

Abstract
Glutamate receptors mediate the majority of excitatory synaptic transmission in the CNS. The AMPA-subtype has rapid kinetics, with activation, deactivation and desensitization proceeding on the millisecond timescale or faster. Crystallographic, biochemical, and functional studies suggest that GluR2 Cys mutants which form intermolecular disulfide cross-links between the lower D2 lobes of the ligand binding cores can be trapped in a conformation that represents the desensitized state. We used multi-channel rapid perfusion techniques to examine the state dependence of cross-linking in these mutants. Under reducing conditions, both wild-type GluR2 and the G725C and S729C mutants have normal activation and desensitization kinetics, but the Cys mutants can be efficiently trapped in nonconducting states when oxidized. In contrast the I664C mutant is only partially inactivated under oxidizing conditions. For S729C, disulfide cross-links form rapidly when receptors are desensitized in the presence of glutamate, but receptors also become trapped at rest, in the absence of agonist. We assessed such spontaneous trapping in various conditions, including CNQX, a competitive antagonist; kainate, a weak partial agonist; or when desensitization was blocked by the L483Y mutation that stabilizes the D1 dimer interface. These experiments suggest that trapping in the absence of glutamate is due to two motions: Spontaneous breaking of the D1 dimer interface and hyperextension of the lower lobes of the ligand binding core. These data show that the glutamate binding domains are surprisingly mobile in the absence of ligand, which could influence receptor activity in the brain.
Introduction
At excitatory synapses in the brain, rapid signaling is mediated by AMPA subtype glutamate receptors. The fast kinetics of AMPA receptors enable postsynaptic currents to follow the release of glutamate at rates approaching several kilohertz (Taschenberger and von Gersdorff, 2000), but desensitization limits their temporal sensitivity and underlies some forms of short-term depression. This type of synaptic depression shapes information transfer at some synapses (Rozov et al., 2001) and contributes to neuronal computation (Rothman et al., 2009). Thus, understanding the molecular mechanisms of desensitization is important for modeling synaptic function.
Structural work gives insight into the gating mechanism of AMPA receptors (Madden, 2002; Armstrong et al., 2006; Mayer, 2006; Hansen et al., 2007), but the nature of the desensitized state is not well understood. In the consensus model for desensitization, two subunits in a ligand binding domain dimer assembly undergo a large conformational change, involving a pivoting motion around the dimer axis of symmetry (Sun et al., 2002). As a result, in the desensitized state the upper lobes (D1) of the two ligand binding domains move apart, and the lower lobes (D2) come closer together. This allows the channel to enter a nonconducting conformation, even though the individual ligand binding domains remain in the glutamate-bound active conformation that initially triggers ion channel gating. The most direct evidence for this model came from studies on AMPA receptor GluR2 Cys mutants, which trap receptors in an inactive conformation (Armstrong et al., 2006). These studies used a GluR2 construct in which the 400 amino acid N terminal was deleted, and focused on three mutants in the D2 dimer surface: I664C in the loop connecting helices F and G, and the pair G725C and S729C adjacent to the second interdomain β-strand linking domains 1 and 2. Following heterologous expression in Xenopus oocytes, steady-state responses to glutamate showed 3–4-fold potentiation by DTT for I664C and G725C, and 56-fold potentiation for the S729C mutant. Biochemical studies revealed spontaneous dimer formation for S729C, and crystallographic studies of both the S729C and G725C mutants revealed dimers trapped in a relaxed conformation that is likely either the desensitized state, or an intermediate in the desensitization pathway (Armstrong et al., 2006).
We performed a quantitative characterization of full-length AMPA receptors carrying these mutations to analyze important aspects of receptor function, using rapid perfusion techniques to characterize the state dependence of intermolecular disulfide cross-linking. We find that the ligand binding cores of AMPA receptors are highly mobile in the absence of bound ligands, allowing spontaneous formation of inter subunit disulfide bonds in the absence of glutamate. These results suggest spontaneous intramolecular movements within single receptor subunits, and also intermolecular motions between subunits. This dynamic picture of a resting AMPA receptor was unexpected because it means that the domain closure that triggers gating of the ion channel, and dimer dissociation that leads to desensitization, occur without binding of glutamate, albeit less frequently.
Materials and Methods
Molecular biology.
For this study we used the pRK5 expression vector encoding the flip splice variant of rat GluR2, also named GluRB or GluA2, unedited at the QR site and edited at the RG site. The three cysteine mutants, I664C, G725C and S729C were generated using overlap PCR. The L483Y S729C double mutant was constructed by ligating a 700 bp XbaI/XmaI fragment from the GluR2 S729C mutant into the GluR2 L483Y construct digested with the same enzymes. The presence of the mutant codons and the absence of PCR errors was confirmed by double-stranded DNA sequencing of the amplified region. The numbering refers to the mature polypeptide chain.
Cell culture and electrophysiology.
Wild-type and mutant AMPA receptors were expressed by transient expression in HEK-293 cells for outside-out patch recording as previously described (Plested and Mayer, 2007). The external solution contained (in mm): 150 NaCl, 0.1 MgCl2, 0.1 CaCl2, 5 HEPES, titrated to pH 7.3 with NaOH, to which we added drugs, and either oxidizing or reducing agents as required. Drugs were obtained from Tocris Bioscience, Ascent Scientific or Sigma-Aldrich. Copper phenanthroline was made up at a 1:3 molar ratio of CuCl2 dissolved in water and 1–10-Phenanthroline dissolved in EtOH; values reported in the paper indicate the CuCl2 concentration; the final concentration of ethanol was 0.001% v/v. The pipette (internal) solution contained (in mm): 115 NaCl, 10 NaF, 0.5 CaCl2, 1 MgCl2, 5 Na4BAPTA, 5 HEPES and 10 Na2ATP, pH 7.3. Outside-out patches were voltage clamped at –30 to –60 mV unless otherwise stated. Patch currents (filtered at 1–10 kHz 8-pole Bessel) were recorded directly to the hard disk of an Apple Macintosh using the Axograph X program (Axograph Scientific) via an Instrutech ITC-16 interface (HEKA Instruments). The sampling rate was 10–25 kHz; the slower sampling rates were used for records of duration 40 s and longer.
Fast perfusion.
We applied ligands and drugs to outside out patches via custom-made three-barrel glass tubing (Vitrocom). The glass was pulled to a final width of ∼500 μm, and the tip was briefly etched in hydrofluoric acid. The perfusion tool was mounted on a piezo-electric wafer (a gift from James Howe, Yale University, New Haven, CT) that in turn was mounted on a micro manipulator. The piezo wafer displacement was computer controlled via a 100 V amplifier (EX-100 Winston Electronics) and the command voltage was typically filtered at 100 Hz (8-pole Bessel) to reduce oscillation of the piezo. This allowed us to make rapid and reproducible solution exchanges due to the fine solution interfaces which formed at the outflow of the etched tube; typical 10–90% rise times were 300 μs, as measured at the end of the experiment from junction potentials at the open tip of the patch pipette.
Capacitive coupling between the piezo wafer and the patch pipette resulted in transients in the current record during charging and discharging of the piezo. We reduced these below the typical level of noise in the patch (∼1 pA RMS at 3 kHz) by placing a grounded foil between the piezo wafer and the patch pipette. However, when the responses were averaged for small agonist activated currents, the piezo transients summed and distorted the rising edge of the current waveform. We used two techniques to eliminate these transients from the current record. First, we applied equal positive and negative potentials to the patch pipette (typically +60 and –60 mV). Because the transients were identical in each case, but the sign of the glutamate-activated currents reversed, subtracting the average of these responses eliminated the transients. Because holding patches at two different potentials was impractical for some longer experiments, we also recorded the filtered piezo command voltage and in some experiments subtracted its scaled first derivative from glutamate-evoked responses to remove the artifact. This process is illustrated in supplemental Figure 1, available at www.jneurosci.org as supplemental material. The scaling was calibrated according to the charging transient at the end of the ligand application, after desensitization was complete, when the transient could be recorded in isolation from the response to glutamate.
Data analysis.
To measure recovery from desensitization, we used a two-pulse protocol with a variable inter pulse interval. Monoexponential recovery did not describe the recovery well, as previously reported (Bowie and Lange, 2002; Robert and Howe, 2003; Zhang et al., 2006), but the rate of recovery (krec) was well fit by a Hodgkin-Huxley type function: N = N0 + (1 − N0) · [1 − exp(−krect)]n, where N is the active fraction of receptors at time t following the first pulse, and N0 was the active fraction at the end of the conditioning pulse. The slope (n) was typically between 2 and 4. To measure the rate of onset of desensitization and its extent at steady-state at low glutamate concentrations (3–200 μm), which produce only limited activation of macroscopic responses (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), the patch was held in control solution and then switched into a prepulse solution containing 3–200 μm glutamate; following a variable prepulse duration (6 ms–3 s), the fraction of active receptors was assayed by jumping into test pulse of a saturating concentration of 10 mm glutamate. The concentration dependence of the extent of desensitization was well fitted with a Langmuir binding isotherm. The rate of onset of desensitization (kdes) was estimated by fitting a monoexponential function to the amplitude of test pulse responses (normalized to the peak current measured without a prepulse): N = N∞ + (1 − N∞) · exp(−kdest), where N was the active fraction of receptors at time t following the beginning of the prepulse, and N∞ was the active fraction at steady-state. The rate of desensitization was weakly dependent on glutamate concentration, and was fitted with the following empirical relation:
 |
where kdes,max was the maximum rate of desensitization, c was the glutamate concentration, n was a slope factor and K was a constant.
To measure state-dependent trapping in oxidizing conditions, we used a protocol where we initially determined a baseline for activation by glutamate in the continuous presence of 1 mm DTT. We measured four responses to 100 ms pulses of 10 mm glutamate at 1 Hz and then waited 1–2 s for complete recovery to the resting state following unbinding of glutamate. We then applied 10 μm CuPhen in the presence or absence of various AMPA receptor ligands via the third barrel of the perfusion tool for variable time intervals. Immediately following this application, we resumed application of 100 ms pulses of 10 mm glutamate at 1 Hz to monitor the untrapping of receptors in DTT. By extrapolating the envelope of these responses fitted with a mono-exponential function back to the end of the CuPhen application, we were able to estimate the proportion of receptors that were trapped. We performed this extrapolation because in certain conditions, a pulse of 10 mm glutamate did not generate a measurable response, e.g., when all channels were desensitized or bound with an antagonist, and also as a form of averaging because of the stochastic variability of the small responses (for instance, see Fig. 4). We plotted the active fraction against the interval to determine the rate and extent of trapping for the different cross-linking conditions. Figures were prepared with Kaleidagraph (Synergy Software). Results are reported as the mean ± SEM.
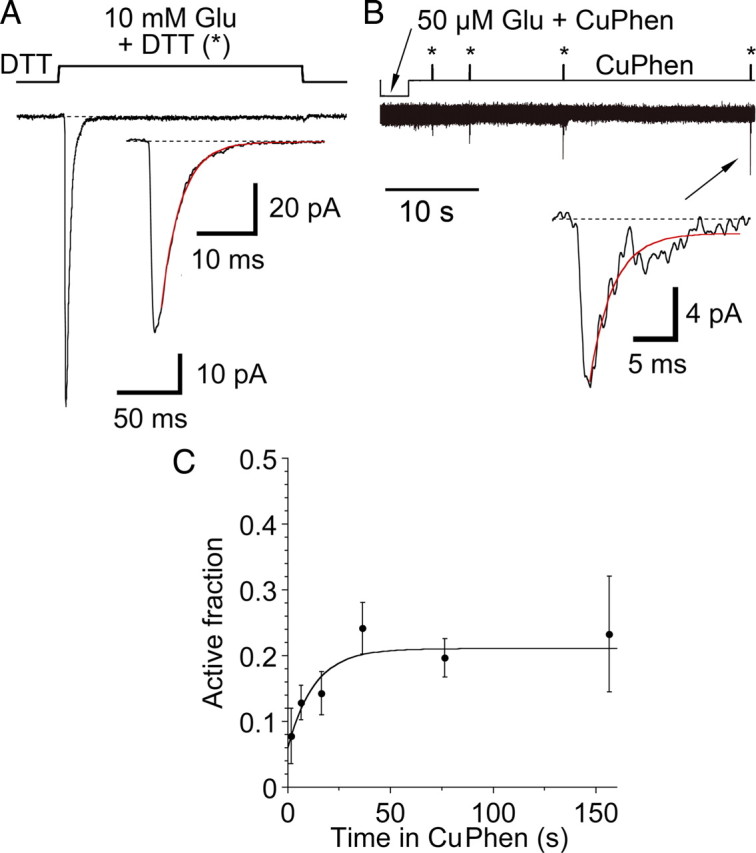
Figure 4.

Disulfide trapping of the S729C mutant. A, Test responses to 10 mm glutamate are inhibited following a 3.2 s exposure to 50 μm glutamate and 10 μm CuPhen (*); the response to the first application of 10 mm glutamate after trapping is too small to measure, but in the continuous presence of DTT full recovery occurs at a rate of 0.3 s−1, as shown by a single exponential fit to the envelope of the peak amplitude of subsequent responses to glutamate (filled red circles); extrapolation to the end of the application of 50 μm glutamate and 10 μm CuPhen (open red circle) was used to estimate the extent of trapping; the upper trace shows the piezo command voltage. B, The 1st and 22nd responses to 10 mm glutamate in DTT from panel A are essentially indistinguishable, as shown in the inset for which the traces are overlaid. These traces were chosen because of their similar amplitude, and in both, desensitization was well described by a single exponential function (red curves, kdes = 332 s–1). The upper trace is the open tip response recorded at the end of the experiment. C, Trapping in the absence of ligand is much slower than in the presence of glutamate. Exposure to 10 μm CuPhen for 12.8 s (*) traps 54% of receptors. The peak currents during recovery in DTT were fitted as in panel A (red curve, 0.3 s–1) and the trapping estimated from the fitted curve (open circle). D, The active fraction of receptors in the presence of 50 μm glutamate (filled circles), or at rest (open circles), was plotted against the period of trapping in 10 μm CuPhen; with 50 μm glutamate there was complete trapping; in contrast, for receptors at rest trapping remained incomplete at 360 s. The arrows represent the time intervals for the representative traces shown in B and C. For trapping in resting conditions, the solid line is a biexponential fit (τ1 = 3.5 ± 0.6 s (amplitude 76 ± 5%), τ2 = 50 ± 20 s; active fraction at t∞ = 32 ± 2%); the dashed line shows a monoexponential fit (τ = 7 ± 1 s).
Results
Wild-type AMPA receptors are not redox sensitive
Our experiments focused on Cys mutants in domain 2 of the AMPA receptor GluR2 ligand binding domain (Fig. 1A). In crystal structures of the glutamate bound and apo conformations of the GluR2 ligand binding domain dimer (Armstrong and Gouaux, 2000), the CA atoms of the mutants are separated by distances of 42 and 40 Å for I664C, 16 and 12 Å for G725C, and 12 and 13 Å for S729C, respectively, too far apart to form disulfide bonds as indicated in a diagram representation of the active dimer complex (Fig. 1B). However, during desensitization the D1 dimer interface breaks, and as a result the D2 dimer surfaces move together, closing the channel and bringing the G725C and S729C mutant Cys thiols close enough to form disulfide bonds (Sun et al., 2002; Armstrong et al., 2006). Wild type GluR2 subunits contain 11 native Cys residues, with one disulfide bond in the N terminal domain (Jin et al., 2009), and one in the ligand binding domain (Armstrong and Gouaux, 2000). Because oxidation of native Cys residues could interfere with analysis of the effects of Cys mutants we first tested the effects of DTT and CuPhen in wild-type GluR2 (Fig. 1C). In five patches, wild-type channels desensitized with indistinguishable kinetics in normal saline, kdes = 148 ± 12 s–1; in 10 μm CuPhen, kdes = 140 ± 9 s–1; or in 1 mm DTT, kdes = 144 ± 13 s–1. These results are in accord with those of Armstrong et al. (2006), who found no effect of cysteine reactive reagents on equilibrium responses to glutamate recorded in Xenopus oocytes for a GluR2 ATD (–) construct, in which the N terminal domain was deleted, and 4 Cys residues in the ligand binding domain and transmembrane segments mutated to non-reactive amino acids.
Figure 1.

Redox sensitivity of wild-type GluR2 and the G725C and S729C mutants. A, Crystal structure of the GluR2 ligand binding dimer glutamate complex (PDB 1FTJ) in which domains 1 and 2 in each subunit are colored blue and pink, respectively; orange spheres separated by 16 Å indicate the wide separation of the CA atoms of the G725C mutant in the glutamate bound active state. B, Diagram representation of the ligand binding domain and ion channel in the glutamate bound open state; the introduced cysteine residues are shown as orange bars. C, Activation and desensitization of wild-type GluR2 is not affected by redox potential; the colored lines show single exponential fits to the decay of the response to glutamate; the rise time and rate of desensitization for glutamate responses in control conditions, kdes 129 s−1 (blue), was indistinguishable from that in 10 μm CuPhen, kdes 116 s−1 (red), or 1 mm DTT, kdes 131 s−1 (cyan). D, Activation and desensitization of the G725C mutant in reducing conditions, kdes 280 s−1 (blue) had similar kinetics to wild-type, but when the patch was bathed in oxidizing conditions a rapid but reversible loss of current was observed; upon recovery in DTT, kdes 250 s−1 (cyan), responses were generally smaller than control. E, Similar redox sensitivity was observed for the S729C mutant, control kdes 298 s−1 (blue), but full recovery occurred on return to DTT, kdes 304 s−1 (cyan). F, Bar plot of desensitization rates in response to 10 mm glutamate in reducing conditions (1 mm DTT); values are mean ± SEM of 5 – 9 observations per construct.
Cys mutants gate normally but are inactivated following oxidation
We next studied the three cysteine mutants with glutamate responses that are potentiated by DTT in Xenopus oocyte experiments (Armstrong et al., 2006). In the presence of 1 mm DTT the GluR2 G725C and S729C mutants both activate rapidly, and desensitize with fast kinetics, kdes 180 ± 20 s–1 (n = 5) for G725C and 300 ± 30 s–1 (n = 9) for S729C, only slightly faster than wild type (Fig. 1E). However, in the presence 10 μm CuPhen, we observed a complete loss of the response to glutamate; this was readily reversible upon return to DTT-containing solution (Fig. 1D,E). Upon averaging patch currents recorded during 10 mm glutamate applications in oxidizing conditions, residual currents were not observed, consistent with essentially complete disulfide trapping in a state resistant to activation by glutamate.
In contrast, under oxidizing conditions the I664C mutant continued to show activation by 10 mm glutamate, and the peak current was only fivefold larger in 1 mm DTT than in 10 μm CuPhen (Fig. 2A). There were also small but significant redox-dependent changes in the steady-state current and the desensitization rate (Fig. 2A), but these effects were difficult to quantify because the I664C mutant showed substantial rundown, possibly reflecting the formation of non-specific cross-links that were much more resistant to reduction than for the other mutants tested. Because crystal structures have not been solved for I664C cross-linked dimers we did not perform further experiments with this mutant, but our results raise the possibility that it traps a different conformation than the G725C and S729C mutants.
Figure 2.
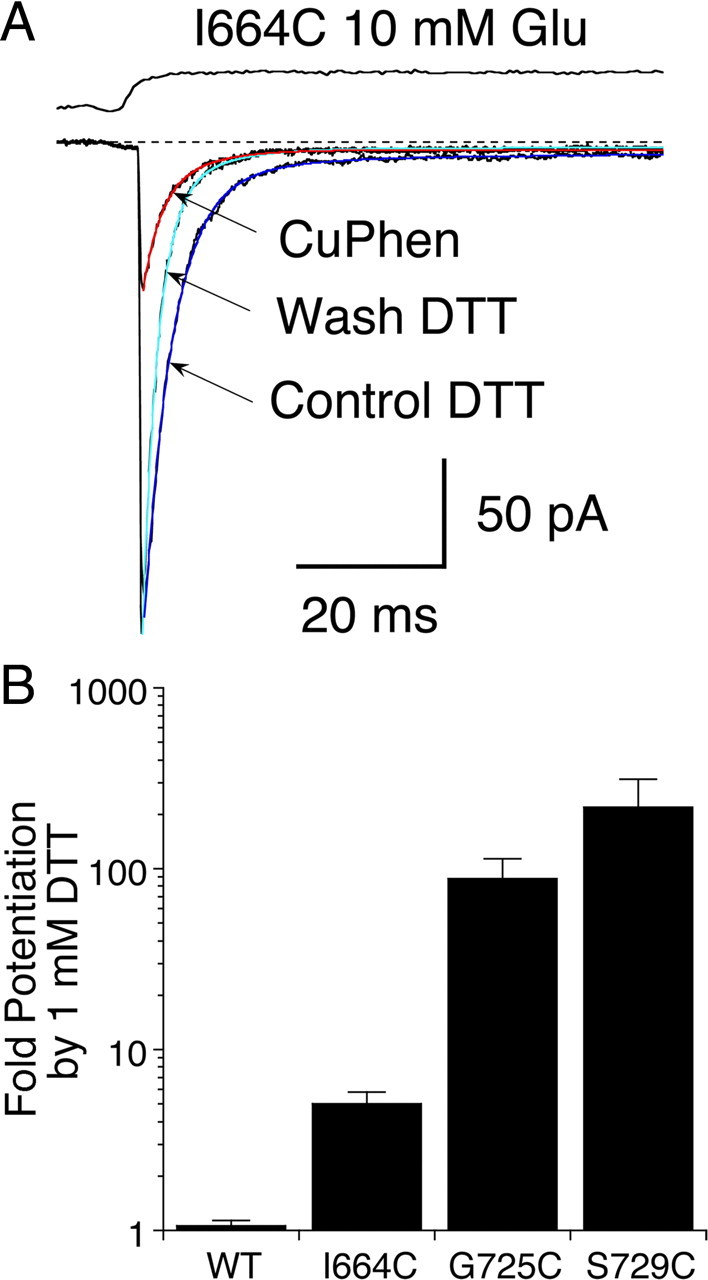
The GluR2 I664C mutant shows partial inactivation under oxidizing conditions. A, Responses to 10 mm glutamate recorded in oxidizing and reducing conditions; the peak current was reduced fourfold in CuPhen but the rate of decay was similar to control conditions: control kdes 210 s–1 (blue), CuPhen kdes 220 s–1 (red); upon return to DTT, the peak amplitude was restored and the decay was faster, kdes 300 s–1 (cyan). B, Bar plot showing the increase in peak current amplitude upon switching from CuPhen to 1 mm DTT for wild-type GluR2 and the three Cys mutants; values are mean ± SEM of 5 observations per construct.
To calculate a lower limit for potentiation by DTT of glutamate responses for the three cysteine mutants, we divided the peak glutamate activated current measured in 1 mm DTT by the current in oxidizing conditions. In the absence of a visually identifiable response to glutamate for the S729C and G725C mutants in the presence of 10 μm CuPhen, we calculated the root-mean-square deviation of the patch current for a 10 ms stretch of the record encompassing the start of the glutamate application. We used this estimate as a conservative upper limit of the glutamate response amplitude. The RMSD was calculated after subtracting electrical transients generated by the solution application system (see Materials and Methods). The effects of DTT are summarized in Figure 2B; for the G725C mutant the mean fold-potentiation was 88 ± 25 (n = 5), ∼20-fold higher than previously estimated (Armstrong et al., 2006); in the case of the S729C mutant, the ratio was higher still, 219 ± 93 (n = 5) and in one patch we measured a maximum potentiation of 565-fold. In contrast, for the I664 mutant, there was only 5 ± 0.8-fold potentiation (n = 5). For the G725C and S729C mutants, we believe that these ratios are limited by the low expression of each mutant rather than by incomplete trapping. On average, peak currents in reducing conditions were smaller for the G725C mutant, and this is reflected in the smaller ratio. The lack of obvious glutamate activated currents in the presence of CuPhen strongly suggests that in the above conditions, the G725C and S729C mutations, which gave rise to similar D2 cross-linked crystal structures, both introduce disulfide bridges that can efficiently trap full-length receptors in a state where glutamate cannot activate the channel.
Kinetics of onset and recovery from desensitization for GluR2 S729C in reducing conditions
Because the GluR2 S729C mutant showed better expression, and less rundown than G725C, further experiments designed to assess the state dependence of cross-linking were performed using S729C. Before proceeding with cross-linking experiments, we thoroughly characterized the desensitization properties of both wild-type GluR2 and the S729C mutant in reducing conditions. First, we measured the rate of recovery from desensitization produced by a conditioning pulse of 10 mm glutamate for both wild-type and mutant channels using a single component Hodgkin-Huxley type mechanism (see Materials and Methods). In control conditions, wild-type channels recovered from desensitization at a rate of 28 ± 3 s–1 (n = 10). The slope was 2.3 ± 0.5, suggesting that, consistent with previous reports, multiple rate limiting steps are involved in recovery from desensitization (Bowie and Lange, 2002; Zhang et al., 2006). We did not observe a separate slow component, and recovery was complete to within 99.9% of the original current over the time course of our analysis which included test intervals up to 2 s. The rate of recovery was slightly, but consistently faster in the presence of 1 mm DTT, 44 ± 6 s–1, slope 3.5 ± 1.0 (n = 5). Because oxidizing conditions inactivate the S729C mutant, we could only measure recovery from desensitization in the presence of 1 mm DTT; the rate was twofold slower for S729C than for wild-type, 22 ± 1 s–1, slope 3.0 ± 0.4, (n = 6), consistent a small increase in stability of the desensitized state. Overall, these results indicate that wild-type GluR2 and the S729C mutant show remarkably similar kinetics of recovery from desensitization when the formation of disulfide bonds by Cys mutants is prevented.
We next examined the concentration dependence of the rate of desensitization for wild-type GluR2 and the S729C mutant at four different prepulse concentrations of glutamate in the presence of DTT to prevent disulfide bond formation. This experiment had two objectives. First, to determine whether the mutant and wild-type receptors had different sensitivity to glutamate. Second, to determine the optimal concentration of glutamate for producing relatively complete desensitization of the S729C mutant, and the time course over which this steady-state level was reached. For both wild-type GluR2 and the S729C mutant, rates of entry into the desensitized state became faster as the prepulse concentration of glutamate was increased, and the extent of desensitization at steady state became more profound. For example, a prepulse of 3 μm glutamate caused wild-type receptors to desensitize at a rate of 7 s–1, and at equilibrium the amplitude of the test response to glutamate was 91% of control, corresponding to 9% desensitization; for a prepulse of 50 μm glutamate the rate of desensitization increased to 30 s−1 and the test pulse amplitude fell to 37% of the control, corresponding to 63% desensitization at steady state. During the prepulse, the S729C mutant desensitized slightly faster and more profoundly than wild-type GluR2; e.g., at 50 μm glutamate the rate of rate of desensitization was 70 s−1 and the test pulse amplitude fell to 14% of the control, corresponding to 86% desensitization at steady state (Fig. 3). The increase in extent of desensitization with concentration was well described by a simple binding isotherm with IC50s of 22 ± 1 μm for wild-type and 5.8 ± 0.4 μm for the S729C mutant (Fig. 3G), but the rate of desensitization had a much shallower concentration dependence (Fig. 3H).
Figure 3.
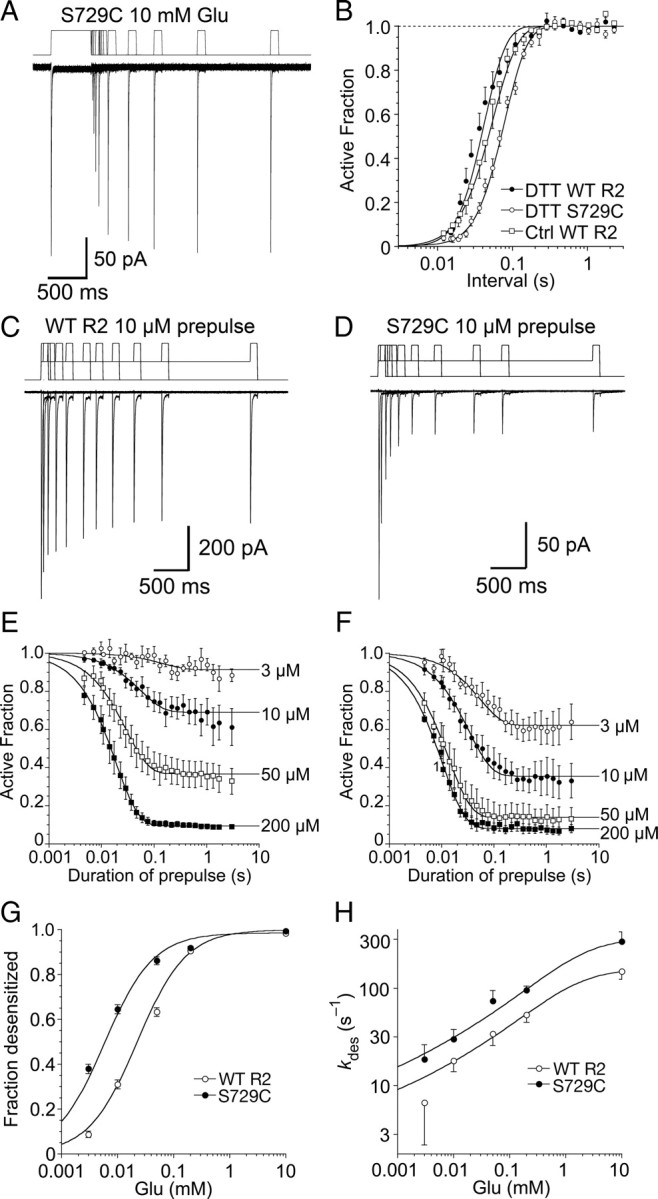
Concentration dependence and kinetics of desensitization for wild-type GluR2 and the S729C mutant. A, Paired pulses of 10 mm glutamate in the presence of 1 mm DTT were applied to the S729C mutant to measure the kinetics of recovery from desensitization; the inter pulse interval for the 11 responses shown ranged from 10 ms to 2 s; the upper trace is the piezo stimulus protocol. B, Kinetics of recovery from desensitization for wild-type GluR2 in normal conditions and in 1 mm DTT, and for the S729C mutant in 1 mm DTT; the solid lines show Hodgkin-Huxley type fits (see Materials and Methods) C, The trace shows 11 superimposed responses to10 mm glutamate for wild-type GluR2, preceded by 10 μm prepulses of duration 6 ms to 2.5 s; the upper trace is the piezo command, showing the resting level, the prepulse, and the final step to the 10 mm test pulse. D, The same protocol as in C for the S729C mutant; desensitization is faster and the steady-state desensitization by 10 μm glutamate more profound that for wild-type. E, The amplitude of responses for wild-type GluR2 following prepulses (3 – 200 μm glutamate) plotted on a log time scale; data points show mean ± SEM for 5–7 patches fit with a mono-exponential function to estimate kdes; the rates and extent of desensitization at steady state were: 3 μm, 7 s−1, 9 ± 1%; 10 μm, 18 s−1, 31 ± 2%; 50 μm, 30 s−1, 63 ± 2%; and 200 μm, 52 s−1, 94.6 ± 0.3%. F, The same analysis for the S729C mutant (n = 5); the rates and extent of desensitization at steady state were: 3 μm, 18 s−1, 38 ± 2%; 10 μm, 30 s−1, 65 ± 2%; 50 μm, 70 s−1, 86 ± 2%; and 200 μm, 100 s−1, 92 ± 1%. G, Concentration response analysis for equilibrium desensitization of wild-type GluR2 and the S729C mutant; wild-type receptors are half-maximally desensitized by 22 ± 1 μm glutamate (n = 5–7 per point); the desensitization of the S729C mutant shows a stronger sensitivity to glutamate, with half-maximal extent of desensitization at 5.8 ± 0.4 μm glutamate. H, Concentration dependence of the rate of onset of desensitization measured as in C and D; the rates were fitted with a binding isotherm raised to a fractional power (see Materials and Methods, slope factors were 0.11 ± 0.04 for wild-type receptors and 0.10 ± 0.02 for the S729C mutant); the shallowness of the concentration dependence, and the lack of precision in estimates of the entry rate at low concentrations precluded accurate determination of the half-maximal rate.
State-dependent trapping by D2 disulfide bonds
Armed with the knowledge that the desensitization properties of the GluR2 S729C mutant make them good surrogates for wild-type AMPA receptors, we next examined the state dependence of trapping in oxidizing conditions. Crystal structures of the resting and DNQX-bound dimer assemblies of wild-type GluR2 (Armstrong and Gouaux, 2000) indicate that the α carbon atoms of the mutant Cys residues will be separated by 13–16 Å for G725C and S729C, and by 40 Å for I664C, too large a distance for the mutant thiols to react without a conformational change in the ligand binding domain. We reasoned that there were two contrasting routes by which the inactivating D2 disulfide bond could form. Breaking of the D1 dimer interface as occurs during desensitization, a molecular rearrangement in which the ligand binding cores in a dimer relax as rigid bodies with respect to each other, allowing the mutant thiols in domain 2 to come within bonding distance. A second route to formation of D2 cross-links would be that the ligand binding core is flexible enough to permit D2 to sample the necessary conformation for disulfide bond formation without D1 dimer disruption. This might especially be true in the absence of ligand, and would mean that the D2 cross-linked structure may form in conditions that are not related to desensitization in the full-length receptor, such as the resting state. We sought to identify conditions where we could isolate these two possible motions to assess their relative importance to formation of the D2 cross-link in the S729C mutant.
First, we compared the kinetics and extent of trapping in the presence of a full agonist, for which we know that a large fraction of receptors will be desensitized at steady state, with trapping in the absence of ligand, where we expected few, if any, receptors to be desensitized. We found that 50 μm glutamate, which produced 86% equilibrium desensitization under reducing conditions, promoted full trapping at a rate of 0.6 s–1. The onset of trapping was well fit by a single exponential function, but was 100-fold slower than the rate of desensitization, which was 70 s–1 at 50 μm glutamate (Fig. 3F). This means that on average, individual receptors spend a long time in the desensitized state before they become trapped, probably making multiple visits. As expected, trapping in the presence of glutamate was much more profound than observed in the absence of ligand, and after a 1.6 s exposure to 50 μm glutamate under oxidizing conditions, ∼70% of receptors were trapped in an inactive state, while only 15% became trapped in the absence of agonist (Fig. 4). However, the fraction trapped in the apo state was substantial, and to our surprise, we found that trapping in the absence of glutamate increased over longer intervals, reaching 69 ± 6% after an 80 s exposure to oxidizing conditions (n = 7 patches), but did not increase further with longer applications, 68 ± 6% after 360 s (n = 3). In contrast to responses triggered by glutamate, the rate of trapping of receptors at rest was slow and not described well by a mono-exponential function. We used a biexponential function to fit the envelope of responses, giving a weighted trapping rate of 0.08 ± 0.02 s−1 (data from 3 to 13 patches per time interval included in the fit), ∼10-fold slower than in the presence of glutamate.
In the presence of 1 mm DTT the time course of recovery following trapping was remarkably consistent, ∼0.3 s–1, independent of the trapping conditions used, or how long the receptor complex was exposed to oxidizing conditions. This common lifetime argues that the receptors are trapped in a similar conformation, regardless of whether a ligand is bound, and that long exposures to CuPhen did not drive receptors into a range of nonspecific conformations. The fact that the trapping in the absence of ligand was incomplete suggests either that there is an extremely slow component of trapping that we were not able to detect, or that the trapping reaction is reversible over the timescale of our experiments. As we will show, other data we obtained favor the latter interpretation. The incomplete trapping we observed is also consistent with partial cross-linking observed in biochemical experiments on Xenopus oocytes where no glutamate was added (Armstrong et al., 2006).
Trapping in the presence of a partial agonist and an antagonist
The substantial trapping observed in the absence of agonist suggests that the formation of disulfide cross-links is not restricted to the desensitized state, and can occur through either spontaneous relaxation of the D1 dimer interface, or hyperextension of the ligand binding domains, bringing the D2 surfaces within reaction distance for the mutant thiol groups. We reasoned that saturation of the binding site of GluR2 by a competitive antagonist would lock the lower lobes of the ligand binding domains into a fixed conformation, and that this would in turn slow trapping, if domain cross-links can form through motion of D2 alone.
The rate of trapping was indeed slowed in the presence of 30 μm CNQX, relative to trapping in the absence of ligand, but only by ∼3-fold. The S729C mutant was trapped at a rate of 0.03 ± 0.01 s–1 (n = 4 – 10 patches) when exposed to oxidizing conditions in the presence of 30 μm CNQX, but the maximal extent of trapping at steady state, 31 ± 4%, was substantially less than the value of 68% obtained in the absence of ligand (Fig. 5). The rapid recovery of AMPA receptor currents following unbinding of weak competitive antagonists like CNQX (Benveniste and Mayer, 1991) suggests that this class of ligand does not promote desensitization, so it seems reasonable that the D1 dimer interface would not be broken more frequently in antagonist-bound receptor complexes than in the resting state. However, it is conceivable that antagonist binding stabilizes the D1 dimer assembly, or that D2 disulfide bonds cannot form in the antagonist bound state as a result of steric exclusion. We cannot completely rule out steric effects, but a rigid body translation of one of the monomers in the GluR2 DNQX crystal structure onto the structure of the S729C mutant cross-linked dimer can bring the pair of D2 domains into close enough apposition for the disulfide bonds to form without any steric clashes. This suggests that the extended conformation of the antagonist-bound ligand binding domains compared with the glutamate-bound monomer is not a barrier to disulfide formation.
Figure 5.
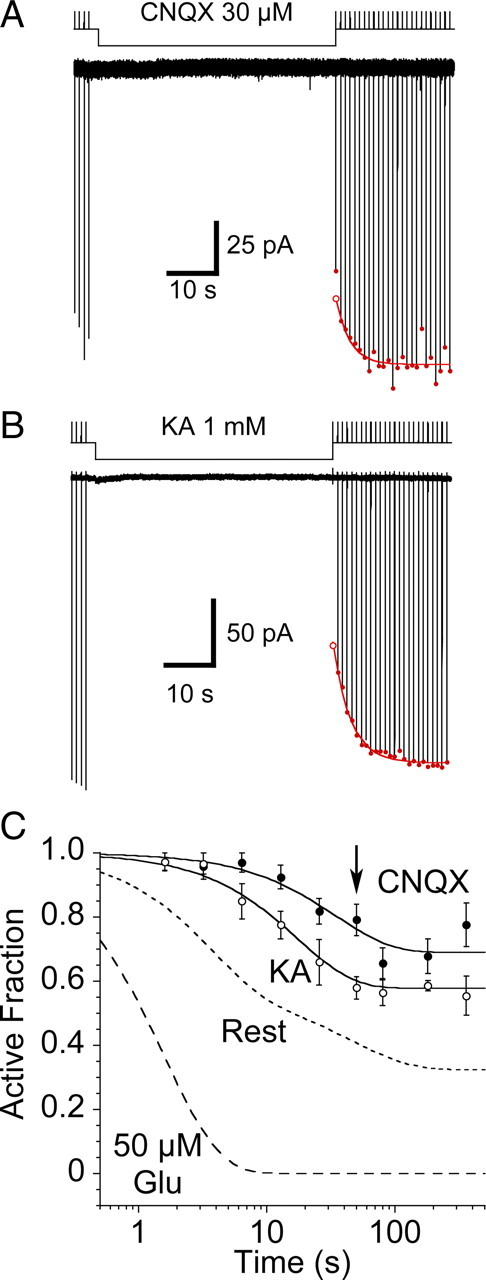
Disulfide trapping of the S729C mutant in the presence of CNQX or kainate. A, Coapplication of 30 μm CNQX slowed the onset of, and reduced the extent of trapping by 10 μm CuPhen; the first response following the end of the antagonist application was reduced in amplitude because CNQX remained bound during the rise of the current evoked by the test application of 10 mm glutamate. B, Trapping in the presence of a saturating concentration of the weak partial agonist kainate also slowed the onset, and reduced the extent of trapping by 10 μm CuPhen, but to a lesser extent than CNQX; the first response following the end of the partial agonist application was reduced in amplitude because kainate remained bound during the rise of the current evoked by the test application of 10 mm glutamate. C, Data summarizing trapping in the presence of 1 mm kainate (n = 6) and 30 μm CNQX (n = 4–10 patches per data point), compared with trapping at rest and in the presence of 50 μm glutamate (dashed lines); the arrow indicates the trapping interval for the representative examples shown in A and B. The rate of trapping of the S729C mutant in the presence of kainate (0.06 ± 0.01 s–1) is slower than for receptors at rest, even though kainate causes weak desensitization (supplemental Fig. 2, available at www.jneurosci.org as supplemental material); trapping by CNQX is slower still (0.03 ± 0.01 s–1); note also the inverse correlation between rate and extent of trapping.
To further investigate the origin of movements that underlie formation of D2 disulfide bonds, we next examined trapping due to a partial agonist that causes intermediate domain closure. For GluR2, domain closure by kainate is limited to 12° compared with 20° for full agonists (Armstrong et al., 1998; Armstrong et al., 2003). Kainate is a very weak agonist at wild-type GluR2 (Armstrong et al., 2003), and also produces less desensitization than full agonists (see supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Exposing the GluR2 S729C mutant to oxidizing conditions, in the presence of a saturating concentration of kainate (1 mm), again slowed and reduced the extent of trapping compared with receptors at rest, but the reduction was less profound, rate 0.06 ± 0.01 s–1, 42 ± 1% trapped at steady state (n = 6), than for CNQX.
Restraint of the D1 dimer interface blocks trapping effectively
The finding that immobilization of D2 by CNQX and kainate reduces the rate of trapping of the GluR2 S729C mutant, relative to the resting state was unexpected. That this reduction occurs even when kainate, a ligand that causes some desensitization (supplemental Fig. 3, available at www.jneurosci.org as supplemental material), is bound was doubly surprising, but strongly suggests that mobility of D2 plays a role. To assay the extent to which an alternative mechanism, spontaneous D1 dimer interface rupture, contributes to trapping by the S729C mutant we used the L483Y mutation, whose properties have been extensively studied (Stern-Bach et al., 1998; Sun et al., 2002). This mutation has been demonstrated to massively stabilize the D1 dimer interface for the isolated ligand binding domain, by 105-fold compared with wild-type GluR2, holding it in a near-native active conformation when bound to glutamate. First, we confirmed that for brief applications of glutamate desensitization was as strongly blocked in reducing conditions for the GluR2 L483Y–S729C double mutant as for the L483Y single mutant (Fig. 6A). With longer applications of 10 mm Glu in the presence of 1 mm DTT, the L483Y–S729C double mutant desensitized by 9 ± 1% at a rate of 5 ± 1 s–1 (Fig. 6B, n = 4 patches). The similarity of these values to those for previously reported experiments on the GluR2 L483Y single mutant (Sun et al., 2002) suggests that under reducing conditions the S729C mutation has essentially no effect on stabilization of the dimer by the L483Y mutation.
Figure 6.

The GluR2 L483Y mutant strongly reduces trapping. A, In the presence of 1 mm DTT, the L483Y-S729C double mutant does not desensitize in response to a 25 ms application of 10 mm glutamate. B, In response to a 500 ms application of glutamate the extent (9 ± 1%) and rate of desensitization (kdes = 4.5 ± 1.5 s–1, n = 3 patches) for the L483Y-S729C double mutant are similar those previously found for the L483Y single mutant (data from Sun et al., 2002). C, At rest the L483Y-S729C double mutant shows no detectable trapping following an 80 s exposure to 10 μm CuPhen, although as shown below with much longer applications weak trapping was observed. D, In contrast the L483Y-S729C double mutant can be efficiently trapped by application of 300 μm glutamate at a rate of 0.13 ± 0.03 s–1, n = 5 patches, estimated from the fit of a single exponential (white dashed line). E, Rate and extent of trapping by glutamate in oxidizing conditions was concentration dependent. The dashed line shows the faster trapping of the single S729C mutant in glutamate. We detected trapping when the receptor was exposed to oxidizing conditions for long intervals (>100 s) in the absence of any ligand, and this was apparently abolished, at least on the timescale of our measurements, when domain 2 was restrained by binding of CNQX (30 μm).
If the S729C disulfide trapped state is accessed mainly by D1 dimer breaking, then restraint of the D1 dimer interface should slow trapping even more strongly than immobilization of D2 by CNQX and kainate. Consistent with this, stabilizing the D1 interface with the L483Y mutation strongly reduces the rate of trapping in oxidizing conditions in the absence of ligand to 0.007 ± 0.003 s–1 (n = 5 patches). This rate is fivefold slower than the trapping of the S729C mutant in the presence of CNQX, suggesting that, in the absence of ligands that cause strong desensitization, spontaneous D1 dimer breaking is the major mechanism by which the D2 cross-link forms. However, despite profound dimer stabilization by the L483Y point mutant (Sun et al., 2002), the L483Y–S729C double mutant can still be fully trapped, if held in oxidizing conditions in the presence of glutamate.
Trapping occurs in the presence of glutamate because the GluR2 L483Y mutant desensitizes for ∼10% of the time at steady state in a high concentration of glutamate (Fig. 6B) (Sun et al., 2002). In oxidizing conditions and the presence of glutamate, we were able to track the trapping of complexes simply by fitting the observed decay of the current (Fig. 6D). The rate of trapping for the L483Y–S729C double mutant in the presence of 100 μm glutamate was at least tenfold slower than for the S729C point mutant, reflecting the greatly reduced rate of breaking of the D1 dimer interface, but again showed agonist concentration dependence. Trapping by 10 μm CuPhen in 100 μm glutamate, which generated a current ∼65% of that activated by 10 mm Glu, occurred at a rate of 0.06 ± 0.01 s–1 (n = 5), but reached only 81 ± 6% at steady state. In 300 μm glutamate, which evoked a response 78% of the amplitude of that to 10 mm glutamate, trapping was twofold faster, 0.13 ± 0.03 s–1 (n = 5) and proceeded to a greater extent, 97 ± 2% at equilibrium. This demonstrates that trapping at rest for the L483Y S729C double mutant is blocked because the D1 dimer interface is held intact, rather than because the introduction of the L483Y mutation prevents formation of the disulfide bond between subunits by steric exclusion or some other nonspecific mechanism.
Finally, we wanted to see whether D2 mobility was contributing to the trapping that remained for the L483Y S729C double mutant in the absence of ligand. At rest, the fraction of receptors trapped after a 6 min exposure to 10 μm CuPhen was 22 ± 4% (n = 7). We tested whether this was due to D2 mobility by also tracking the extent to which disulfide trapping occurred in the presence of a saturating concentration of CNQX (Fig. 6E). We expected that this experimental condition would represent the limit of our ability to restrain the conformation of the ligand binding cores. When the L483Y-S729C mutant was exposed to oxidizing conditions for 6 min, which due to limited patch stability with rapid perfusion represented the longest exposure we could reliably make, trapping was negligible, 3 ± 3% (n = 5), and probably beyond the limit of our resolving power. The specific effect of CNQX, which we expect to restrain domain 2, suggests that even in the virtual absence of D1 dimer breaking at rest, which is ensured by the L483Y mutation, trapping can occur through mobility of D2. This trapping can only occur if there is appreciable motion of D2 in the absence of bound ligand.
D2 disulfide bonds are not stable
We noted a clear correlation between the rate of trapping (Fig. 7A) and the extent of trapping at equilibrium (Fig. 7B). This would occur if the disulfide bonds that were formed by the S729C mutation were not stable on the time course of the experiment. When we plotted the residual active receptor fraction at steady state against the trapping time constant, we were able to describe the data adequately with a simple isotherm where the lifetime of the disulfide bond was 25 s (Fig. 7C), ∼10-fold longer than in 1 mm DTT. Following a trapping period of 1.6 s in 50 μm glutamate, receptors should be 95% trapped, estimated from the fitted curve in Figure 4D, whereas at rest the steady-state trapping is 68 ± 2%. Because at steady-state the fraction of receptors trapped at rest is less than the fraction trapped in the presence of glutamate, we hypothesized that we could monitor breaking of the disulfide bonds in oxidizing conditions by measuring the relaxation from a glutamate trapped state to a resting state in the presence of 10 μm CuPhen (Fig. 8). Using this approach we were able to measure recovery from glutamate-evoked trapping under oxidizing conditions, providing direct evidence that the disulfide bonds that are formed between Cys 729 residues in adjacent subunits are unstable in the full-length receptor.
Figure 7.
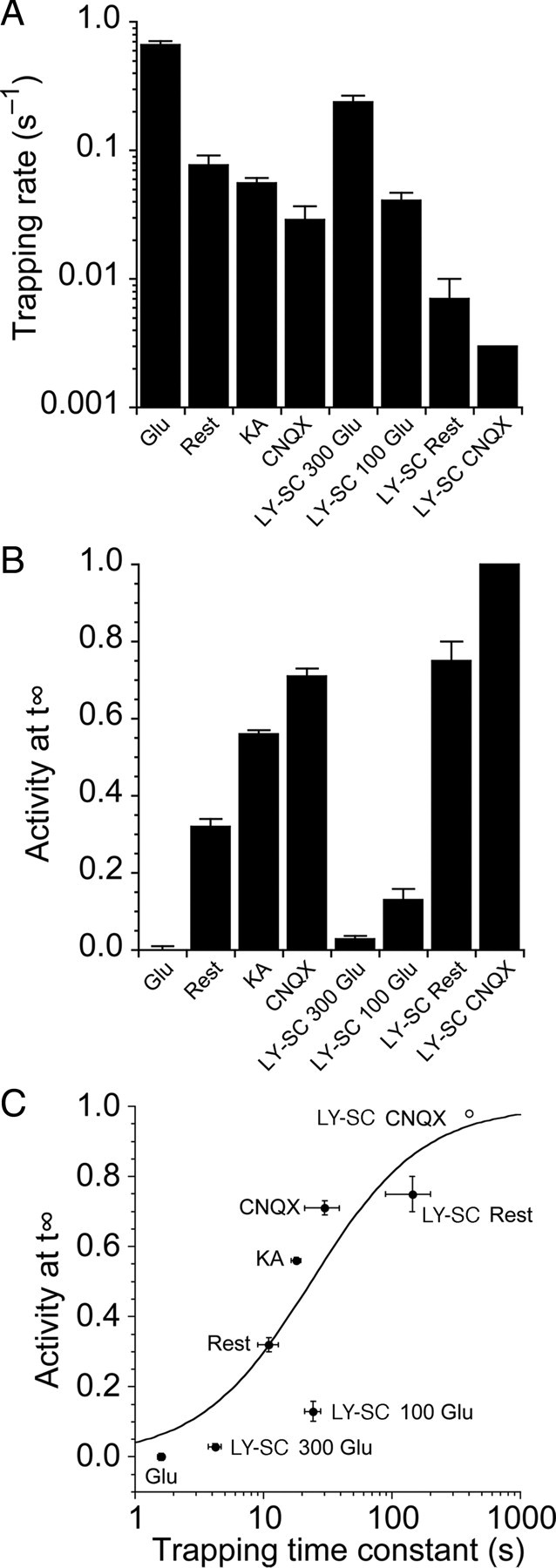
The relationship between the rate and extent of trapping suggests that the S729C mutant disulfide bond has a limited lifetime. A, Bar plot summarizing the rate of trapping for the S729C and L483Y-S729C mutants in different conditions. Trapping is fastest in the presence of 50 μm glutamate. The L483Y mutant slows trapping, and in the presence of CNQX trapping was eliminated; the bar for LY-SC CNQX is an upper estimate of the trapping rate given that we observed no trapping after 360 s. B, Bar plot summarizing the extent of trapping in the steady state for the same conditions as in A. C, The proportion of receptors that were not trapped in steady-state conditions showed a strong negative correlation to the trapping rate. These data were reasonably well described by a simple isotherm where the lifetime of the disulfide trapped conformation was 25 s. The LY-SC CNQX point (open circle) was not included in the fit. Although two disulfide bonds presumably form in a fully trapped tetrameric receptor, we do not know the activity of a singly trapped receptor, and so we did not include this complexity in our fitting.
Figure 8.
Disulfide bond breakage in oxidizing conditions. A, Average of 5 responses of the S729C mutant to 10 mm glutamate in reducing conditions (1 mm DTT); inset shows single exponential fit (red), kdes 287 s–1. B, In the same patch, a time-dependent relief of trapping occurred in the continuous presence of CuPhen. Four separate traces are overlaid with 1, 3, 16 and 36 s exposures to CuPhen at rest, following the application of 50 μm glutamate; DTT was present only during the test pulses of 10 mm Glu indicated by *. Relief from trapping in DTT is slow enough (0.3 s–1) that there is negligible change in the rise time and peak amplitude of the test pulse. The inset shows the final pulse, after 36 s of exposure to CuPhen at rest; single exponential fit (red) to the current during the 20 ms application of 10 mm Glu (red), kdes = 337 s–1. C, The relaxation of the trapped receptors at rest in CuPhen, normalized to the mean of prerelaxation and postrelaxation responses in DTT, was fitted with a monoexponential recovery with a lifetime of 13 ± 6 s (data from 5 patches). The steady-state level of nontrapped receptors was 21 ± 4%, similar to the level of trapped receptors at rest without pretrapping in glutamate (32 ± 2%, Fig. 4).
Discussion
We show here that GluR2 disulfide-trapping D2 mutants gate with close to normal kinetics under reducing conditions, but are inactivated by application of glutamate under oxidizing conditions. These observations are consistent with the cross-linked form corresponding to a desensitized state, or a state from which a full agonist has zero efficacy. The extent of redox-sensitive changes in peak current amplitude is larger than that obtained previously for GluR2 ATD (–) mutant constructs expressed in Xenopus oocytes, for which the I664C and G725C mutants were only weakly potentiated by DTT (Armstrong et al., 2006). Even for the S729C mutant, which showed 56-fold potentiation when expressed in Xenopus oocytes, we observed much larger effects of DTT. This difference is almost certainly due to differences in the rate of application of glutamate, since in two electrode voltage-clamp experiments on Xenopus oocytes it is not possible to resolve rapid desensitization, while we used outside-out patches and rapid perfusion techniques.
The rate of AMPA receptor desensitization is concentration dependent
Using a triple-barrel perfusion system to make precisely timed glutamate applications with ms resolution, we measured the rate of onset of desensitization with prepulse concentrations ranging from 3 to 200 μm, which produced a 7.4-fold increase in kdes, from 7 to 52 s−1. This avoids several complications in interpretation that arise when measuring concentration dependence of the rate of desensitization from the decay of the response to a test application of agonist. First, the rate of desensitization could be slower than the observed current relaxation because, on average, only those receptors fully bound by glutamate generate substantial current flow during the test pulse. Second, when desensitization is measured in the presence of a competitive antagonist, so that receptors are not fully bound by glutamate, it is unknown whether receptors bound with a mixture of glutamate and antagonist molecules desensitize at the same rate as receptors partially occupied by glutamate alone. It is probable that these differences, and the shallowness of the concentration dependence we measure, account for the report that the rate of desensitization for wild type GluR2 appears to be independent of receptor occupancy (Robert and Howe, 2003). This shallow concentration dependence represents a new constraint on models of AMPA-receptor activation and warrants further investigation in other AMPA receptor subtypes.
State dependence of trapping
We interpret the rate of cross-linking measured in oxidizing conditions as being proportional to the fraction of time receptors spend in conformations that permit intermolecular disulfide cross-links to form (Fig. 9). The best evidence for this is the dependence of the trapping rate on glutamate concentration for the L483Y S729C double mutant. The faster and more complete trapping observed with a higher concentration of glutamate is consistent with the intuitive notion that a receptor population fully bound by glutamate will be desensitized more frequently than at lower glutamate concentrations, since it seems unlikely that different glutamate concentrations will drive the receptor into mutually exclusive sets of conformations, only some of which support cross-linking. However, in the absence of ligands, or for complexes with kainate or CNQX, it is conceivable that the differences in rate and extent of trapping are due to the receptor adopting a range of conformations only a subset of which is permissive for disulfide bond formation.
Figure 9.
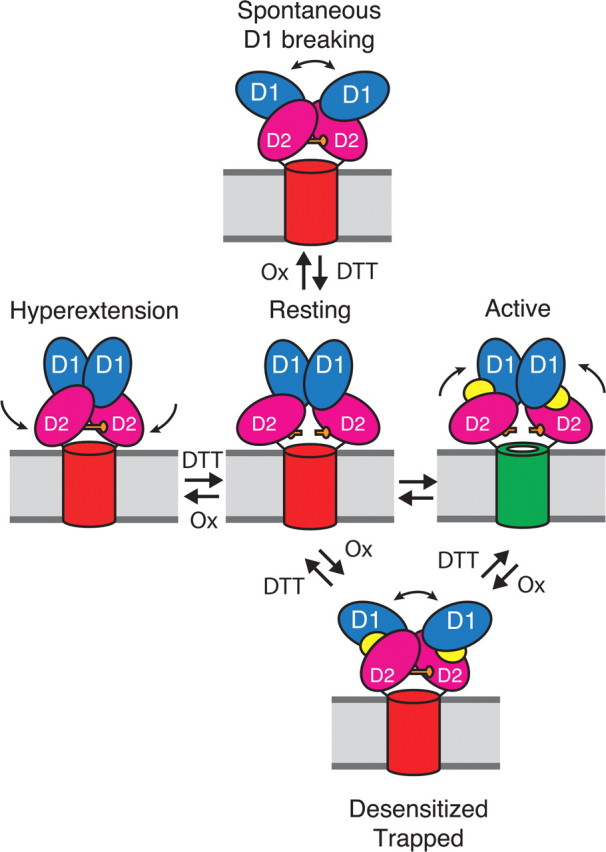
Diagram showing pathways for formation of D2 cross-links in AMPA receptors. One dimer of the two dimers in the receptor complex is depicted, with a cylinder representing the membrane-associated ion channel domain. The extracellular ligand binding clamshell domains have two lobes (D1, blue and D2, magenta). At rest, the ion channel is closed (red cylinder). The closure of the clamshell following the binding of glutamate (yellow) opens the channel (green); subsequent desensitization leads to disulfide bond cross-linking of the lower lobes (D2) between the introduced cysteine residues (orange bars). From the resting state two further conformational changes also lead to spontaneous D2 cross-linking. The first is dissociation of the active D1 dimer interface (top). The second is spontaneous hyperextension of the lower lobe of the ligand binding domain (left).
Breaking of the D1 dimer interface in the absence of ligand is not equivalent to desensitization, because the bound agonist is indispensable in stabilizing the desensitized receptor complex. Indeed, part of the process of recovery from desensitization is the unbinding of ligand. We suspect that any glutamate receptors that have the D1 dimer interface broken at rest would immediately desensitize following glutamate binding. Thus these receptors would be functionally silent. Recombinant AMPA receptors, and also native channels that probably incorporate auxiliary proteins such as TARPS, are weakly potentiated by cyclothiazide which stabilizes the D1 dimer interface. This observation suggests that a minor fraction of receptors desensitize in this way before channels can be opened by glutamate. This phenomenon might be more pronounced for AMPA receptor subtypes that recover from desensitization more slowly, such as GluR1. Thus, the inherent lack of stability of the receptor will tend to render some complexes refractory to activation by synaptically released glutamate.
The state dependence is broadly compatible with the structure of the S729C mutant representing either the desensitized state or a desensitization intermediate. The latter could explain why trapping of receptors in the presence of glutamate was so much slower than the rate of desensitization at the same concentration. Receptors that repeatedly desensitize would still spend only a small fraction of the time in such an intermediate state. Other possible explanations for the slowness of the trapping relaxation are the instability of the disulfide trap, and also the possible limited availability of molecular oxygen, essential for the oxidation of disulfide bonds by CuPhen (Kobashi, 1968), in our solutions which were necessarily degassed for rapid solution exchange experiments.
Implications for receptor structure and function
To our surprise, cross-linking occurred spontaneously in the absence of ligand, and was faster than in the presence of either kainate, a partial agonist that produces weak desensitization, or CNQX, a competitive antagonist that likely produces no desensitization. This suggests that, depending on the experimental conditions, cross-linking reports at least two different molecular processes (Fig. 9). Desensitization, synonymous with the breaking of the D1 dimer interface, is one. The other is the inherent flexibility and motion of the lower lobe of the ligand binding core, when there is no interdomain stabilization from a bound ligand. Limited information is available about the conformation and mobility of the ligand binding cores, either as isolated proteins or when part of a full-length receptor. However, several published studies on isolated ligand binding cores support the idea that D2 might be comparatively unrestrained and able to sample a range of conformations in the absence of agonist. Small angle X-Ray scattering (SAXS) data from soluble GluR2 ligand binding core monomers in the absence of ligands yielded a scattering envelope interpreted as corresponding to the time average of the agonist-bound closed state and antagonist-bound open state crystal structures (Madden et al., 2005). However, molecular dynamics simulations of the GluR2 ligand binding cores, and analysis of the same SAXS dataset, yielded a different interpretation, that the ligand binding core can sample hyper extended open conformations (Lau and Roux, 2007), like those we propose contribute to trapping in the absence of ligands. Additional evidence for conformational flexibility of the iGluR ligand binding cores comes from studies of additional ligand binding domain antagonist crystal structures, which also reveal hyper extension compared with the GluR2 apo crystal structure (Kasper et al., 2006; Mayer et al., 2006; Ahmed et al., 2009). These experiments were all performed using soluble ligand binding domains, but our experiments provide the first evidence that this flexibility is also present in the full-length receptor, where the ligand binding cores are constrained by attachment to the ion channel and to the large extracellular N terminal domain.
Redox chemistry of the introduced disulfide bridges
We expected that disulfide bonds formed during oxidizing conditions in our experiments would essentially be irreversible, until we reapplied reducing conditions. The incomplete trapping that we observed in certain conditions, even with comparatively long exposures to oxidizing agents (6 min), similar to the timescale of biochemical experiments, was therefore surprising, suggesting that trapping was reversible on the timescale of the experiment. The extent of trapping was not increased with higher concentrations of CuPhen (data not shown). The apparent instability of the disulfide bonds in oxidizing conditions was mirrored by the fast rate of reduction of the bonds in 1 mm DTT (0.3 s–1).
Our results are entirely consistent with those of Armstrong et al. (2006) who reported incomplete trapping in biochemical experiments, and a standing current in the absence of DTT in oocytes. Thus, in full-length AMPA receptors, the introduced disulfides at S729 are inherently unstable and, on average, only partially oxidized under the conditions of our experiments.
Although protein disulfide bonds are typically stable, surrounding amino acids can act as nucleophiles and attack the bond. Disulfide isomerases contain unstable disulfide bonds in their active sites (Zapun et al., 1993, 1995), and disulfide bonds resistant to oxidation by CuPhen have been observed in crystal structures (Heras et al., 2004). Although for the GluR2 ligand binding domain continuous electron density was observed for the disulfide bond between the Cys residues at position 729 (Armstrong et al., 2006), the cross-linked dimers may have been “selected” from a mixed solution population by the crystal form.
Conclusions
Our results reveal an unexpected degree of mobility in the ligand binding cores of AMPA receptor ion channels. The molecular framework for interpreting our data was made possible by recent progress in the structural biology of glutamate receptors. Whether similar conformational flexibility occurs also in kainate and NMDA receptors, and for heteromeric AMPA receptor assemblies which predominate in the brain, is worth investigating.
Footnotes
This work was supported by the intramural research program of the National Institute of Child Health and Human Development, National Institutes of Health, and Department of Health and Human Services. A.J.R.P performed experiments and analyzed data; A.J.R.P and M.L.M. designed experiments and wrote the paper. We thank Carla Glasser for preparing cDNAs; Drs. James Howe and Wei Zhang (Yale University) for advice and the gift of piezo wafers; Dr. J. Diamond [National Institute of Neurological Disorders and Stroke (NINDS)] for the loan of the EX100 amplifier; and the NINDS DNA sequencing facility and Dr. P. Seeburg for the gift of wild-type cDNAs.
References
- Ahmed AH, Thompson MD, Fenwick MK, Romero B, Loh AP, Jane DE, Sondermann H, Oswald RE. Mechanisms of antagonism of the GluR2 AMPA receptor: structure and dynamics of the complex of two willardiine antagonists with the glutamate binding domain. Biochemistry. 2009;48:3894–3903. doi: 10.1021/bi900107m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong N, Gouaux E. Mechanisms for activation and antagonism of an AMPA-sensitive glutamate receptor: Crystal structures of the GluR2 ligand binding core. Neuron. 2000;28:165–181. doi: 10.1016/s0896-6273(00)00094-5. [DOI] [PubMed] [Google Scholar]
- Armstrong N, Sun Y, Chen GQ, Gouaux E. Structure of a glutamate-receptor ligand-binding core in complex with kainate. Nature. 1998;395:913–917. doi: 10.1038/27692. [DOI] [PubMed] [Google Scholar]
- Armstrong N, Mayer M, Gouaux E. Tuning activation of the AMPA-sensitive GluR2 ion channel by genetic adjustment of agonist-induced conformational changes. Proc Natl Acad Sci U S A. 2003;100:5736–5741. doi: 10.1073/pnas.1037393100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong N, Jasti J, Beich-Frandsen M, Gouaux E. Measurement of conformational changes accompanying desensitization in an ionotropic glutamate receptor. Cell. 2006;127:85–97. doi: 10.1016/j.cell.2006.08.037. [DOI] [PubMed] [Google Scholar]
- Benveniste M, Mayer ML. Structure-activity analysis of binding kinetics for NMDA receptor competitive antagonists: the influence of conformational restriction. Br J Pharmacol. 1991;104:207–221. doi: 10.1111/j.1476-5381.1991.tb12409.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie D, Lange GD. Functional stoichiometry of glutamate receptor desensitization. J Neurosci. 2002;22:3392–3403. doi: 10.1523/JNEUROSCI.22-09-03392.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KB, Yuan H, Traynelis SF. Structural aspects of AMPA receptor activation, desensitization and deactivation. Curr Opin Neurobiol. 2007;17:281–288. doi: 10.1016/j.conb.2007.03.014. [DOI] [PubMed] [Google Scholar]
- Heras B, Edeling MA, Schirra HJ, Raina S, Martin JL. Crystal structures of the DsbG disulfide isomerase reveal an unstable disulfide. Proc Natl Acad Sci U S A. 2004;101:8876–8881. doi: 10.1073/pnas.0402769101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin R, Singh SK, Gu S, Furukawa H, Sobolevsky AI, Zhou J, Jin Y, Gouaux E. Crystal structure and association behavior of the GluR2 amino terminal domain. EMBO J. 2009;28:1812–1823. doi: 10.1038/emboj.2009.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasper C, Pickering DS, Mirza O, Olsen L, Kristensen AS, Greenwood JR, Liljefors T, Schousboe A, Wätjen F, Gajhede M, Sigurskjold BW, Kastrup JS. The structure of a mixed GluR2 ligand-binding core dimer in complex with (S)-glutamate and the antagonist (S)-NS1209. J Mol Biol. 2006;357:1184–1201. doi: 10.1016/j.jmb.2006.01.024. [DOI] [PubMed] [Google Scholar]
- Kobashi K. Catalytic oxidation of sulfhydryl groups by o-phenanthroline copper complex. Biochim Biophys Acta. 1968;158:239–245. doi: 10.1016/0304-4165(68)90136-0. [DOI] [PubMed] [Google Scholar]
- Lau AY, Roux B. The free energy landscapes governing conformational changes in a glutamate receptor ligand-binding domain. Structure. 2007;15:1203–1214. doi: 10.1016/j.str.2007.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madden DR. The structure and function of glutamate receptor ion channels. Nat Rev Neurosci. 2002;3:91–101. doi: 10.1038/nrn725. [DOI] [PubMed] [Google Scholar]
- Madden DR, Armstrong N, Svergun D, Pérez J, Vachette P. Solution X-ray scattering evidence for agonist- and antagonist-induced modulation of cleft closure in a glutamate receptor ligand-binding domain. J Biol Chem. 2005;280:23637–23642. doi: 10.1074/jbc.M414523200. [DOI] [PubMed] [Google Scholar]
- Mayer ML. Glutamate receptors at atomic resolution. Nature. 2006;440:456–462. doi: 10.1038/nature04709. [DOI] [PubMed] [Google Scholar]
- Mayer ML, Ghosal A, Dolman NP, Jane DE. Crystal structures of the kainate receptor GluR5 ligand binding core dimer with novel GluR5-selective antagonists. J Neurosci. 2006;26:2852–2861. doi: 10.1523/JNEUROSCI.0123-06.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plested AJ, Mayer ML. Structure and mechanism of kainate receptor modulation by anions. Neuron. 2007;53:829–841. doi: 10.1016/j.neuron.2007.02.025. [DOI] [PubMed] [Google Scholar]
- Robert A, Howe JR. How AMPA receptor desensitization depends on receptor occupancy. J Neurosci. 2003;23:847–858. doi: 10.1523/JNEUROSCI.23-03-00847.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman JS, Cathala L, Steuber V, Silver RA. Synaptic depression enables neuronal gain control. Nature. 2009;457:1015–1018. doi: 10.1038/nature07604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozov A, Jerecic J, Sakmann B, Burnashev N. AMPA receptor channels with long-lasting desensitization in bipolar interneurons contribute to synaptic depression in a novel feedback circuit in layer 2/3 of rat neocortex. J Neurosci. 2001;21:8062–8071. doi: 10.1523/JNEUROSCI.21-20-08062.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern-Bach Y, Russo S, Neuman M, Rosenmund C. A point mutation in the glutamate binding site blocks desensitization of AMPA receptors. Neuron. 1998;21:907–918. doi: 10.1016/s0896-6273(00)80605-4. [DOI] [PubMed] [Google Scholar]
- Sun Y, Olson R, Horning M, Armstrong N, Mayer M, Gouaux E. Mechanism of glutamate receptor desensitization. Nature. 2002;417:245–253. doi: 10.1038/417245a. [DOI] [PubMed] [Google Scholar]
- Taschenberger H, von Gersdorff H. Fine-tuning an auditory synapse for speed and fidelity: developmental changes in presynaptic waveform, EPSC kinetics, and synaptic plasticity. J Neurosci. 2000;20:9162–9173. doi: 10.1523/JNEUROSCI.20-24-09162.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zapun A, Bardwell JC, Creighton TE. The reactive and destabilizing disulfide bond of DsbA, a protein required for protein disulfide bond formation in vivo. Biochemistry. 1993;32:5083–5092. doi: 10.1021/bi00070a016. [DOI] [PubMed] [Google Scholar]
- Zapun A, Missiakas D, Raina S, Creighton TE. Structural and functional characterization of DsbC, a protein involved in disulfide bond formation in Escherichia coli. Biochemistry. 1995;34:5075–5089. doi: 10.1021/bi00015a019. [DOI] [PubMed] [Google Scholar]
- Zhang W, Robert A, Vogensen SB, Howe JR. The relationship between agonist potency and AMPA receptor kinetics. Biophys J. 2006;91:1336–1346. doi: 10.1529/biophysj.106.084426. [DOI] [PMC free article] [PubMed] [Google Scholar]