

Abstract
SU5416 is a novel small molecule tyrosine kinase inhibitor of the VEGF receptors 1 and 2. A phase I dose escalation study stratified by concurrent use (stratum II) or absence (Stratum I) of enzyme-inducing anticonvulsant drugs was undertaken to estimate the maximum-tolerated dose (MTD) and to describe the toxicity profile of SU5416 in pediatric patients with refractory brain tumors. Dose escalations were conducted independently for stratum I starting at 110mg/m2 while stratum II started at 48mg/m2. Thirty-three eligible patients were treated on stratum I (n=23) and stratum II (n=10). Tumor types included 23 glial tumors, 4 neural tumors, 4 ependymomas and 2 choroid plexus carcinomas. The MTD in Stratum I was initially estimated to be 110mg/m2. The protocol was amended to determine the MTD after excluding transient AST elevation. Re-estimation of the MTD began at the 145mg/m2 dose level but due to development of SU5416 being stopped by the sponsor, the trial was closed before completion. The most serious drug-related toxicities were grade 3 liver enzyme abnormalities, arthralgia and hallucinations. The plasma pharmacokinetics of SU5416 was not significantly affected by the concurrent administration of enzyme-inducing anticonvulsant drugs. Mean values of the total body clearance, apparent volume of distribution, and terminal phase half-life of SU5416 for the 19 patients in Stratum I were 26.1 ± 12.5 liter/h/m2, 41.9 ± 21.4 liter/m2, and 1.11 ± 0.41 h, respectively. The plasma pharmacokinetics of SU5416 in children was similar to previously reported findings in adult cancer patients. Prolonged disease stabilization was observed in 4 of 16 stratum 1 patients.
Keywords: VEGF, Anti-angiogenesis, Brain tumor, SU5416
Introduction
In order to expand their mass, tumors must up-regulate the angiogenic cascade so that sufficient nutrients and oxygen are available to sustain tumor growth [1]. While many angiogenic cytokines have been identified in brain tumors [2], VEGF appears to play a dominant role, especially in malignant gliomas [3]. The degree of neovascularization is in fact one of the pathologic criteria on which tumor grade is assigned for astrocytic tumors and for which there is prognostic correlation [4]. In animal models, inhibition of VEGF or its receptors (VEGFR-1, 2) have resulted in decreased growth of CNS tumors [5, 6].
SU5416 (semaxanib) is a small organic molecule that exerts a biologic effect by inhibiting VEGF mediated VEGFR receptor auto-phosphorylation which is necessary for receptor signaling [7, 8]. This agent interferes with endothelial cell proliferation thus preventing both neovascularization and endothelial turnover. SU5416 mediated inhibition of proliferation has been observed for endothelial cells and for cell lines transduced for VEGF activated VEGFR-2 receptor signaling; SU5416 may function through the PI3/AKT kinase pathway [9]. The potent in vivo activity of SU5416 has been demonstrated in murine and human tumor cell line implant studies [10]. It does not penetrate the CSF, which is not required given its target within the vasculature [11]. SU5416 has demonstrated activity in orthotopicaly implanted CNS tumor models, supporting the notion that CNS penetration through the intact blood brain barrier (BBB) is not required [6, 12]. It has demonstrated synergistic activity when combined with traditional therapeutic modalities including irradiation [12] and cytotoxic chemotherapy [13, 14] as well as other angiogenic pathway inhibitors [15].
Adult clinical trials of SU5416 have provided important information regarding the toxicity, delivery methods and activity of this agent both alone and in combination with chemotherapy [16–23]. Some studies have failed to demonstrate activity for this agent, which may relate to the different angiogenic cascades in addition to VEGF available to tumors [24–26]. By contrast, tumors that are highly dependent on the VEGF pathway, such as those in patients with Von Hippel-Lindau, have demonstrated responses [27–29] while others have identified alterations in VEGF expression [30–32]. The MTD for SU5416 in adults with solid tumors was 145mg/m2 when given intravenously twice weekly. Dose-limiting toxicities included headache, nausea, and vomiting [16, 18, 23–25, 30]. Episodes of tumor associated bleeding or thrombosis have also been reported [26].
We here report the first experience of a small molecule inhibitor of the VEGF pathway in pediatric patients with central nervous system tumors.
Patients and Methods
Patient eligibility
Patients had to be < 22 years of age with a brain tumor refractory to standard therapy. Karnofsky or Lansky performance of at least 60%, a life expectancy of greater than 8 weeks and neurological deficits that were stable for a minimum of 1 week prior to study entry were required. Recovery from prior irradiation (3 months for craniospinal or 2 weeks from focal therapy) and chemotherapy (a minimum of 3 weeks; 6 weeks for nitrosourea) and off growth factors for 1 week was also required. Patients taking dexamethasone had to be on a stable dose for at least one week. Hematopoeitic function required ANC > 1,000/μl and platelets > 75,000/μl (transfusion independent); hemoglobin >9g/dl. In addition, creatinine ≤ 1.5 times normal or GFR > 70 ml/min/1.73m2, bilirubin within normal range; SGPT (ALT) and SGOT (AST) < 2.5X normal range for age were required. PT/PTT had to be ≤ 120% upper limit of normal and there could be no overt renal, hepatic, cardiac or pulmonary disease. Patients needed weight for height greater than the 3rd percentile and an albumin of greater than 3g/dl. Patients had to initiate therapy within fourteen (14) days of registration. Exclusion criteria included the concurrent use of any other anticancer or experimental drug therapy, the presence of uncontrolled infection, pregnancy or breast-feeding, a deep venous or arterial thrombosis (including pulmonary embolism) within the last three months prior to study entry, a history of myocardial infarction, severe or unstable angina, or severe peripheral vascular disease. Patients with known allergies to paclitaxel or any other agent that uses cremophor as the excipient found in SU5416 were excluded. Patients could not have any prior cerebral bleeds or prior use of stereotactic radiosurgery.
The institutional review board (IRBs) of each respective PBTC institution approved the protocol before patient enrollment and continuing approval was maintained throughout the study. Patients or their legal guardians gave written informed consent, and assent was obtained as appropriate at the time of enrollment.
Treatment plan
Patients were stratified according to those not receiving enzyme inducing anticonvulsant drugs (EIACD) (Stratum I) or those receiving EIACD (Stratum II). The dose of SU5416 was escalated independently in the two groups using a traditional 3+3 phase I design. SU5416 was given by IV infusion through a central venous catheter. Dose escalations were performed in approximately 33% increments. The observation time for determining dose-limiting toxicity (DLT) was one course (6 weeks). Patients who came off therapy for reasons other than toxicity before the DLT observation period was complete were replaced for purposes of determining the MTD. Patients missing more than 4 doses for reasons other than toxicity were also replaced. Planned dose levels were 110 (dose level 1 in stratum I), 145, 190, 250 and 330mg/m2/dose given by intravenous infusion twice weekly. Patients in stratum II started at dose level –3 (48mg/m2/dose) with planned escalations at 65 and 85mg/m2/dose before escalating at the same levels as in stratum I. Patients with a body surface area (BSA) less than 0.6m2 were dosed per kilogram by dividing the dose above by a factor of 30. As a result of the known toxicities of cremophor, a component of the vehicle used to solubilize SU5416 for parenteral administration, all patients also received diphenhydramine (or equivalent) and ranitidine (or equivalent) as well as an initial dose of 0.5mg/kg (max 10mg) dexamethasone for the first three infusions. This was weaned in patients tolerating the infusion. Dexamethasone was given orally 12 and 6 hours prior to the infusion of SU5416.
SU5416 was supplied by Sugen, Inc. (South San Francisco, CA), through the Cancer Therapy Evaluation Program of the National Cancer Institute Bethesda, MD. SU5416 was given at a concentration of 1.5mg/cc by intravenous administration at 100cc/hr twice weekly with at least 3 days between doses without breaks. A course was defined as 6 weeks of therapy, and the maximum duration of planned therapy was two years.
Pharmacokinetic analysis
The pharmacokinetic analysis evaluated the plasma concentration-time profile of SU5416 in pediatric brain tumor patients on day 1 and day 42 in consenting patients. Determination of changes in pharmacokinetic behavior with increasing and repeated dosing of SU5416 was also studied. We also sought to determine the interpatient variability in pharmacokinetic variables in patients treated at the MTD. Since many brain tumor patients require concurrent anti-seizure medication, we sought to evaluate whether the concurrent use of enzyme inducing anticonvulsant drugs alters the elimination of SU5416. Finally, we sought to establish the existence of correlations between pharmacokinetic variables and pretreatment laboratory values, toxicity, and indications of biological response.
The concentrations of SU5416 and its two major oxidative metabolites, the 5′-hydroxymethyl (SU9838) and 5′-carboxylic acid (SU6595) derivatives [33], were measured in plasma by high performance liquid chromatography with ultraviolet detection, as previously described, with minor modifications [34]. The lower range of the calibration curves was 5ng/mL (0.021 μM) for the parent drug, 5ng/mL (0.020 μM) for SU9838 and 10ng/mL (0.037 μM) for SU6595. Between-day accuracy and precision of the assay were calculated using data from a series of 12 independent calibration curves used for the analysis of study samples over a period of 3 months. At the lower range of the standard curves, accuracy expressed as the percentage of the difference between the mean calculated and nominal concentration was 8.6% for SU5416, 14.1% for SU9838, and 5.8% for SU6595. Corresponding values of the precision were 5.4% for SU5416, 9.5% for SU9838, and 10.3% for SU6595.
Actual sample times were calculated from the beginning of the drug infusion to the sample collection time. Plasma concentration-time profiles of SU5416 and its metabolites were analyzed individually for each patient by non-compartmental methods using the WinNonlin Version 4.0.1 software package (Pharsight Corp., Cary, NC). Area under the curve from time zero to the last sample with a measurable concentration AUC(0-t) was estimated using the linear/log trapezoidal method. Area under the curve from time zero to infinity (AUC) for SU5416 was calculated by adding the extrapolated area, given as the quotient of the observed plasma concentration at the last sample time to the estimated value of the slope of the log-linear terminal phase, to AUC(0-t). The relative extent of metabolism was calculated as the ratio of the AUC(0-t) for each metabolite to the parent drug expressed as a percentage [14, 35]. Values of the pharmacokinetic parameters at each dose level are reported as the geometric mean ± SD of the values for the individual patients [36]. Standard deviations for the geometric mean values were estimated by the jackknife method [37]. Parametric statistical tests of pharmacokinetic variables were performed after logarithmic transformation of the data [36, 38].
Response and Toxicity
Neuro-imaging evaluation included 3-dimensional MRI, single voxel MRS, and echoplanar perfusion/diffusion MRI, and FDG-PET. The imaging studies were analyzed by exploratory methods. Studies were conducted pre-therapy (within 2 weeks prior to the initiation of therapy), and at the end of course 1, 2, 4, 6 and 8 (at 6, 12, 24, 36 and 48 weeks of therapy) and were centrally reviewed through the PBTC Neuroimaging Center (NIC). MRI response criteria were defined as:
Complete Response (CR)
Complete disappearance on MR/CT of all enhancing tumor and mass effect, and off all corticosteroids (or receiving only adrenal replacement doses), accompanied by a stable or improving neurologic examination, and maintained for at least 6 weeks. If CSF was positive it must become negative.
Partial Response (PR)
Greater than or equal to 50% reduction in tumor size by bi-dimensional measurement or volumetric MR/CT, on a stable or decreasing dose of corticosteroids, accompanied by a stable or improving neurologic examination, and maintained for at least 6 weeks.
Stable Disease (SD)
Neurologic exam is at least stable and maintenance corticosteroid dose not increased, and MR/CT imaging meets neither the criteria for PR nor the criteria for Progressive Disease. For “Stable Disease” to be reported as a possible clinical benefit, it must have been maintained for a clinically appropriate interval (e.g. 12 weeks for recurrent high-grade gliomas).
Progressive Disease (PD)
Progressive neurologic abnormalities or worsening neurologic status not explained by causes unrelated to tumor progression (e.g., anticonvulsant or corticosteroid toxicity, electrolyte disturbances, sepsis, hyperglycemia, etc.), OR a greater than 25% increase in the bi-dimensional measurement or volumetric measurement on MR/CT, OR increasing doses of corticosteroids required to maintain stable neurologic status or imaging. Note: patients who met the above classic criteria for progressive disease were coded as having progressed but could remain on therapy unless the tumor size increased by > 50% if they had not previously responded (CR or PR) to this therapy. Since the biologic effect of anti-angiogenic agents may not occur immediately, and tumors will continue to grow until enough vascular supply is damaged to limit further growth, this approach allowed for a more appropriate evaluation of response.
Dose-limiting toxicity was based on the Common Toxicity Criteria (CTC), version 2.0 and included any grade 3 or 4 non-myelotoxicity, grade 4 neutropenia of any duration, grade 3 thrombocytopenia of any duration, or any grade 3 hepatic toxicity except liver transaminase elevation < 10x upper limit of normal for one week or less. One dose of SU5416 could be held while waiting for liver transaminase levels to return to ≤ grade 1. If the level did not return to ≤ grade 1 before two scheduled doses, the event was considered a DLT. All grade 3 hypersensitivity reactions occurring with maximal pre-medication and all grade 4 reactions resulted in discontinuation of therapy. These were not included in the definition of MTD and these patients were replaced on the study. The MTD was defined at the dose at which one or none of six patients experienced a DLT and the next highest dose level was too toxic (two or more of three or six patients experienced a DLT).
Statistical Considerations
Progression-free survival (PFS) in this phase I trial could only be measured from the date of treatment initiation to the off treatment date or last disease assessment if within 30 days of the last treatment. A failure in the Kaplan-Meier estimate of PFS was either classical disease progression or death. Patients not failing in the time interval for measuring PFS were censored as of their last date of contact in this interval. All eligible patients who received any SU5416 are included in the estimates of PFS. Cox proportional hazards models were used to investigate possible associations between biology and imaging covariates and PFS; provided at least 10 failures were available per covariate being explored. This restriction was imposed due to the well known spurious results when fewer events are analyzed with Cox models. Due to distributions of the variables being skewed, log transformations of the variables were analyzed in the models.
Results
A total of 33 eligible patients were enrolled on this phase I dose escalation study; 23 in stratum I and 10 in stratum II (see Table I). Three patients in stratum I were not evaluable for dose finding: one patient died prior to receiving therapy, one patient withdrew from the study before completing the toxicity observation period and one patient experienced hypersensitivity reaction to the drug. Two patients in stratum II were not evaluable for dose finding: one patient died (unrelated to treatment) on day four of treatment and one patient experienced hypersensitivity reaction on initial treatment.
Table I.
Patient Characteristics
| Stratum I (N=23) | Stratum II (N=10) | |||
|---|---|---|---|---|
| Age at Study Entry (Years) | ||||
| Median (Minimum, Maximum) | 8.5 (2.2, 21.0) | 10.6 (1.5,16.4) | ||
| Number | % | Number | % | |
| SEX | ||||
| Males | 15 | 65.2 | 5 | 50.0 |
| Females | 8 | 34.8 | 5 | 50.0 |
| RACIAL CATEGORY | ||||
| Asian | 0 | 0.0 | 1 | 10.0 |
| Black | 4 | 17.4 | 3 | 30.0 |
| Unknown | 0 | 0.0 | 1 | 10.0 |
| White, Hispanic | 2 | 8.7 | 0 | 0.0 |
| White, Non-Hispanic | 17 | 73.9 | 5 | 50.0 |
| DIAGNOSIS | ||||
| Astrocytoma, Anaplastic | 3 | 13.0 | 2 | 20.0 |
| Astrocytoma, Nos | 1 | 4.3 | 0 | 0.0 |
| Brain Stem Glioma | 3 | 13.0 | 0 | 0.0 |
| Choroid Plexus Carcinoma | 1 | 4.3 | 1 | 10.0 |
| Ependymoma, Nos | 4 | 17.4 | 0 | 0.0 |
| Ganglioglioma | 1 | 4.3 | 1 | 10.0 |
| Glioblastoma Multiforme | 4 | 17.4 | 2 | 20.0 |
| Glioma, Malignant | 2 | 8.7 | 0 | 0.0 |
| Pilocytic Astrocytoma | 3 | 13.0 | 1 | 10.0 |
| Pineoblastoma | 0 | 0.0 | 2 | 20.0 |
| Primitive Neuroectodermal Tumor | 1 | 4.3 | 0 | 0.0 |
| Rhabdoid Sarcoma | 0 | 0.0 | 1 | 10.0 |
The dose levels studied and the numbers of patients treated at each dose level for each stratum are shown in Table II. Two of five patients treated at 145mg/m2 experienced DLTs (one patient experienced grade 3 transaminase elevations (SGOT & SGPT) and one patient experienced grade 3 hallucinations) thus establishing the MTD at 110mg/m2 in stratum I As planned in the study design, six additional patients were treated at 110mg/m2 to better describe the toxicity profile of SU5416.
Table II.
Dose Escalation History And DLTs During Course I
| Strata | Dose (mg/m2) | No. of Patients Entered | No. of Patients Evaluable | No. of DLTs | DLTs |
|---|---|---|---|---|---|
| I | 110 | 3 | 3 | 1 | Grade 3 SGOT(AST) |
| 110 | 4 | 3 | 0 | ||
| 145 | 3 | 3 | 1 | Grade 3 Hallucinations | |
| 145 | 3 | 2 | 1 | Grade 3 SGOT(AST) and SGPT(ALT) | |
| MTD Established at 110 mg/m2 | |||||
| 110 | 6 | 0 | 0 | Six additional patients were treated at the MTD to better describe toxicity | |
| Protocol modified to 1) investigate the relationship between the occurrence of hallucinations and SU5416 and 2) to eliminate transient liver transaminase levels as DLT – accrual re-opens at the 145 dose level | |||||
| 145 | 4 | 3 | 0 | ||
| Protocol closed to accrual due to drug availability | |||||
| II | 48 | 3 | 3 | 0 | |
| 65 | 3 | 2 | 1 | Grade 3 SGPT(ALT) | |
| Protocol modified to 1) investigate the relationship between the occurrence of hallucinations and SU5416 and 2) to eliminate transient liver transaminase levels as DLT – accrual of additional patients at the 65 dose level | |||||
| 65 | 4 | 3 | 1 | Grade 3 Headache | |
| Protocol closed to accrual due to drug availability | |||||
Based on the reversible nature of the transaminase elevations in the patients treated with SU5416, the protocol was amended to allow transient elevation of AST or ALT to <10 times normal for ≤ 1 week duration. Furthermore, the patient who experienced grade 3 hallucinations at 145mg/m2 was also receiving dexamethasone and morphine that have been associated with hallucinations. Thus, given the unclear role of SU5416 in what had been called a DLT for grade 3 hallucinations and the reversible nature of elevated transaminases, the protocol was amended in October 2001 and accrual reopened. Three of four additional patients treated in stratum I at 145mg/m2 were evaluable and none experienced DLTs. Three of four patients were treated in stratum II at 65mg/m2 and one developed a DLT (grade 3 headache).
In June 2002, the PBTC was notified by the National Cancer Institute Division of Cancer Treatment and Diagnosis that based on results characterized by a relative lack of clinical activity in several single agent phase I/II studies sponsored by SUGEN, INC/Pharmacia Corporation discontinued the development of the agent. Thus this trial was closed.
Significant adverse events experienced during the dose-finding period (first course) for all eligible patients are reported in Table II. Significant adverse events observed during subsequent cycles are also included and are similar to those observed in adults (Table III).
Table III.
Frequency of Grade 3 or 4 Adverse Events (Number of Patients) Attributed to SU5416 Observed in All Eligible Patients (n=33).
| Stratum I | Stratum II | |||
|---|---|---|---|---|
| Course 1 | Course 2–10 | Course 1 | Course 2–10 | |
| Lymphopenia | 1 (1) | 7 (1) | ||
| Allergic reaction/Hypersensitivity | 1 (1) | |||
| SGOT (AST) | 2 (2) | |||
| SGPT (ALT) | 1 (1) | 1 (1) | ||
| Hypophosphatemia | 3 (2) | |||
| Hallucinations | 1 (1) | |||
| Arthralgia (joint pain) | 1 (1) | 1 (1) | ||
| Headache | 1 (1) | 3 (2) | ||
| Rash | 1 (1) | |||
Tumor Response
Radiographic response was evaluated after course 1 in patients who completed the first course and were evaluated for response are reported in Table IV. No objectives responses were observed in either stratum. However, 4 of 16 [25% (95% CI: 9%–50%)] patients evaluable for response in stratum I (three at completion of the first on therapy scan, and 1 on the 2nd scan) had sustained stable disease for a sufficient period to be considered of possible clinical benefit of treatment (Table V). The stable disease durations were 16.9 weeks (GBM treated at 110 mg/m2); 63.1 weeks (GBM treated at 110 mg/m2); 40.4 weeks (AA treated at 110 mg/m2); 14.4 weeks (Astrocytoma, NOS treated at 110 mg/m2). Of the 6 evaluable patients in stratum II, a single patient [17% (95% CI: 1%–60%)] experienced stable disease at a sufficient duration to be considered to represent a possible clinical benefit (Table V). This patient had a choriod plexus carcinoma; stable disease was maintained for 56.1 weeks.
Table IV.
Radiographic response after course 1
| Stratum I | Stratum II | |
|---|---|---|
| CR or PR | 0 | 0 |
| SD | 6 (38%) | 1 (17%) |
| PD | 10 (63%) | 5 (83%) |
| Total Evaluable | 16 | 6 |
| Total Not Included in Response Assessment | 7 | 4 |
Six patients in stratum I were not evaluable for response because of absence of measurable tumor at baseline (n=1), withdrawal prior to initiation of therapy (n=1), off therapy for DLT before the first radiographic assessment (n=2), off therapy for allergic reaction to the drug (n=1), development of a secondary cancer – lung tumor (n=1) and inability to perform the disease assessment (n=1). Four stratum II patients were inevaluable for response for off therapy for DLT prior to the first radiographic assessment (n=2), off therapy for allergic reaction to drug (n=1) and death unrelated to treatment or primary cancer (n=1).
Table V.
Percent change in tumor size in patients with prolonged stable disease
| Histology | Tumor Location | Stable Disease Duration (months) | Timing of the Scan (Months from Treatment) | Optimal sequences measured | Tumor size | Change from baseline (%) | |
|---|---|---|---|---|---|---|---|
| Pt 1 | Glioblastoma Multiforme | Spinal Cord | 5.3 | Baseline | Enhancement | 4.4a | |
| 1.4 | 4.4 | 0 | |||||
| 2.6 | 4.6 | ↑ 5 | |||||
| 5.3 | 4.6 | ↑ 5 | |||||
| Pt 3 | Choroid Plexus Carcinoma | Cerebrum | 14.3 | Baseline | Enhancement | 1.21a | |
| 1.3 | 1.30 | ↑ 7 | |||||
| 3.1 | 1.46 | ↑ 21 | |||||
| 5.9 | 1.48 | ↑ 22 | |||||
| 7.0 | 1.31 | ↑ 8 | |||||
| 11.4 | 1.22 | ↑ 1 | |||||
| 14.3 | 1.33 | ↑ 10 | |||||
| Pt 8 | Glioblastoma Multiforme | Parietal lobe | 15.9 | Baseline | FLAIR | 32.7b | |
| 2.7 | 35.5 | ↑ 9 | |||||
| 8.1 | 29.1 | ↓ 11 | |||||
| 9.7 | 23.9 | ↓ 27 | |||||
| 11.1 | 30.5 | ↓ 7 | |||||
| 15.9 | 28.9 | ↓ 12 | |||||
| Pt 19 | Astrocytoma, Anaplastic | Frontal lobe | 10.6 | Baseline | Enhancement | 104.2b | |
| 1.3 | 95.3 | ↓ 9 | |||||
| 2.7 | 75.1 | ↓ 28 | |||||
| 3.9 | 76.0 | ↓ 27 | |||||
| 5.4 | 75.9 | ↓ 27 | |||||
| 6.8 | 65.8 | ↓ 37 | |||||
| 9.5 | 80.0 | ↓ 23 | |||||
| Pt 26 | Astrocytoma, NOS | Supratentorial, NOS | 5.9 | Baseline | Enhancement | 28.1a | |
| 2.6 | 31.9 | ↑ 14 | |||||
| 4.4 | 34.7 | ↑ 23 | |||||
| 5.9 | 34.7 | ↑ 23 |
Two-dimensional measurement (cm2);
Three-dimensional measurement (cm3)
Pharmacokinetics
Pharmacokinetic data for the first dose of SU5416 was available for 19 patients in the non-EIACD arm (stratum I) treated with doses of 110mg/m2 (13 patients) and145 mg/m2 (6 patients). Mean plasma concentration-time profiles of SU5416 and its two major phase I metabolites, SU9838 and SU6595, for the group of patients receiving the initial weekly dose of 110mg/m2 are shown in Figure 1. The concentration of SU6595, the carboxylic acid metabolite, became similar to the parent drug shortly after the end of the infusion and remained as such throughout the sampling interval, which concluded at 7 hours relative to the starting time of the infusion. Plasma levels of SU9838, the hydroxymethyl metabolite that is a precursor to SU6595, were more than 10-times lower than SU6595 at all sample times. Mean pharmacokinetic parameters of SU5416 and the relative extent of its metabolism to SU9838 and SU6595, calculated as the ratio of the AUC of the metabolite to parent drug expressed as a percentage, are presented in Table VI. Results of the statistical comparisons that were performed to assess the effect of dose, duration of treatment, and use of EIACDs on the pharmacokinetics and metabolism of SU5416 are summarized in Supplemental Table SI. Analysis of the data for the first weekly dose of SU5416 identified no significant differences between the mean values of the pharmacokinetic parameters or the relative extent of metabolism to SU9838 and SU6595 at the two dose levels evaluated in the non-EIACD stratum. Pharmacokinetic data for the first dose of SU5416 was obtained from only 9 patients in the EIACD stratum at doses of 35 (1 patient), 45 (2 patients) and 65 mg/m2 (6 patients). The anticonvuslants used in these patients were carbamazepine (4 patients), phenytoin (2 patients), carbamazepine and phenytoin (1 patient), oxcarbazepine (2 patients), and phenobarbital (1 patient). Pharmacokinetic data was available from an insufficient number of patients at the two lower doses to permit statistical comparisons between the three dose levels. Nevertheless, in consideration of the relatively small number of patients in the EIACD stratum, mean values of the pharmacokinetic data from all 9 patients were calculated to enhance the power of statistical comparisons to assess the influence of concurrently administered EIACDs on the disposition and metabolism of SU5416. Mean values of the pharmacokinetic parameters and relative extent of metabolism for patients in the EIACD stratum were very similar to those for the non-EIACD stratum, with no statistically significant differences between the two treatment groups. Finally, for patients in the non-EIACD stratum, differences between the mean data for the sixth weekly dose (13 patients) and the first dose of SU5416 did not achieve statistical significance.
Figure 1.
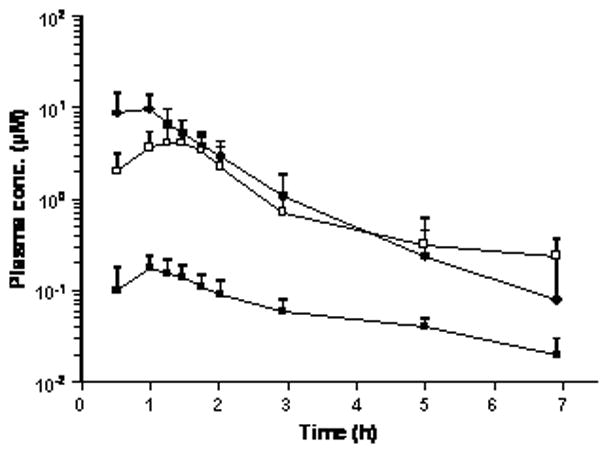
Mean plasma concentration-time profiles of SU5416 (●) and its two major metabolites, SU9838 (■) and SU6595 (□), for the initial weekly dose of 110 mg/m2 SU5416 in the group of 13 patients who were not taking enzyme inducing anticonvulsant drugs. Error bars depict the SD of the mean concentrations.
Table VI.
Pharmacokinetic parameters of SU5416 and its relative extent of metabolism.
| SU5416 |
Relative extent of metabolism (%) |
|||||||
|---|---|---|---|---|---|---|---|---|
| Dose (mg/m2) | Week no. | No. of patients | CL (liter/h/m2) | Vz (liter/m2) | t1/2,z (h) | |||
| Stratum | SU9838 | SU6595 | ||||||
| I | 110 | 1 | 13 | 28.7 ± 12.8a | 43.3 ± 20.3 | 1.04 ± 0.39 | 3.95 ± 1.59 | 57.4 ± 36.4 |
| 145 | 1 | 6 | 21.3 ± 11.5 | 39.1 ± 24.8 | 1.27 ± 0.41 | 3.98 ± 2.13 | 37.4 ± 25.3 | |
| All | 1 | 19 | 26.1 ± 12.5 | 41.9 ± 21.4 | 1.11 ± 0.41 | 3.96 ± 1.68 | 50.1 ± 33.2 | |
| I | 110 | 6 | 10 | 38.0 ± 28.7 | 65.5 ± 26.7 | 1.15 ± 0.59 | 4.41 ± 2.28 | 72.9 ± 43.9 |
| 145 | 6 | 3 | 26.1 ± 17.1 | 37.2 ± 33.5 | 0.99 ± 0.38 | 5.24 ± 0.81 | 72.4 ± 26.1 | |
| All | 6 | 13 | 34.9 ± 24.9 | 57.5 ± 34.6 | 1.11 ± 0.53 | 4.59 ± 2.11 | 72.8 ± 39.1 | |
| II | 35 | 1 | 1 | 33.1 | 23.5 | 0.49 | 2.44 | 55.2 |
| 48 | 1 | 2 | 48.0 ± 5.1 | 74.3 ± 71.7 | 1.07 ± 0.90 | 4.07 ± 1.37 | 26.4 ± 1.1 | |
| 65 | 1 | 6 | 27.8 ± 11.4 | 45.5 ± 26.7 | 1.13 ± 0.62 | 5.72 ± 3.41 | 57.3 ± 20.4 | |
| All | 1 | 9 | 32.0 ± 13.1 | 47.1 ± 30.6 | 1.02 ± 0.60 | 4.82 ± 2.69 | 48.1 ± 21.3 | |
Values are mean ± SD.
Discussion
A number of studies of SU5416 utilized in adults, either alone or in combination with traditional therapy, have been published [16–20, 24, 25, 30, 31]. We here report the results of the first pediatric phase I study of a small molecule inhibitor of VEGF in patients with central nervous system tumors. Due to the known pathways involved in the metabolism of this compound, patients were stratified by the presence or absence of the use of enzyme inducing anticonvulsant drugs (EIACDs).
Dose-limiting toxicities were observed at 145mg/m2/dose in stratum I, consisting predominantly of grade 3 transaminase elevations, in addition to a single grade 3 episode of hallucinations. These toxicities may have been related to cremophor rather than SU5416. The starting dose of 110mg/m2/dose was based on initiating pediatric clinic trials at 80% of the adult MTD, which has been identified at 145mg/m2/dose.
The pharmacokinetics of SU5416 in adult cancer patients has been characterized in several phase I clinical trials [17, 20, 34]. These studies have shown that SU5416 is rapidly eliminated from plasma and that pharmacokinetic parameters determined for the first infusion of drug appear to be independent of the administered dose. At doses ranging from 20 to 145 mg/m2, mean values of the apparent terminal phase half-life ranged from 0.58 to 0.87 h and the total body clearance ranged from 25.8 to 44.8 liter/h/m2 in the studies that have been reported. The clearance of SU5416 increased by more than 50% upon repeated administration when the interval between successive doses was less than 7 days [20]. Hepatic metabolism by cytochrome P450 (CYP) enzymes is an important route of elimination for SU5416, with CYP3A4 being the major isozyme catalyzing the initial oxidation of the compound [33, 39]. The increased clearance observed with daily and twice weekly dosing results from the induction of CYP3A4 and may be due to either the SU5416 itself or the co-administration of dexamethasone used to prevent hypersensitivity reactions [20].
The pharmacokinetic behavior of SU5416 has not been previously studied in children or patients with CNS malignancies. The total body clearance for the initial dose of SU5416 in pediatric cancer patients who were not receiving EIACDs, 26.1 ± 12.5 liter/h/m2, was within the range of reported values for adults. Similarly, as observed in the clinical studies of the drug in adult patients, repeated administration at a dosing interval of 7 days did not result in any significant changes in the pharmacokinetics of SU5416. Moreover, there was no evidence to suggest that the pharmacokinetics of SU5416 was affected by the concomitant use of EIACDs. The absence of a clinically significant pharmacokinetic interaction between these anticonvulsants and SU5416 is indeed surprising in consideration of the presumably prominent role of CYP3A4 in the elimination of SU5416, as evidenced by the autoinduction of its clearance upon frequent repeated administration. Increased systemic clearance of other chemotherapeutic agents that are metabolized by CYP3A4 in brain cancer patients receiving these anticonvulsants includes the epipodophyllotoxins, vinca alkaloids, taxanes, and the camptothecins [40].
The toxicity profile of SU5416 is similar to that reported in adult clinical trials with this agent, as well as other trials with drugs that are dissolved in cremaphor. Severe grade 3 headache, which was prevalent in the adult protocols assessing SU5416, was less frequent in the pediatric population, and was only reported in the first two cycles, especially in those not receiving appropriate pre-medication. A number of toxicities were the result of the cremophor used to solubilize SU5416, although only two significant allergic reactions were reported, in contrast to what was observed in adults. The significant toxicity of cremophor may have prevented a significant dose escalation of SU5416, thus limiting the latter’s activity as evaluated in this phase I trial. In spite of this potential limitation, SU5416 demonstrated potentially interesting activity as measured by stable disease of sufficient duration to suggest clinical benefit in several of the patients treated. Such findings may be meaningful in a phase I dose escalation study of this nature with a heterogeneous population of patients and tumor types. The clinical benefit observed in this protocol was related to prolonged disease stabilization, which is an effect similar to that with SU5416 in pre-clinical models.
The clinical development of SU5416 has been halted, in part due to the limited single agent activity of this highly specific VEGFR inhibitor, as well as the significant toxicity related to the SU5416 dependant co-administration in cremaphor. The results reported herein represent the first demonstration of a small molecule inhibitor of angiogenesis used in pediatric patients. We were able to demonstrate that pediatric patients tolerate dose levels of this drug in a range that is similar to that observed in adults. Furthermore, there was significant overlap in the pharmacokinetic and toxicity profiles between the two populations. As new VEGF signal inhibitory molecules become available, these results will be used to guide these newer trials in terms of identifying activity.
Supplementary Material
Acknowledgments
This work was supported in part by NIH grant U01 CA81457 for the Pediatric Brain Tumor Consortium (PBTC) and American Lebanese Syrian Associated Charities.
References
- 1.Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285(21):1182–1186. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 2.Harrigan MR. Angiogenic factors in the central nervous system. Neurosurgery. 2003;53(3):639–660. doi: 10.1227/01.neu.0000079575.09923.59. discussion 660–631. [DOI] [PubMed] [Google Scholar]
- 3.Zhou YH, Tan F, Hess KR, et al. The expression of PAX6, PTEN, vascular endothelial growth factor, and epidermal growth factor receptor in gliomas: relationship to tumor grade and survival. Clin Cancer Res. 2003;9(9):3369–3375. [PubMed] [Google Scholar]
- 4.Kleihues P, Cavenee W. World Health Organization Classification of Tumours. Lyon: IARC Press; 2000. Pathology and Genetics of Tumours of the Nervous System. [Google Scholar]
- 5.Sandstrom M, Johansson M, Andersson U, et al. The tyrosine kinase inhibitor ZD6474 inhibits tumour growth in an intracerebral rat glioma model. Br J Cancer. 2004;91(6):1174–1180. doi: 10.1038/sj.bjc.6602108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takamoto T, Sasaki M, Kuno T, et al. Flk-1 specific kinase inhibitor (SU5416) inhibited the growth of GS-9L glioma in rat brain and prolonged the survival. Kobe J Med Sci. 2001;47(4):181–191. [PubMed] [Google Scholar]
- 7.Smith JK, Mamoon NM, Duhe RJ. Emerging roles of targeted small molecule protein-tyrosine kinase inhibitors in cancer therapy. Oncol Res. 2004;14(4–5):175–225. doi: 10.3727/000000003772462298. [DOI] [PubMed] [Google Scholar]
- 8.Fong TA, Shawver LK, Sun L, et al. SU5416 is a potent and selective inhibitor of the vascular endothelial growth factor receptor (Flk-1/KDR) that inhibits tyrosine kinase catalysis, tumor vascularization, and growth of multiple tumor types. Cancer Res. 1999;59(1):99–106. [PubMed] [Google Scholar]
- 9.Zhong XS, Zheng JZ, Reed E, et al. SU5416 inhibited VEGF and HIF-1alpha expression through the PI3K/AKT/p70S6K1 signaling pathway. Biochem Biophys Res Commun. 2004;324(2):471–480. doi: 10.1016/j.bbrc.2004.09.082. [DOI] [PubMed] [Google Scholar]
- 10.Litz J, Sakuntala Warshamana-Greene G, Sulanke G, et al. The multi-targeted kinase inhibitor SU5416 inhibits small cell lung cancer growth and angiogenesis, in part by blocking Kit-mediated VEGF expression. Lung Cancer. 2004;46(3):283–291. doi: 10.1016/j.lungcan.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 11.Renbarger J, Aleksic A, McGuffey L, et al. Plasma and cerebrospinal fluid pharmacokinetics of SU5416 after intravenous administration in nonhuman primates. Cancer Chemother Pharmacol. 2004;53(1):39–42. doi: 10.1007/s00280-003-0683-z. [DOI] [PubMed] [Google Scholar]
- 12.Lund EL, Olsen MW, Lipson KE, et al. Improved effect of an antiangiogenic tyrosine kinase inhibitor (SU5416) by combinations with fractionated radiotherapy or low molecular weight heparin. Neoplasia. 2003;5(2):155–160. doi: 10.1016/s1476-5586(03)80007-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhong X, Li QQ, Reed E. SU5416 sensitizes ovarian cancer cells to cisplatin through inhibition of nucleotide excision repair. Cell Mol Life Sci. 2003;60(4):794–802. doi: 10.1007/s00018-003-3002-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ma J, Li S, Reed K, et al. Pharmacodynamic-mediated effects of the angiogenesis inhibitor SU5416 on the tumor disposition of temozolomide in subcutaneous and intracerebral glioma xenograft models. J Pharmacol Exp Ther. 2003;305(3):833–839. doi: 10.1124/jpet.102.048587. [DOI] [PubMed] [Google Scholar]
- 15.Strieth S, Eichhorn ME, Sutter A, et al. Antiangiogenic combination tumor therapy blocking alpha(v)-integrins and VEGF-receptor-2 increases therapeutic effects in vivo. Int J Cancer. 2006;119(2):423–431. doi: 10.1002/ijc.21838. [DOI] [PubMed] [Google Scholar]
- 16.Fiedler W, Mesters R, Tinnefeld H, et al. A phase 2 clinical study of SU5416 in patients with refractory acute myeloid leukemia. Blood. 2003;102(8):2763–2767. doi: 10.1182/blood-2002-10-2998. [DOI] [PubMed] [Google Scholar]
- 17.Cooney MM, Tserng KY, Makar V, et al. A phase IB clinical and pharmacokinetic study of the angiogenesis inhibitor SU5416 and paclitaxel in recurrent or metastatic carcinoma of the head and neck. Cancer Chemother Pharmacol. 2005;55(3):295–300. doi: 10.1007/s00280-004-0871-5. [DOI] [PubMed] [Google Scholar]
- 18.Peterson AC, Swiger S, Stadler WM, et al. Phase II study of the Flk-1 tyrosine kinase inhibitor SU5416 in advanced melanoma. Clin Cancer Res. 2004;10(12 Pt 1):4048–4054. doi: 10.1158/1078-0432.CCR-03-0766. [DOI] [PubMed] [Google Scholar]
- 19.Giles FJ, Stopeck AT, Silverman LR, et al. SU5416, a small molecule tyrosine kinase receptor inhibitor, has biologic activity in patients with refractory acute myeloid leukemia or myelodysplastic syndromes. Blood. 2003;102(3):795–801. doi: 10.1182/blood-2002-10-3023. [DOI] [PubMed] [Google Scholar]
- 20.Stopeck A, Sheldon M, Vahedian M, et al. Results of a Phase I dose-escalating study of the antiangiogenic agent, SU5416, in patients with advanced malignancies. Clin Cancer Res. 2002;8(9):2798–2805. [PubMed] [Google Scholar]
- 21.Mesters RM, Padro T, Bieker R, et al. Stable remission after administration of the receptor tyrosine kinase inhibitor SU5416 in a patient with refractory acute myeloid leukemia. Blood. 2001;98(1):241–243. doi: 10.1182/blood.v98.1.241. [DOI] [PubMed] [Google Scholar]
- 22.Lockhart AC, Cropp GF, Berlin JD, et al. Phase I/pilot study of SU5416 (semaxinib) in combination with irinotecan/bolus 5-FU/LV (IFL) in patients with metastatic colorectal cancer. Am J Clin Oncol. 2006;29(2):109–115. doi: 10.1097/01.coc.0000199882.53545.ac. [DOI] [PubMed] [Google Scholar]
- 23.O’Donnell A, Padhani A, Hayes C, et al. A Phase I study of the angiogenesis inhibitor SU5416 (semaxanib) in solid tumours, incorporating dynamic contrast MR pharmacodynamic end points. Br J Cancer. 2005;93(8):876–883. doi: 10.1038/sj.bjc.6602797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heymach JV, Desai J, Manola J, et al. Phase II study of the antiangiogenic agent SU5416 in patients with advanced soft tissue sarcomas. Clin Cancer Res. 2004;10(17):5732–5740. doi: 10.1158/1078-0432.CCR-04-0157. [DOI] [PubMed] [Google Scholar]
- 25.Stadler WM, Cao D, Vogelzang NJ, et al. A randomized Phase II trial of the antiangiogenic agent SU5416 in hormone-refractory prostate cancer. Clin Cancer Res. 2004;10(10):3365–3370. doi: 10.1158/1078-0432.CCR-03-0404. [DOI] [PubMed] [Google Scholar]
- 26.Fury MG, Zahalsky A, Wong R, et al. A Phase II study of SU5416 in patients with advanced or recurrent head and neck cancers. Invest New Drugs. 2007;25(2):165–172. doi: 10.1007/s10637-006-9011-x. [DOI] [PubMed] [Google Scholar]
- 27.Jennens RR, Rosenthal MA, Lindeman GJ, et al. Complete radiological and metabolic response of metastatic renal cell carcinoma to SU5416 (semaxanib) in a patient with probable von Hippel-Lindau syndrome. Urol Oncol. 2004;22(3):193–196. doi: 10.1016/j.urolonc.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 28.Girmens JF, Erginay A, Massin P, et al. Treatment of von Hippel-Lindau retinal hemangioblastoma by the vascular endothelial growth factor receptor inhibitor SU5416 is more effective for associated macular edema than for hemangioblastomas. Am J Ophthalmol. 2003;136(1):194–196. doi: 10.1016/s0002-9394(03)00101-6. [DOI] [PubMed] [Google Scholar]
- 29.Aiello LP, George DJ, Cahill MT, et al. Rapid and durable recovery of visual function in a patient with von hippel-lindau syndrome after systemic therapy with vascular endothelial growth factor receptor inhibitor su5416. Ophthalmology. 2002;109(9):1745–1751. doi: 10.1016/s0161-6420(02)01159-4. [DOI] [PubMed] [Google Scholar]
- 30.Zangari M, Anaissie E, Stopeck A, et al. Phase II study of SU5416, a small molecule vascular endothelial growth factor tyrosine kinase receptor inhibitor, in patients with refractory multiple myeloma. Clin Cancer Res. 2004;10(1 Pt 1):88–95. doi: 10.1158/1078-0432.ccr-0221-3. [DOI] [PubMed] [Google Scholar]
- 31.Lara PN, Jr, Quinn DI, Margolin K, et al. SU5416 plus interferon alpha in advanced renal cell carcinoma: a phase II California Cancer Consortium Study with biological and imaging correlates of angiogenesis inhibition. Clin Cancer Res. 2003;9(13):4772–4781. [PubMed] [Google Scholar]
- 32.Loges S, Tinnefeld H, Metzner A, et al. Downregulation of VEGF-A, STAT5 and AKT in acute myeloid leukemia blasts of patients treated with SU5416. Leuk Lymphoma. 2006;47(12):2601–2609. doi: 10.1080/10428190600948253. [DOI] [PubMed] [Google Scholar]
- 33.Antonian L, Zhang H, Yang C, et al. Biotransformation of the anti-angiogenic compound SU5416. Drug Metab Dispos. 2000;28(12):1505–1512. [PubMed] [Google Scholar]
- 34.Kuenen BC, Rosen L, Smit EF, et al. Dose-finding and pharmacokinetic study of cisplatin, gemcitabine, and SU5416 in patients with solid tumors. J Clin Oncol. 2002;20(6):1657–1667. doi: 10.1200/JCO.2002.20.6.1657. [DOI] [PubMed] [Google Scholar]
- 35.Rivory LP, Haaz MC, Canal P, et al. Pharmacokinetic interrelationships of irinotecan (CPT-11) and its three major plasma metabolites in patients enrolled in phase I/II trials. Clin Cancer Res. 1997;3(8):1261–1266. [PubMed] [Google Scholar]
- 36.Mizuta E, Tsubotani A. Preparation of mean drug concentration--time curves in plasma. A study on the frequency distribution of pharmacokinetic parameters. Chem Pharm Bull (Tokyo) 1985;33(4):1620–1632. doi: 10.1248/cpb.33.1620. [DOI] [PubMed] [Google Scholar]
- 37.Miller R. The Jackknife - a review. Biometrika. 1974;61:1–15. [Google Scholar]
- 38.Lacey LF, Keene ON, Pritchard JF, et al. Common noncompartmental pharmacokinetic variables: are they normally or log-normally distributed? J Biopharm Stat. 1997;7(1):171–178. doi: 10.1080/10543409708835177. [DOI] [PubMed] [Google Scholar]
- 39.Sugen I. Investigator Brochure. on file. [Google Scholar]
- 40.Vecht CJ, Wagner GL, Wilms EB. Interactions between antiepileptic and chemotherapeutic drugs. Lancet Neurol. 2003;2(7):404–409. doi: 10.1016/s1474-4422(03)00435-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.