

Abstract
At a number of synapses, long-term potentiation (LTP) can be expressed by an increase in presynaptic strength, but it is unknown whether presynaptic LTP is expressed solely through an increase in the probability that a single vesicle is released or whether it can increase multivesicular release (MVR). Here, we show that presynaptic LTP decreases inhibition of AMPA receptor EPSCs by a low-affinity antagonist at parallel fiber–molecular layer interneuron (PF–MLI) synapses. This indicates that LTP induction results in larger glutamate concentration transients in the synaptic cleft, a result indicative of MVR, and suggests that MVR can be modified by long-term plasticity. A similar decrease in inhibition was observed when release probability (PR) was increased by forskolin, elevated extracellular Ca2+, and paired-pulse facilitation. Furthermore, we show that MVR may occur under baseline physiological conditions, as inhibition increased when PR was lowered by reducing extracellular Ca2+ or by activating presynaptic adenosine receptors. These results suggest that at PF–MLI synapses, MVR occurs under control conditions and is increased when PR is elevated by both short- and long-term plasticity mechanisms.
Introduction
Long-term potentiation (LTP) is an essential component of many models of learning and memory. LTP can be mediated by changes on either side of the synaptic cleft, either presynaptically through an increase in transmitter release or postsynaptically through the insertion of additional glutamatergic receptors (Nicoll and Malenka, 1995). LTP expressed presynaptically has been observed at cerebellar parallel fiber (PF) synapses, hippocampal mossy fiber synapses, and corticothalamic synapses (Zalutsky and Nicoll, 1990; Salin et al., 1996; Castro-Alamancos and Calcagnotto, 1999; Rancillac and Crépel, 2004). The induction of presynaptic LTP at these synapses is independent of postsynaptic NMDA receptor activation and is instead mediated by a rise in presynaptic Ca2+, activation of adenylyl cyclase, and the protein kinase A (PKA) pathway (Weisskopf et al., 1994; Salin et al., 1996), and modification of release machinery proteins RIM1α and Rab3a (Castillo et al., 1997, 2002; Lonart et al., 2003). Because presynaptic LTP does not affect basal- or activity-dependent presynaptic Ca2+ dynamics (Regehr and Tank, 1991; Chen and Regehr, 1997; but see Qiu and Knöpfel, 2007), it is thought that the changes in RIM1α and Rab3a result in enhanced coupling between Ca2+ and release (Lonart et al., 2003).
Expression of presynaptic LTP may result from an increase in the number of synapses that release a single vesicle, but it could also increase the probability that multiple vesicles are released from individual synapses. Multivesicular release (MVR) has been observed at synapses with initially high release probability (PR) and when PR is transiently increased during short-term plasticity (Tong and Jahr, 1994; Wadiche and Jahr, 2001; Oertner et al., 2002; Foster et al., 2005; Biró et al., 2006; Christie and Jahr, 2006). It is unknown whether long-term plasticity such as presynaptic LTP can enhance MVR.
We studied presynaptic LTP at PF–molecular layer interneuron (MLI) synapses in rat cerebellum. We assayed MVR by measuring the inhibition of AMPA receptor (AMPAR)-mediated EPSCs by the low-affinity antagonist γ-d-glutamylglycine (γ-DGG). We found that presynaptic LTP is expressed, in part, by an increase in MVR. A similar increase in MVR was observed when PR was increased by forskolin, elevated extracellular Ca2+, and short-term facilitation, suggesting that an increase in PR underlies the enhancement of MVR after presynaptic LTP induction. The degree of MVR covaried with PR; MVR decreased when we lowered PR by reducing extracellular Ca2+ or by applying an adenosine receptor agonist. Thus, at PF–MLI synapses, MVR occurs under control conditions and is increased when PR is elevated by both short- and long-term plasticity mechanisms.
Materials and Methods
Coronal cerebellar slices (300 μm) were cut from Sprague Dawley rats aged postnatal day 15–19 in accordance with Oregon Health & Science University Institutional Animal Care and Use Committee guidelines. Rats were anesthetized, decapitated, and the brain was rapidly removed in ice-cold cutting solution containing (in mm) 110 choline chloride, 25 glucose, 25 NaHCO3, 11.5 sodium ascorbate, 7 MgCl2, 3 sodium pyruvate, 2.5 KCl, 1.25 NaH2PO4, 0.5 CaCl2. Slices were cut (VT1000S; Leica) from the vermis and incubated at 34°C for 30 min, then at room temperature in artificial CSF (ACSF) containing (in mm) 124 NaCl, 26 NaHCO3, 10 glucose, 3 KCl, 2 CaCl2, 1.3 MgCl2, 1 NaH2PO4, 0.010 3-((R)-2-carboxypiperazin-4-yl)-propyl-1-phosphonic acid (R-CPP). When the extracellular [Ca2+] was changed to 1 and 3 mm Ca2+, the extracellular Mg2+ concentration was changed to 2.3 and 0.3 mm, respectively. The internal solution contained (in mm) 130 CsMeSO3, 10 HEPES, 10 Cs-BAPTA, 4 NaCl, 4 Mg-ATP, 0.4 Na-GTP, adjusted to pH 7.30 with CsOH (295 mOsm). Cells were held at −80 mV (not corrected for junction potential).
MLIs were identified by their location in the outer third of the molecular layer and their high input resistance (>1 GΩ). PFs were stimulated with patch electrodes filled with 2 m NaCl, positioned 50–100 μm lateral to the recorded cell. Whole-cell recordings were made with 4–6 MΩ pipettes using an Axopatch 1C amplifier (Molecular Devices). Recordings were filtered at 5 kHz and digitized at 20 kHz (Instrutech; ITC-18) using custom programs running in IgorPro (Wavemetrics). Cells were not analyzed if the series resistance changed >15%. Recordings were made at 32–34°C except for a few recordings in 1 and 2 mm Ca2+ which were made at room temperature (21–23°C). There was no difference in inhibition by γ-DGG at the two temperatures.
Assessing changes in γ-DGG block.
AMPAR-mediated EPSCs were evoked in MLIs by stimulating PFs (5–70 V, 10 μs) in the presence of R-CPP and picrotoxin to block NMDA and GABAA receptors, respectively. Pairs of EPSCs separated by 20 ms were elicited every 10 s. After a baseline period, γ-DGG (1 mm) was bath applied for 5–10 min. Cells were analyzed if the EPSCs recovered to at least 75% of baseline after washout. The degree of inhibition by a low-affinity, competitive antagonist, such as γ-DGG, is an indication of the size of the concentration transient of transmitter released into the synaptic cleft (Clements et al., 1992; Tong and Jahr, 1994; Wadiche and Jahr, 2001). A larger glutamate transient will compete more effectively than a smaller transient, resulting in a smaller percentage inhibition of the EPSC. The high-affinity competitive antagonist NBQX has a bound time much longer than the duration of the glutamate transient and will act in a noncompetitive manner.
Drugs.
R-CPP and N6-cyclopentyladenosine (CPA) (Tocris Bioscience) were dissolved in ACSF. γ-DGG (Tocris Bioscience) was dissolved in 10 μm NaOH and ACSF. Picrotoxin (Sigma) and forskolin (Tocris Bioscience) were dissolved in DMSO, at 500 and 50 mm, respectively.
Coefficient of variation analysis.
Coefficient of variation (CV2) was determined for cells that had a stable baseline (no significant regression) for at least 40 stimuli in each condition. CV was calculated as [(σ)2 − (σ of noise)2]/mean2.
Analysis of quantal amplitude.
Asynchronous events were identified using a template function in Axograph X and then sorted manually. Average cumulative probability histograms were made by normalizing the amplitude of events to the mean during the baseline for each pathway. Data were binned in deciles, and histograms were compared using the Kolmogorov–Smirnov (KS) test.
Statistics.
A paired t test was used to compare paired experimental manipulations. A single-factor ANOVA and Fisher's post hoc test were used when comparing more than two data sets. Significance was considered as p < 0.05.
Results
MVR increased after presynaptic LTP induction
Presynaptic LTP is well characterized at PF synapses (Salin et al., 1996; Lonart et al., 2003; Rancillac and Crépel, 2004) and is expressed by an increase in release probability, but whether LTP increases MVR has not been explored. To test this, AMPAR-mediated EPSCs were evoked by stimulating two PF pathways (Fig. 1A). Pathway independence was verified by stimulating the two pathways in quick succession (20 ms) to test for paired-pulse facilitation (PPF) (Fig. 1B). The pathways were considered independent if no facilitation of the second stimulated pathway occurred.
Figure 1.
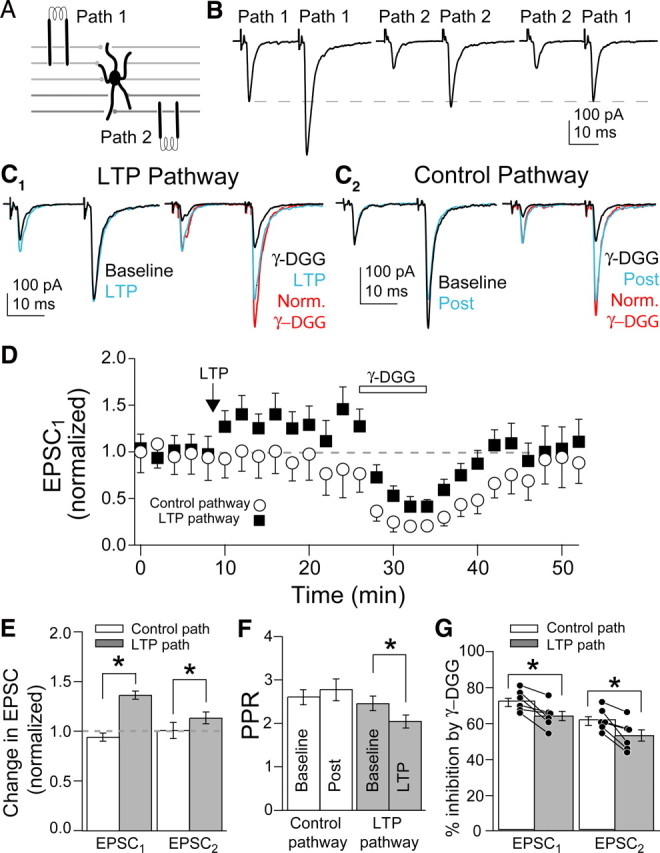
MVR increased after presynaptic LTP induction. A, Schematic of independent PF pathway stimulation onto MLI. Stimulation electrodes were placed at different depths in the cerebellar slice. B, Left, PF EPSCs evoked by paired stimuli on Path 1. Middle, EPSCs evoked by stimuli on Path 2. Right, Path 1 was stimulated 20 ms after Path 2 to check for facilitation of Path 1. Dashed line, Amplitude of the first Path 1 EPSC. C1, Left, EPSCs before (baseline) and after LTP induction (LTP). Right, Effect of γ-DGG on EPSCs after LTP induction. C2, In the same cell as C1, EPSCs evoked on the control pathway. Left, EPSCs before (baseline) and after (post) LTP was induced on the other pathway. Right, Effect of γ-DGG on EPSCs evoked on control pathway. D, Average time course of the two pathway experiments. E, F, Summary of the changes for EPSCs and PPR on both pathways. G, Percentage inhibition by γ-DGG for EPSCs compared between pathways within a single cell. Bars are SEM. Asterisks, p < 0.05.
LTP was induced by stimulating one pathway (chosen randomly) at 8 Hz for 30 s. We observed significant LTP in 6 of 10 cells, similar to previous reports (Rancillac and Crépel, 2004). In those cells, LTP induction increased EPSC1 to 134 ± 4% of baseline and EPSC2 to 112 ± 6% of baseline, whereas the control pathway was unchanged (Fig. 1C,E). Paired-pulse ratio (PPR) decreased in the LTP pathway (p < 0.04; n = 6) (Fig. 1C,F), consistent with presynaptic expression. To test for changes in MVR, we compared the inhibition of the PF–MLI EPSC in the control and potentiated pathway by the low-affinity AMPAR antagonist γ-DGG (1 mm). γ-DGG inhibited both EPSC1 and EPSC2 less in the LTP pathway than in the control pathway (p < 0.02 and p < 0.01, respectively) (Fig. 1G); the degree of potentiation of EPSC1 was inversely related to the amount of inhibition by γ-DGG (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). In the four cells in which LTP could not be induced (EPSC1 107 ± 5%; EPSC2 100 ± 2%), γ-DGG inhibition was not different from the control pathway or the control pathway of the six cells in which LTP was induced (supplemental Table 1, available at www.jneurosci.org as supplemental material). These results indicate that presynaptic LTP is at least partly expressed by an increase in MVR.
MVR increased after forskolin
Previous studies have shown that presynaptic LTP is mediated by the activation of PKA and is mimicked by application of the adenylyl cyclase activator forskolin (Salin et al., 1996; Chen and Regehr, 1997). Thus, forskolin should increase MVR similarly to that of LTP induction. Forskolin (10 μm) increased EPSC1 to 176 ± 13% and EPSC2 to 131 ± 11% of baseline (Fig. 2A) and decreased PPR from 2.72 ± 0.30 to 1.99 ± 0.17 (p < 0.04) (Fig. 2B). After forskolin, γ-DGG inhibited EPSC1 less than in control cells (p < 0.003; forskolin, n = 6; control, n = 14) (Fig. 2B). Inhibition of EPSC2 did not change significantly (p = 0.11). Forskolin did not change the inhibition by the high-affinity competitive antagonist NBQX (n = 3) (Fig. 2C). These results show that forskolin-induced potentiation increased MVR to a similar extent as presynaptic LTP induction, consistent with the idea that these two phenomena share a common mechanism (Salin et al., 1996; Lonart et al., 1998).
Figure 2.

MVR increased with forskolin application. A, Time course of average EPSC1 amplitudes, in control, forskolin, and γ-DGG. B, Percentage inhibition of EPSCs by γ-DGG before and after forskolin application. Control refers to cells in 2 mm Ca2+. C, Percentage inhibition of EPSCs by NBQX before and after forskolin application.
Paired-pulse facilitation and elevated extracellular [Ca2+] increased MVR at PF–MLI synapses
At synapses where MVR occurs, the degree of MVR is dependent on PR (Tong and Jahr, 1994; Wadiche and Jahr, 2001; Foster et al., 2005; Biró et al., 2006; Christie and Jahr, 2006), but this has not been tested at PF–MLI synapses. The association of MVR and PR raises the possibility that the increase in MVR associated with LTP and forskolin results exclusively from increasing PR. To test this, we compared γ-DGG inhibition in those conditions to short-term plasticity and elevated extracellular Ca2+. In control (2 mm Ca2+), γ-DGG inhibited EPSC2 9.6% less than EPSC1, increasing PPR (p < 0.001) (Fig. 3A). Raising extracellular Ca2+ to 3 mm decreased PPR (p < 0.05; n = 5) (Fig. 3A) and the amount of inhibition by γ-DGG for both EPSC1 and EPSC2 (p < 0.001 and p < 0.03, respectively) (Fig. 3B), although γ-DGG still inhibited EPSC2 less than EPSC1 (p < 0.04) (Fig. 3B). NBQX inhibited all EPSCs equally in both 2 and 3 mm extracellular Ca2+ (Fig. 3C). Therefore, at PF–MLI synapses, the transient increase in PR underlying (PPF) (Zucker and Regehr, 2002) and the increase in PR caused by elevated extracellular Ca2+ increased MVR. These manipulations modified MVR to a similar extent as LTP, arguing that the change in MVR after LTP can be attributed to its effect on PR.
Figure 3.
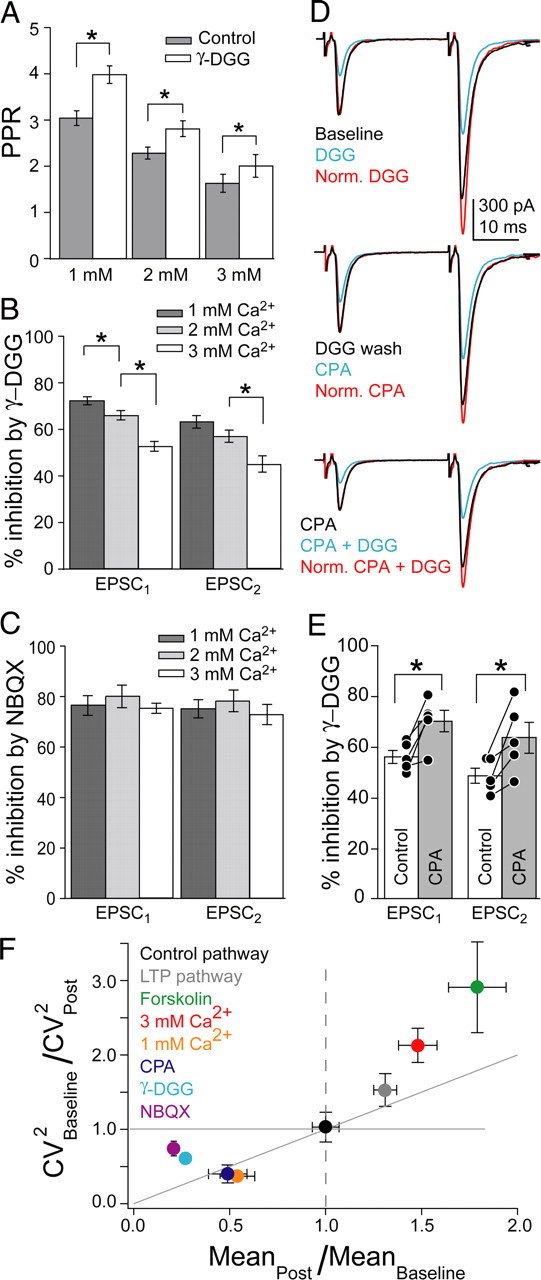
MVR covaried with PR. A, PPR in control conditions and during γ-DGG application in 1, 2, and 3 mm Ca2+. B, Percentage inhibition by γ-DGG in 1, 2, and 3 mm Ca2+ for EPSCs. C, Percentage inhibition by NBQX in 1, 2, and 3 mm Ca2+. D, Top, EPSCs before and after γ-DGG application. Middle, EPSCs in the same cell after γ-DGG wash and subsequent CPA application. Bottom, γ-DGG was applied again in the presence of CPA. E, Percentage inhibition of EPSCs by γ-DGG before and during CPA application within single cells. F, CV analysis, plotted as the ratio of CV−2 versus the mean amplitude ratio, normalized to baseline. For increases in mean amplitude (mean ratio, >1), points that fall on or above the diagonal signify a presynaptic change, whereas for decreases in mean amplitude (mean ratio, <1), presynaptic changes are reflected in points that fall on or below the diagonal.
MVR covaried with PR at PF–MLI synapses
Our data indicate that MVR increased when PR was elevated, but it was not clear whether MVR only occurs when PR is elevated or whether more MVR occurs in these conditions. We tested this by lowering PR. First, we lowered PR by decreasing extracellular Ca2+ to 1 mm from 2 mm. This increased PPR (p < 0.001; n = 14) (Fig. 3A) and increased γ-DGG-mediated inhibition of EPSC1 (p < 0.02) (Fig. 3B). Inhibition of EPSC2 did not increase compared with 2 mm Ca2+ (p = 0.08) (Fig. 3B) and was still less than that of EPSC1 (p < 0.001) (Fig. 3B). NBQX inhibited EPSCs to the same extent regardless of [Ca2+] (Fig. 3C). Second, we also lowered PR by applying an adenosine type 1 (A1) receptor agonist, CPA (10 nm). A1 receptors decrease PR at PF synapses by reducing action potential-evoked Ca2+ influx (Kreitzer and Regehr, 2000). CPA decreased EPSC1 to 53 ± 3% and EPSC2 to 64 ± 2% of baseline levels, resulting in increased PPR (p < 0.02; n = 5) (Fig. 3D). Inhibition of both EPSCs by γ-DGG increased (EPSC1, p < 0.05; EPSC2, p < 0.03) (Fig. 3E). The level of MVR was decreased by lowering extracellular Ca2+ and activation of A1 receptors, indicating that MVR occurs under control conditions and that it can be dynamically altered by changes in PR.
Coefficient of variation analysis
To verify that dynamic changes in PR underlie the corresponding changes in MVR, we analyzed the CV of the EPSC amplitude (Bekkers and Stevens, 1990; Malinow and Tsien, 1990; Faber and Korn, 1991). Increasing extracellular Ca2+ from 2 to 3 mm changed (CV)−2 in a manner consistent with increased PR (n = 5) (Fig. 3F), whereas decreasing extracellular Ca2+ from 2 to 1 mm or applying the adenosine agonist CPA decreased PR (1 mm, n = 4; CPA, n = 3) (Fig. 3F). In contrast, partial block of AMPA receptors by application of γ-DGG or NBQX changed (CV)−2 only slightly, indicative of a postsynaptic effect (Bekkers and Stevens, 1990; Faber and Korn, 1991) (γ-DGG, n = 6; NBQX, n = 7) (Fig. 3F). CV analysis indicated a presynaptic change after LTP induction, whereas the control pathway (CV)−2 was unaffected (n = 6) (Fig. 3F). Forskolin also increased (CV)−2 (n = 5) (Fig. 3F). Thus, CV analysis confirmed that LTP induction and forskolin increased PR to a similar extent as raising extracellular Ca2+ to 3 mm, arguing that increased PR is responsible for the observed enhancement of MVR for all these manipulations.
Quantal amplitude does not change after presynaptic LTP induction
We have attributed the changes in inhibition by γ-DGG to changes in MVR, although the γ-DGG manipulation actually reports changes in the glutamate concentration transient. It is possible, then, that instead of enhancing MVR, LTP increases glutamate release by increasing quantal size through changes in vesicle loading or compound fusion between vesicles before exocytosis (He et al., 2009). To test this, we examined asynchronous, presumably quantal (Atluri and Regher, 1998), events occurring up to 400 ms after the paired stimuli (Fig. 4A). There was no difference in average amplitude of these events before and after LTP (baseline, −103.8 ± 10.6 pA; LTP, −108.5 ± 10.2 pA; n = 4, paired t test; p = 0.23), nor was there a difference in the cumulative amplitude probability distributions (Fig. 4B) (KS test; p = 0.49). These results suggest that presynaptic LTP did not change quantal size and that it potentiated evoked EPSCs by increasing the number of vesicles released.
Figure 4.

Quantal size is not altered by LTP induction. A, Asynchronous events (box) after paired stimulation of PFs. B, Cumulative probability plots of the amplitudes of aEPSCs in control and after inducing LTP. Inset, Averages of aEPSCs in the two conditions. All non-overlapping aEPSCs after the decay of the second evoked EPSC were included in the analysis.
Discussion
We find that all manipulations that increase PR at PF–MLI synapses also increase the synaptic glutamate transient. This suggests that MVR can be regulated by both short- and long-term plasticity mechanisms. Spillover from adjacent synapses can also augment the glutamate transient, but the peak amplitude of the postsynaptic response is usually unaffected by transmitter spillover, even at closely spaced synapses with little glial investiture (Wadiche and Jahr, 2001; DiGregorio et al., 2002; Christie and Jahr, 2006; Balakrishnan et al., 2009). At PF–MLI synapses, spillover and pooling of glutamate occurs only at high stimulus strength when many adjacent PFs simultaneously fire action potentials (Carter and Regehr, 2000). This would be an unlikely occurrence with the low stimulus strengths used in the present experiments. Compound fusion may also increase the synaptic glutamate transient. We find, however, that the amplitudes of asynchronous quantal events are not altered after LTP induction. This argues that compound fusion is not a common event at this synapse, unlike recent findings at the calyx of Held where asynchronous events increase in size after intense stimulation (He et al., 2009). Therefore, presynaptic LTP at PF–MLI synapses is at least partly expressed by an increase in the frequency of MVR at individual synapses rather than being mediated solely by an increase in the number of synapses that release a single vesicle. That the low-affinity antagonist technique is not particularly sensitive to changes in the glutamate transient (Wadiche and Jahr, 2001) suggests that MVR plays a prominent role in LTP expression.
At both PF synapses and mossy fiber synapses, the resting intracellular Ca2+ concentration and the action potential-evoked Ca2+ influx into presynaptic terminals are unchanged by presynaptic LTP induction (Regehr and Tank, 1991; Chen and Regehr, 1997). Instead, it is thought that presynaptic LTP is expressed through modification of release machinery proteins RIM1α and Rab3a that enhance coupling between Ca2+ and release (Lonart et al., 1998; Lonart et al., 2003). Thus, increased Ca2+ transients, as occurs during PPF and with elevated extracellular Ca2+, as well as modification of the release machinery, increase both PR and MVR indicating that however PR is altered, changes in MVR follow. As the mechanisms of presynaptic LTP seem to be shared by all the synapses where it has been observed, including PF, mossy fiber and corticothalamic inputs in somatosensory cortex (Salin et al., 1996; Castro-Alamancos and Calcagnotto, 1999), MVR is probably enhanced at all of these synapses after LTP induction. Indeed, optical quantal analysis at mossy fiber synapses suggested that MVR may occur after LTP (Reid et al., 2004). In addition, induction of presynaptically expressed long-term depression (LTD) can also decrease MVR (Lei and McBain, 2004). At PFs synapses, LTD is primarily expressed through postsynaptic mechanisms (Hartell, 2002; Shen et al., 2002), but it was recently found that block of presynaptic LTP reveals a form of presynaptically expressed LTD (Qiu and Knöpfel, 2009). At this and other synapses where presynaptic LTD decreases PR (Lovinger, 2008), LTD may be expressed in part by a decrease in MVR.
PF synapses onto Purkinje cells display an increase in MVR with PPF and with increasing extracellular Ca2+ (Foster et al., 2005), similar to our present findings. We additionally show that the level of MVR that occurs at PF–MLI synapses is altered by lowering PR; MVR is decreased by lowering extracellular Ca2+ to 1 mm or by application of an adenosine agonist, CPA. This indicates that a basal level of MVR occurs in control conditions (2 mm extracellular Ca2+) even at this relatively low PR synapse and suggests that MVR may occur more readily than previously thought.
Our results suggest that both long- and short-term forms of plasticity can influence MVR through their effects on PR. In some conditions, it is possible to observe the interaction between long-term changes and short-term plasticity resulting from PPF. More MVR always occurs on EPSC2 than EPSC1 at this facilitating synapse, suggesting that PR is always higher on EPSC2 relative to EPSC1. However, forskolin does not increase MVR for EPSC2 over control levels. Forskolin increases PR overall, but at the same time decreases PPF, resulting in no net change in MVR for EPSC2. Similarly, in 1 mm Ca2+, EPSC2 does not show a decrease in MVR relative to control. In this case, the decrease in PR caused by low extracellular Ca2+ is counteracted by the enhancement of PR resulting from increased PPF. These results show that the amount of MVR occurring during a given release event is a result of the interaction between long-term and short-term effects on PR. Thus, the magnitude of MVR may be simply a function of PR, regardless of the underlying mechanisms defining PR at any given time.
Footnotes
This work was supported by National Institutes of Health (NIH) grants (MH074989), NIH training grants (DK007680 and NS007381; V.A.B. and J.R.P., respectively), and a Tartar Trust Fellowship (V.A.B.). We thank the Jahr laboratory members and Dr. Kevin J. Bender for discussions and critical readings of this manuscript.
References
- Atluri PP, Regehr WG. Delayed release of neurotransmitter from cerebellar granule cells. J Neurosci. 1998;18:8214–8227. doi: 10.1523/JNEUROSCI.18-20-08214.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishnan V, Kuo SP, Roberts PD, Trussell LO. Slow glycinergic transmission mediated by transmitter pooling. Nat Neurosci. 2009;12:286–294. doi: 10.1038/nn.2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekkers JM, Stevens CF. Presynaptic mechanism for long-term potentiation in the hippocampus. Nature. 1990;346:724–729. doi: 10.1038/346724a0. [DOI] [PubMed] [Google Scholar]
- Biró AA, Holderith NB, Nusser Z. Release probability-dependent scaling of the postsynaptic responses at single hippocampal GABAergic synapses. J Neurosci. 2006;26:12487–12496. doi: 10.1523/JNEUROSCI.3106-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AG, Regehr WG. Prolonged synaptic currents and glutamate spillover at the parallel fiber to stellate cell synapse. J Neurosci. 2000;20:4423–4434. doi: 10.1523/JNEUROSCI.20-12-04423.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo PE, Janz R, Südhof TC, Tzounopoulos T, Malenka RC, Nicoll RA. Rab3A is essential for mossy fibre long-term potentiation in the hippocampus. Nature. 1997;388:590–593. doi: 10.1038/41574. [DOI] [PubMed] [Google Scholar]
- Castillo PE, Schoch S, Schmitz F, Südhof TC, Malenka RC. RIM1alpha is required for presynaptic long-term potentiation. Nature. 2002;415:327–330. doi: 10.1038/415327a. [DOI] [PubMed] [Google Scholar]
- Castro-Alamancos MA, Calcagnotto ME. Presynaptic long-term potentiation in corticothalamic synapses. J Neurosci. 1999;19:9090–9097. doi: 10.1523/JNEUROSCI.19-20-09090.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Regehr WG. The mechanism of cAMP-mediated enhancement at a cerebellar synapse. J Neurosci. 1997;17:8687–8694. doi: 10.1523/JNEUROSCI.17-22-08687.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie JM, Jahr CE. Multivesicular release at Schaffer collateral-CA1 hippocampal synapses. J Neurosci. 2006;26:210–216. doi: 10.1523/JNEUROSCI.4307-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements JD, Lester RA, Tong G, Jahr CE, Westbrook GL. The time course of glutamate in the synaptic cleft. Science. 1992;258:1498–1501. doi: 10.1126/science.1359647. [DOI] [PubMed] [Google Scholar]
- DiGregorio DA, Nusser Z, Silver RA. Spillover of glutamate onto synaptic AMPA receptors enhances fast transmission at a cerebellar synapse. Neuron. 2002;35:521–533. doi: 10.1016/s0896-6273(02)00787-0. [DOI] [PubMed] [Google Scholar]
- Faber DS, Korn H. Applicability of the coefficient of variation method for analyzing synaptic plasticity. Biophys J. 1991;60:1288–1294. doi: 10.1016/S0006-3495(91)82162-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster KA, Crowley JJ, Regehr WG. The influence of multivesicular release and postsynaptic receptor saturation on transmission at granule cell to Purkinje cell synapses. J Neurosci. 2005;25:11655–11665. doi: 10.1523/JNEUROSCI.4029-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartell NA. Parallel fiber plasticity. Cerebellum. 2002;1:3–18. doi: 10.1080/147342202753203041. [DOI] [PubMed] [Google Scholar]
- He L, Xue L, Xu J, McNeil BD, Bai L, Melicoff E, Adachi R, Wu LG. Compound vesicle fusion increases quantal size and potentiates synaptic transmission. Nature. 2009;459:93–97. doi: 10.1038/nature07860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG. Modulation of transmission during trains at a cerebellar synapse. J Neurosci. 2000;20:1348–1357. doi: 10.1523/JNEUROSCI.20-04-01348.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei S, McBain CJ. Two loci of expression for long-term depression at hippocampal mossy fiber-interneuron synapses. J Neurosci. 2004;24:2112–2121. doi: 10.1523/JNEUROSCI.4645-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonart G, Janz R, Johnson KM, Südhof TC. Mechanism of action of rab3A in mossy fiber LTP. Neuron. 1998;21:1141–1150. doi: 10.1016/s0896-6273(00)80631-5. [DOI] [PubMed] [Google Scholar]
- Lonart G, Schoch S, Kaeser PS, Larkin CJ, Südhof TC, Linden DJ. Phosphorylation of RIM1alpha by PKA triggers presynaptic long-term potentiation at cerebellar parallel fiber synapses. Cell. 2003;115:49–60. doi: 10.1016/s0092-8674(03)00727-x. [DOI] [PubMed] [Google Scholar]
- Lovinger DM. Presynaptic modulation by endocannabinoids. Handb Exp Pharmacol. 2008:435–477. doi: 10.1007/978-3-540-74805-2_14. [DOI] [PubMed] [Google Scholar]
- Malinow R, Tsien RW. Presynaptic enhancement shown by whole-cell recordings of long-term potentiation in hippocampal slices. Nature. 1990;346:177–180. doi: 10.1038/346177a0. [DOI] [PubMed] [Google Scholar]
- Nicoll RA, Malenka RC. Contrasting properties of two forms of long-term potentiation in the hippocampus. Nature. 1995;377:115–118. doi: 10.1038/377115a0. [DOI] [PubMed] [Google Scholar]
- Oertner TG, Sabatini BL, Nimchinsky EA, Svoboda K. Facilitation at single synapses probed with optical quantal analysis. Nat Neurosci. 2002;5:657–664. doi: 10.1038/nn867. [DOI] [PubMed] [Google Scholar]
- Qiu DL, Knöpfel T. An NMDA receptor/nitric oxide cascade in presynaptic parallel fiber-Purkinje neuron long-term potentiation. J Neurosci. 2007;27:3408–3415. doi: 10.1523/JNEUROSCI.4831-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu DL, Knöpfel T. Presynaptically expressed long-term depression at cerebellar parallel fiber synapses. Pflugers Arch. 2009;457:865–875. doi: 10.1007/s00424-008-0555-9. [DOI] [PubMed] [Google Scholar]
- Rancillac A, Crépel F. Synapses between parallel fibres and stellate cells express long-term changes in synaptic efficacy in rat cerebellum. J Physiol. 2004;554:707–720. doi: 10.1113/jphysiol.2003.055871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regehr WG, Tank DW. The maintenance of LTP at hippocampal mossy fiber synapses is independent of sustained presynaptic calcium. Neuron. 1991;7:451–459. doi: 10.1016/0896-6273(91)90297-d. [DOI] [PubMed] [Google Scholar]
- Reid CA, Dixon DB, Takahashi M, Bliss TV, Fine A. Optical quantal analysis indicates that long-term potentiation at single hippocampal mossy fiber synapses is expressed through increased release probability, recruitment of new release sites, and activation of silent synapses. J Neurosci. 2004;24:3618–3626. doi: 10.1523/JNEUROSCI.3567-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salin PA, Malenka RC, Nicoll RA. Cyclic AMP mediates a presynaptic form of LTP at cerebellar parallel fiber synapses. Neuron. 1996;16:797–803. doi: 10.1016/s0896-6273(00)80099-9. [DOI] [PubMed] [Google Scholar]
- Shen Y, Hansel C, Linden DJ. Glutamate release during LTD at cerebellar climbing fiber-Purkinje cell synapses. Nat Neurosci. 2002;5:725–726. doi: 10.1038/nn895. [DOI] [PubMed] [Google Scholar]
- Tong G, Jahr CE. Multivesicular release from excitatory synapses of cultured hippocampal neurons. Neuron. 1994;12:51–59. doi: 10.1016/0896-6273(94)90151-1. [DOI] [PubMed] [Google Scholar]
- Wadiche JI, Jahr CE. Multivesicular release at climbing fiber-Purkinje cell synapses. Neuron. 2001;32:301–313. doi: 10.1016/s0896-6273(01)00488-3. [DOI] [PubMed] [Google Scholar]
- Weisskopf MG, Castillo PE, Zalutsky RA, Nicoll RA. Mediation of hippocampal mossy fiber long-term potentiation by cyclic AMP. Science. 1994;265:1878–1882. doi: 10.1126/science.7916482. [DOI] [PubMed] [Google Scholar]
- Zalutsky RA, Nicoll RA. Comparison of two forms of long-term potentiation in single hippocampal neurons. Science. 1990;248:1619–1624. doi: 10.1126/science.2114039. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]