant, which showed a marked decrease in Km for cortisol and a dramatic increase in Km for cortisone (Table II). In the case of the human enzyme, the Km for the F278E mutant was virtually unchanged compared with wild type in both directions, although the kcat increased approximately four- and twofold for the dehydrogenase and reductase reactions, respectively (Table I). The constants for the L266E mutant were virtually identical to the F278E mutant, except that there was a ∼50% increase in Km for cortisone. In contrast, both the L279E and I275E mutants showed an increase in Km and kcat for dehydrogenase and reductase reactions (Table I).
Table I.
Kinetic Analysis of Human 11β-HSD1 Wild-Type and Mutant Proteins
| Dehydrogenase |
Reductase |
|||
|---|---|---|---|---|
| Mutant | Km (μM) | kcat (min−1) | Km (μM) | kcat (min−1) |
| Wild type | 7.05 ± 2.15 | 0.31 ± 0.04 | 8.95 ± 0.80 | 0.18 ± 0.00 |
| L266E | 6.63 ± 0.50 | 1.29 ± 0.03 | 15.26 ± 1.58 | 0.31 ± 0.01 |
| I275E | 11.68 ± 1.13 | 0.98 ± 0.04 | 27.93 ± 1.80 | 0.22 ± 0.01 |
| F278E | 6.16 ± 0.44 | 1.27 ± 0.03 | 10.61 ± 1.49 | 0.31 ± 0.01 |
| L279E | 12.57 ± 1.05 | 0.65 ± 0.00 | 18.93 ± 2.26 | 0.14 ± 0.01 |
| F278E/R137C | 4.82 ± 0.67 | 0.48 ± 0.02 | 15.42 ± 5.93 | 0.09 ± 0.01 |
Assays were carried out as per Materials and Methods. Km values are given in μM with kcat values in min−1 (mean ± SEM). Results were analyzed using nonlinear regression by VisualEnzymics (Softzymics).
Table II.
Kinetic Analysis of Guinea Pig 11β-HSD1 Wild-Type and Mutant Proteins
| Dehydrogenase |
Reductase |
|||
|---|---|---|---|---|
| Mutant | Km (μM) | kcat (min−1) | Km (μM) | kcat (min−1) |
| Wild Type | 459 ± 0.49 | 1.02 ± 0.04 | 4.09 ± 0.35 | 0.57 ± 0.01 |
| Y266E | 2.86 ± 0.22 | 0.47 ± 0.01 | 3.67 ± 0.58 | 0.20 ± 0.01 |
| I275E | 1.35 ± 0.40 | 0.24 ± 0.02 | 31.1 ± 9.8 | 0.37 ± 0.04 |
| F278E | 5.73 ± 1.13 | 1.12 ± 0.08 | 3.13 ± 0.63 | 0.48 ± 0.02 |
| L279E | 3.23 ± 0.29 | 0.53 ± 0.01 | 5.27 ± 1.33 | 0.41 ± 0.03 |
Assays were carried out as per Materials and Methods. Km values are given in μM with kcat values given in min−1 (mean ± SEM). Results were analyzed using nonlinear regression by VisualEnzymics (Softzymics).
Effect of naturally occurring mutations, R137C and K187N, on 11β-HSD1 activity
Recent sequencing efforts (Walker, personal communication) have revealed two naturally occurring mutations in the coding region of the human HSD11B1 gene, which result in K187N and R137C substitutions in the 11β-HSD1 protein sequence. Expression of the R137C and K187N mutants in the wild-type protein background did not yield any soluble protein in the recombinant E. coli system [Fig. 4(A)]. This was also true for the active-site mutant K187N in the human F278E background [Fig. 4(B)]. However, expression of the R137C mutation in the human F278E background did result in soluble protein, albeit with an 80% reduction in protein yield. Kinetic analysis of the R137C/F278E mutant showed a pronounced reduction of the kcat of the enzyme (compared with the F278E control), with little effect on the Km (Table I).
Figure 4.
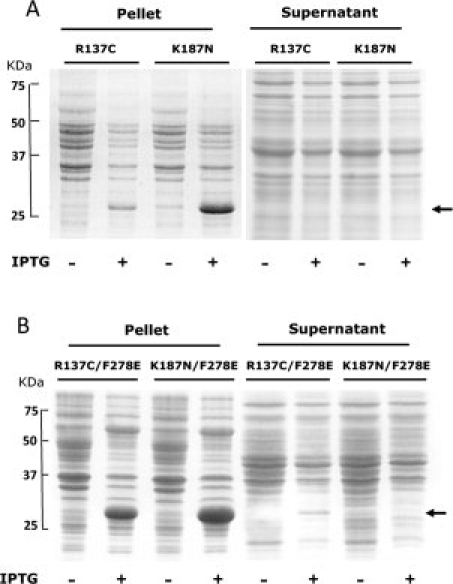
SDS-Page analysis of expression in E. coli of the human 11β-HSD1 mutants (R137C and K187N) in both wild-type (A) and F278E (B) backgrounds. (A) Insoluble (pellets) and soluble (supernatant) fractions from lysates of cultures expressing R137C and K187N mutants in the wild-type background. Expression levels of both recombinant proteins were low, and neither mutant was expressed in a soluble form. (B) Insoluble (pellets) and soluble (supernatant) fractions from lysates of cultures expressing the double mutants R137C/F278E and K187N/F278E. Arrows indicate position of the recombinant proteins. Although the protein produced was still largely insoluble, some soluble protein was evident in the supernatant for the F278E/R137C mutant.
Effect of mutations on aggregation state
Guinea pig and human F278E and WT proteins were analyzed by sedimentation analytical ultracentrifugation (AUC) to assess their aggregation state in vitro (see Fig. 5). Both the guinea pig and human wild-type proteins exhibited a rather broad peak with a maximum at around 200 kDa. Because the 11β-HSD1 dimer has a size of 68 and 64 kDa for the guinea pig and human enzymes, respectively, these data suggest that the recombinant wild-type proteins aggregate significantly in solution, with a size averaging around a hexamer. Interestingly, it was observed that the F278E mutation in the guinea pig enzyme converted the main species to a much narrower peak with a size indicative of a tetramer. This is shown by a peak at ∼120–140 kDa. The human F278E mutant at 0.5 mg mL−1 also showed a main peak at 130 kDa, but in addition had a second major peak at 65 kDa, indicating the majority of protein existed as tetramer with a lesser amount as a dimeric protein. To probe the equilibrium between the dimeric and tetrameric forms, three different concentrations of guinea pig and human F278E were analyzed by AUC (see Fig. 5). Dilution of the guinea pig F278E protein did not affect its aggregation state, with all dilutions showing one peak at around 140 kDa, indicating a stable tetrameric form. The human F278E mutant, however, showed a significant increase in the ratio of dimer to tetramer as the concentration of protein was decreased, indicating a concentration-dependent equilibrium between dimeric and tetrameric forms.
Figure 5.
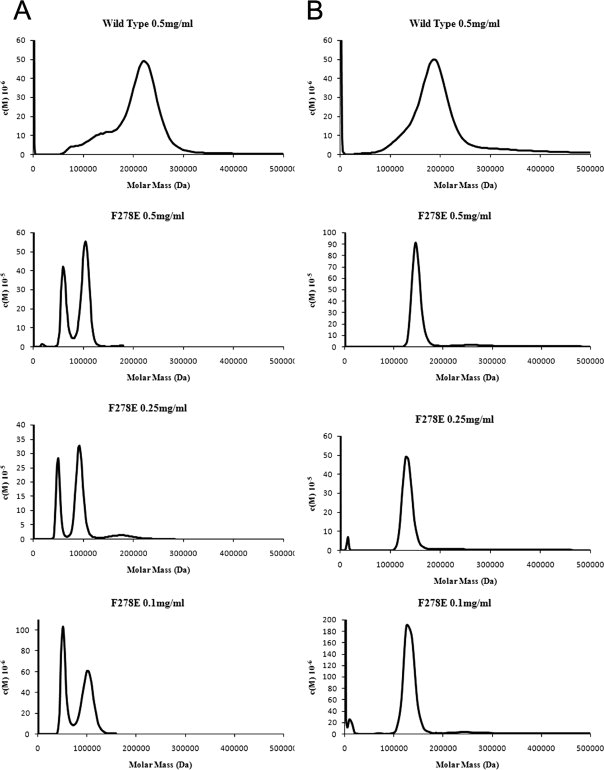
AUC analysis of the aggregation states of the human (A) and guinea pig (B) wild-type and F278E proteins; also shown is the effect of dilution on the oligomerization state of the human and guinea pig F278E enzymes. Wild-type proteins demonstrate marked heterogeneity and aggregation, whereas both F278E mutants appear far less aggregated, with the guinea pig enzyme resolving to a monodisperse tetramer, and the human protein existing as a concentration-dependent equilibrium of dimeric and tetrameric forms. Data were analyzed using SedFit27 with density, viscosity, and v-bar measurements calculated by Sednterp.
Crystal structure of guinea pig F278E
Because of a large yield of protein, we were able to determine the crystal structure of guinea pig F278E mutant protein to check for changes in structure. Crystals were grown as per Materials and Methods with the data collection and refinements statistics given in Table III. Each monomer in the guinea pig F278E structure had the same overall Rossman fold as the other published 11β-HSD1 structures14,24,28 and had a quaternary assembly of a tetramer made up of a dimer-of-dimers [Fig. 6(A)]. Analysis using the PISA server30 suggested tetramer to be the most probable quaternary structure for the guinea pig F278E protein, whereas the previously published wild-type structure (1XSE)24 was suggested to exist as a dimer. The structure of the guinea pig F278E dimer is largely identical to 1XSE with the dimers superimposable with a root mean square displacement (RMSD) of 0.48 Å RMS. Slight differences in the backbone exist in the loop region Glu221 to Pro234 and in the final helix from Leu267 to Ala291. One clear difference is evident between chains A and D and chains B and C in the guinea pig F278E structure [Fig. 6(B)]. In chains A and D, Tyr123 points toward the active site bound NADP+, whereas in chains B and C, the Tyr123 points away from the active site. Both orientations have good electron density in both the 2fofc map and the omit map calculated by PHENIX. Interestingly, strong electron density can be seen for NADP+ [Fig. 6(C)] even though none was added at any point in the purification or crystallization process. Our structure also shows that the C-terminal residue, Trp299, in each chain has bound into a hydrophobic pocket consisting of residues Phe129, Val152, Met155, Leu197, and Phe201. The position of this Trp residue has not been reported in any other 11β-HSD1 structure.
Table III.
X-Ray Data and Refinement Statistics
| Data collection statisticsa | |
|---|---|
| Cell parameters a, b, c (Å) | 78.1, 85.9, 176.3 |
| Space group | P212121 |
| Resolution (Å) | 2.2 (2.33–2.20) |
| Completeness (%) | 99.2 (96.6) |
| Multiplicity | 4.7 |
| I/σI | 11.43 (2.8) |
| No. of observations | 288,334 (39,957) |
| No. of unique observations | 60,915 (9432) |
| Rsym | 9.5 (54.5) |
| Refinement statistics | |
| Average B factor | 43.8 |
| No. of nonhydrogen atoms/waters | 9153/452 |
| RMSD bond (Å)/angle (°) | 0.005/0.923 |
| Ramachandran (%)b | 90.1/9.0/0.7/0.2 |
| R/Rfreec | 20.0/25.5 |
| PDB code | 3DWF |
Statistics as reported by XDS.
Percentage of nonglycine, nonproline residues in the core/allowed/generously allowed/disallowed regions of the Ramachandran plot.
Five percent of reflections set aside for cross-validation.
Figure 6.
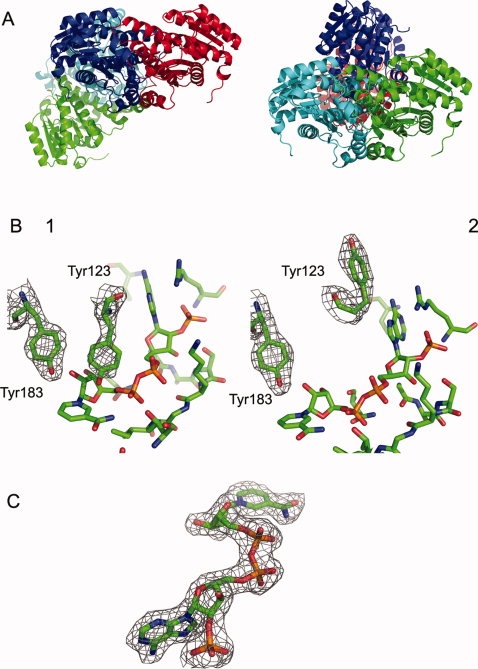
Crystal structure of the guinea pig F278E enzyme. (A) Tetrameric arrangement of the two 11βHSD1 dimers; the three-dimensional structure can be superimposed on the previously published guinea pig structure to a value of 0.48 Å RMSD. Two views are shown to illustrate the orientation of the two dimers. (B) The two orientations of Tyr123 seen in the active site of the guinea pig F278E mutant; one shows the Tyr123 residue pointing toward the active site, which would block substrate binding; the other shows the Tyr123 residue pointing away from the active site toward bulk solvent, which would allow substrate to bind. (C) The bound NADP+ molecule seen in all monomers of the guinea pig F278E tetramer. Map shown in (B) and (C) is the 2fofc map produced by Refmac5 to a value of 1.0 sigma. Image produced using PyMOL.29
Estimation of proportion of active molecules in the human F278E and wild-type enzymes
Because the crystal structure indicated the enzyme contained bound cofactor, we used fluorescence to monitor the single turnover of cofactor on addition of excess steroid to a known amount of protein, as a means to estimate the proportion of molecules that contained bound cofactor and were active. Addition of cortisol to enzyme resulted in a rapid rise in fluorescence because of conversion of endogenous NADP+ to NADPH, which reached a plateau from which the original concentration of NADP+ was estimated. By contrast, addition of cortisone resulted in no change in fluorescence, confirming that the cofactor was present in the enzyme in the oxidized form. Measurements on several preparations of the human F278E enzyme indicated that between 62 and 69% of the protein molecules contained NADP+ and were active. In contrast, measurements on wild-type enzyme yielded values of 20–48%.
Discussion
It has been hypothesized24 that because of the unusual charge distribution across the C-terminal helices in the 11β-HSD1 structures, this area could be involved in “membrane dipping,” with residues in this region forming a nonpolar plateau encircled by positively charged residues (see Fig. 1). This plateau could exist in the nonpolar center of the membrane, with the charged residues forming salt bridges with the displaced phospholipid and sulfolipid head groups. This conformation might increase accessibility of the enzyme to membrane-embedded steroid substrates. One way to test this hypothesis is to mutate the nonpolar residues in this region of 11β-HSD1 to the hydrophilic residue glutamic acid and observe the effect on activity of full-length protein in vivo. First, however, the effect of such mutations on enzyme activity and protein structure in vitro, in the absence of membranes, had to be established. For this purpose, we chose to introduce mutations into an N-terminally truncated, His-tagged enzyme, produced in the bacterial expression system we had previously developed and validated.17 C-terminal mutations introduced into the human enzyme were L266E, I275E, F278E, and L279E, whereas the equivalent guinea pig mutants created were Y266E, I275E, F278E, and L279E. On expression in E. coli, all mutations increased the yield of soluble 11β-HSD1, with human F278E and guinea pig I275E and F278E showing the most dramatic increases compared with wild-type protein (see Fig. 3). Presumably, the pronounced surface hydrophobicity of the C-terminal region of the wild-type protein enhances the chance of misfolding and aggregation under the high-level expression conditions of the bacterial expression system. The lack of membrane association via the N-terminal anchor may also compound the problem. The mutations to glutamate introduced here not only decrease hydrophobicity but also promote repulsion between these regions, and thus aid correct folding and solubility. This was evidenced by a reduced formation of insoluble inclusion bodies in addition to an increase in soluble protein production. A similar increase in solubility has been previously been engineered into several other proteins by mutation of surface hydrophobic residues31–33 often by substitution with negatively charged amino acids,34,35 indicating the general applicability of this approach to improving protein expression. Also of interest is that this study indicates that the wild-type guinea pig enzyme is naturally more soluble than its human counterpart. This may also be related, at least in part, to surface hydrophobicity of the C-terminal region, in that the guinea pig enzyme has a number of residues with reduced hydrophobicity in this area when compared with the human protein (e.g., Y266 vs. L266, R262 vs. L262).
Sedimentation AUC analysis of purified wild-type and mutant proteins endorsed the concept that aggregation was reduced by the C-terminal mutations (see Fig. 5). Both human and guinea pig wild-type recombinant proteins existed in solution as a collection of aggregated species, with broad peaks averaging ∼200 kDa. In contrast, the F278E mutants showed a much lower degree of aggregation and a greater degree of monodispersity, with the guinea pig F278E protein appearing to be a stable monodisperse tetramer and the human F278E protein existing as a concentration-dependent equilibrium of dimer and tetramer.
All the mutant proteins had similar kinetics (kcat and Km) for the steroid substrate as the wild type, in both dehydrogenase and reductase directions—a prerequisite for the use of these proteins in the analysis of the membrane-dipping hypothesis. The F278E mutants, in particular, seem promising for future studies. The guinea pig F278E protein showed unaltered substrate affinity or turnover rate in either reaction direction, while the human F278E protein also showed unaltered Km values, although, in common with the other mutations to this enzyme, the apparent kcat was significantly higher than wild type. Because it had previously been shown that bacterially expressed wild-type human 11β-HSD1 may have only ∼20% active enzyme molecules,36 it seemed likely that the increased specific activity seen with these mutations was due to an increased proportion of active molecules in these preparations. To get an estimate of the proportion of active molecules in our enzyme preparations, we used fluorescence to monitor the single turnover of enzyme-bound NADP+ to NADPH on addition of an excess of cortisol. This indicated that 62–69% of the human F278E protein molecules contained bound cofactor and were functional, whereas the proportion for the wild type was lower at 20–48%. This supports the hypothesis that the increased observed kcat of this mutant is at least in part because of an increased proportion of active molecules in the preparation. It is nevertheless possible that the decreased aggregation status of the mutant also contributes to the increase in turnover rates.
To investigate the effect of the C-terminal mutations on the structure of 11β-HSD1, the guinea pig F278E protein was crystallized. The structure of guinea pig F278E reported here is of a higher resolution with a lower Rfree than that of the previously published wild-type guinea pig structure (PDB code: 1XSE24). Of importance, the conditions required to crystallize the guinea pig F278E protein were different from those used previously; in particular, it was not necessary to include guanidinium hydrochloride to enhance monodispersity of the enzyme.24 The crystal structure we obtained indicated that the enzyme exists as a tetramer made up of a dimer-of-dimers [Fig. 6(A)]. This contrasts with the 1XSE structure, which is seen to exist as a single dimer. Analysis of our structure using the PISA server30 showed that the interface for the two dimers is sufficient to make the tetramer the most likely quaternary assembly, which is in agreement with the AUC findings that the guinea pig F278E protein exists as a stable tetramer in solution. Apart from this quaternary difference, the structure of each of the guinea pig F278E dimers is largely identical to 1XSE. Interestingly, all monomers of the F278E protein contained a well-defined NADP+ molecule, even though none was added during any step of the purification or crystallization [Fig. 6(C)]. This supports previous suggestions that cofactor is required for correct protein folding and conformational stability.36
The only significant difference that exists between the previously published 1XSE and the guinea pig F278E structure reported here is the orientation of the residue Tyr123. In the 1XSE structure, the Tyr123 in both chains A and B is pointed toward the bound NADP+ molecule. This would not be expected because in the active site of the murine 11β-HSD1 structure (PDB code: 1Y5R), the equivalent Gln123 residue is pointing away from the active site allowing the steroid substrate to bind.20 If the 1XSE structure is superimposed on 1Y5R, it can be seen that the Tyr123 in this orientation would sterically prevent glucocorticoid binding. To explain this orientation, Ogg et al. hypothesized that a conformational change must be a prerequisite for substrate binding.24 In our structure, two orientations of Tyr123 can be seen. In chains A and D, the Tyr123 is in the same orientation as the 1XSE structure, whereas in chains B and C, the Tyr123 is pointing away from the active site, toward bulk solvent [Fig. 6(B)]. These two conformations of Tyr123 have also been seen in an unpublished wild-type guinea pig 11β-HSD1 structure (Loh and Ding, personal communication). Each dimer, therefore, has one Tyr123 pointing into the active site and one Tyr123 pointing toward bulk solvent. Although this conformation is highly suggestive of cooperative kinetics for 11β-HSD1,13 the data from this study and others37–39 do not support this concept. The fact that the protein is not in its physiological environment could mean that the kinetics seen here and elsewhere are not indicative of the in-cell situation.
11β-HSD1 is involved in a rare form of polycystic ovary syndrome (PCOS) known as cortisone reductase deficiency (CRD), with two mutations having recently been found in the gene encoding 11β-HSD1 (HSD11B1) in patients presenting with CRD (Walker, personal communication). These mutations resulted in K187N or R137C substitutions in the protein sequence and, when expressed in a mammalian cell line, led to a reduction in enzyme activity. However, no kinetic data on these mutations have been reported. To assess the effect of these mutations on the kcat and Km of 11β-HSD1, these mutations were engineered into both the human WT and F278E backgrounds for expression and purification from bacteria. Expression of either mutant in the WT background did not yield any soluble protein, with bands only seen in the insoluble (pellet) fractions on SDS-PAGE analysis of bacterial lysates (see Fig. 4). Attempts to express the active-site mutant K187N in the human F278E background also resulted in a protein that would not express in a soluble form in the recombinant E. coli system. Because the K187 residue is known to be involved in cofactor binding,14,20,24,28 these results again suggest that the interaction with NADP(H) is required for correct folding and stability, at least in E. coli. This is supported by the observation that high-level expression of human 11β-HSD1 in a soluble form in bacteria requires addition of an inhibitor compound,36 for example, the steroid analogue carbenoxolone used in these studies, which also presumably requires prior cofactor binding.
Expression of the R137C mutation in the human F278E background resulted in an 80% reduction in expression with pronounced effects on the apparent kcat of the enzyme, but with little effect on the Km (Table I). This can perhaps be explained by the interference with the salt bridges between adjacent monomers. We assume that the disruption of the salt bridges between R137 of one monomer and E141 of the other leads to a certain amount of incorrect folding, leaving a protein of unchanged affinity but a reduced apparent kcat.
In summary, we have shown that mutations to the C-terminus of recombinant 11β-HSD1 can increase yield of soluble protein without adversely affecting activity. We have also shown, using one of the mutants as an example (guinea pig F278E), that these mutations have no adverse effects on structure compared with wild-type protein. We have also shown that the human F278E enzyme is a suitable background for analyzing the effects of naturally occurring mutations on enzyme activity/stability, which is not possible with the wild-type protein. Hence, we have engineered 11β-HSD1 mutants that will be useful, not only for probing membrane interactions but also for many other future biochemical and biophysical studies of the enzyme.
Materials and Methods
Cloning and expression of recombinant 11β-HSD1
A bacterial expression construct containing residues 24–292 of the human 11β-HSD1 protein was generated in pET28b(+) (Novagen) by PCR amplification from a previous construct.17 A guinea pig expression construct containing residues 24–300 was a kind gift from Dr. P Rejto (Pfizer, La Jolla, CA). Both constructs incorporated an N-terminal His6-tag to aid purification. QuikChange site-directed mutagenesis (Stratagene, La Jolla, CA) was used to make the mutations L266E, I275E, F278E, and L279E for the human protein and Y266E, I275E, F278E, and L279E for the guinea pig protein. In addition, mutant constructs were generated containing two previously identified naturally occurring mutations in the human 11β-HSD1 protein, namely R137C and K187N (Lavery, personal communication). These additional mutations were also generated in the human F278E construct, giving the double mutants F278E/R137C and F278E/K187N.
After verification, constructs were used to transform the BL21(DE3) E. coli expression strain (Novagen). E. coli BL21(DE3) cells carrying the human gene were cotransformed using the pBAD-ESL plasmid (gift of Dr. P Lund40), which contains the genes for the E. coli chaperonins GroEL/ES. For the expression of the guinea pig protein, cells were grown in LB broth supplemented with 30 μg/mL kanamycin with shaking (220 rpm) at 37°C. Cells were induced with isopropyl-β-d-thiogalactoside (IPTG,1 mM) when A600 had reached a value of 0.8–1. Cells were grown with shaking for a further 30 min at 37°C, then transferred to 15°C, and grown with shaking overnight. For the expression of the human protein, cells were grown with shaking in LB broth supplemented with 30 μg/mL kanamycin and 50 μg/mL ampicillin at 37°C. Arabinose (0.1% w/v) was added to the cultures at an A600 of 0.8–1 to induce the expression of the chaperonin proteins. Cells were grown with shaking for 1 h at 37°C, before addition of 1 mM IPTG. The 11β-HSD inhibitor carbenoxolone (CBX, 0.1 mM) was also added at this stage. Following a further incubation of 30 min at 37°C, cultures were transferred to 15°C and grown with shaking for 16 h.
Purification of 11β-HSD1
Cells were pelleted (3000g, 15 min) and then resuspended in Bugbuster reagent (Novagen) containing protease inhibitors (Mini-Complete EDTA free, Roche Molecular Biochemicals) and Benzonase DNase (Novagen). Cells were incubated with shaking at room temperature for 40 min and then cell debris was pelleted at 38,000g for 30 min at 4°C. Supernatant was loaded onto a His-Select (Sigma Aldrich) column and washed with buffer containing 25 mM sodium phosphate, 300 mM NaCl, 5% glycerol, 2 mM TCEP, and 0.005% Anapoe X-100 (Anatrace), pH 8.0. Loosely bound protein was washed off with three column volumes of the same buffer containing 17.5 mM imidazole. Protein was then eluted with three volumes of buffer containing 175 mM imidazole. Fractions containing protein were separated by size-exclusion chromatography on a Superdex-200 HR10/30 column (Pharmacia) running at 0.4 mL min−1 in 25 mM sodium phosphate, 5% glycerol, and 0.005% Anapoe X-100, pH 8.0.
Measurement of 11β-HSD1 activity
Dehydrogenase activity of 11β-HSD1 was assayed at 37°C in 25 mM sodium phosphate, pH 8.0, with 200 μM NADP+ and cortisol concentrations ranging from 0.5 to 32 μM, using a Perkin-Elmer LS-5 spectrofluorimeter. Excitation and emission wavelengths were 340 and 456 nm, respectively. Amount of enzyme added was adjusted so that a linear rate of reaction was obtained for 5 min after cortisol addition. A calibration curve was constructed using 0–1 μM NADPH. Reductase activity of 11β-HSD1 was assayed by measuring the cortisone to cortisol ratio using HPLC. In this reaction, an enzyme concentration that gave no more than 20% substrate conversion was incubated for 5 min at 37°C in 25 mM sodium phosphate, pH 8.0, containing 200 μM NADPH and an NADPH-regenerating system (6 mM MgCl2, 1 U/mL glucose-6-phosphate dehydrogenase (Sigma-Aldrich), 1 mM glucose-6-phosphate). Reactions were started by addition of 1–128 μM cortisone and incubated for 20 min. Reactions were terminated by addition of 3 mL dichloromethane. Samples were then centrifuged for 5 min at 1000g and the aqueous layer removed. After evaporation of the dichloromethane, samples were redissolved in 60 μL of 50% (v/v) aqueous acetonitrile and loaded onto an RP-HPLC system consisting of a Prevail Select C-18, 5 μm column (Grace), a GP50 gradient pump, and a UVD170S detector (Dionex). Samples were eluted with a gradient of 54–69% aqueous methanol over a period of 15 min. Substrate and product were monitored by UV absorbance at 242 nm with retention times of ∼11 and 12 min, respectively.
Measurement of turnover of enzyme-bound NADP+
To estimate the proportion of protein molecules with bound NADP+, human F27E or wild-type enzyme (1 μM) was incubated in 25 mM sodium phosphate pH 8.0 at 37°C, and the fluorescence of cofactor monitored as described earlier. An excess of cortisol was then swiftly added to a final concentration of 50 μM, and the single turnover of the NADP+ in the enzyme preparation monitored. The fluorescence increased and reached a plateau from which the original concentration of NADP+ could be estimated. The system was calibrated by subsequent addition of a known substoichimetric amount of NADP+ and monitoring the fluorescence increase. Control reactions with cortisone in place of cortisol showed no change in fluorescence.
Analytical ultracentrifugation
Sedimentation velocity measurements were made using a Beckman XLI analytical ultracentrifuge. Samples were centrifuged at 40,000 rpm in an X rotor at 4°C for 8 h. The concentration of protein within the cells was analyzed by scanning each cell at 280 nm, a total 200 scans being made during each run. These data were then processed using the C(M) model implemented in SEDFIT27 using parameters for protein and buffer determined using SEDNTERP.
Structure determination of the F278E mutant
Crystals were grown using the sitting-drop vapor diffusion method with a 10 mg/mL protein stock solution in 25 mM sodium phosphate, 5% glycerol, 0.005% Anapoe X-100, pH 8.0, equilibrated against a reservoir containing 0.1M Tris-HCl, 1.75M ammonium sulfate, and 5% glycerol (pH 8.0). The sitting drop contained 2 μL of protein solution and 2 μL of reservoir. No NADP+ was added during purification or crystallization. Crystals of the F278E 11β-HSD1 were soaked for 10 min in each of a series of artificial mother liquor solutions containing glycerol in 5% increments. In the final solution containing 25% glycerol, crystals were soaked for 45 min before being flash-cooled by plunging into liquid nitrogen. Data were collected at 100 K at the European Synchrotron Radiation Facility, Grenoble, France, using beam line ID14-1. All data were indexed, integrated, and scaled using XDS.41 Full data collection statistics are shown in Table III. The structure was determined by molecular replacement using the program Phaser with 1XSE as the search molecule. After initial refinement, the model was improved using Arp/wArp42 and then refined using phenix.refine.43 The refinement statistics are listed in Table III.
Acknowledgments
This work was funded by an MRC studentship (to AJL). The authors acknowledge Christine Loh, Yuan-Hua Ding, and Paul Rejto (Pfizer, La Jolla, CA) for the kind gift of the guinea pig expression construct, helpful initial discussions, and personal communications. They also acknowledge ESRF for help in data collection and for access to facilities.
References
- 1.Tomlinson JW, Walker EA, Bujalska IJ, Draper N, Lavery GG, Cooper MS, Hewison M, Stewart PM. 11 β-Hydroxysteroid dehydrogenase type 1: a tissue-specific regulator of glucocorticoid response. Endocr Rev. 2004;25:831–866. doi: 10.1210/er.2003-0031. [DOI] [PubMed] [Google Scholar]
- 2.Krozowski Z, Stuchbery S, White P, Monder C, Funder JW. Characterization of 11 β-hydroxysteroid dehydrogenase gene expression: identification of multiple unique forms of messenger ribonucleic acid in the rat kidney. Endocrinology. 1990;127:3009–3013. doi: 10.1210/endo-127-6-3009. [DOI] [PubMed] [Google Scholar]
- 3.Draper N, Stewart PM. 11 β-Hydroxysteroid dehydrogenase and the pre-receptor regulation of corticosteroid hormone action. J Endocrinol. 2005;186:251–271. doi: 10.1677/joe.1.06019. [DOI] [PubMed] [Google Scholar]
- 4.Tomlinson JW, Stewart PM. Mechanisms of disease: selective inhibition of 11 β-hydroxysteroid dehydrogenase type 1 as a novel treatment for the metabolic syndrome. Nat Clin Pract Endocrinol Metab. 2005;1:92–99. doi: 10.1038/ncpendmet0023. [DOI] [PubMed] [Google Scholar]
- 5.Masuzaki H, Paterson J, Shinyama H, Morton NM, Mullins JJ, Seckl JR, Flier JS. A transgenic model of visceral obesity and the metabolic syndrome. Science. 2001;294:2166–2170. doi: 10.1126/science.1066285. [DOI] [PubMed] [Google Scholar]
- 6.Masuzaki H, Yamamoto H, Kenyon CJ, Elmquist JK, Morton NM, Paterson JM, Shinyama H, Sharp MG, Fleming S, Mullins JJ, Seckl JR, Flier JS. Transgenic amplification of glucocorticoid action in adipose tissue causes high blood pressure in mice. J Clin Invest. 2003;112:83–90. doi: 10.1172/JCI17845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paterson JM, Morton NM, Fievet C, Kenyon CJ, Holmes MC, Staels B, Seckl JR, Mullins JJ. Metabolic syndrome without obesity: hepatic overexpression of 11 β-hydroxysteroid dehydrogenase type 1 in transgenic mice. Proc Natl Acad Sci USA. 2004;101:7088–7093. doi: 10.1073/pnas.0305524101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kotelevtsev Y, Holmes MC, Burchell A, Houston PM, Schmoll D, Jamieson P, Best R, Brown R, Edwards CR, Seckl JR, Mullins JJ. 11 β-Hydroxysteroid dehydrogenase type 1 knockout mice show attenuated glucocorticoid-inducible responses and resist hyperglycemia on obesity or stress. Proc Natl Acad Sci USA. 1997;94:14924–14929. doi: 10.1073/pnas.94.26.14924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morton NM, Holmes MC, Fievet C, Staels B, Tailleux A, Mullins JJ, Seckl JR. Improved lipid and lipoprotein profile, hepatic insulin sensitivity, and glucose tolerance in 11 β-hydroxysteroid dehydrogenase type 1 null mice. J Biol Chem. 2001;276:41293–41300. doi: 10.1074/jbc.M103676200. [DOI] [PubMed] [Google Scholar]
- 10.Alberts P, Nilsson C, Selen G, Engblom LO, Edling NH, Norling S, Klingstrom G, Larsson C, Forsgren M, Ashkzari M, Nilsson CE, Fiedler M, Bergqvist E, Ohman B, Björkstrand E, Abrahmsen LB. Selective inhibition of 11 β-hydroxysteroid dehydrogenase type 1 improves hepatic insulin sensitivity in hyperglycemic mice strains. Endocrinology. 2003;144:4755–4762. doi: 10.1210/en.2003-0344. [DOI] [PubMed] [Google Scholar]
- 11.Bhat BG, Hosea N, Fanjul A, Herrera J, Chapman J, Thalacker F, Stewart PM, Rejto P. Demonstration of proof of mechanism and pharmacokinetics and pharmacodynamic relationship with PF-915275, an inhibitor of 11{β}HSD1, in cynomolgus monkeys. J Pharmacol Exp Ther. 2007;5:5. doi: 10.1124/jpet.107.128280. [DOI] [PubMed] [Google Scholar]
- 12.Ozols J. Lumenal orientation and post-translational modifications of the liver microsomal 11 β-hydroxysteroid dehydrogenase. J Biol Chem. 1995;270:10360. [PubMed] [Google Scholar]
- 13.Maser E, Volker B, Friebertshauser J. 11 β-Hydroxysteroid dehydrogenase type 1 from human liver: dimerization and enzyme cooperativity support its postulated role as glucocorticoid reductase. Biochemistry. 2002;41:2459–2465. doi: 10.1021/bi015803t. [DOI] [PubMed] [Google Scholar]
- 14.Hosfield DJ, Wu Y, Skene RJ, Hilgers M, Jennings A, Snell GP, Aertgeerts K. Conformational flexibility in crystal structures of human 11 β-hydroxysteroid dehydrogenase type I provide insights into glucocorticoid interconversion and enzyme regulation. J Biol Chem. 2005;280:4639–4648. doi: 10.1074/jbc.M411104200. [DOI] [PubMed] [Google Scholar]
- 15.Mziaut H, Korza G, Hand AR, Gerard C, Ozols J. Targeting proteins to the lumen of endoplasmic reticulum using N-terminal domains of 11 β-hydroxysteroid dehydrogenase and the 50-kDa esterase. J Biol Chem. 1999;274:14122–14129. doi: 10.1074/jbc.274.20.14122. [DOI] [PubMed] [Google Scholar]
- 16.Odermatt A, Arnold P, Stauffer A, Frey BM, Frey FJ. The N-terminal anchor sequences of 11 β-hydroxysteroid dehydrogenases determine their orientation in the endoplasmic reticulum membrane. J Biol Chem. 1999;274:28762–28770. doi: 10.1074/jbc.274.40.28762. [DOI] [PubMed] [Google Scholar]
- 17.Walker EA, Clark AM, Hewison M, Ride JP, Stewart PM. Functional expression, characterization, and purification of the catalytic domain of human 11-β-hydroxysteroid dehydrogenase type 1. J Biol Chem. 2001;276:21343–21350. doi: 10.1074/jbc.M011142200. [DOI] [PubMed] [Google Scholar]
- 18.Filling C, Nordling E, Benach J, Berndt KD, Ladenstein R, Jornvall H, Oppermann U. Structural role of conserved Asn179 in the short-chain dehydrogenase/reductase scaffold. Biochem Biophys Res Commun. 2001;289:712–717. doi: 10.1006/bbrc.2001.6032. [DOI] [PubMed] [Google Scholar]
- 19.Blum A, Martin HJ, Maser E. Human 11 β-hydroxysteroid dehydrogenase type 1 is enzymatically active in its nonglycosylated form. Biochem Biophys Res Commun. 2000;276:428–434. doi: 10.1006/bbrc.2000.3491. [DOI] [PubMed] [Google Scholar]
- 20.Filling C, Berndt KD, Benach J, Knapp S, Prozorovski T, Nordling E, Ladenstein R, Jornvall H, Oppermann U. Critical residues for structure and catalysis in short-chain dehydrogenases/reductases. J Biol Chem. 2002;277:25677–25684. doi: 10.1074/jbc.M202160200. [DOI] [PubMed] [Google Scholar]
- 21.Oppermann UC, Filling C, Jornvall H. Forms and functions of human SDR enzymes. Chem Biol Interact. 2001;130-132:699–705. doi: 10.1016/s0009-2797(00)00301-x. [DOI] [PubMed] [Google Scholar]
- 22.Wimley WC, White SH. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat Struct Biol. 1996;3:842–848. doi: 10.1038/nsb1096-842. [DOI] [PubMed] [Google Scholar]
- 23.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 24.Ogg D, Elleby B, Norstrom C, Stefansson K, Abrahmsen L, Oppermann U, Svensson S. The crystal structure of guinea pig 11 β-hydroxysteroid dehydrogenase type 1 provides a model for enzyme-lipid bilayer interactions. J Biol Chem. 2005;280:3789–3794. doi: 10.1074/jbc.M412463200. [DOI] [PubMed] [Google Scholar]
- 25.Williams PA, Cosme J, Sridhar V, Johnson EF, McRee DE. Mammalian microsomal cytochrome P450 monooxygenase: structural adaptations for membrane binding and functional diversity. Mol Cell. 2000;5:121–131. doi: 10.1016/s1097-2765(00)80408-6. [DOI] [PubMed] [Google Scholar]
- 26.Pernecky SJ, Larson JR, Philpot RM, Coon MJ. Expression of truncated forms of liver microsomal P450 cytochromes 2B4 and 2E1 in Escherichia coli: influence of NH2-terminal region on localization in cytosol and membranes. Proc Natl Acad Sci USA. 1993;90:2651–2655. doi: 10.1073/pnas.90.7.2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang J, Osslund TD, Plant MH, Clogston CL, Nybo RE, Xiong F, Delaney JM, Jordan SR. Crystal structure of murine 11 β-hydroxysteroid dehydrogenase 1: an important therapeutic target for diabetes. Biochemistry. 2005;44:6948–6957. doi: 10.1021/bi047599q. [DOI] [PubMed] [Google Scholar]
- 29.DeLano WL. The PyMOL molecular graphics System. Palo Alto, CA: DeLano Scientific; 2002. [Google Scholar]
- 30.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 31.Sim J, Sim TS. Amino acid substitutions affecting protein solubility: high level expression of Streptomyces clavuligerus isopenicillin N synthase in Escherichia coli. J Mol Catal B. 1999;6:133–143. [Google Scholar]
- 32.Daujotyte D, Vilkaitis G, Manelyte L, Skalicky J, Szyperski T, Klimasauskas S. Solubility engineering of the HhaI methyltransferase. Protein Eng. 2003;16:295–301. doi: 10.1093/proeng/gzg034. [DOI] [PubMed] [Google Scholar]
- 33.Mosavi LK, Peng ZY. Structure-based substitutions for increased solubility of a designed protein. Protein Eng. 2003;16:739–745. doi: 10.1093/protein/gzg098. [DOI] [PubMed] [Google Scholar]
- 34.Dale GE, Broger C, Langen H, Darcy A, Stuber D. Improving protein solubility through rationally designed amino-acid replacements—solubilization of the trimethoprim-resistant type S1 dihydrofolate-reductase. Protein Eng. 1994;7:933–939. doi: 10.1093/protein/7.7.933. [DOI] [PubMed] [Google Scholar]
- 35.Nieba L, Honegger A, Krebber C, Pluckthun A. Disrupting the hydrophobic patches at the antibody variable/constant domain interface: improved in vivo folding and physical characterization of an engineered scFv fragment. Protein Eng. 1997;10:435–444. doi: 10.1093/protein/10.4.435. [DOI] [PubMed] [Google Scholar]
- 36.Elleby B, Svensson S, Wu X, Stefansson K, Nilsson J, Hallen D, Oppermann U, Abrahmsen L. High-level production and optimization of monodispersity of 11 β-hydroxysteroid dehydrogenase type 1. Biochim Biophys Acta. 2004;1700:199–207. doi: 10.1016/j.bbapap.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 37.Arampatzis S, Kadereit B, Schuster D, Balazs Z, Schweizer RA, Frey FJ, Langer T, Odermatt A. Comparative enzymology of 11 β-hydroxysteroid dehydrogenase type 1 from six species. J Mol Endocrinol. 2005;35:89–101. doi: 10.1677/jme.1.01736. [DOI] [PubMed] [Google Scholar]
- 38.Castro A, Zhu JX, Alton GR, Rejto P, Ermolieff J. Assay optimization and kinetic profile of the human and the rabbit isoforms of 11 β-HSD1. Biochem Biophys Res Commun. 2007;357:561–566. doi: 10.1016/j.bbrc.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 39.Shafqat N, Elleby B, Svensson S, Shafqat J, Jornvall H, Abrahmsen L, Oppermann U. Comparative enzymology of 11 β -hydroxysteroid dehydrogenase type 1 from glucocorticoid resistant (Guinea pig) versus sensitive (human) species. J Biol Chem. 2003;278:2030–2035. doi: 10.1074/jbc.M210135200. [DOI] [PubMed] [Google Scholar]
- 40.Walker A. 2000. In vivo functions and applications of molecular chaperones. PhD Thesis, University of Birmingham. [Google Scholar]
- 41.Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J Appl Cryst. 1993;26:795–800. [Google Scholar]
- 42.Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nat Struct Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 43.Afonine PV, Grosse-Kunstleve RW, Adams PD. 2005. The phenix refinement framework. CCP4 Newsletter 42 (Contribution 8)