

Abstract
Bimolecular rate constants have been measured for reactions that involve hydrogen atom transfer (HAT) from hydroxylamines to nitroxyl radicals, using the stable radicals TEMPO• (2,2,6,6-tetramethylpiperidine-1-oxyl radical), 4-oxo-TEMPO• (2,2,6,6-tetramethyl-4-oxo-piperidine-1-oxyl radical), di-tert-butylnitroxyl (tBu2NO•), and the hydroxylamines TEMPO-H, 4-oxo-TEMPO-H, 4-MeO-TEMPO-H (2,2,6,6-tetramethyl-N-hydroxy-4-methoxy-piperidine), and tBu2NOH. The reactions have been monitored by UV-vis stopped-flow methods, using the different optical spectra of nitroxyl radicals. The HAT reactions all have |ΔGo| ≤ 1.4 kcal mol−1 and therefore are close to self-exchange reactions. The reaction of 4-oxo-TEMPO• + TEMPO-H → 4-oxo-TEMPO-H + TEMPO• occurs with k2H,MeCN = 10 ± 1 M−1 s−1 in MeCN at 298 K (K2H,MeCN = 4.5 ± 1.8). Surprisingly, the rate constant for the analogous deuterium atom transfer reaction is much slower: k2D,MeCN = 0.44 ± 0.05 M−1 s−1 with k2H,MeCN/k2D,MeCN = 23 ± 3 at 298 K. The same large kinetic isotope effect (KIE) is found in CH2Cl2, 23 ± 4, suggesting that the large KIE is not caused by solvent dynamics or hydrogen bonding to solvent. The related reaction of 4-oxo-TEMPO• with 4-MeO-TEMPO-H(D) also has a large KIE, k3H/k3D = 21 ± 3 in MeCN. For these three reactions, the EaD – EaH values, between 0.3 ± 0.6 and 1.3 ± 0.6 kcal mol−1, and the log(AH/AD) values, between 0.5 ± 0.7 and 1.1 ± 0.6, indicate that hydrogen tunneling plays an important role. The related reaction of tBu2NO• + TEMPO-H(D) in MeCN has a large KIE, 16 ± 3 in MeCN, and very unusual isotopic activation parameters, EaD – EaH = −2.6 ± 0.4 and log(AH/AD) = 3.1 ± 0.6. Computational studies, using POLYRATE, also indicate substantial tunneling in the (CH3)2NO• + (CH3)2NOH model reaction for the experimental self-exchange processes. Additional calculations on TEMPO(•/H), tBu2NO(•/H), and Ph2NO(•/H) self-exchange reactions reveal why the phenyl groups make the last of these reactions several orders of magnitude faster than the first two. By inference, the calculations also suggest why tunneling appears to be more important in the self-exchange reactions of dialkylhydroxylamines than of arylhydroxylamines.
Introduction
Hydrogen transfer reactions are among the most fundamental of chemical reactions.1 Tunneling of the proton is emphasized in many treatments of hydrogen transfer,1 2 3-4 and an increasing number of these reactions are being found to have large hydrogen/deuterium kinetic isotope effects (KIEs) and activation parameters that indicate the importance of tunneling.5 While a number of these reactions are understood in detail, there is limited intuition about why some hydrogen-transfer reactions display the hallmarks of tunneling and others do not. Described here are experimental and computational studies of hydrogen-atom transfer (HAT) pseudo self-exchange reactions between dialkylnitroxyl radicals and dialkylhydroxylamines (eq 1) that indicate the occurrence of substantial hydrogen tunneling. These are simple and unusual cases of tunneling in oxygen-to-oxygen HAT. The contrast between these reactions and the closely related reactions of arylnitroxyl radicals which do not show the experimental markers of tunneling is also examined.
| (1) |
Hydrogen atom transfer (HAT) has been studied for over a century6 and is the simplest chemical reaction that involves the transfer of two particles, a proton and an electron. It can therefore be considered to be a type of ‘proton-coupled electron transfer’ (PCET).7,8 HAT is important in combustion and selective oxidation of alkanes, in the formation and reactivity of protein-based radicals and reactive oxygen species (ROS), and many other processes.9 For example, HAT from the double allylic C–H bond in linoleic acid to the iron(III)-hydroxide active site in lipoxygenases has received particular attention because its H/D KIE of up to ∼80 indicates the importance of tunneling.5a,10 HAT involving of nitroxyl radicals and hydroxylamines is important in much of the chemistry of nitroxyl radicals,11 such as their role as catalysts and co-catalysts in oxidation of organic substrates.12-13 14 15 N-hydroxyphthalimide (NHPI) has been widely explored as a co-catalyst in Co/Mn-catalyzed autoxidations of alkylaromatics, with the active species being the corresponding phthalimide N-oxyl radical (PINO•).16 HAT reactions from benzylic C-H bonds to PINO• in acetic acid have large deuterium KIEs (17−28 at 298 K)17 and the pseudo self-exchange reaction between PINO• and 4-Me-NHPI in acetic acid has kH/kD = 11.0 (kH = 677 ± 24 M−1 s−1).18 Reactions of nitroxyl radicals with arylhydroxylamines, however, exhibit much smaller KIEs, with kH/kD = 1.5−1.9 at ambient temperatures (see Table 2 below).13,14
Table 2.
Driving Forces, Rate Constants, and Kinetic Isotope Effects for Nitroxyl plus Hydroxylamine Reactions.a
| Reaction | Solvent | ΔG°b | kH (M−1 s−1)a | kD (M−1 s−1)a,c | kH/kDa,c | Ref. |
|---|---|---|---|---|---|---|
| 4-oxo-TEMPO• + TEMPO-Hd | MeCN | −0.9 ± 0.2 | 10 ± 1 | 0.44 ± 0.05 | 23 ± 3 | e |
| 4-oxo-TEMPO• + TEMPO-H | CH2Cl2 | −1.2 ± 0.2 | 48 ± 4 | 2.1 ± 0.3 | 23 ± 4 | e |
| 4-oxo-TEMPO• + TEMPO-H | CCl4 | – | 300 ± 30 | 17 ± 4 | 18 ± 5 | e |
| 4-oxo-TEMPO• + 4-MeO-TEMPO-H | MeCN | −0.6 ± 0.2 | 7.8 ± 0.7 | 0.37 ± 0.05 | 21 ± 3 | e |
| tBu2NO• + TEMPO-H | MeCN | 1.3 ± 0.2 | 1.9 ± 0.2 | 0.12 ± 0.02 | 16 ± 3 | e |
| tBu2NO• + TEMPO-H | CH2Cl2 | 1.4 ± 0.2 | 4.6 ± 0.4 | 0.35 ± 0.04 | 13 ± 2 | e |
| PINO• + 4Me-NHPIf | AcOH(D) | −0.4 ± 0.1 | 677 ± 24 | 61.3 ± 2.1 | 11.0 ± 0.5 | 18 |
| 4-Me-PINO• + NHPIf | AcOH(D) | 0.4 ± 0.1 | 354 ± 23 | 31.8 ± 2.0 | 11.1 ± 1.0 | 18 |
| tBu2NO• + tBu2NOH | CCl4 | 0 | 320 ± 40 | – | – | 13 |
| tBu2NO• + tBu2NOH | C6H5Cl | 0 | 240 ± 60 | – | – | 13 |
| tBu(Ar)NO• + tBu(Ar)NOHg | CCl4 | 0 | (2.0 ± 0.4) × 103 | (1.3 ± 0.2) × 103 | 1.5 ± 0.4 | 13 |
| tBu(Ar)NO• + tBu(Ar)NOHg | C6H5Cl | 0 | (5.2 ± 0.4) × 102 | – | – | 13 |
| tBu(Ar)NO• + tBu(Ar)NOHg | CH2Cl2 | 0 | < 20 | – | – | 13 |
| Ph2NO• + Ph2NOH | CCl4 | 0 | > 107 | – | – | 13 |
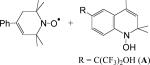 |
hexane | −0.2 ± 0.1 | (4.3 ± 0.2) × 104 | (2.5 ± 0.1) × 104 | 1.7 ± 0.1 | 14 |
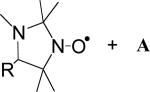 |
hexane | 2.0 ± 0.1 | (4.5 ± 0.2) × 103 | (2.9 ± 0.2) × 103 | 1.6 ± 0.1 | 14 |
| B + A (R = CPh3) | hexane | 0.9 ± 0.1 | (1.4 ± 0.1) × 104 | (7.3 ± 0.4) × 103 | 1.9 ± 0.2 | 14 |
| B (R = Ph) + A | hexane | 1.6 ± 0.1 | (8.6 ± 0.4) × 103 | (5.7 ± 0.3) × 103 | 1.5 ± 0.1 | 14 |
| TEMPO• + A | hexane | −0.6 ± 0.1 | (7.6 ± 0.4) × 104 | – | – | 14 |
| TEMPO• + A (R = CPh3) | hexane | −1.4 ± 0.1 | (1.5 ± 0.1) × 105 | – | – | 14 |
| 4-oxo-TEMPO• + A | hexane | −1.3 ± 0.1 | (6.4 ± 0.3) × 104 | – | – | 14 |
kcal mol−1.
Not corrected for the incomplete (98±1%) deuterium enrichment; the true kH/kD values are roughly a factor of two higher; see text.
Ref. 20.
This work.
PINO• = phthalimide N-oxyl radical, NHPI = N-hydroxyphthalimide.
Ar = 2,6-dimethoxyphenyl.
We have focused on HAT self-exchange reactions, such as the nitroxyl/hydroxylamine reactions examined here (eq 1), both because of their relative simplicity and because of our finding that the Marcus cross relation usually predicts HAT rate constants within an order of magnitude or two.19 20-21 This treatment is a new approach to understanding HAT rate constants22 and has been found to hold for both organic reactions and examples involving transition metal complexes. For instance, the cross relation predicts and explains the inverse temperature dependence of the rate of HAT from [FeII(H2bip)3]2+ to the stable nitroxyl radical TEMPO• (2,2,6,6-tetramethylpiperidine-1-oxyl radical).20 Of the various HAT reactions involving TEMPO• / TEMPO-H and transition metal complexes that we have examined,19,20,23,24 the Marcus approach appears to be least accurate for RuII(acac)2(py-imH) + TEMPO• → TEMPO-H and RuIII(acac)2(py-im), which has a large KIE.24b These results prompted our examination of nitroxyl/hydroxylamine self-exchange reactions; the kinetics of 4-oxo-TEMPO• plus TEMPO-H were briefly mentioned in a preliminary communication about the [FeII(H2bip)3]2+ reaction.20
Results
I. Equilibrium Constants
The reaction of 4-oxo-TEMPO• and TEMPO-H in CD3CN forms an equilibrium mixture with 4-oxo-TEMPO-H and TEMPO• (eq 2), with all four species observed by 1H NMR spectroscopy. All of the resonances have been assigned for the
 |
(2) |
paramagnetic and diamagnetic species, even though 4-oxo-TEMPO-H has not been isolated. Equilibrium is rapidly established (see below) and integration of each species using Lorentzian line fitting gave K2H,CD3CN = 4.5 ± 1.8 at 298 K.25 In CD2Cl2, K2H,CD2Cl2 = 7.6 ± 2.4 was measured similarly. As expected, the equilibrium constants for deuterium atom transfer in CD3CN and CD2Cl2 (K2D), measured using TEMPO-D, are within experimental error of being the same as the K2H values (Table 1). The equilibrium constants are the averages of three independent measurements. All errors reported herein are ± 2σ.
Table 1.
Thermodynamic Parameters for Pseudo Self-Exchange Reactions of Nitroxyl Radicals and Hydroxylamines.a
| Reaction | Solvent | K (298 K) | ΔH° | ΔS° |
|---|---|---|---|---|
| (2) 4-oxo-TEMPO• + TEMPO-H | CD3CN | 4.5 ± 1.8 | −1.7 ± 0.7 | −2.6 ± 2.1 |
| 4-oxo-TEMPO• + TEMPO-D | CD3CN | 4.7 ± 1.8 | −1.7 ± 0.7 | −2.5 ± 2.4 |
| 4-oxo-TEMPO• + TEMPO-H | CD2Cl2 | 7.6 ± 2.4 | −2.6 ± 0.8 | −4.8 ± 2.6 |
| 4-oxo-TEMPO• + TEMPO-D | CD2Cl2 | 6.6 ± 2.2 | −2.6 ± 0.8 | −5.1 ± 2.8 |
| (3) 4-oxo-TEMPO• + 4-MeO-TEMPO-H | CD3CN | 2.8 ± 1.2 | −2.2 ± 0.4 | −5.8 ± 2.3 |
| 4-oxo-TEMPO• + 4-MeO-TEMPO-D | CD3CN | 3.1 ± 1.2 | −2.4 ± 0.7 | −5.8 ± 2.3 |
| (4) tBu2NO• + TEMPO-H | CD3CN | 0.11 ± 0.02 | 2.7 ± 0.9 | 4.6 ± 2.8 |
| tBu2NO• + TEMPO-D | CD3CN | 0.11 ± 0.03 | 2.4 ± 0.9 | 3.7 ± 2.9 |
| tBu2NO• + TEMPO-H | CD2Cl2 | 0.10 ± 0.02 | 3.1 ± 0.8 | 5.6 ± 2.6 |
| tBu2NO• + TEMPO-D | CD2Cl2 | 0.10 ± 0.02 | 3.1 ± 0.8 | 5.8 ± 2.4 |
Measurements at 278−318 K; ΔH° in kcal mol−1, ΔS° in cal mol−1 K−1.
HAT equilibria are also rapidly established between 4-oxo-TEMPO• and 4-MeOTEMPO-H (eq 3) and between tBu2NO• and TEMPO-H (eq 4; Table 1). The measured uphill free energy for reaction 4, ΔGo4H,CD3CN = 1.3 ± 0.1 kcal mol−1, is very close to the 1.5 kcal mol−1 difference in O–H bond dissociation enthalpies (ΔBDE) of TEMPO-H (69.7 kcal mol−1) and tBu2NOH (68.2 kcal mol−1) reported by Bordwell in DMSO.26
 |
(3) |
 |
(4) |
The equilibrium constants for H and D transfer were determined at temperatures from 278−318 K for reactions 2−4 in CD3CN (Figure 1) and for reactions 2 and 4 in CD2Cl2 (Figure S1; Table 1). Van't Hoff analysis yields small ground state reaction enthalpies and entropies, |ΔHo| ≤ 3.1 kcal mol−1 and |ΔSo| ≤ 5.8 cal mol−1 K−1. These values support the hypothesis that reactions 2−4 can be regarded as pseudo self-exchange reactions. Small values of ΔSo are also typical of organic HAT reactions, even for those that are not self-exchange reactions.23
Figure 1.

Van't Hoff plot for reactions 2−4 in CD3CN at 278−318 K.
II. Kinetic Measurements
Attempts to directly measure the rate of HAT self-exchange between TEMPO• and TEMPO-H by 1H NMR line broadening were unsuccessful.27 Therefore, we have studied the pseudo self-exchange reactions in eqs 2-4 using stopped-flow optical measurements. The reaction between 4-oxo-TEMPO• and TEMPO-H (eq 2), for instance, is readily monitored by UV-vis spectroscopy because the spectrum of 4-oxo-TEMPO• in MeCN (λmax = 440 nm, ε = 5.5 M−1 cm−1) is different from that of TEMPO• (λmax = 460 nm, ε = 10.3 M−1 cm−1), particularly in ε.
The reaction kinetics have been measured by UV-vis stopped flow techniques under pseudo first order conditions, with an excess of TEMPO-H (59−292 mM, ≥10 equiv) over 4-oxo-TEMPO• (5.9−12 mM) in MeCN (Figure 2a). Under these conditions the reaction proceeds essentially to completion (K2H,MeCN = 4.5). The optical data were fitted across the whole spectral region (350−550 nm) using SPECFIT global analysis software.28 The data fit well to a simple first-order A → B model (Figure 2b), and the pseudo first-order rate constants kobs are independent of the initial concentration of 4-oxo-TEMPO•. Plotting kobs versus [TEMPO-H] yields a straight line (Figure 3), indicating a first order dependence on [TEMPO-H] and a bimolecular rate constant for the forward reaction in eq 2 of k2H,MeCN = 10 ± 1 M−1 s−1 at 298 K. Reactions with less TEMPO-H, under second order approach-to-equilibrium conditions A + B ⇄ C + D, gave the same value of k2H,MeCN within error.
Figure 2.

(a) Overlay of UV-vis spectra for the reaction of 8.8 mM 4-oxo-TEMPO• with 88 mM TEMPO-H (eq 2) in MeCN over 5 s at 298 K. (b) Absorbance at 460 nm showing the raw data (○) and first order A → B fit using SPECFIT (—).
Figure 3.

Plot of pseudo first order kobs versus [TEMPO-H(D)] for reaction 2 in MeCN (kH/kD = 23 ± 3) and in CH2Cl2 (kH/kD = 23 ± 4) at 298 K.
To investigate the role of solvent, the same kinetic experiments were performed in CH2Cl2 and CCl4 solutions. Analysis as above gave k2H,CH2Cl2 = 48 ± 4 M−1 s−1 (Figure 3) and k2H,CCl4 = 300 ± 30 M−1 s−1 at 298 K. These rate constants are, respectively, 4.8 and 30 times larger than in MeCN.
The KIE for reaction 2 was examined using TEMPO-D prepared by TEMPO• reduction with Na2S2O4 in acetone-d6/D2O. This TEMPO-D, which was 98 ± 1% D by 1H NMR integration, reacts with 4-oxo-TEMPO• in MeCN with k2D,MeCN = 0.44 ± 0.05 M−1 s−1 (Figure 3). This rate constant is 23 ± 3 times slower than that for the same reaction of TEMPO-H at 298 K. Within experimental error, essentially the same apparent isotope effects are found in CH2Cl2, 23 ± 4, and in CCl4, 18 ± 5. Thus solvent polarity and hydrogen bonding to solvent do not significantly affect the KIEs. The ratio of the rate constants, however, is only a lower limit to the true KIE because the residual 2 ± 1% H in the TEMPO-D contributes significantly to the reactions. If the TEMPO-D was 99% D, the true KIE would be 30, and if it was 97% D, the true KIE would be 72.29 These are the experimental bounds on the KIE.
The kinetics of 4-oxo-TEMPO• plus excess 4-MeO-TEMPO-H (eq 3) in MeCN were measured and analyzed in a similar fashion, to give k3H = 7.8 ± 0.7 M−1 s−1 at 298 K (Figure S2). This is about 20% lower than the rate constant for reaction 2 under the same conditions, which is consistent with the equilibrium constant for reaction 3, K3H,MeCN = 2.8 ± 1.2, being a little smaller than K2H,MeCN = 4.5 ± 1.8 for reaction 2.
Similar to the case for reaction 2, the rate constant for reaction of the deuterated hydroxylamine, 4-MeO-TEMPO-D, at 298 K is much slower, k3D = 0.37 ± 0.05 M−1 s−1 (Figure S2), than that for the reaction of the undeuterated compound. Since the 4-MeO-TEMPO-D is also 98 ± 1% deuterated, the k3H/k3D = 21 ± 3 at 298 K is also a lower limit to the true KIE. Rate constants and kH/kD values at 298 K for the forward reactions 2−4 are given in Table 2, along with the rate constants for other nitroxyl plus hydroxylamine reactions.
The reaction of tBu2NO• and TEMPO-H (eq 4) in MeCN is uphill in free energy (K4H,MeCN = 0.11), so the kinetics were measured by UV-vis stopped flow techniques under second order approach-to-equilibrium conditions. The optical spectra of tBu2NO• (λmax = 454 nm, ε = 8.9 M−1 cm−1) and TEMPO• (λmax = 460 nm, ε = 10.3 M−1 cm−1) are similar, so the overall change of the absorbance is small (Figure S3). Under the experimental conditions ([tBu2NO•] =12−139 mM, [TEMPO-H] = 118−237 mM), spectra of reaction mixtures at short times show absorbances ∼10% higher than equimolar solutions of tBu2NO•, suggesting partial formation of a complex between tBu2NO• and TEMPO-H. Consistent with this suggestion, adding 2,4,6-tri-tert-butylphenol to solutions of tBu2NO• caused similar changes in optical spectra, even though HAT does not occur (ΔBDE = +14 kcal mol−1).30 These changes in reaction 4 are subtle, however, and are too small to enable determination of a value for the equilibrium constant. There are also subtle differences between the final reaction spectrum and that of TEMPO•, suggesting that the products could also be, in part, hydrogen-bonded.
The forward bimolecular rate constant for eq 4 under these conditions, k4H,MeCN, is 1.9 ± 0.2 M−1 s−1 at 298 K, as determined by fitting the data to an opposing second order equilibrium model (A + B ⇄ C + D) with a fixed K4H,MeCN = 0.11, using SPECFIT28 (Figure S3). When TEMPO-D is used, the initial spectra of the reaction mixtures are much closer to those of solutions of pure tBu2NO•, suggesting that adduct formation is isotopically sensitive, and is less favorable for TEMPO-D than for TEMPO-H. The rate constant for D-atom transfer, k4D,MeCN = 0.12 ± 0.02 M−1 s−1 indicates k4H,MeCN/k4D,MeCN = 16 ± 3 at 298 K. Reaction 4 behaves similarly in CH2Cl2 solvent, showing a similar isotope effect: k4H,CH2Cl2/k4D,CH2Cl2 = 13 ± 2.
Rate constants for reactions 2−4 have been measured as a function of temperature over 35−40 K temperature ranges. Selected Eyring plots are shown in Figure 4, and all Eyring and Arrhenius activation parameters are listed in Table 3. Table 4 shows the difference between the H versus D activation energies (EaD – EaH) and pre-exponential factors, log(AH/AD).
Figure 4.

Eyring plots for (a) 4-oxo-TEMPO• + TEMPO-H (reaction 2) and (b) tBu2NO• + TEMPO-H (reaction 4), both in MeCN and CH2Cl2.
Table 3.
Eyring and Arrhenius Parameters for H- and D-Atom Transfer Reactions.a
| Reaction | Solvent | T (K) | ΔH‡ | ΔS‡ | Ea | log A |
|---|---|---|---|---|---|---|
| 4-oxo-TEMPO• + TEMPO-Hb | MeCN | 278−318 | 3.0 ± 0.5 | −43 ± 3 | 3.6 ± 0.5 | 3.8 ± 0.6 |
| 4-oxo-TEMPO• + TEMPO-D | MeCN | 278−318 | 4.3 ± 0.4 | −46 ± 2 | 4.9 ± 0.4 | 3.3 ± 0.4 |
| 4-oxo-TEMPO• + TEMPO-H | CH2Cl2 | 273−308 | 3.0 ± 0.4 | −41 ± 2 | 3.6 ± 0.4 | 4.3 ± 0.4 |
| 4-oxo-TEMPO• + TEMPO-D | CH2Cl2 | 273−308 | 3.3 ± 0.4 | −46 ± 2 | 3.9 ± 0.4 | 3.2 ± 0.4 |
| 4-oxo-TEMPO• + 4-MeO-TEMPO-H | MeCN | 278−318 | 4.8 ± 0.3 | −38 ± 2 | 5.4 ± 0.3 | 4.9 ± 0.4 |
| 4-oxo-TEMPO• + 4-MeO-TEMPO-D | MeCN | 288−328 | 5.4 ± 0.3 | −42 ± 2 | 6.0 ± 0.3 | 4.0 ± 0.4 |
| tBu2NO• + TEMPO-H | MeCN | 278−318 | 7.7 ± 0.3 | −31 ± 2 | 8.3 ± 0.3 | 6.4 ± 0.4 |
| tBu2NO• + TEMPO-D | MeCN | 278−318 | 5.1 ± 0.3 | −45 ± 2 | 5.7 ± 0.3 | 3.3 ± 0.4 |
| tBu2NO• + TEMPO-H | CH2Cl2 | 273−308 | 7.1 ± 0.3 | −32 ± 2 | 7.6 ± 0.3 | 6.3 ± 0.4 |
| tBu2NO• + TEMPO-D | CH2Cl2 | 273−308 | 4.0 ± 0.3 | −47 ± 2 | 4.5 ± 0.3 | 2.9 ± 0.4 |
| PINO• + 4Me-NHPIc | AcOH | 290−309 | 9.8 ± 0.2 | −13 ± 1 | 10.4 ± 0.2 | 10.4 ± 0.2 |
| 4-Me-PINO• + NHPIc | AcOH | 290−309 | 10.0 ± 0.2 | −13 ± 1 | 10.6 ± 0.2 | 10.4 ± 0.2 |
Table 4.
Kinetic Isotope Effects and Differences in Protio and Deutero Arrhenius Parameters.a
| Reaction | solvent | kH/kD | EaD – EaH | log(AH/AD) |
|---|---|---|---|---|
| 4-oxo-TEMPO• + TEMPO-H | MeCN | 23 ± 3 | 1.3 ± 0.6 | 0.5 ± 0.7 |
| 4-oxo-TEMPO• + TEMPO-H | CH2Cl2 | 23 ± 4 | 0.3 ± 0.6 | 1.1 ± 0.6 |
| 4-oxo-TEMPO• + 4-MeO-TEMPO-H | MeCN | 21 ± 3 | 0.6 ± 0.4 | 0.9 ± 0.6 |
| tBu2NO• + TEMPO-H | MeCN | 16 ± 3 | −2.6 ± 0.4 | 3.1 ± 0.6 |
| tBu2NO• + TEMPO-H | CH2Cl2 | 13 ± 2 | −3.1 ± 0.4 | 3.4 ± 0.6 |
kH/kD at 298 K, EaD – EaH in kcal mol−1.
III. Computational Studies
In order to investigate the role of tunneling in self-exchange reactions between nitroxyl radicals and hydroxylamines, we performed multi-dimensional tunneling calculations, using Gaussrate31 as the interface between Gaussian 0332 and Polyrate.33 Our computations were carried out using both the MPW1K34 and the more recently developed MO5−2X35 and MO636 functionals. The 6−31+G(d,p) basis set37 was employed for all of these calculations. Tunneling rates were computed, using the small-curvature tunneling (SCT) approximation.38
Because of the computational demands of the SCT tunneling calculations, calculations on the tBu2NO• + tBu2NOH and TEMPO• + TEMPO-H systems were too big to be practical. Therefore, we began by performing calculations on (CH3)2NO• + (CH3)2NOH. The calculated rate constants, activation parameters, and H/D kinetic isotope effects for this reaction are given in Table 5.
Table 5.
Computed canonical variational transition state theory (CVT) and small curvature tunneling (SCT) rate constants and Arrhenius activation parameters for at the MPW1K/6−31+G(d,p) level of theory.
| Reaction | Method | kH | kD | kH/kD | Ea,H | log(AH) | Ea,D | log(AD) |
|---|---|---|---|---|---|---|---|---|
| Me2NO• + HONMe2 | CVT | 5.65 × 10−4 | 8.67 × 10−5 | 6.5 | 14.8 | 7.6 | 15.8 | 7.6 |
| Me2NO• + HONMe2 | SCT | 4.85 × 101 | 2.47 × 10−1 | 196 | 6.1 | 6.2 | 7.3 | 4.8 |
| MeHNO• + HONHMe | SCT | 1.83× 101 | 7.65 × 10−2 | 239 | 6.1 | 5.8 | 7.4 | 4.3 |
a kH, kD, M−1 s−1; Ea,H, Ea,D, kcal mol−1.
The transition structure (TS) for (CH3)2NO• + (CH3)2NOH, shown in Figure 5a, has C2h symmetry. The SCT calculations start at this TS and descend down a minimum energy path toward the reactants/products. Unfortunately, starting from the TS and moving along this reaction coordinate, the Cs plane of symmetry was maintained throughout the reaction. Consequently, the SCT calculations did not lead to the hydrogen-bonded reactant complex (CH3)2NOH...•ON(CH3)2, which has C1 symmetry (Figure 5b). Instead, the calculations led to the Cs TS that connects the two enantiomeric geometries of the reactant complex. Destroying the Cs plane in the C2h TS by substituting two CD3 for two CH3 groups failed to coax the hydrogen-bonded complex to depart from the ridge on the potential energy surface that connects the two enantiomers.
Figure 5.

(a) Transition structure for hydrogen transfer between (CH3)2NOH and (CH3)2NO•. (b) H-bonded complex between (CH3)2NOH and (CH3)2NO•. Bond lengths are in Ångstroms.
However, we were able to follow the reaction path back from the TS to the reactants for the self-exchange reaction between the monomethylnitroxyl radical and the monomethylhydroxylamine. The calculated values of kH, Ea, A, and kH/kD for this reaction, (CH3)HNO• + HONH(CH3), are also given in Table 5. The two reactions are close enough to lead us to believe that the computational results for the dimethyl reaction are reliable, despite our being unable to follow the reaction path all the way back to the reactant complex in this case. Most of the tunneling seems to originate from regions along the reaction path that are closer to the TS, where the reaction barrier is narrow, than from regions close to the reactants, where the barrier is much wider and the deviation from Cs symmetry is significant.
The formation of the (CH3)2NOH...•ON(CH3)2 reactant complex (Figure 5B) from the separated reactants is enthalpically favorable: ΔH = −4.6 kcal mol−1 [calculated with MPW1K/6−31+ G(d,p)]. However, the entropy of complex formation [ΔS = −26.7 cal K−1 mol−1] is so unfavorable that the calculated value of the gas phase equilibrium constant [K = 1.4 × 10−4 M−1] at 298 K is small. Since at 298 K the reactants are lower in free energy than the reactant complex, the computed rate constants, A factors, and activation energies reported in Table 5 are for reactions starting from the separated reactants.
We used all three functionals -- MPW1K, MO5−2X, and MO6 -- to calculate the barrier height for the hydrogen self-exchange reaction, (CH3)2NO• + (CH3)2NOH, starting from the C1 reactant complex. All three functionals gave enthalpies of activation for passage over the reaction barrier that were the same to within 1.0 kcal mol−1. Therefore, we elected to do the tunneling calculations with just one of them, MPW1K.
The MPW1K rate constants at 298 K for the hydrogen and deuterium self-exchange reactions were computed by canonical variational transition state theory (CVT) for passage over the barrier and by small curvature tunneling (SCT) calculations for passage through the barrier. The results are contained in Table 5. The Arrhenius activation parameters, obtained from the temperature dependences of the calculated rate constants around 298 K, are also given in Table 5.
The kH values in Table 5 show that the SCT rate constant for hydrogen tunneling through the barrier is computed to be larger than the CVT rate constant for passage over it by a factor of about 105. Tunneling reduces Ea by 8.7 kcal mol−1 for hydrogen, with a decrease in log A of only 1.4. The large reduction in Ea and the small decrease in log A shows that our SCT calculations predict that tunneling is very efficient in (CH3)2NO• + (CH3)2NOH. The width of the barrier to this reaction is computed to be only 0.42 Å, which is presumably why tunneling is computed to be so effective at increasing the reaction rate.
Table 5 also contains the results of our calculations for deuterium self-exchange in (CH3)2NO• + (CH3)2NOD. Comparison of the CVT and SCT rate constants for D shows that tunneling is also predicted to dominate the reaction involving deuterium. However, a very large H/D KIE of kH/kD = 196 is predicted for tunneling. Of this ratio, a factor of 7.4 is due to H tunneling with a 1.2 kcal mol−1 lower Ea, than D. An even larger factor is due to log AH being about 1.4 larger than log AD. Even though H is calculated to tunnel through the barrier at an average energy of about 1.2 kcal mol−1 lower than D, H is calculated to tunnel with a higher probability than D by a factor of 26.5.39
Our SCT value of H/D KIE of 196 for (CH3)2NO• + (CH3)2NOH/D is about a factor of 5 larger than the experimental values for reaction of TEMPO-H with a variety of nitroxide radicals (Table 2). However, as already noted, the true H/D KIEs are substantially larger than the values given in Table 2, because of the incomplete D enrichment. For TEMPO-H/D + 4-oxo-TEMPO•, the measured kH/kD of 23 at 98 ± 1% D corresponds to a true KIE of about 40 (the 98 ± 1% range gives values of 30−70).
The calculated bimolecular SCT rate constant of k = 48.5 M−1 s−1 is in excellent agreement with the rate constants in Table 2 for the TEMPO-H reactions. The closest experimental analogy to this gas phase rate constant is the k of 300 M−1 s−1 for 4-oxo-TEMPO• + TEMPO-H in CCl4, which has ΔGo ≅ −1 kcal mol−1. In contrast, the CVT rate constant of k = 5.65 × 10−4 M−1s−1 for (CH3)2NO• + (CH3)2NOH underestimates the experimental rate constants (Table 2) by a factor of 104 − 106. Thus, our calculations on both the rate of and H/D KIE for this model reaction strongly support the conclusion that the dialkylnitroxyl radical reactions in Table 2 all occur by tunneling through the reaction barrier, rather than by passage over it.
Although we were unable to perform SCT calculations on larger systems, we did carry out CVT calculations on the tBu2NO(•/H), TEMPO(•/H) and Ph2NO(•/H) self-exchange reactions. For each one, the TS was located and confirmed to have one imaginary frequency by a vibrational analysis. Key features of the geometries of the nitroxide reactants and the self-exchange TSs are given in Figure 6, along with the calculated Ea values from CVT for each reaction.
Figure 6.

Bond lengths (Å), atomic spin densities (in green), and activation energies (kcal mol−1) for the nitroxyl radicals, (a) tBu2NO•, (b) TEMPO•, and (c) Ph2NO•, and the transition structures and CVT activation energies for their hydrogen self-exchange reactions with the corresponding hydroxylamines.
As shown in Figure 6, the geometries of the TSs for all three reactions are very similar. In each TS the O-H bonds approximately bisect the C-N-C angles, strongly suggesting that the electron and the proton are transferred together between the same AO on each oxygen. This inference is confirmed by inspection of the SOMOs for the three TSs, which are shown in Figure 7. Thus, all three reactions proceed by a mechanism in which a hydrogen atom is transferred between a pair of oxygen AOs, rather than by a mechanism in which a proton is transferred between one pair of oxygen AOs and an electron is transferred between a different pair of oxygen AOs (which in some contexts has been called proton-coupled electron transfer, PCET).40
Figure 7.

SOMOs of the transition structures for hydrogen exchange reactions: (a) tBu2NO• + tBu2NOH, (b) TEMPO• + TEMPO-H , and (c) Ph2NO• + Ph2NOH.
The CVT activation energies for the dialkylnitroxyl reactions, Ea = 15.7 and 14.5 kcal mol−1 for tBu2NO• + tBu2NOH and TEMPO• + TEMPO-H, respectively, are much larger than the corresponding energy for Ph2NO• + Ph2NOH, 5.6 kcal mol−1. This is in pleasing agreement with the experimental finding that the rate constant for Ph2NO• + Ph2NOH13 is at least five orders of magnitude faster than those for tBu2NO• + tBu2NOH13 and TEMPO• + TEMPO-H. Comparisons of the geometries and spin densities of the nitroxide reactants and TSs in Figure 6 indicates that the phenyl groups lower the barrier to reaction by providing greater delocalization of the unpaired electron in the TS for Ph2NO• + Ph2NOH reaction than in the Ph2NO reactant.
In the nitroxyl radicals, the unpaired spin density of 0.88 in the N-O group of Ph2NO• is about 0.1 smaller than the unpaired spin densities of 0.98 and 1.00 in the NO groups of TEMPO• and tBu2NO•, respectively. The C-N bond lengths in Ph2NO• are calculated to be 0.07 and 0.08 Å shorter than those in TEMPO and tBu2NO•, respectively. This is a larger difference than the ∼0.04 Å difference in covalent radii between sp2 and sp3 carbon atoms. These results indicate that the phenyl groups in Ph2NO• delocalize the unpaired electron in the two-center, three-electron N-O π bond.
On going from the reactants to the TS, the four C-N bond lengths in Ph2NO• shorten by an average of 0.02 Å; whereas the C-N bond lengths in TEMPO• and tBu2NO• shorten by less than 0.01 Å. In addition, the spin densities in the NO groups of the Ph2NO• + Ph2NOH TS are 0.08 smaller than the spin density in the NO group of Ph2NO•; whereas, the corresponding decrease in the spin densities in the NO groups is only about 0.03 in both TEMPO• + TEMPO-H and tBu2NO• + tBu2NOH. The larger decreases in the C-N bond lengths and in the NO spin densities between the Ph2NO• reactants and the TS for Ph2NO• + Ph2NOH are both consistent with the TS for this reaction being stabilized by electron delocalization into the phenyl groups. We believe that this is the reason why the hydrogen self-exchange reaction of this diarylnitroxyl radical is many orders of magnitude faster than those of the dialkylnitroxyl radicals in Table 2.
Discussion
Nitroxyl radical plus hydroxylamine reactions have been studied experimentally and computationally. The experimental reactions (eqs 2-4) are close to isoergic in both MeCN and CH2Cl2, with |ΔGo| ≤ 1.4 kcal mol−1 (Table 2), and involve reagents that are sterically quite similar. Thus these reactions are good approximations of self-exchange reactions. Self-exchange reactions are inherently simpler to analyze because of their symmetry, which requires that the transition structure, or more generally the seam on the potential energy surface, be symmetrically placed between the reactant and product.
Thermochemical data show that the reactions must occur by concerted rather than stepwise transfer of the proton and electron. For self-exchange reactions, the potential stepwise mechanisms with initial electron or proton transfer are the microscopic reverse of each other.41 For the TEMPO• + TEMPO-H self-exchange reaction, both stepwise pathways proceed through a TEMPO− + TEMPO-H•+ intermediate state. Based on the known E1/2 and pKa values, this state is about 60 kcal mol−1 higher than TEMPO• + TEMPO-H (2.6 V or 44 pKa units).42 For the pseudo self-exchange reactions in eqs 2-4, the data are not available to make a full analysis, but the E1/2 and pKa values are not very different for the various compounds,26 so these reactions will also have potential intermediate states lying ca. 60 kcal mol−1 uphill. This is dramatically higher than the Eyring barriers ΔG‡ < 20 kcal mol−1 found for reactions 2−4. Thus the stepwise pathways are not possible and the reaction must occur by concerted H+/e− transfer.
I. Solvent Effects
The experimental rate constants are faster in less polar and less hydrogen-bonding solvents (Table 2). For 4-oxo-TEMPO• + TEMPO-H, the rate constants are 10, 48, and 300 M−1 s−1 in MeCN, CH2Cl2, and CCl4, respectively. Litwinienko, Ingold, et al. have shown that for HAT reactions of phenols, solvent effects on the rate constants are predominantly due to the formation of a hydrogen bond between the H-atom donor and the solvent.43 Only the fraction of the phenol that is not hydrogen bonded is reactive, and the reactivity of the non-hydrogen bonded phenol is not significantly affected by solvent. Qualitatively, this explains why the TEMPO-H reactions are slower in MeCN, a good hydrogen bond acceptor, than in chlorinated solvents. The formation of TEMPO-H•••NCMe hydrogen bonds is indicated by the TEMPO-H O–H stretching frequency in CD3CN (3495 cm−1), being ∼100 cm−1 lower than ν(TEMPO-H) in CD2Cl2 (3583 cm−1) and CCl4 (3597 cm−1). A recent crystal structure of TEMPO-H shows hydrogen bonds between TEMPO-H molecules with O...O distances of 2.83 and 2.88 Å.23
Quantitatively, the Litwinienko and Ingold model predicts that the solvent effect will be independent of the H-atom acceptor.44,45 However, the experimental ratios of rate constants in CH2Cl2 vs. MeCN are 4.8 ± 0.6 for 4-oxo-TEMPO• + TEMPO-H and 2.4 ± 0.3 for tBu2NO• + TEMPO-H.44 The 4-oxo-TEMPO• + TEMPO-H reaction (eq 2) appears to be affected by solvent polarity as well as hydrogen bonding, as it is 6 times faster in CCl4 than in CH2Cl2 (Table 2). A more dramatic effect has been reported for the tBuArNO•/tBuArNOH self-exchange reaction (Ar = 2,6-dimethoxyphenyl), which is more than a hundred times faster in CCl4 than in CHCl3 and CH2Cl2: 2 × 103 versus < 20 M−1 s−.13 A reviewer has suggested that these anomalous kinetic solvent effects may be due to changes in the nitroxyl radical spin density with solvent,46 which are known to affect their rate constant for reaction with alkyl radicals.47
II. Kinetic Isotope Effects and Evidence for Tunneling
Hydrogen tunneling is suggested by the large kH/kD values of 23 ± 3, 21 ± 3, and 16 ± 3 at 298 K for reactions 2−4 in MeCN. A one-dimensional semi-classical transition state theory model, taking ΔG‡D – ΔG‡H to be at most the difference in zero-point energies, predicts a maximum kH/kD of 9 at 298 K using the measured OH and OD stretches.3,4,48 A more complete semi-classical model including bending modes gives a maximum KIE of about 13 for cleavage of an O–H bond.3 The computed CVT H/D KIEs at 298 K, without tunneling, are 6.5, 4.9, 4.9, and 4.7 for the self-exchange reactions involving Me2NO•, tBu2NO•, TEMPO•, and Ph2NO•. These computed H/D KIEs are all significantly lower than the measured values for reactions 2−4, but they are ca. three times larger than the KIEs in Table 2 for reactions involving monoarylhydroxylamines, including the tBu(Ar)NO• + tBu(Ar)NOH self-exchange [Ar = 2,6-(MeO)2C6H3].
The experimental activation energies also provide evidence for tunneling. According to Bell,3 tunneling is indicated when EaD – EaH is larger than the difference in zero-point energies, 1.3 kcal mol−1 in this case,48 and/or when there are significant differences in the pre-exponential terms, AH/AD < 0.7 or AH/AD > 1.4, or |log(AH/AD)| > 0.15. All of the reactions studied here have log(AH/AD) larger than this semi-classical limit, except perhaps for reaction 2 in MeCN which has log(AH/AD) = 0.5 ± 0.7 (Table 4). For reactions 2 and 3, the positive values of log(AH/AD), the low values of both AH and AD (<105 M−1 s−1), and low Ea (<6 kcal mol−1) values all suggest that there is significant tunneling in the reactions of both the H and D isotopomers.5
The small-curvature tunneling calculations on (CH3)2NO• + (CH3)2NOH also show that tunneling is the dominant pathway in this model reaction for a self-exchange involving a dialkylnitroxyl radical reacting with a dialkylhydroxylamine. Tunneling through the barrier is computed to be about 105 times faster than passage over the barrier for H, and more than 103 times faster, even for D. The experimental KIEs vary little with solvent – between MeCN, CH2Cl2, and CCl4 for reaction 2, and between MeCN and CH2Cl2 for reaction 4 – indicating that the solvent plays little role in the tunneling process.49
The tBu2NO• + TEMPO-H reaction (eq 4) is found to have very unusual activation parameters. These data derive from quite small changes in absorbance (see above), but they are based on three separate measurements of rate constants at each temperature, with consistent multiple stopped-flow runs in each measurement. The resulting AH is a thousand times larger than AD both in MeCN and CH2Cl2, and EaD is substantially smaller than EaH: EaD – EaH = −2.5 ± 0.4 in MeCN and −3.1 ± 0.4 kcal mol−1 in CH2Cl2. That EaD < EaH is indicated by the increase in k4H/k4D values with temperature, in MeCN from 11 ± 1 at 278 K to 17 ± 2 at 308 K and 21 ± 6 at 318 K. These values are surprising both because they are so different than those for the very similar reactions 2 and 3,50 and because EaD < EaH is opposite to the predictions of both semi-classical and tunneling models.3,5 While similar unusual activation parameters have been reported for two other HAT/PCET reactions,51 we are not sure of the origin in this case. It is interesting that the formation of the hydrogen-bonded precursor complex in this reaction, TEMPO-H/D...•ONtBu2, also appears to be isotopically sensitive (see above).17,52
In light of the importance of tunneling for the TEMPO•/TEMPO-H self-exchange reaction, the success of the Marcus cross relation for reactions involving TEMPO• is perhaps surprising. The cross relation is a semiclassical treatment that does not include hydrogen tunneling. Still, the cross relation is essentially interpretative, and will still hold if the self-exchange and cross reactions are accelerated a comparable amount by tunneling.
III. Comparison with related reactions
The rate constants for nitroxyl + hydroxylamine self-exchange and pseudo self-exchange reactions show a remarkable range, from 2 to >107 M−1 s−1 (Table 2). Other XO• + XOH reactions show similar variation: tBu2C=NO• + tBu2C=NOH in benzene, 1.3 M−1 s−1;53 2,4,6-tBu3C6H2O• + 2,4,6-tBu3C6D2OH in CCl4, 220 M−1 s−;13b tBuOO• + sBuOOH in isopentane, 490 M−1 s−1;54 tBuO• + tBu3COH in tBuOOtBu, ∼3 × 104 M−1 s−1;55 and PhO• + 2-naphthol (ΔGo ≈ −2 kcal mol−1) in MeCN, 4.5 × 106 M−1 s−1.52,56 The most striking comparison is from the measurements of R2NOH + R2NO• self-exchange reactions by Kreilick and Weissman in CCl4: 320 M−1 s−1 for R = tBu vs. >107 M−1 s−1 for R = Ph.13 The dichotomy between the dialkyl- and arylhydroxylamine reactions is also evident in the HAT kinetic isotope effects. The more rapid arylhydroxylamine reactions have kH/kD < 2, while the slower dialkylnitroxyl reactions have kH/kD > 10 (Table 2).
Reactions of the acylnitroxyl PINO• display yet another pattern of reactivity.17,18 The pseudo self-exchange reaction of PINO• with the hydroxyphthalate Me-NHPI has values of Ea (10 kcal mol−1) and AH (1010.4 M−1 s−1) that are significantly larger than those of the TEMPO•/4-oxo-TEMPO•/4-MeO-TEMPO• reactions (eqs 2 and 3): Ea ≤ 6 kcal mol−1 and AH ≤ 105 M−1 s−1 (Table 3). The reactions of PINO• with p-xylene and toluene appear to involve tunneling, but in contrast to reactions 2−4, they show large values of EaD – EaH and negative values of log(AH/AD) (3.0 ± 0.3 kcal mol−1, −0.8 ± 0.3 for p-xylene), suggesting that tunneling is more pronounced for H than for D.17,18 The related HAT reaction of (CF3)2NO• with toluene, with kH/kD = 13 and log(AH/AD) ≈ 0 (AH ≈ AD ≈ 104), was not suggested to involve significant tunneling.57
The diversity of behavior is remarkable for such similar reactions. All of the nitroxyl/hydroxylamine reactions discussed here are close to isoergic (|ΔGo| ≤ 2 kcal mol−1, Table 2) so the driving forces are not the cause of these differences. The differences between the dialkyl- and the arylhydroxylamine reactions could conceivably be largely due to a steric effect. The bulkier tertiary alkyl groups might make assembly of the precursor complex more difficult and could keep the oxygen atoms farther apart. Such a difference in TS geometries would lead to higher barriers and longer H-transfer distances, enhancing the importance of tunneling in the dialkylnitroxyl reactions. However, the results of our calculations show that there is very little difference between the geometries of the TSs for the dialkyl- and diarylnitroxyl self-exchange reactions. Instead, our calculations find that the much faster Ph2NO• + Ph2NOH self-exchange reaction is due to the greater electronic delocalization provided by the phenyl groups in the transition structure. The CVT barrier for Ph2NO(•/H) self-exchange reaction, 5.6 kcal mol−1, is 9−10 kcal mol−1 lower than the CVT barriers for the tBu2NO(•/H) and TEMPO(H) self-exchange reactions (15.7, 14.5 kcal mol−1, respectively).
Because the reaction barriers for the arylhydroxylamines are lower than those for the dialkylhydroxylamines, the former reactions can proceed by passage over, or close to the top of the barriers, without the need for substantial tunneling. In contrast, the higher reaction barriers on the potential energy surfaces for the dialkylhydroxylamine reactions lead to both H and D preferentially tunneling through the barriers. Tunneling produces the low Ea and A values and the high H/D KIEs that we have both measured and calculated for the dialkylnitroxyl radical + dialkylhydroxylamine reactions.
While many discussions of tunneling emphasize the distance over which the H or D transfers must occur, in this case the O-H bond distances in the reactant complexes and transition structures are calculated to be quite similar for the reactions of dialkylhydroxylamines vs. those of arylhydroxylamines. The results of our calculations suggest that the former reactions show the experimental indications of tunneling while the latter do not because of the different barrier heights for degenerate HAT in these two types of hydroxylamines. These studies thus appear to be an experimental example of the well-known theoretical result that barrier height is as key a factor as barrier width in determining the probability of tunneling in a chemical reaction.3
Conclusions
Hydrogen atom transfer from dialkylhydroxylamines to dialkylnitroxyl radicals predominantly involves hydrogen tunneling. This has been shown through a combination of experimental and computational studies. Experimentally, the kinetics of three pseudo-self exchange reactions have been examined. For the reactions of 4-oxo-TEMPO• with TEMPO-H (eq 2) and 4-MeO-TEMPO-H (eq 3), the activation parameters and H/D kinetic isotope effects (Tables 2-4) suggest that tunneling is important in both H and D transfers. The measured ratios of kH/kD = 21−23 ± 4 correspond to intrinsic KIEs of ca. 40 at 298 K, given the 98 ± 1% deuterium enrichment of the hydroxylamines. Computational studies of the (CH3)2NO• + (CH3)2NOH model reaction, using the small-curvature tunneling (SCT) approximation, also find substantial tunneling by both H and D. The reaction of tBu2NO• with TEMPO-H (eq 4) has very unusual activation parameters, with Ea(D) < Ea(H) and log(AH/AD) ≈ 3.
The properties of these pseudo self-exchange reactions of dialkylhydroxylamines contrast with related reactions of arylhydroxylamines. Aryl-substituted hydroxylamines generally react with higher rate constants and very small KIEs (< 2). Calculations indicate that the Ph2NO• + Ph2NOH self-exchange has a 9−10 kcal mol−1 lower barrier than the tBu2NO• + tBu2NOH and TEMPO + TEMPO-H self-exchange reactions, due to greater electron delocalization in the [Ph2NO..H..ONPh2]• transition structure. The picture that emerges from these studies is that the self-exchange reactions of the dialkylhydroxylamines involve tunneling of both H and D, while the related reactions of arylhydroxylamines proceed over or close to the top of the reaction barriers. This dichotomy is due to the higher barriers for the dialkylhydroxylamine reactions, rather than to differences in the geometries of the reactant complexes or transition structures between the two types of hydroxylamines.
Experimental
Physical Techniques and Instrumentation
1H (500 MHz) and 13C{1H} (126 MHz) NMR were recorded on Bruker Avance spectrometers, referenced to a residual solvent peak, and reported as: δ (multiplicity, assignment, number of protons). The error for NMR integration is estimated to be ±10%. Lorentzian line fitting for accurate integration was done using NUTS.25 Electrospray ionization mass spectra (ESI/MS) were obtained on a Bruker Esquire-LC ion trap mass spectrometer and reported as m/z, with samples infused as MeCN solutions. UV-vis spectra were acquired with a Hewlett-Packard 8453 diode array spectrophotometer, and reported as λmax/nm (ε/M−1 cm−1). IR spectra were obtained as CD3CN, CD2Cl2, or CCl4 solutions in a NaCl solution cell, using a Bruker Vector 33 or Perkin Elmer 1720 FT-IR spectrometer, and reported in cm−1. UV-vis stopped-flow measurements were obtained on an OLIS RSM-1000 stopped-flow spectrophotometer. Elemental analyses were performed by Atlantic Microlab (Norcross, GA). All reactions were performed in the absence of air using standard glove box/vacuum line techniques.
Materials
All reagent grade solvents were purchased from Fisher Scientific, EMD Chemicals, or Honeywell Burdick & Jackson (for anhydrous MeCN). Deuterated solvents were obtained from Cambridge Isotope Laboratories. CD3CN was dried over CaH2, vacuum transferred to P2O5, and over to CaH2, then to a dry glass flask. CD2Cl2 was dried over CaH2, and vacuum transferred to a dry glass flask. TEMPO•, 4-oxo-TEMPO•, 4-MeO-TEMPO•, and tBu2NO• were purchased from Aldrich, and were sublimed onto a cold-finger apparatus before use. UV-vis (MeCN): TEMPO•, 460 (10.3); 4-oxo-TEMPO•, 440 (5.5); 4-MeO-TEMPO•, 460 (10.4); tBu2NO•, 454 (8.9). UV-vis (CH2Cl2): TEMPO•, 460 (11.1); 4-oxo-TEMPO•, 446 (6.0); 4-MeO-TEMPO•, 462 (11.0); tBu2NO•, 450 (9.0). UV-vis (CCl4): TEMPO•, 471 (12.2); 4-oxo-TEMPO•, 450 (7.1). Anal. Calcd (Found) for TEMPO• (C9H18NO): C, 69.18 (69.14); H, 11.61 (11.65); N, 8.96 (9.09); for 4-oxo-TEMPO• (C9H16NO2): C, 63.50 (63.61); H, 9.47 (9.51); N, 8.23 (8.19); for 4-MeO-TEMPO• (C10H20NO2): C, 64.48 (64.64); H, 10.82 (10.97); N, 7.52 (7.47); for tBu2NO• (C8H18NO): C, 66.62 (66.87); H, 12.58 (12.79); N, 9.71 (9.61).
TEMPO-H was prepared according to literature procedures.58 1H NMR (CD3CN): δ 1.06 (s, CH3, 12H), 1.45 (s, CH2, 6H; the C3/C5 and C4 signals are coincident), 5.34 (br s, OH, 1H). 1H NMR (CD2Cl2): δ 1.10 (s, CH3, 12H), 1.46 (s, CH2, 6H), 4.31 (br s, OH, 1H). 13C{1H} NMR (CD3CN): δ 17.78 (C4), 26.11 (CH3), 40.16 (C3), 58.78 (C2). Anal. Calcd (Found) for TEMPO-H (C9H19NO): C, 68.74 (69.01); H, 12.18 (12.39); N, 8.91 (8.82). TEMPO-D was prepared analogously to TEMPO-H, using (CD3)2CO/D2O (99.9% D in D2O) as the solvent; it was 98 ± 1% OD by NMR integration. Anal. Calcd (Found) for TEMPO-D (C9H18DNO): C, 68.30 (67.42); H, 12.10 (12.21); N, 8.85 (8.68). IR: νOH/νOD = 3495/2592 (CD3CN), 3583/2648 (CD2Cl2), 3597/2658 (CCl4).
Preparation of 4-MeO-TEMPO-H
A suspension of 4-MeO-TEMPO• (2.00 g, 10.7 mmol) and Na2S2O4 (3.60 g, 20.7 mmol) in Me2CO/H2O (15 mL each) was stirred for 30 min at room temperature under N2. The solvent was then partially evacuated under vacuum to remove Me2CO. The leftover aqueous layer was extracted with Et2O (3 × 20 mL), and the solvent was evacuated to dryness to give the crude product, which was purified by sublimation to a cold finger, yielding 951 mg of white powder (48%). 1H NMR (CD3CN): δ 1.09, 1.10 (s, CH3, 6H each); 1.25, 1.90 (m, CH2, 2H each); 3.25 (s, OCH3, 3H); 3.46 (m, 4-CH, 1H); 5.37 (br s, OH, 1H). 1H NMR (CD2Cl2): δ 1.16, 1.20 (s, CH3, 6H each); 1.33, 1.90 (m, CH2, 2H each); 3.32 (s, OCH3, 3H); 3.44 (m, 4-CH, 1H); 4.51 (br s, OH, 1H). 13C{1H} NMR (CD3CN): δ 20.83, 32.74 (CH3); 45.25 (C3); 55.77 (OCH3); 59.22 (C2); 72.57 (C4). ESI/MS+: 188 [M + H]+, 170 [M – OH]+. Anal. Calcd (Found) for 4-MeO-TEMPO-H (C10H21NO2): C, 64.13 (64.28); H, 11.30 (11.31); N, 7.48 (7.49). 4-MeO-TEMPO-D was prepared analogously using (CD3)2CO/D2O (99.9% D in D2O) and was 98 ± 1% OD by NMR integration. Anal. Calcd (Found) for 4-MeO-TEMPO-D (C10H20DNO2): C, 63.79 (63.92); H, 11.24 (11.37); N, 7.44 (7.55). IR (CD3CN): νOH/νOD = 3492/2584.
1H NMR Equilibrium Measurements
A typical experiment involved a J-Young sealable NMR tube being loaded with 4-oxo-TEMPO• (48 mg, 0.28 mmol) and TEMPO-H(D) (76 mg, 0.48 mmol) in 0.5 mL CD3CN or CD2Cl2 to form an equilibrium mixture with 4-oxo-TEMPOH(D) and TEMPO•. 1H NMR spectra of the sample were obtained at 278−318 K. All chemical species have resolvable peaks, whose integrations were determined by Lorentzian line fitting using NUTS.25 An equilibrium constant was calculated at each temperature from the ratios of the peak areas, corrected for the number of protons for each peak. The experiment was repeated with 4-oxo-TEMPO•/TEMPO-H(D) = 32 mg/51 mg, and 24 mg/38 mg and the reported K at each temperature is the average of three runs. The errors on K are 2σ of the variation of measured values. The errors on ΔHo and ΔSo are 2σ errors from the least-squares linear fit using KaleidaGraph59 to the Van't Hoff equation.
1H NMR of TEMPO• in CD3CN: −29.74 (3,5-CH2, 4H), −16.51 (CH3, 12H), 15.33 (4-CH2, 2H); in CD2Cl2: −27.97 (3,5-CH2, 4H), −15.14 (CH3, 12H), 15.19 (4-CH2, 2H). 1H NMR of 4-oxo-TEMPO• in CD3CN: −7.78 (CH3, 12H), 1.80 (3,5-CH2, broad, 4H, overlaps with residual solvent [CH2DCN] peak); in CD2Cl2: −7.12 (CH3, 12H), 2.22 (3,5-CH2, 4H). 1H NMR of 4-MeO-TEMPO• in CD3CN: −33.89, −20.42 (3,5-CH2, 2H each); −29.43, −1.74 (CH3, 6H each); 3.07 (OCH3, 3H); 8.67 (4-CH, 1H). 1H NMR of tBu2NO• (s, tBu) in CD3CN: −6.67; in CD2Cl2: −6.20. 4-oxo-TEMPO-H and tBu2NOH were not isolated but were generated in situ in reactions 2 and 4, respectively. 1H NMR of 4-oxo-TEMPO-H in CD3CN: 1.22 (s, CH3, 12H); 2.39 (s, CH2, 4H); in CD2Cl2: 1.24 (s, CH3, 12H); 2.49 (s, CH2, 4H). 1H NMR of tBu2NOH (s, tBu) in CD3CN: 1.23; in CD2Cl2: 1.31.
UV-Vis Stopped-Flow Kinetic Measurements
Solutions of 4-oxo-TEMPO• (12−24 mM) and TEMPO-H(D) (118−584 mM) in MeCN or CH2Cl2 were prepared and loaded into gas-tight syringes inside a N2 glovebox. The stopped-flow apparatus was flushed with the solvent, and a background spectrum was acquired. The syringes were immediately loaded onto the stopped-flow apparatus to minimize air exposure. The stopped-flow drive syringes were flushed with the reagents, then filled and allowed to thermally equilibrate. A minimum of six kinetic runs were performed for each set of concentrations at 278−318 K in MeCN or at 273−308 K in CH2Cl2 for reaction 2. The contents of the two syringes were rapidly mixed at equal volume resulting in half of the original concentrations (5.9−12 mM 4-oxo-TEMPO• and 59−292 mM TEMPO-D). Kinetic data were analyzed using SPECFIT global analysis software28 to determine the rate constants. Under pseudo-first order conditions (≥ 10 equiv TEMPO-H(D)), kobs values were derived from fitting an A → B model at each [TEMPO-H(D)], and second order rate constants were obtained from plotting kobs versus [TEMPO-H(D)] (Figure 3). Under second order conditions, the data were fit to an opposing second order equilibrium model, A + B ⇄ C + D (A and C colored), with a fixed equilibrium constant (Table 1). Reaction 2 was also performed in CCl4 at 298 K under pseudo-first order conditions (≥ 10 equiv TEMPO-H(D)). Temperature dependent measurements of reaction 3 used similar amounts of 4-oxo-TEMPO• and 4-MeOTEMPO-H(D) in MeCN; for reaction 4, 12−139 mM tBu2NO• and 118−237 mM TEMPO-H(D) in MeCN or CH2Cl2 were used (as the initial concentrations right after stopped-flow mixing). The errors on k are 2σ of the variation of measured values. The errors on the activation parameters are 2σ from the least-squares linear fit using KaleidaGraph59 to the Eyring or Arrhenius equation.
Supplementary Material
Acknowledgment
We thank J. J. Warren for insightful discussions and the US National Institutes of Health (GM50422) and the University of Washington for financial support. The research at the University of North Texas was supported by the National Science and Robert A. Welch Foundations. Some of the results reported here were obtained on computers, purchased with funds provided by the National Science Foundation under grant CHE-0741936. E.A.M. gratefully acknowledges funding from the Natural Science and Engineering Research Council of Canada (NSERC PGS D2).
Footnotes
Supporting Information Available: Figures S1-S3, the complete author lists for references 32 and 33, the optimized geometries, energies, frequencies and thermal corrections for nitroxyl radicals (RR′NO.), hydroxylamines (RR'NOH), the corresponding hydrogen bonded complexes and hydrogen atom transfer transition structures, for R = R′ = Me, R = H/R′ = Me, R = R′ = tbutyl, R = R′ = phenyl, and TEMPO (34 pages). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hynes JT, Klinman JP, Limbach H-H, Schowen RL, editors. Hydrogen-Transfer Reactions. Wiley-VCH; Weinheim: 2007. [Google Scholar]
- 2.Hammes-Schiffer S. pp. 479–502.Fernandez-Ramos A, Ellingson BA, Garrett BC, Truhlar DG. In: Reviews in Computational Chemistry. Cundari TR, Lipkowitz KB, editors. Vol. 23. Wiley-VCH; Hoboken, NJ: 2007. pp. 125–232. In ref. 1
- 3.Bell RP. The Tunnel Effect in Chemistry. Chapman and Hall; London: 1980. pp. 77–105. [Google Scholar]
- 4.Carpenter BK. Determination of Organic Reaction Mechanisms. John Wiley & Sons; 1984. pp. 83–111. [Google Scholar]
- 5. cf.; a Klinman JP. Phil. Trans. R. Soc. B. 2006;361:1323. doi: 10.1098/rstb.2006.1870. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Sutcliffe MJ, Masgrau L, Roujeinikova A, Johannissen LO, Hothi P, Basran J, Ranaghan KE, Mulholland AJ, Leys D, Scrutton NS. Phil. Trans. R. Soc. B. 2006;361:1375. doi: 10.1098/rstb.2006.1878. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Barroso M, Arnaut LG, Formosinho SJ. J. Phys. Org. Chem. 2009;22:254–263. [Google Scholar]; d Meyer TJ, Huynh MHV. Inorg. Chem. 2003;42:8140–8160. doi: 10.1021/ic020731v. [DOI] [PubMed] [Google Scholar]; e Reinaudt OM, Theopold KH. J. Am. Chem. Soc. 1994;116:6979–6980. [Google Scholar]; f Mahapatra S, Halfen JA, Tolman WB. J. Am. Chem. Soc. 1996;118:11575–11586. [Google Scholar]; g Zheng H, Lipscomb JD. Biochemistry. 2006;45:1685–1692. doi: 10.1021/bi051605g. [DOI] [PubMed] [Google Scholar]
- 6.Kochi JK, editor. Free Radicals. Wiley; New York: 1973. [Google Scholar]
- 7.a Huynh MHV, Meyer TJ. Chem. Rev. 2007;107:5004. doi: 10.1021/cr0500030. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Meyer TJ, Huynh MHV. Inorg. Chem. 2003;42:8140. doi: 10.1021/ic020731v. [DOI] [PubMed] [Google Scholar]; c Hodgkiss JM, Rosenthal J, Nocera DG. In: Hydrogen-Transfer Reactions. Hynes JT, Klinman JP, Limbach H-H, Schowen RL, editors. Vol. 2. Wiley-VCH; Weinheim: 2007. pp. 503–562. [Google Scholar]; d Stubbe J, Nocera DG, Yee CS, Chang MCY. Chem. Rev. 2003;103:2167. doi: 10.1021/cr020421u. [DOI] [PubMed] [Google Scholar]; e Cukier RI, Nocera DG. Annu. Rev. Phys. Chem. 1998;49:337. doi: 10.1146/annurev.physchem.49.1.337. [DOI] [PubMed] [Google Scholar]; f Partenheimer W. Catal. Today. 1995;23:69. [Google Scholar]
- 8.a Mayer JM. Annu. Rev. Phys. Chem. 2004;55:363. doi: 10.1146/annurev.physchem.55.091602.094446. [DOI] [PubMed] [Google Scholar]; b Mayer JM, Rhile IJ. Biochim. Biophys. Acta. 2004;1655:51. doi: 10.1016/j.bbabio.2003.07.002. [DOI] [PubMed] [Google Scholar]; c Mayer JM, Rhile IJ, Larsen FB, Mader EA, Markle TF, DiPasquale AG. Photosynth. Res. 2006;87:3. doi: 10.1007/s11120-005-8164-3. [DOI] [PubMed] [Google Scholar]; d Mayer JM, Mader EA, Roth JP, Bryant JR, Matsuo T, Dehestani A, Bales BC, Watson EJ, Osako T, Valliant-Saunders K, Lam W-H, Hrovat DA, Borden WT, Davidson ER. J. Mol. Catal. A: Chem. 2006;251:24. [Google Scholar]
- 9.a Knapp MJ, Meyer M, Klinman JP. In: Hydrogen-Transfer Reactions. Hynes JT, Klinman JP, Limbach H-H, Schowen RL, editors. Vol. 4. Wiley-VCH; Weinheim: 2007. pp. 1241–1284. [Google Scholar]; b Stubbe J, van der Donk WA. Chem. Rev. 1998;98:705. doi: 10.1021/cr980059c. [DOI] [PubMed] [Google Scholar]; c Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine. Oxford University Press; Oxford: 1999. [Google Scholar]
- 10.a Knapp MJ, Rickert K, Klinman JP. J. Am. Chem. Soc. 2002;124:3865. doi: 10.1021/ja012205t. [DOI] [PubMed] [Google Scholar]; b Lewis ER, Johansen E, Holman TR. J. Am. Chem. Soc. 1999;121:1395. [Google Scholar]
- 11.Likhtenshtein G, Yamauchi J, Nakatsuji S, Smirnov AI. Nitroxides: Applications in Chemistry, Biomedicine, and Materials Science. Wiley-VCH; New York: 2008. [Google Scholar]
- 12.Sheldon RA, Arends IWCE. J. Mol. Catal. A: Chem. 2006;251:200.Sheldon RA, Arends IWCE. Adv. Synth. Catal. 2004;346:1051.Sheldon RA, Arends IWCE, Brink G-JT, Dijksman A. Acc. Chem. Res. 2002;35:774. doi: 10.1021/ar010075n.Vasbinder MJ, Bakac A. Inorg. Chem. 2007;46:2322. doi: 10.1021/ic062010s. For other applications of nitroxyl radicals, see refs. 5-20 in
- 13.a Kreilick RW, Weissman SI. J. Am. Chem. Soc. 1966;88:2645. [Google Scholar]; b Arick MR, Weissman SI.J. Am. Chem. Soc 19689016544295237 [Google Scholar]
- 14.a Malievskii AD, Shapiro AB. Kinet. Catal. 2005;46:472. [Google Scholar]; b Malievskii AD, Koroteev SV, Shapiro AB. Kinet. Catal. 2005;46:812. [Google Scholar]; c Malievskii AD, Koroteev SV, Gorbunova NV, Brin EF. Kinet. Catal. 1997;38:485. [Google Scholar]
- 15.Dijksman A, Marino-González A, Mairata i Payeras A, Arends IWCE, Sheldon RA. J. Am. Chem. Soc. 2001;123:6826. doi: 10.1021/ja0103804. [DOI] [PubMed] [Google Scholar]
- 16.Ishii Y, Sakaguchi S, Iwahama T. Adv. Synth. Catal. 2001;343:393. [Google Scholar]
- 17.a Koshino N, Saha B, Espenson JH. J. Org. Chem. 2003;68:9364. doi: 10.1021/jo0348017. [DOI] [PubMed] [Google Scholar]; b Koshino N, Cai Y, Espenson JH. J. Phys. Chem. A. 2003;107:4262. [Google Scholar]; c Amorati R, Lucarini M, Mugnaini V, Pedulli GF. J. Org. Chem. 2003;68:1747. doi: 10.1021/jo0342931. [DOI] [PubMed] [Google Scholar]
- 18.Cai Y, Koshino N, Saha B, Espenson JH. J. Org. Chem. 2005;70:238. doi: 10.1021/jo048418t. [DOI] [PubMed] [Google Scholar]
- 19.Roth JP, Yoder JC, Won T-J, Mayer JM. Science. 2001;294:2524. doi: 10.1126/science.1066130. [DOI] [PubMed] [Google Scholar]
- 20.Mader EA, Larsen AS, Mayer JM. J. Am. Chem. Soc. 2004;126:8066. doi: 10.1021/ja049246k. H2bip = 2,2′-bi-1,4,5,6-tetrahydropyrimidine.
- 21.Warren JJ, Mayer JM. manuscript in preparation.
- 22.Ingold KU, In Kochi JK, editors. Free Radicals. Vol. 1. Wiley; New York: 1973. p. 69ff.Russel GA. pp. 275–331.O'Neal HE, Benson SW, In Kochi JK, editors. Free Radicals. Vol. 2. Wiley; New York: 1973. p. 302ff.Tedder JM. Angew. Chem. Int. Ed. Engl. 1982;21:401. HAT rate constants have traditionally been analyzed using a correlation of Arrhenius activation energy Ea with enthalpic driving force ΔH (the Bell-Evans-Polanyi relation), together with ‘polar effects’ and other influences. In ref. 6
- 23.a Mader EA, Davidson ER, Mayer JM. J. Am. Chem. Soc. 2007;129:5153. doi: 10.1021/ja0686918. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Mader EA, Manner VW, Markle TF, Wu A, Franz JA, Mayer JM. J. Am. Chem. Soc. 2009;131:4335–4345. doi: 10.1021/ja8081846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu A, Masland J, Swartz RD, Kaminsky W, Mayer JM. Inorg. Chem. 2007;46:11190. doi: 10.1021/ic7015726.Wu A, Mayer JM. J. Am. Chem. Soc. 2008;130:14745–14754. doi: 10.1021/ja805067h. acac = 2,4-pentanedionato; py-imH = 2-(2′-pyridyl)imidazole.
- 25.NUTS – NMR Utility Transform Software, 1D version. Acorn NMR Inc.; Livermore, CA: 2003. [Google Scholar]
- 26.Bordwell FG, Liu W-Z. J. Am. Chem. Soc. 1996;118:10819–10823. The close agreement between ΔG°4H and the reported ΔBDE does not show that entropic effects are unimportant, since ΔS°(XOH) – ΔS°(XO•) ≅ 0 is implicit in the Bordwell derivation of BDEs. For the bond dissociation free energy of TEMPO–H, and a discussion of entropies, see reference 23b.
- 27.Mader EA. unpublished results. The CH resonances for TEMPO-H broaden linearly with increasing TEMPO• concentrations but the derived rate constants did not vary exponentially with temperature, suggesting that the self-exchange reaction is not the only source of the line broadening.
- 28.SPECFIT/32, versions v3.0.26 and v3.0.36. Spectrum Software Associates; Marlbourough, MA: 2000. [Google Scholar]
- 29.Sorokin A, Robert A, Meunier B. J. Am. Chem. Soc. 1993;115:7293. The ‘true’ kH/kD, at complete deuteration, is equal to χ(D)/[(kH/kD)obs−1 – χ(H)], where χ(H) and χ(D) are the mole-fraction in H and D, respectively. For a discussion of the residual protio effect on the observed kH/kD, see
- 30.ΔBDE of 2,4,6-tri-tert-butylphenol (82.3 kcal mol−1) and tBu2NOH (68.2 kcal mol−1).26
- 31.Corchado JC, Chuang Y-Y, Coitino EL, Truhlar DG. GAUSSRATE–version 9.5. University of Minnesota; Minneapolis, MN: 2007. [Google Scholar]
- 32.Frisch MJ, et al. Gaussian 03, revision D.02. Gaussian, Inc.; Wallingford, CT: 2004. [Google Scholar]
- 33.Corchado JC, et al. POLYRATE–version 9.5. University of Minnesota; Minneapolis, MN: 2007. [Google Scholar]
- 34.Lynch BJ, Fast PL, Harris M, Truhlar DG. J. Phys. Chem. A. 2000;104:4811. [Google Scholar]
- 35.Zhao Y, Schultz NE, Truhlar DG. J. Chem. Theory Comput. 2006;2:364–382. doi: 10.1021/ct0502763. [DOI] [PubMed] [Google Scholar]
- 36.Zhao Y, Truhlar DG. Theor. Chem. Accounts. 2008;120:215–241. [Google Scholar]
- 37.a Frisch MJ, Pople JA, Binkley JS. J. Chem. Phys. 1984;80:3265–3269. [Google Scholar]; b Clark T, Chandrasekhar J, Spitznagel GW, Schleyer P. v. R. J. Comp. Chem. 1983;4:294–301. [Google Scholar]; c Hehre WJ, Ditchfield R, Pople JA. J. Chem. Phys. 1972;56:2257–2261. [Google Scholar]
- 38.Fernandez-Ramos A, Ellingson BA, Garrett BC, Truhlar DG. In: Reviews in Computational Chemistry. Lipkowitz KB, Cundari TR, editors. Vol. 23. Wiley-VCH; Hoboken, NJ: 2007. pp. 125–232. [Google Scholar]
- 39.Shelton GR, Hrovat DA, Borden WT. J. Am. Chem. Soc. 2007;129:164. doi: 10.1021/ja0664279. When the tunneling distance is large, there is a temperature range in which D tunnels considerably closer to the top of the barrier than H, so that the probability of passing through or over the barrier is actually greater for D than for H. As a result, in this temperature range, Ea,D – Ea,H > 1.3 kcal/mol, but log(AH/AD) < 0. See, for example
- 40.Mayer JM, Hrovat DA, Thomas JL, Borden WT. J. Am. Chem. Soc. 2002;124:11142–11147. doi: 10.1021/ja012732c.Litwinienko G, Ingold KU. Acc. Chem. Res. 2007;40:222–230. doi: 10.1021/ar0682029.Waidmann CR, Zhou X, Tsai EA, Kaminsky W, Hrovat DA, Borden WT, Mayer JM. J. Am. Chem. Soc. 2009;131:4729–4743. doi: 10.1021/ja808698x.In some circumstances, it is valuable to distinguish between an ‘HAT mechanism’ where the e− and H+ come from/transfer to the same bond, vs. a ‘PCET mechanism’ in which the two particles are separated in the reactants and/or products. See the discussion in
- 41.Roth JP, Lovell S, Mayer JM. J. Am. Chem. Soc. 2000;122:5486. [Google Scholar]
- 42.Mori Y, Sakaguchi Y, Hayashi H. J. Phys. Chem. A. 2000;104:4896.Semmelhack MF, Chou CS, Cortes DA. J. Am. Chem. Soc. 1983;105:4492–4494. Chantooni MK, Jr., Kolthoff IM. J. Phys. Chem. 1976;80:1306–1310. (a) ΔG°(ET) = −23.1[E(TEMPO•) – E(TEMPO-H)] ≈ 60 kcal mol−1. ΔG°(PT) = −1.37[pKa(TEMPO-H•+) – pKa(TEMPO-H)] = ΔG°(ET). E(TEMPO•) ≈ −1.91 V versus Cp2Fe+/0: E(TEMPO-H) ≈ 0.71 V versus Cp2Fe+/0: (d) pKa(TEMPO-H) ≈ 41 in MeCN is estimated from pKa(TEMPO-H) = 31.0 in DMSO.26 pKa conversion from DMSO to MeCN: (f) pKa(TEMPO-H•+) = pKa(TEMPO-H) + 23.1[E(TEMPO•) – E(TEMPO-H)]/1.37 ≈ −3.
- 43.a Avila DV, Ingold KU, Lusztyk J, Green WH, Procopio DR. J. Am. Chem. Soc. 1995;117:2929–30. [Google Scholar]; b Valgimigli L, Banks JT, Ingold KU, Lusztyk J. J. Am. Chem. Soc. 1995;117:9966–71. [Google Scholar]; c Snelgrove DW, Lusztyk J, Banks JT, Mulder P, Ingold KU. J. Am. Chem. Soc. 2001;123:469–477. [Google Scholar]
- 44.Abraham MH, Grellier PL, Prior DV, Morris JJ, Taylor PJ. J. Chem. Soc., Perkin Trans. 2. 1990:521.Abraham MH, Grellier PL, Prior DV, Duce PP, Morris JJ, Taylor PJ. J. Chem. Soc., Perkin Trans. 2. 1989:699.Warren JJ. unpublished results. (a) In the Litwinienko/Ingold/Abraham model,43,44b,c the CH2Cl2/MeCN kinetic solvent effect is given by log(kCH2Cl2/kMeCN) = 8.3α2H(β2HMeCN – β2HCH2Cl2). Using α2H (the solute constant) = 0.29 for dialkylhydroxylamines and β2H (the hydrogen-bond acceptor solvent constant) = 0.44 for MeCN and 0.05 for CH2Cl2,45 this model predicts kCH2Cl2/kMeCN = 8.7, in modest agreement with the experimental values of 4.8 ± 0.6 and 2.4 ± 0.3 for reactions 2 and 4. However, in our experience44e this model is not very accurate for reactions in CH2Cl2 (see reference 45). The Ingold/Abraham model may be incomplete for this example because the interaction of TEMPO (β2H = 0.46) with CH2Cl2 (α2H = 0.15) may be as important as the interaction of TEMPOH with CH2Cl2. The radical-solvent interaction is absent in many radical reactions because α2H(solvent) or β2H(RO•) is negligible.
- 45.Galian RE, Litwinienko G, Pérez-Prieto J, Ingold KU. J. Am. Chem. Soc. 2007;129:9280–9281. doi: 10.1021/ja071716y.Foti MC, Daquino C, Mackie ID, DiLabio GA, Ingold KU. J. Org. Chem. 2008;73:9270–9282. doi: 10.1021/jo8016555. (a) The β2H is given as 0.05 for CH2Cl2 by Abraham et al.44b,c A different value of 0.15 has been suggested using the KSE model,45b which was later revised to 0.20.45c As pointed out by a reviewer, the agreement with the KSE model is better using these revised values. However, we feel that the KSE model becomes more circular and less valuable when the β2H values are not independent parameters but rather extracted from the radical reaction rates.
- 46.a Knauer BR, Napier JJ. J. Am. Chem. Soc. 1976;98:4395–4400. [Google Scholar]; b Improta R, Barone V. Chem. Rev. 2004;104:1231–1254. doi: 10.1021/cr960085f. [DOI] [PubMed] [Google Scholar]
- 47.Beckwith ALJ, Bowry VW, Ingold KU. J. Am. Chem. Soc. 1992;114:4983–4992. [Google Scholar]
- 48.The experimental difference in zero-point energies for TEMPO-H(D) in MeCN is estimated as 0.5[νOH – νOD] = 452 cm−1 = 1.3 kcal mol−1.
- 49.Horng ML, Gardecki JA, Papazyan A, Maroncelli M. J. Phys. Chem. 1995;99:17311. The KIEs are not sensitive to solvent even though the characteristic solvent reorganization times are about a factor of two longer for CH2Cl2 than for MeCN:
- 50.Cotton FA, Felthouse TR. Inorg. Chem. 1982;21:2667. Andersen B, Andersen P. Acta Chem. Scand. 1966;20:2728.Bordeaux PD. Acta Crystallogr., Sect. B. 1974;30:790. (a) tBu2NO• is slightly more crowded than the cyclic analogs, as reflected in its slightly larger CNC angle of 129.6(3)° versus 123.5(2)° for 4-oxo-TEMPO•. Two molecules of tBu2NO• were co-crystallized with Rh2(O2CCF3)4(H2O)2. ∠CNC = 136(3)° of tBu2NO• in the gas phase was determined by electron diffraction.
- 51.Cape JL, Bowman MK, Kramer DM. J. Am. Chem. Soc. 2005;127:4208. doi: 10.1021/ja043955g.Ludlow MK, Soudackov AV, Hammes-Schiffer S. J. Am. Chem. Soc. 2009;131:7094–7102. doi: 10.1021/ja9001184.Yoder JC, Roth JP, Gussenhoven EM, Larsen AS, Mayer JM. J. Am. Chem. Soc. 2003;125:2629. doi: 10.1021/ja0273905. EaD – EaH = −2.8 kcal mol−1 and log(AH/AD) = 2.2 have been reported for e−/H+ from 2,3-dimethoxy-5-methyl-1,4-benzoquinol to the excited state of [Ru(2,2′-bipyridine)2{2-(2-pyridyl)benzimidazolate}+]: This result has very recently been rationalized in terms of being close to the Marcus inverted region for PCET: HAT self-exchange between [FeII(H2bip)3]2+ and [FeIII(H2bip)2(Hbip)]2+ (H2bip = 2,2'-bi-1,4,5,6-tetrahydropyrimidine) also appears to have negative EaD – EaH = −1.2 ± 0.8 kcal mol−1 and positive log(AH/AD) = 0.9 ± 1.2 values:
- 52.Foti M, Ingold KU, Lusztyk J. J. Am. Chem. Soc. 1994;116:9440. [Google Scholar]
- 53.Mendenhall GD, Ingold KU. J. Am. Chem. Soc. 1973;95:627–628. [Google Scholar]
- 54.Chenier JHB, Howard JA. Can. J. Chem. 1975;53:623. [Google Scholar]
- 55.Griller D, Ingold KU. J. Am. Chem. Soc. 1974;96:630. [Google Scholar]
- 56.Bordwell FG, Cheng J. J. Am. Chem. Soc. 1991;113:1736. [Google Scholar]
- 57.a Doba T, Ingold KU. J. Am. Chem. Soc. 1984;106:3958. [Google Scholar]; b Malatesta V, Ingold KU. J. Am. Chem. Soc. 1981;103:3094. [Google Scholar]
- 58.Ozinskas AJ, Bobst AM. Helv. Chim. Acta. 1980;63:1407. [Google Scholar]
- 59.KaleidaGraph, version 3.5. Synergy Software; 2000. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.