

Abstract
A novel hierarchical MS2/MS3 database search algorithm has been developed to analyze MS2/MS3 phosphopeptides proteomic data. The algorithm is incorporated in an automated database search program, MassMatrix. The algorithm matches experimental MS2 spectra against a supplied protein database to determine candidate peptide matches. It then matches the corresponding experimental MS3 spectra against those candidate peptide matches. The MS2 and MS3 spectra are used in concert to arrive at peptide matches with overall higher confidence rather than combining MS2 and MS3 data searched separately. Receiver operating characteristic analysis showed that hierarchical MS2/MS3 database searches with MassMatrix had better sensitivity and specificity than the two-stage MS2/MS3 database searches obtained with MassMatrix, Mascot and X!Tandem. A greater number of true peptide matches at a given false rate were identified by use of this new algorithm for data collected on both LCQ and LTQ-FTICR mass spectrometers. The additional MS3 spectral data also improved the overall reliability and the number of true positives due to the fact that the true positives of the MS2/MS3 search results had higher scores than those of the MS2.
Keywords: Tandem Mass Spectrometry, Hierarchical MS2/MS3 Database Search, Phosphoproteomics
1 INTRODUCTION
Tandem mass spectrometry has been widely used in protein identification and characterization. In tandem mass spectrometry, the MS/MS or MS2 spectra produced by fragmentation of peptide ions contain product ion signatures that can be used to sequence the peptides and characterize their post-translational modifications (PTMs).[1] However, phosphopeptide MS2 spectra often do not contain sufficient sequencing information to identify the peptide. Poor sequencing of phosphopeptide ions is due to the labile nature of the phosphate group resulting in MS2 spectra that are dominated by the neutral loss of the phosphate moiety. As a result, phosphopeptides are not identified as reliably as non-phosphorylated peptides in LC-MS/MS experiments. To overcome the shortcomings of MS2 experiments for phosphopeptides, a third stage of mass spectrometry (MS3) can be performed on ions in the MS2 scans resulting from the neutral loss of phosphate. These fragment ions produce MS3 spectra with sufficient fragment ions to not only identify the peptide but often determine the site of phosphorylation.[2]
There are several de novo sequencing-based algorithms that have been developed for analysis of MS2 and MS3 spectral data.[3, 4] However, these algorithms can not be used in high-throughput data analysis due to their high computational expenses. Therefore, the analysis of MS2/MS3 experimental data for phosphopeptides relies primarily on database search algorithms, such as Mascot, SEQUEST, X!Tandem and OMSSA.[5–9] Often data analysis by database search programs is performed in two stages (Figure 1). In the first stage, MS2 spectral data are searched against a supplied protein database to obtain a set of peptide and protein identifications for the MS2 data. In the second stage, MS3 spectral data are also searched against the same protein database to obtain an additional set of peptide and protein identifications. Integration of these two sets of results is necessary to give the overall protein and peptide identifications. Ulintz et al has recently published an algorithm to integrate scores for the two matches of a set of MS2 and MS3 spectra.[2]
Figure 1.
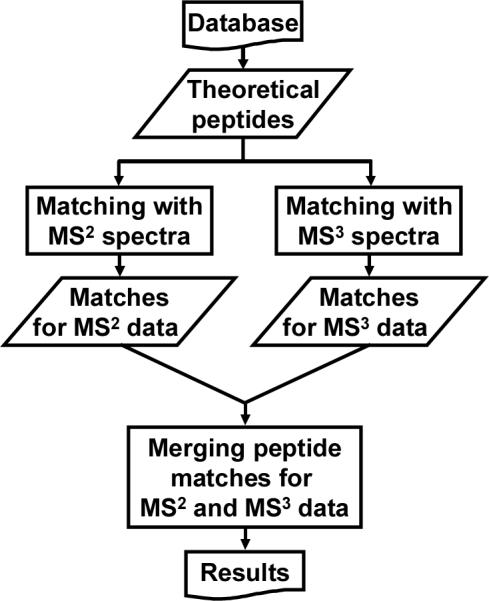
Diagram of two-stage MS2/MS3 database search process. The MS2 and MS3 data are searched in two parallel independent database searches. In the first database search stage, MS2 spectral data are searched against a protein database to give a set of peptide and protein identifications for the MS2 data. In the second stage, MS3 spectral data are searched against the same protein database to give the other set of peptide and protein identifications for the MS3 data. Peptide matches for the MS2 and MS3 data are then merged to give the final list of peptide matches for the data set by use of software tools.
Overall the integrated two-stage approach results in better results than analysis of MS2 and MS3 data separately. However, this approach does not take advantage of the inherent hierarchical nature of MS2 and MS3 data. In the two-stage search process, the fact that MS2 and MS3 spectra are created from the same peptide precursor ion following two consecutive fragmentations is ignored by the database search algorithm. Therefore, the MS2 and MS3 spectra for the same precursor may result in different peptide matches. Furthermore, for typical data sets collected on high mass accuracy capable mass spectrometers, the MS2 precursor ions are measured at high mass accuracy and the MS3 precursor ions are often measured at a much lower mass accuracy. In this case, MS3 spectral data do not fully exploit the benefit from the high mass accuracy of the original precursor ions.
In this manuscript we describe a novel algorithm for performing hierarchical MS2/MS3 database searches. This algorithm performs MS2/MS3 pattern matching analysis and returns peptide/protein identifications for each set of MS2/MS3 spectra. This approach does not use post-search merging of results from the MS2 and MS3 data that can lead to confounding peptide and protein matches. The algorithm first searches MS2 spectral data against a supplied protein database, and then searches the associated MS3 spectral data against candidate peptide matches obtained in the prior MS2 search. In this manner, MS2 and MS3 data are used in concert to arrive at peptide identifications. The MS2/MS3 search algorithm described herein takes full advantage of the hierarchical nature of the MS2/MS3 data. The hierarchical search process eliminates the discrepancy between the MS2 peptide matches and MS3 peptide matches that may occur in the two-stage search process. Furthermore, the high mass accuracy of the precursor ion for the MS2 experiment can be inherited by the MS3 data analysis in the hierarchical search algorithm resulted in overall improved confidence in peptide and protein identifications.
2 MATERIALS AND METHODS
2.1 Sample Preparation and Mass Spectrometry
α-Casein from bovine milk was purchased from Sigma-Aldrich (St. Louis, MO). The α-Casein was digested by trypsin in 25 mM ammonium biocarbonate buffer (pH = 8.0) at 37 °C for 1 hour. Enzymes were used in 50:1 ratio (substrate:enzyme). The tryptic digests were then dried and dissolved in HPLC water with 0.1% formic acid (pH = 3.0) to a final concentration of 1.0 μg/μl. The phosphopeptides in the solution were then enriched by use of a zirconium dioxide coated NuTips (Glugen Corp., Columbia, MD) as described by Kweon and Hakansson.[10] The resulted peptides were identified by use of data-dependent LC-MS3 on a LCQ Deca XP ion trap and a LTQ-FTICR mass spectrometer (Thermo Fisher, San Jose, CA, USA). 2.0 μL of enriched peptides with a total concentration of 1.0 μg/μL before enrichment was injected into the LC-MS system and eluted off the capillary HPLC column into the mass spectrometer with a linear gradient of 5% – 50% of mobile phase B over 28 minutes at a overall flow rate of ~250 nL/min. Solvent A was water with 0.1% formic acid and solvent B was acetenitrile with 0.1% formic acid. Ions were fragmented by use of collision induced dissociation (CID). The MS3 scan was targeted at phosphorylation neutral loss ions in the MS2 scan with a mass difference of 98.0 Da, 80.0 Da, 49.0 Da, 40 Da, 32.7 Da or 26.7 Da from the precursor mass.
2.2 Database Search and Search Parameters
The .RAW data files obtained from the LCQ Deca XP ion trap and LTQ-FTICR mass spectrometers were converted to mzXML files by use of ReAdW (http://tools.proteomecenter.org/ReAdW.php). For low mass accuracy data collected on the LCQ Deca XP ion trap mass spectrometer, LC-MS/MS spectra that were not derived from singly charged precursor ions were extracted as both doubly and triply charged precursors. For high mass accuracy data collected on the LTQ-FTICR mass spectrometer, isotope distributions for the precursor ions of the MS2 spectra were deconvoluted to obtain the charges and monoisotopic m/z values of the precursor ions by use of ReAdW. However, we found that the precursor m/z values for some MS3 spectra in the mzXML files created by ReAdW in this way were incorrect. Therefore, a Perl script, ReAdW_patch (www.massmatrix.net), was developed to address this problem with mzXML files. The ReAdW_patch uses an associated mzData file, which is created from the .RAW data file by use of Xcalibur program (Thermo Fisher, San Jose, CA) and contains correct precursor m/z values for all MS3 spectra, to fix the incorrect MS3 precursor m/z values in the mzXML file.
The hierarchical MS2/MS3 database searches of the mzXML files were performed by use of the online version of MassMatrix (www.massmatrix.net) with the following options: i) Variable modifications: Sodium adduct of Aspartic acid and Glutamic acid, Phosphorylation of Serine, Theronine and Tyrosine; ii) Enzyme: trypsin; iii) Missed Cleavages: 2; iv) Peptide Length: 6 to 42 amino acid residues; v) Mass tolerances of 2.0 Da and 10 ppm for the precursor ions on LCQ Deca XP ion trap and LTQ-FTICR mass spectrometers respectively; and vi) Mass tolerances of 0.8 Da for the product ions. The standard data sets were searched against a protein database containing both the target protein database (α-Casein) and a decoy reversed National Center for Biotechnology Information non-redundant (NCBInr) human database (96,997 decoy protein sequences). The hierarchical search algorithm was automatically enabled in the searches by MassMatrix when MS3 spectral data were detected in the input mzXML files.
The two data sets were also evaluated by use of two-stage MS2/MS3 database searches in MassMatrix, Mascot (www.matrixscience.com) and X!Tandem (www.thegpm.org/TANDEM/), in which the MS2 and MS3 data for a data set were searched in two parallel and separate database search processes against the same database. The MS2 and MS3 spectral data of an mzXML were extracted to two separate MGF files by use of the tools available at www.massmatrix.net. For MS2 data, precursor ion m/z values and charges in the mzXML were preserved during the extraction. For MS3 data, precursor ion m/z values in the mzXML were preserved and precursor ion charges for the MS3 were assumed to be the same as the precursor ion charges of their precursor MS2 spectra. This assumption is valid here because the MS3 experiments were targeted at phosphorylation neutral loss ions and those ions had the same charge state as their precursor ions. It was also found that best search results were obtained by use of this extraction approach. The MGF files containing MS2 and MS3 data for each experiment were then searched separately in MassMatrix, Mascot and X!Tandem against the same protein database as the one used in the hierarchical MS2/MS3 database searches. The search parameters for searches of the MS2 data were identical to those in the hierarchical MS2/MS3 data searches. For the MS3 data, the mass tolerances of precursor ions for both LCQ and LTQ-FTICR data sets were set to be 0.8 Da due to fact that the precursor ions of MS3 spectra were measured with the same mass accuracy as those product ions of MS2 spectra. In other words, both precursor and product ions of MS3spectra were measured with low mass accuracy even on the LTQ-FTICR mass spectrometer. Additional variable modifications, water loss of Serine and Theronine, were added to MS3 searches due to the facts that MS3 spectra were created from phosphorylation neutral loss ions and those ions have a mass difference of −18.0 Da from the original peptide. All other parameters in the MS3 searches were identical to those in the hierarchical MS2/MS3 data searches.
Results from MassMatrix and Mascot were output as html files. Results from X!Tandem were output as pepXML format. The scan number, charge, calculated mass, observed mass, mass difference, missed cleavages, score(s) and peptide sequence for each match from the three programs were extracted from the original output files into tab delimited TXT files by use of Perl scripts. All outputted peptide matches without any filtering from the three search programs were considered. The hierarchical MS2/MS3 searches in MassMatrix were performed in a single stage and thus merging of results was not required. For the two-stage MS2/MS3 searches in the three programs, the lists of peptide matches from the MS2 and MS3searches were merged by use of a Perl script. In brief, for each set of MS2 and MS3 spectra, a MS3 spectral peptide match was considered consistent to a MS2 spectral peptide match if the MS3 peptide sequence was the same as or a subsequence of the MS2 peptide sequence. Consistent MS2and MS3 peptide matches for the same set of MS2/MS3 spectra were combined as one peptide match with a score equal to the sum of scores of the MS2 and MS3matches for MassMatrix and Mascot or the product of expectation values of the MS2 and MS3 matches for X!Tandem. A MS2 or MS3 spectral peptide match without a consistent counterpart was also considered as a match for the set of MS2/MS3 spectra with its original score. Under the circumstances that there were multiple peptide matches for a set of MS2/MS3 from a search program after merging, the one with the highest score was considered as the best match. All the Perl scripts used for result extraction and merging have been made available at www.massmatrix.net.
Receiver operating characteristic (ROC) analysis was used to evaluate the search algorithms. The pp score in MassMatrix, score in Mascot, expectation value in X!Tandem were used for ROC analysis. The decoy reversed human database creates ~10,000 times more theoretical peptides as the target α-Casein protein sequences. Therefore, false positive matches from the α-Casein proteins were assumed to be negligible. Thus, the peptide matches returned from the target α-Casein proteins were considered as true positives (TPs) while those from the decoy reversed human proteins were considered as false positives (FPs). [11]
3 RESULTS AND DISCUSSION
3.1 Hierarchical MS2/MS3 Database Search Algorithm in MassMatrix
Figure 2 shows the diagram of the hierarchical MS2/MS3 database search algorithm in MassMatrix. Theoretical peptide ions are created from the protein database by in silico digestion, addition of posttranslational modifications with specified PTMs and fragmentation. MassMatrix then matches the experimental MS2 spectra to the theoretical peptide spectra to obtain candidate peptide matches. In this step, all peptide matches with theoretical precursor m/z values that match to the experimental MS2 precursor ions are considered as valid candidate peptide matches even if their MS2 product ion spectral quality is extremely poor and their scores are as low as 0. In the next step of the search process, MassMatrix matches the corresponding MS3 experimental spectra to the candidate peptide matches returned from the MS2 search. During this step, the precursor m/z value of a MS3 spectrum is matched against all product ions in the theoretical MS2 spectrum of a candidate peptide sequence. All potential MS3 precursor ions are then fragmented in silico and matched to the experimental MS3 spectrum. In some cases, multiple product ions in the theoretical MS2 spectrum, such as b/y product ions and b/y product ions with neutral losses, may match a particular MS3 precursor. The best match between the theoretical MS3 spectra and the experimental one is determined to be the one(s) with the highest statistical score(s).[12]
Figure 2.
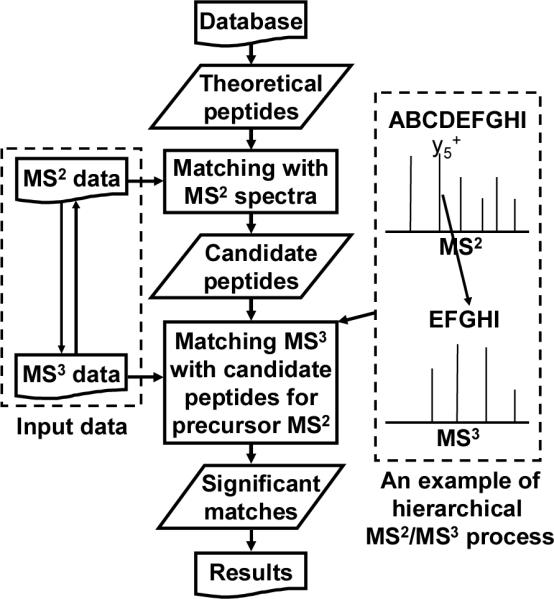
Diagram of hierarchical MS2/MS3 database search algorithm in MassMatrix. Theoretical peptides are created from the protein database by in silico digestion and modification with specified PTMs. MassMatrix first matches experimental MS2 spectra to the theoretical peptides to obtain candidate peptide matches. MassMatrix then matches the corresponding MS3 spectra against each candidate peptide matches from the MS2 search results. In this manner, the MS2 and MS3 spectral data are used in concert to obtain the peptide matches.
The pp score for a MS2 or MS3 spectrum match is defined as rhe negative common logarithm of the probability that the match is random for either the MS2 or MS3 spectrum.[12] The pp score for a match from a hierarchical MS2/MS3 search is defined as the negative common logarithm of the probability that the match is random with regard to both MS2 and MS3 spectrum, and is calculated by
| (2) |
The quality of a peptide match for a set of MS2/MS3 spectra is measured based on its final ppMS2/MS3 score instead of either ppMS2 or ppMS3 scores independently. Therefore, a set of MS2/MS3 spectra with either a MS2 or MS3 spectrum of good quality will still return a significant peptied.
The algorithm also accounts for the modification site localizations for peptides with PTMs. Under the circumstances where there are several potential peptide matches with the same sequence but different modification site localizations for a set of MS2/MS3 spectra, all peptides are considered as potential peptide matches in the MS2 step due to the fact that they have the same theoretical precursor MS2 m/z value. These peptide matches are then searched in the MS3 search step. During the MS3 search step, those matches may or may not generate the same sets of theoretical MS3 spectra. For those MS3 spectra from phosphorylation neutral loss ions, those matches generate different sets of theoretical MS3 spectra due to their different phosphate neutral loss site locations. For those MS3 spectra from a subsequence that does not contain the potential modification sites, those peptide matches will have the same sets of theoretical MS3 spectra. The theoretical MS3 spectra are matched to the experimental MS3 and scores between the matches and the MS3 spectrum are calculated. MassMatrix infers the specific modification site locations based on the final ppMS2/MS3 scores if the best match has much higher pp scores (Δpp > 6.0) than all other candidates. An example is shown in Figure 3 where a phosphopeptide containing multiple potential phosphorylation sites was identified with the specific phosphorylation site by the hierarchical MS2/MS3 search in MassMatrix. However, there are circumstances where several peptide matches may have very similar scores due to close proximity of modification sites or spectra resulting from a mixture of phosphopeptides. The user can specify whether the software will return all peptide matches or just the match with the highest ppMS2/MS3 score.
Figure 3.

Identification of a phosphorylation site on α-Casein-S2 by hierarchical MS2/MS3 database search. The figure demonstrates the capability of hierarchical search to localize the phosphorylation on a phosphopeptide containing multiple possible phosphorylation sites. The MS2 (top) and MS3 (bottom) spectra are shown to highlight the effect of neutral loss on the MS2 spectra. The detected phosphorylation site was previously reported by Kweon and Hakansson [10]. Neutral losses in the figure are labeled as follows: ~ (loss of phosphate moiety),* (loss of ammonia), ' (loss of water).
We must draw a careful distinction between our hierarchical MS2/MS3 approach and other two-stage approaches. At present there are no database search programs that directly handle MS3 spectral data. Rather researches have to treat MS3 spectral as an additional set of MS2 data, search MS3 spectral data separately in the same way as that for MS2, and then merge the results from MS2 and MS3 searches (a two-stage approach). However, in the two-stage analysis, there are two lists of candidate peptide matches for each pair of MS2/MS3 spectra. Each list is from the MS2 search or the MS3 search. The two lists of candidates output from the database search program can be different even when multiple candidate peptide matches are allowed for each spectrum due to either the low quality of the MS2 or the low quality of the MS3 spectrum.
Hierarchical MS2/MS3 is a natural and naive approach for analyzing DDNL MS3 spectra. The hierarchical approach does not simply use the MS2 precursor to narrow down the list of candidate peptides for MS3 spectra. In this algorithm, MS2 and MS3 spectra are searched in concert to obtain peptide matches with overall higher confidence instead of searched separately against the original protein database. Therefore, each pair of MS2/MS3 spectra has a single list of candidate peptide matches. The type of the ion in the MS2 spectrum that undergo fragmentation to create the MS3 spectrum can also be determined in addition to the peptide sequence. For peptides with MS2 spectra of poor quality due to the predominant neutral losses, such as phosphopeptides, their MS3 data may contain the necessary sequencing information to sequence the peptides and differentiate true and false positive matches.
The hierarchical MS2/MS3 search algorithm described herein is automated in MassMatrix and triggered by the program when MS3 spectra are detected in the input data file. This database search process is performed in a single stage and the match information for both MS2 and MS3 are reported in a single result file. The algorithm is generic and can be used to search the data from all types of MS2/MS3 experiments. The performance of the algorithm on MS2/MS3 proteomic data for phosphopeptides were evaluated and discussed in details in the following sections.
3.2 Evaluation of Hierarchical MS2/MS3 Database Search
The hierarchical MS2/MS3 database search algorithm was evaluated by searching two data sets for tryptic digests of α-Casein from an LCQ Deca XP mass spectrometer and a LTQ-FTICR mass spectrometer. The data sets were searched against a database with target α-Casein protein sequences and the reversed human database appended as decoy sequences. From the LCQ data set, 480 out of 1310 spectra were scored with potential peptide matches. From the LTQ-FTICR data set, 1382 out of 4747 spectra were scored with potential peptide matches. The complete lists of peptide matches for the two data sets are provided in supplementary tables 1 & 2.
The search results were evaluated and compared with those from the two-stage MS2/MS3 searches in MassMatrix, Mascot and X!Tandem by use of receiver operating characteristic (ROC) analysis.[8, 13, 14] Because false positive peptide matches from the target α-Casein proteins were considered negligible due to the large decoy database, peptide matches from α-Casein proteins were considered as TPs and those from the decoy database were considered as FPs.[11] ROC curves were created by plotting TP against FP as the score threshold decreased in the search results. The ROC curves for the hierarchical MS2/MS3 search results and those from the two-stage searches in MassMatrix, Mascot and X!Tandem are displayed in Figure 4.
Figure 4.

ROC analysis of search results from the hierarchical MS2/MS3 searches in MassMatrix and two stage MS2/MS3 searches in MassMatrix, Mascot and X!Tandem for a tryptic of αCasein: (a) all the peptide matches for the LCQ data set, (b) all the peptide matches for the LTQ-FTICR data set, (c) phosphopeptide matches for the LCQ data set, and (d) phosphopeptide matches for the LTQ-FTICR data set.
An ideal database search results should return all true positives with scores higher than all false positives and a ROC curve with a right angle. A ROC curve toward the left indicates higher specificity and a curve toward the top indicates higher sensitivity. It can be seen from Figure 4a that the hierarchical MS2/MS3 search had better overall sensitivity than the two-stage MS2/MS3 searches in MassMatrix, Mascot and X!Tandem for the LCQ proteomic data. Figure 4c shows the ROC results for the phosphopeptides only. After enrichment, phosphopeptides were of higher abundance than non-phosphorylated peptides. Due to the fact that only a small portion of the peptide matches from the LCQ data set were non-phosphorylated peptides, The ROC results for phosphopeptides in the LCQ data set (Figure 4c) were very similar to those for all the peptides (Figure 4a). For the LTQ-FTICR data set, there are many non-phosphorylated peptide matches of relatively poorer quality than phosphopeptides. The sensitivity and specificity of phosphopeptides in the data set were much higher than those of all the peptides as indicated by the ROC analysis (Figure 4d vs. Figure 4b). Since the MS3 experiments were targeted at phosphorylation neutral loss ions in the MS2 spectra, all the MS3 spectral data were presumably due to phosphopeptides. The improvement of the hierarchical MS2/MS3 search over the two-stage MS2/MS3 search in MassMatrix was not significant in the ROC analysis of all the peptides as shown in Figure 4b. However, the hierarchical MS2/MS3 search gained higher sensitivity and specificity than the two-stage MS2/MS3 searches in all three programs for the phosphopeptides as shown in Figure 4d.
Overall, the hierarchical MS2/MS3 searches performed in MassMatrix had improved sensitivities and specificities for the phosphopeptides than the two-stage MS2/MS3 searches in MassMatrix, Mascot and X!Tandem. At a false rate of 5%, the hierarchical MS2/MS3 search in MassMatrix returned 119 true positive phosphopeptides and the two stage MS2/MS3 searches in MassMatrix, Mascot and X!Tandem returned 112, 46 and 20 true phosphopeptides respectively for the LCQ data set. At the same false rate for the LTQ-FTICR data set, the hierarchical MS2/MS3 search in MassMatrix returned 394 true positive phosphopeptides and the two-stage MS2/MS3 searches in MassMatrix, Mascot and X!Tandem returned 305, 320, and 310 true phosphopeptides respectively. These results suggest the advantage of hierarchical MS2/MS3 database search algorithm over the two-stage MS2/MS3 search processes, especially for the data collect on LTQ-FTICR mass spectrometers.
There are two main factors that contribute to the higher sensitivities and specificities of the hierarchical MS2/MS3 search algorithm than the two-stage MS2/MS3 search approach. The first is that the hierarchical search process eliminates the discrepancy between the MS2 peptide matches and MS3 peptide matches that may occur in the two-stage search process. In the hierarchical search process, a set of MS2 and MS3 spectra is always used in concert to arrive at a peptide match (Figure 2). However, in the two-stage search process, the fact that a set of MS2 and MS3 spectra is created from the same peptide by use of two consecutive fragmentations is ignored during database searching (Figure 1). Therefore, the MS2 and MS3 spectra from the same original precursor ion may return two different peptide matches in the final merge of results. The other main advantage of the hierarchical search process is that the high mass accuracy of the precursor ion for the MS2 experiment is inherited by the MS3 data analysis. In a typical MS2/MS3 experiment performed on high mass accuracy capable mass spectrometers (LTQ-FTICR and LTQ-Orbitrap mass spectrometers), precursor ions for the MS2 spectra are measured with high mass accuracy (< 10 ppm), whereas the product ions for the MS2 spectra (including the MS3 precursor) and product ions for the MS3 spectra are all measured with a relative lower mass accuracy (0.5 ~ 1.0 Da). In the two-stage process as shown in Figure 1, the MS3 data have to be searched with low mass accuracies for both precursor and product ions and the advantage of high mass accuracy is lost for MS3 data analysis. However, the high mass accuracy for the MS2 precursor ions is inherited during the search process of MS3 spectral data in the hierarchical MS2/MS3 search process due to its hierarchical nature. In the hierarchical MS2/MS3 search process, MS3 spectra are only matched against the peptide candidates for their precursor MS2 spectra. In other words, those peptides must have masses that matched the higher mass accuracy MS2 precursor ion. In this way, the high accuracy of the MS2 precursor ion is naturally inherited during the MS3 database searching (Figure 2). Due to the second factor, the improvement of the hierarchical MS2/MS3 search over the two-stage MS2/MS3 search was more significant for the high mass accuracy LTQ-FTICR data than that for the low mass accuracy LCQ data (Figure 4c vs. Figure 4d).
3.3 Effect of MS3 on Score Distribution
Figures 5a & 5b show the pp score distributions of TPs and FPs for the hierarchical MS2/MS3 searches and the searches without considering any MS3 data in MassMatrix. The additional MS3 data had little effect on the score distributions of FPs due to the randomness of FPs. However, the score distributions of TPs shifted to higher values after including the MS3 data in the hierarchical search mode. This separation of score resulted in improved sensitivities and specificities of the search results. It also improved the overall reliability and the number of the true positive peptide matches due to the fact that they had higher statistical pp scores.
Figure 5.

Score distributions of TPs and FPs in the hierarchical MS2/MS3 searches and the searches of MS2 data only: (a) all the peptide matches returned for the LCQ data set, (b) all the peptide matches returned for the LTQ-FTICR data set, (c) phospopeptide matches for the LCQ data set, and (d) phosphopeptide matches for the LTQ-FTICR data set. The score distributions for TPs were split into two groups as labeled “1” and “2”.
The pp score distributions for TPs were split into two groups in the hierarchical MS2/MS3 search compared with those for TPs in the search without considering MS3 data (Figure 5). Group 1 represents peptides with good quality MS3 spectral matches (pp score ≥ 6.0) where their pp scores were improved significantly by including the MS3 data. Peptide matches in this group are well separated from FPs and can be identified with high sensitivity, specificity and reliability. Group 2 represents those peptides with moderate or poor quality MS3 spectral matches (pp score < 6.0) and those without any MS3 spectral matches. Their pp scores were only slightly improved or not improved at all.
Figures 5c & 5d show the pp score distributions for the phosphopeptides only. The pp score distributions for phosphopeptides from the LCQ data set (Figure 5a) were similar to those for all the peptides (Figure 5c). For the LTQ-FTICR data set, there were many non-phosphorylated peptide matches of relatively poorer quality than phosphopeptides. Because the MS3 targeted phosphopeptide ions, the score distribution of TPs for phosphopeptides was different from that for all the peptides as shown in Figures 5b & 5d. A great portion of the true positive phosphopeptide matches of the hierarchical MS2/MS3 search fell in group 1. These results suggest that the hierarchical MS3 experiments were effective for targeted phosphopeptide identification on both LCQ and LTQ-FTICR mass spectrometers.
4 CONCLUDING REMARKS
A novel algorithm to analyze hierarchical MS2/MS3 experiments was developed and automated in a database search program, MassMatrix. Due to the fact that the MS2 and MS3 spectral data are collected sequentially from the same peptide precursor ion, the hierarchical MS2/MS3 database search algorithm has advantages over the two-stage search algorithms in which MS2 and MS3 spectral data are searched independently. The hierarchical MS2/MS3 search algorithm takes full advantage of the hierarchical nature of the MS2/MS3 data. The hierarchical search process eliminates the discrepancy between the MS2 peptide matches and MS3 peptide matches that may occur in the two-stage search process. Furthermore, the high mass accuracy of the precursor ion for the MS2 experiment can be inherited by the MS3 data analysis in the hierarchical search algorithm.
The algorithm was evaluated and compared with the two-stage search approaches using the search programs MassMatrix, Mascot and X!Tandem. Receiver operating characteristic analysis showed that the hierarchical MS2/MS3 database search improved sensitivitiesties and specificities for phosphopeptides, especially for the data collect on an LTQ-FTICR mass spectrometer. At a false rate of 5%, the hierarchical MS2/MS3 search in MassMatrix returned 118 true positive phosphopeptides and the two-stage MS2/MS3 searches in MassMatrix, Mascot and X!Tandem returned 112, 39 and 13 true phosphopeptides respectively for the LCQ data set. At the same false rate for the LTQ-FTICR data set, the hierarchical MS2/MS3 search in MassMatrix returned 418 true positive phosphopeptides and the two-stage MS2/MS3 searches in MassMatrix, Mascot and X!Tandem returned 350, 370, and 321 true phosphopeptides respectively. Score distributions indicated that the additional MS3 data improved the overall reliability and the number of true positives due to the fact that the true positives of the MS2/MS3 search results had higher scores than those of the MS2 results.
Supplementary Material
ACKNOWLEDGEMENTS
The study was funded by the Ohio State University, the National Institutes of Health (CA107106, RR023647, CA101956), the V Foundation (AACR Translational Cancer Research Grant) and the Leukemia & Lymphoma Society (SCOR).
Glossary
Abbreviations
- MS/MS or MS2
second stage of tandem mass spectrometry
- MS3
third stage of tandem mass spectrometry
- PTM
post-translational modification
- CID
collision induced dissociation
- ROC
receiver operating characteristic
- TP
true positive
- FP
false positive
REFERENCES
- [1].Aebersold R, Goodlett DR. Mass spectrometry in proteomics. Chem.Rev. 2001;101:269–295. doi: 10.1021/cr990076h. [DOI] [PubMed] [Google Scholar]
- [2].Ulintz PJ, Bodenmiller B, Andrews PC, et al. Investigating MS2/MS3 matching statistics. Mol. Cell. Proteomics. 2008;7:71–87. doi: 10.1074/mcp.M700128-MCP200. [DOI] [PubMed] [Google Scholar]
- [3].Frank A, Pevzner P. PepNovo: de novo peptide sequencing via probabilistic network modeling. Anal. Chem. 2005;77:964–973. doi: 10.1021/ac048788h. [DOI] [PubMed] [Google Scholar]
- [4].Goodlett DR, Keller A, Watts JD, et al. Differential stable isotope labeling of peptide for quantitation and de novo sequence derivation. Rapid Commun. Mass Spectrom. 2001;15:1214–1221. doi: 10.1002/rcm.362. [DOI] [PubMed] [Google Scholar]
- [5].Sadygov RG, Cociorva DC, Yates JR. Large-scale database searching using tandem mass spectra: Looking up the answer in the back of the book. Nature Methods. 2004;1:195–202. doi: 10.1038/nmeth725. [DOI] [PubMed] [Google Scholar]
- [6].Perkins DN, Pappin DJC, Creasy DM, et al. Probability-based protein identification by searching sequence database using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- [7].Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- [8].Geer LY, Markey SP, Kowalak JA, et al. Open mass spectrometry search algorithm. J. Proteome Res. 2004;3:958–964. doi: 10.1021/pr0499491. [DOI] [PubMed] [Google Scholar]
- [9].Craig R, Cortens JP, Beavis R.c. Open source system for analyzing, validating, and storing protein identification data. J.proteome Res. 2004;3:1234–1242. doi: 10.1021/pr049882h. [DOI] [PubMed] [Google Scholar]
- [10].Kweon HK, Hakansson K. Selective zirconium dioxide-based enrichment of phosphorylated peptides for mass spectrometeric analysis. Anal. Chem. 2006;78:1743–1749. doi: 10.1021/ac0522355. [DOI] [PubMed] [Google Scholar]
- [11].Huttlin EL, Hegeman AD, Harms AC, et al. Prediction of error associated with false-positive rate determinantion for peptide identification in large-scale proteomics experiments using a combined reversed and forward peptide sequence database strategy. J. Proteome Res. 2007;6:392–398. doi: 10.1021/pr0603194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Xu H, Freitas AF. MassMatrix: A Database Searching Program for Rapid Characterization of Proteins and Peptides from Tandem Mass Spectrometry Data. BMC Bioinformatics. 2007;8:133. [Google Scholar]
- [13].Tabb DL, Saraf A, Yates JR. GutenTag: high-throughput sequence tagging via an empirically derived fragmentation model. Anal. Chem. 2003;75:6415–6421. doi: 10.1021/ac0347462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Elias JE, Haas W, Faherty BK, et al. Comparative evaluation of mass spectrometry platforms used in large-scale proteomics investigations. Nature Methods. 2005;2:647–648. doi: 10.1038/nmeth785. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.