

Abstract
An important factor in the transition from an open to a closed circulatory system was a change in vessel wall structure and composition that enabled the large arteries to store and release energy during the cardiac cycle. The component of the arterial wall in vertebrates that accounts for these properties is the elastic fiber network organized by medial smooth muscle. Beginning with the onset of pulsatile blood flow in the developing aorta, smooth muscle cells in the vessel wall produce a complex extracellular matrix (ECM) that will ultimately define the mechanical properties that are critical for proper function of the adult vascular system. This review discusses the structural ECM proteins in the vertebrate aortic wall and will explore how the choice of ECM components has changed through evolution as the cardiovascular system became more advanced and pulse pressure increased. By correlating vessel mechanics with physiological blood pressure across animal species and in mice with altered vessel compliance, we show that cardiac and vascular development are physiologically coupled, and we provide evidence for a universal elastic modulus that controls the parameters of ECM deposition in vessel wall development. We also discuss mechanical models that can be used to design better tissue-engineered vessels and to test the efficacy of clinical treatments.
I. INTRODUCTION
In a closed circulatory system, large arteries became an important component of proper cardiac function by serving as elastic reservoirs, enabling the arterial tree to undergo large volume changes with little change in pressure. Without elastic vessels, the tremendous surge of pressure as blood is ejected from the heart would inhibit the heart from emptying, and the pressure in the vessels would fall so rapidly that the heart could not refill. The large elastic arteries are capable of storing a portion of the stroke volume with each systole and discharging that volume with diastole. This phenomenon, known as the windkessel effect (54), helps to decrease the load on the heart and to minimize the systolic flow and maximize diastolic flow in the arterioles. The result is a more even distal flow throughout the cardiac cycle. What makes this possible is a vessel wall containing a specialized ECM uniquely designed to provide elastic recoil.
As we look back through evolution and follow the transition from an open to a closed circulatory system, we see arteries gradually develop mechanical properties that support pulsatile flow. In invertebrates with a highly developed, partially open circulatory system, arteries have distensible elasticity with nonlinear stress-strain curves similar in many ways to vertebrate vessels (50, 67, 253). The mechanical properties of the invertebrate vessel wall are contributed by ECM components, namely, collagen and microfibrils, which can accommodate the relatively low intraluminal and pulse pressures seen in these animals. With the transition to a fully closed circulation, the higher pulse pressure associated with ejection of the entire cardiac output into the aorta during systole required a different ECM able to provide elastic recoil at higher pressure.
The components of the arterial wall in vertebrates that account for the majority of its mechanical properties are the collagen and elastin deposited by smooth muscle cells (SMCs) in the medial layer. Medial elastin is woven into a three-dimensional, interconnecting lamellar network designed to transfer stress throughout the vessel wall. Between the lamellar layers are bundles of collagen that show no definite overall arrangement at low pressure but become circumferentially aligned as pressure increases (54, 229, 303). Previous studies suggest that <10% of collagen fibers are engaged at physiological pressure (93), whereas at higher pressures, the vessel becomes progressively less distensible as collagen fibers are recruited to support passive wall tension and restrict aortic distension. With additional increases in wall strain or stretch ratio, there is little further change in radius as additional collagen fibers are recruited, accounting for the nonlinear nature of vascular elasticity (Fig. 1) (17, 229, 303). Because the stress-strain relationship is nonlinear, a single constant, like Young’s elastic modulus for linear materials, cannot be used to describe the slope of the curve. However, the local slope, or incremental elastic modulus, can be calculated. The incremental elastic modulus is only valid for specified strains because it is a function of strain (Fig. 1). In the range of physiological strain for mammalian arteries, the incremental elastic modulus is less than that of collagen, but much greater than that of elastin alone. This is because the wall acts as a “two phase” material with an incremental modulus similar to elastin at low strains and to collagen at high strains. All vertebrates and invertebrates with closed circulatory systems have arteries with this nonlinear mechanical behavior (253).
FIG. 1.

Nonlinear mechanical behavior of the adult mouse aorta. A: average circumferential stress versus stretch ratio. B: circumferential incremental elastic modulus (Einc) versus stretch ratio. Einc was calculated by determining the local slope of the stress-stretch ratio relationship in A. The physiological region is highlighted in gray for each graph. Note the decreased incremental elastic modulus at low stretch ratios where elastin dominates the vessel mechanical behavior and the increased modulus at high stretch ratios where collagen dominates. The physiological range is at the intersection of these two regions. The sharp increase in modulus just beyond the physiological range prevents distension of, and damage to, the vessel with increased pressure. [Data replotted from Wagenseil et al. (290).]
In addition to providing the structural and mechanical properties required for vessel function, the vascular ECM provides instructional signals that induce, define, and stabilize vascular cell phenotypes. There are many examples of ECM molecules playing critical roles in the regulation of gene expression by interacting with specific matrix receptors on cells and by binding and storing growth factors that influence cellular function. This reciprocal instructive interaction between the cell and its ECM is important in directing the developmental transitions that occur in embryogenesis, postnatal development, and response to injury. How vascular cells interpret these regulatory signals is a major area of research today.
This review will address the relationships between vascular development and vessel mechanics as imparted by the mix of ECM molecules in the vessel wall, with the primary focus on developing mouse aorta. As the vessel wall matures, the vascular cells go through multiple overlapping phenotypic transitions, characterized broadly by cellular proliferation, matrix production, and the assembly of an appropriate contractile apparatus within the cell cytoplasm. Defining the functional characteristics of these transitions is difficult because of the nonspecific and transient nature of many marker proteins that are used to characterize vascular cells. Further complicating our understanding is the phenotypic plasticity the vascular cell exhibits during embryogenesis, vessel maturation, and response to injury (74, 89, 246). Nevertheless, significant progress has been made in elucidating the molecular mechanisms and processes that control differentiation of vascular SMCs during development and repair (207). Several excellent reviews have summarized our current understanding of SMC phenotypes based on expression of cytoskeletal and other marker proteins (92, 126, 207, 208). There are also numerous ultrastructural studies documenting the architecture of the developing vessel wall (3, 81, 102, 143, 195, 211, 212, 277). Extensive information on the vascular SMC and a still timely discussion of questions and issues driving research in vascular biology can be found in a monograph by Schwartz and Mecham (246).
II. ARTERIAL WALL STRUCTURE
A. Tunica Intima
The luminal surface of large vertebrate arteries is lined with endothelial cells, which play a major role in defining the embryonic vascular pattern and in recruiting SMCs to the vascular wall (10, 227, 264). Endothelial cells produce and attach to a basal lamina that is supported by the internal elastic lamina (IEL) or is separated from the IEL by amorphous material and “anchoring and connecting filaments” that consist of fibrillin-containing microfibrils and collagen fibers (48, 80, 244). This region of the wall, called tunica intima, is defined as the endothelial cells and subendothelial area on the luminal side of the IEL (Fig. 2). The ability of endothelial cells to produce elastin suggests that they contribute to the formation of the IEL (25, 26, 46), perhaps in response to a signal from medial cells (186). The subendothelial matrix is normally acellular in smaller animals but contains a population of SMCs in humans and in other larger animals (244, 245). It is not clear if these cells are there by design with unique functions or if they are medial cells that were trapped in these areas during vessel wall formation by localized reduplication and remodeling of the IEL (80). The tunica intima is particularly important in atherosclerosis and restenosis, but contributes little to the mechanical properties of the normal conducting vessel.
FIG. 2.
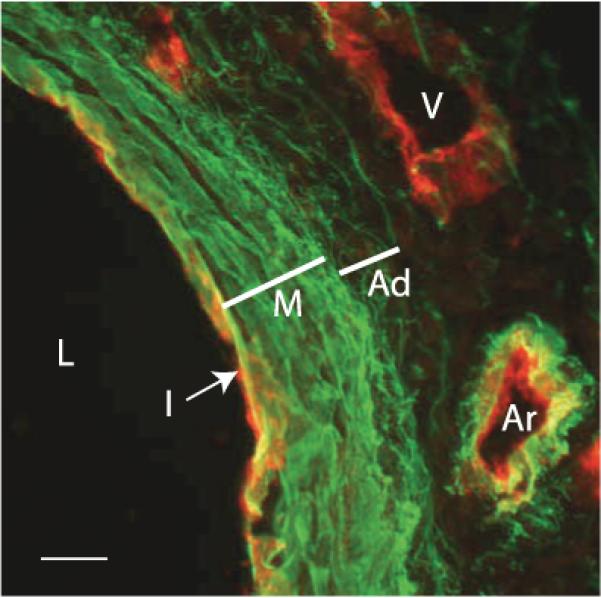
Immunofluorescence micrograph of E17 mouse aorta. Sections through the aorta of an E17 mouse were stained with an antibody for elastin (green) and for flk, a marker for endothelial cells (red). On the left is the lumen (L) of the artery. The intima (I) is evident as a single layer of red-staining endothelial cells. The media (M) contains dense layers of elastin, whereas the elastin in the adventitia (Ad) consists of fine fibers. The vein (V) on the top right shows the presence of endothelial cells but no elastin, whereas the small artery (Ar) directly below shows both. Scale bar = 100 μm. (Micrograph provided by Dr. Sean McLean.)
B. Tunica Media
SMCs and most of the elastin make up the tunica media. The elastin is arranged in fenestrated sheets (lamellae) between which are collagen fibers, thin layers of proteoglycan-rich ECM, and SMCs. Thin elastic fibers connect the lamellae into a three-dimensional continuous network (53, 200) (Fig. 3) and connect the lamellae with the SMCs (47). Elastin, which is distensible and has a low tensile strength, functions primarily as an elastic reservoir and distributes stress evenly throughout the wall and onto collagen fibers (15, 81, 304). Elimination of smooth muscle function does not significantly alter the static mechanical properties of the mature aorta (18), suggesting that these characteristics are mainly due to the elastin and collagen components, which account for ~50% of the vessel’s dry weight (100, 200). The number of lamellar units (generally defined as an elastic lamella and adjacent SMCs) in a vascular segment is related linearly to tensional forces within the wall (37, 161, 304), with the greatest number of elastic layers occurring in the larger, more proximal vessels that experience the highest wall tension (16). Interestingly, the number of lamellar units in a particular vascular segment does not change after birth.
FIG. 3.

Elastic lamellae in human aorta. Top: electron micrograph of human aorta in cross-section showing the arrangement of smooth muscle cell layers separated by the darkly stained elastic lamellae. The lumen of the vessel is at the top. The image on the bottom shows the network of elastin after all cells and other extracellular matrix (ECM) proteins are removed by autoclaving. The circumferential sheets of elastin are joined across the wall by numerous interlamellar elastin connections, which are important for transferring stress across the wall and throughout the elastic fiber network. Scale bar = 20 μm.
C. Tunica Adventitia
The outermost layer of the vessel wall is the tunica adventitia. It is generally defined in large arteries as the area outside of the external elastic lamina and consists of a collagen-rich ECM produced by a heterogeneous population of myofibroblast cells (143, 263). The high relative collagen content of the adventitia helps prevent vascular rupture at extremely high pressures (23). This region also gives rise to the vasa vasorum—small blood vessels that provide nourishment and oxygen to the cells in the vessel wall. Mice and other mammals with 29 or fewer medial lamellar units have no demonstrable vasa vasorum (301). In larger mammals with >29 lamellae, vasa vasorum is found in the outer aspect of the media, but there is always a subintimal avascular zone equal to 29 medial units for which filtration from the lumen (transitional filtration) is adequate for medial nutrition (301). Beyond this zone, medial nutrition must be supplemented by the vasa vasorum. Because of this unique vascular network, the adventitia is a prominent site of vascular inflammation.
Recent studies have shown that the adventitia may be a unique injury-sensing compartment of the vessel wall (243, 263) and, in mature blood vessels, contains residential progenitor cells capable of differentiating into SMCs that repopulate the media and intima (111, 116, 210, 279).
III. CARDIOVASCULAR DEVELOPMENT
Cardiovascular development follows similar steps in most warm-blooded animals, but on widely varying time scales. Total gestation in chickens and in mice is ~21 days, in sheep gestation is ~147 days, and in humans it is ~267 days. In mice, which are the focus of this review, early cardiovascular development occurs between embryonic day (E) 8 and 14, which corresponds approximately to gestational days 20-56 in humans (296). A heartbeat is detectable by high-frequency ultrasound at approximately E8.25, and blood flow is detectable by Doppler ultrasound at ~E8.5 in mice (137). This corresponds with the observed redistribution of blood cells due to limited circulation at E8.25-E8.5, followed by detection of red blood cells throughout the entire circulation by ~E10.5 (180). The right and left ventricle are visually distinct entities between E11-E12 (288), but are connected by interventricular foramen until about E15. Blood flow and blood pressure increase rapidly through the later embryonic and early postnatal stages in mice (Fig. 4). For example, mean left ventricular outflow velocity increases from 10 to 23 mm/s from E10.5 to E14.5 (147), and peak systolic left ventricular pressure increases from 2 to 11 mmHg from E9.5 to E14.5 (132). Cardiac output increases from 5 to 15 ml/min from postnatal day (P) 10 to P35 (297), and mean arterial pressure increases from 30 to 70 mmHg from P0 to P35 (118).
FIG. 4.

Hemodynamic parameters and ECM expression increase sharply in late embryonic and early postnatal development in mice. A: systolic pressure and cardiac output versus age are replotted from published studies (131, 132, 147, 297). Dotted lines were interpolated between different studies, as data on the hemodynamic parameters for the last third of embryonic development in mice has not been published. Age was calculated assuming that embryonic development lasts 21 days and the mice are born on day 0. B: median normalized values for elastin and collagen type I expression are replotted from Kelleher et al. (146). Expression of both proteins steadily increases from E14 through P14-21, then rapidly decreases to low levels by ~P30. C: sum of the median normalized elastin and collagen expression versus age. The sum was calculated by totaling all median normalized gene expression from the start of development to the current age. This graph also illustrates that little new elastin or collagen protein accumulates after expression of the two genes are downregulated. Note the logarithmic scale on the vertical axes of graphs in A and C. The similar developmental timeline of hemodynamic parameters and total ECM expression suggests correlations between these events.
The aortic arch develops concurrently with the heart, with significant patterning changes occurring between E11 and E14 in the mouse. The truncus arteriosus becomes divided into what will become the aorta and pulmonary artery around E11. At this stage the truncus arteriosus leads to numerous symmetric aortic arches that will transform into the aorta, the major branches off the aorta, and the pulmonary artery (61). The complete separation of the extracardial aorta and pulmonary artery occurs around E13.5 (288), and by E14, the definitive vascular pattern is established (61). The ductus arteriosus connects the aorta and pulmonary artery during embryonic development, but is closed off approximately 3 h after birth in the mouse (270) (24-48 h in humans). After the early developmental period, the arteries alter geometry (namely diameter, wall thickness, and length) without further changes in patterning. In addition to geometric changes, the vessel wall undergoes compositional changes in the ECM that are related to changes in mechanical stimuli.
IV. THE VASCULAR SMOOTH MUSCLE CELL
A. Embryonic Origins of Vascular SMCs
The vertebrate vessel wall is built around endothelial tubes that begin to form in the absence of blood flow (126, 227). As blood flow commences, presumptive vascular SMCs are recruited from the surrounding mesenchyme and/or cardiac neural crest. The angiopoietin/Tie receptor pathway (58, 241) is an important player in early stages of this process, but questions remain about what other factors guide smooth muscle differentiation through the various stages of vessel wall formation. A recent review by Majesky (174) summarizes the different embryonic origins of vascular SMCs and notes that different vessels, or even different segments of the same vessel, are composed of SMC populations that arise from distinct sources of progenitors, each with its own unique lineage and developmental history. At least four populations of precursor cells contribute to the smooth muscle cells of large elastic conducting vessels: 1) cells in the base portion of the aorta and pulmonary trunk derive from the secondary heart field; 2) cranial neural crest contributes SMCs to the ascending and arch portions of the aorta, the ductus arteriosus, the innominate and right subclavian arteries, and the right and left common carotid arteries; and 3) somatic and 4) splanchnic mesoderm contribute to the dorsal aorta. There is also accumulating evidence that some SMCs arise from transdifferentiation of fetal (51) or adult (73) endothelial cells, or from macrophages (199) or marrow-derived progenitor cells (136, 233).
It is instructive to compare formation of the vertebrate vessel wall with the process in invertebrates. Invertebrates do not have endothelial cells lining the vessel lumen (101), thereby establishing that endothelial cells are not a conserved feature of cardiovascular tube formation. The invertebrate vessel tubes themselves are also different, containing contractile myoepithelial cells that, in many organisms, facilitate circulation of blood fluid through vessel contraction. Another difference is that the myoepithelial cells orient their basal surface towards the lumen of the vessel where they deposit an extensive basement membrane (101, 153). Thus unlike other internal tubes and cavities whose lumen is contacted by the apical surface of the surrounding epithelium, primitive blood vessels are outlined by the basal surfaces of epithelial cells (153). These invertebrate vascular cells arise from the spanchnopleura mesothelium, which is strikingly similar to the origin of vertebrate smooth muscle cells that populate the vessel wall outside of the region populated by neural crest-derived cells. Similarities extend further to conserved regulatory genes and signaling pathways that specify the vascular and blood progenitors within the splanchnic mesoderm of vertebrates and invertebrates. An excellent phylogenetic perspective on the evolution of the blood vascular system can be found in Hartenstein and Mandal (101).
B. Vascular SMC Differentiation
SMC differentiation has traditionally been studied by monitoring the expression of genes for vascular smooth muscle cytoskeletal and contractile protein. The most commonly examined are smooth muscle α-actin (SMαActin), calponin, smoothelin, SM22, and smooth muscle myosin heavy chain isoforms SM-MHC1 and SM-MHC2 [reviewed in Owens (208)]. An antibody to a smooth muscle α-actinin (antibody 1E12) also specifically labels SMCs from the early stages of development to adulthood and offers the advantage of identifying the primordial cell restricted to the smooth muscle lineage (56, 125, 126). While clearly useful in sorting through cell types in the developing cardiovascular system, these markers are not without their limitations. All are expressed in multiple embryonic muscle cell types (e.g., cardiac and skeletal), and calponin, smoothelin, SM22, and the MHCs appear later than SMαActin in vessel wall development.
The temporal appearance of SMCs in the large vessels has been extensively studied in embryonic chicks and quail where it was shown that neural crest contributes to the media of the aorta, pulmonary artery, and other aortic arch vessels (156). Using SMαActin as a marker, Rosenquist et al. (231) described two phases of SMαActin expression in the developing chick vasculature. The first occurs in the primal vessels before the arrival of the neural crest cells. The second phase occurs after the neural crest cells populate the aortic arches, but not until the vanguard cells arrive at the myocardial cuff of the truncus arteriosus. SMαActin expression then proceeds from the region nearest the heart to downstream vessels and begins in the medial cells near the adventitia and appears later in the cells nearer the lumen. A similar biphasic actin expression pattern was identified by Bergwerff et al. (14) using a different actin marker antibody. The peri-endothelial cells in the ductus arteriosus, the coronary arteries, the pulmonary arteries, and the descending aorta do not lose actin expression during this period.
Comparable fine detail is not available for SMC differentiation in mouse aortic development. Studies using smooth muscle myosin heavy chain (SM-MHC) expression as a marker of SMC differentiation suggest that the process in the mouse is generally similar to what has been described in the bird, but also illustrates some interesting differences. For example, at E10.5, SM-MHC transcripts were first observed in the developing mouse dorsal aorta, whereas the outflow tract was notably devoid of signal. One day later (E11.5), SM-MHC was observed in the outflow tract as well as in the branching arches of the ascending aorta (190). These results illustrate the unique segmental characteristics that arise from the different embryonic origins of the vessel wall cells (174) and suggest that the early developmental program is different depending on vessel location.
As the vessel wall matures, SMCs condense down around the endothelial tubes to eventually form circumferential layers that will define the elastic lamellae of the mature vessel. Interestingly, the number of smooth muscle layers that will be present in the adult vessel is established relatively early in development (~E14 in the mouse). During subsequent vessel wall maturation, the number of SMC layers does not change. What does change, however, is the phenotype of the vessel wall cell as typified by ECM expression.
V. THE VASCULAR EXTRACELLULAR MATRIX
A major function of the vascular SMC in medium to large vessels is to synthesize and organize the unique ECM responsible for the mechanical properties of the wall. Unlike cells in the small muscular and resistance vessels, the SMCs of the mature elastic conducting vessels contribute little to the passive mechanical properties of the wall (67). Hence, the ability to produce ECM can be considered a defining “differentiated” phenotype, and the spectrum of ECM molecules that vascular cells produce provides a molecular signature for a specific state of differentiation (56, 124, 126). Because the formation of a functional ECM must occur in an organized sequence, the “matrix phenotype” is changing throughout the entire period of vessel wall development. The change in the vessel structure from a primarily cellular artery in early development to a mechanically appropriate elastic artery at maturity requires the stepwise and coordinated expression of numerous matrix components. Beginning with maturation of the outflow tract of the heart and the onset of pulsatile blood flow in the aorta, SMCs in the vessel wall produce a complex ECM that will ultimately define the mechanical properties of the adult vascular system. These properties include the following: 1) a highly resilient wall where a large proportion of the energy input during systolic inflation will be recovered by elastic recoil during diastole, 2) low hysteresis (the energy lost during an inflation-deflation cycle), and 3) nonlinear elasticity characterized by stiffening with increasing pressure to protect the wall from rupture.
Because these properties are derived from the combination of matrix components deposited in the wall, turning matrix production off when the correct material properties are met is just as important as turning matrix production on in early development. Blood pressure increases incrementally as the wall strengthens incrementally (or is it possibly the other way around?), and one cannot occur without the other. Once the correct pressure (or vessel wall material property) is obtained, elastin and collagen synthesis are downregulated so as to maintain the appropriate mechanical properties of the wall for optimized physiological function. The nature of the “matrix off” switch is unknown.
Production of a functional matrix requires the coordinated expression, both temporally and spatially, of complex sets of genes that encode ECM proteins as well as the enzymes responsible for their secretion and assembly. For example, building a functional collagen fiber involves activating and regulating genes for collagen α-chains, hydroxylating enzymes, proteases to process propeptide regions, lysyl oxidases for cross-linking, and other chaperones and assembly proteins. Similar complexities are involved in the processing and assembly of most ECM networks, including basement membranes, elastic fibers, and large proteoglycan matrices.
To identify the types of matrix proteins produced by SMCs during vascular development, we performed large-scale gene expression analysis on developing descending thoracic and abdominal mouse aortas using oligonucleotide microarrays. Our dataset begins at E14 and extends through 6 mo of age in the adult mouse (146, 183). Principle component analysis of the array data identified several major patterns for ECM gene expression. The first and most prevalent, which has been labeled the “matrix phenotype,” consists of a major increase in matrix protein expression at E14 followed by a steady rise through the first 7-14 days after birth. This is followed by a decrease in expression over 2-3 mo to low levels that persist in the adult (Fig. 4). Elastin, fibrillar collagens, and most of the structural matrix proteins discussed in this review follow this pattern. A similar pattern for elastin and structural matrix expression has been documented in rats, humans, and other animals (12, 13, 19, 81). The second most prevalent pattern was one of consistent expression throughout the time series and was typical of basement membrane components, fibronectin, most integrins, and some matrix metalloproteinases. The third pattern consists of high expression levels in the embryonic/fetal period followed by decreased expression postnatally. The final and least populated pattern was low expression in fetal and postnatal development with an increase in the adult period. A list of genes in each group can be found in McLean et al. (183). Graphs of the expression data for many ECM proteins can be found in Kelleher et al. (146). In the sections below we summarize the expression profiles of the elastic fiber genes, collagens, and proteoglycans, which together are the major structural matrix proteins of the vessel wall.
A. Elastin
Elastin is the major protein that imparts the property of elasticity to tissues such as the lung, skin, and blood vessels (209). It functions as a cross-linked polymer as part of an elastic fiber, and its assembly outside the cell requires an association with numerous other extracellular proteins. When discussing elastin it is important to distinguish between elastin itself and the elastic fiber. These terms are sometimes used interchangeably when, in fact, they refer to separate entities. Elastic fibers are complex structures that contain elastin as well as microfibrils (Fig. 5). Elastin has an amorphous appearance by transmission electron microscopy and is the major component of mature elastic fibers. Microfibrils, in turn, are 10- to 15-nm filaments that are thought to facilitate elastin assembly and provide overall structure to the growing elastic fiber (65, 234, 235).
FIG. 5.

Electron micrographs of developing elastic fibers. A: electron micrograph showing a developing elastic fiber adjacent to an elastin-producing cell. Bar = 1.0 μm. B: at higher magnification, the elastic fibers are seen to consist of black amorphous elastin deposited within a bundle of microfibrils. Bar = 0.25 μm. C: in cross section, the microfibrils have a tubular appearance. Bar = 0.25 μm. D: an elastic fiber visualized using quick-freeze, deep etch microscopy (104). Unlike standard transmission microscopy, quick-freeze, deep-etch images provide insight into organization of elastin (E) within the fiber (184, 187). The major feature is a densely packed matrix of 5-7 nm tropoelastin molecules that are associated so tightly that little or no etching occurs during sample preparation. Microfibrils (MF) are seen along the periphery of the fiber and at the end. [A-C from Mecham and Davis (184), copyright Elsevier 1994.]
The emergence of elastin in evolution is quite recent, appearing coincident with the closed circulatory system and found exclusively in vertebrates (237). In most animal species, a single gene encodes elastin. The only known exceptions are zebrafish and frogs, where two elastin genes have been identified (33, 191). In mammals, the elastin gene is composed of 36 exons distributed throughout ~40 kbp of genomic DNA (128). Rat and mouse ELN have 37 exons due to an additional short exon inserted after exon 4. The human ELN gene, however, has only 34 exons due to the sequential loss of two exons during primate evolution. The loss of exon 35 occurred at least 35-45 million years ago, when Catarrhini [Old World monkeys and hominoids (apes and humans)] diverged from Platyrrhines (New World monkeys). Loss of exon 34, in contrast, occurred only ~6-8 million years ago, when Homo separated from the common ancestor shared with chimpanzees and gorillas (267). Although still contained within the gene, exon 22 is rarely included in the elastin gene transcript (68). Both exons 34 and 35 are present in all nonprimate vertebrates studied to date, including chicken, which means that they predate the mammalian radiation. Their recent excision in primates was most likely mediated by recombination events driven by Alu-repeats that flank both exons in the primate genome (267). It is unclear what, if any, selective advantage is conferred upon the primate protein by the loss of these two exons and the silencing of a third in primate lineages, but these changes suggest that this relatively new ECM gene is undergoing strong purifying selection (215).
The mammalian elastin gene encodes a protein of 60-70 kDa called tropoelastin. Tropoelastins from all species share a characteristic domain arrangement of hydrophobic sequences alternating with lysine-containing cross-linking motifs (Fig. 6) (193, 272). In the extracellular space, >80% of tropoelastin’s lysine residues are modified to form covalent cross-links between and within elastin molecules. It is this cross-linked polymer that is the functional form of the protein. Cross-linking is initiated by one of the lysyl oxidase family members whose major function is the oxidative deamination of the ε-amino group on lysine side chains (reviewed in Refs. 138, 173). The resultant aldehyde condenses with another aldehyde residue through an aldol condensation reaction or with an unoxidized lysyl amino group through a Schiff base reaction to form the bifunctional cross-links aldol condensation product and dehydrolysinonorleucine, respectively (71, 72). These two cross-links can then interact to form the tetrafunctional cross-links desmosine and isodesmosine (Fig. 6). Several other cross-links of minor abundance have been identified (69, 262, 282, 283).
FIG. 6.
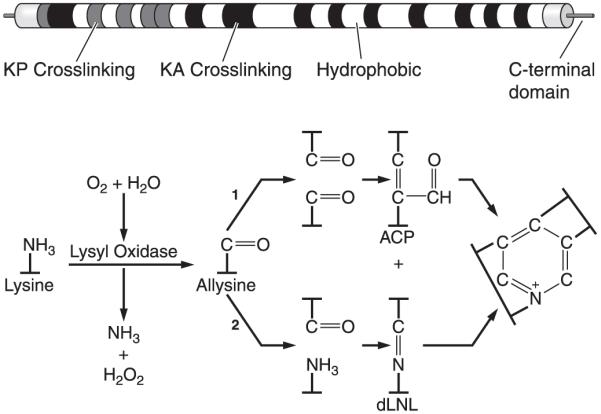
Elastin domain structure and cross-link formation. Top: schematic diagram of exon and domain structure of human tropoelastin. Shaded squares represent lysine cross-linking domains that contain prolines (KP) or are enriched in alanines (KA). White squares are hydrophobic sequences. Bottom: cross-linking of elastin monomers is initiated by the oxidative deamination of lysine side chains by the enzyme lysyl oxidase in a reaction that consumes molecular oxygen and releases ammonia. The aldehyde (allysine) that is formed can condense with another modified side chain aldehyde (1) to form the bivalent aldol condensation product (ACP) cross-link. Reaction with the amine of an unmodified side chain through a Schiff base reaction (2) produces dehydrolysinonorleucine (dLNL). ACP and dLNL can then condense to form the tetrafunctional cross-link desmosine or its isomer isodesmosine.
Lysyl oxidase is also responsible for cross-linking of collagen, where lysine or hydroxylysine residues in the telopeptides (nonhelical portions of the molecule) are converted into aldehydes that then form bifunctional and more complex cross-links (64). One major difference between cross-links in elastin and collagen is the overall number found in each. Whereas collagen contains 1-4 cross-links per collagen unit, elastin contains 15-20. This high degree of cross-linking is important for elastin’s recoil properties and is also responsible for the proteins insolubility and contributes to its longevity. Shapiro et al. (254) estimated the longevity of elastin using aspartic acid racemization and 14C turnover to be the human life span. Studies using sensitive immunological techniques to measure elastin peptides in the blood or desmosine cross-links excreted in the urine suggest that <1% of the total body elastin pool turns over in a year (261). As mentioned above, elastin expression in most tissues occurs over a narrow window of development, beginning in mid gestation and continuing at high levels through the postnatal period (Fig. 4) (12, 13). In the aorta, expression decreases rapidly when the physiological rise in blood pressure stabilizes postnatally, and there is minimal elastin synthesis in the adult animal (39, 57, 145, 254). This explains why repair of elastic fibers is incomplete in the adult period and why the elastin protein must have a long half-life.
Elastin is one of the earliest structural matrix proteins to be expressed by vascular SMCs in large vessels. In situ hybridization studies show that elastin expression in the avian vascular system begins in the truncus arteriosus of the developing chick near the aorta-pulmonary septum and proceeds both towards the heart and peripherally towards distal vessels until expressed in the entire arterial tree (231, 232, 250). Elastin expression is coincident with the condensation of cells around the endothelial tube, which is in agreement with observations by Thompson and Fitzharris (276) that cells near the primary luminal bifurcation are the first to condense into what will be the lamellar layers of the media. In addition to the longitudinal expression gradient down the vascular tree, elastin expression in the larger arteries initiates in the external portion of the media and moves towards the lumen. In later stages, the synthetic activity decreases in opposite ways: in the pulmonary artery, expression decreases from the adventitia towards the lumen, whereas the opposite pattern is seen in the systemic vessels (110, 250). Rosenquist and Beall (231) showed similar longitudinal and radial elastin expression in the developing chick aorta using an antibody to tropoelastin. They also showed that tropoelastin was detectable in the developing aorticopulmonary septum by antibody staining. Based on this finding, the authors proposed that elastogenesis is a critical event in septation (232).
B. Proteins of the Elastic Fiber Microfibril
The relationship between elastin and microfibrils is unique and evolutionarily interesting. Early in elastic fiber formation, microfibrils are observed in the extracellular space prior to the appearance of elastin (38, 45, 65, 234). With time and near the cell surface, tropoelastin associates with the microfibrils to form small globules of amorphous cross-linked elastin. These globules then interact with other elastin globules to form larger structures (44, 152). It is possible to find microfibrils without elastin (such as the ciliary zonule in the eye and the oxytalan fibers in the skin and periodontal ligament), but elastin without microfibrils is rare.
The structural building blocks of the microfibril are the fibrillin molecules (223, 239). These widely distributed proteins are evolutionarily conserved from jellyfish to human (188, 224). Several microfibril-associated proteins have also been described, but their importance to microfibril structure and function is not yet clear. The best characterized are the latent transforming growth factor (TGF)-β binding proteins (LTBP 1-4), emilins, microfibril-associated glycoproteins (MAGP-1 and -2), and members of the fibulin family. A list of other microfibril-associated proteins can be found in Kielty et al. (148).
C. Fibrillin
The human genome contains three fibrillins, but fibrillin-3 appears to have been inactivated in the mouse genome due to chromosome rearrangements (42). There is also suggestion of a fourth fibrillin in zebrafish (79). These 350-kDa glycoproteins are highly homologous, with modular structures consisting of repeating calcium-binding epidermal growth factor (EGF)-like domains inter-spersed between 8-cysteine domains similar to those found in the latent TGF-β-binding protein family (99). It has long been assumed that microfibrils provide a scaffold or template for elastin assembly by binding and aligning tropoelastin monomers so that lysine-containing regions are in register for cross-linking. Recent studies from fibrillin knockout mice (22, 213) confirm that fibrillin is required for assembly of the elastin fiber, although whether it participates early or late in the process is still under investigation.
In the developing mouse aorta, fibrillin-1 has an expression pattern similar to elastin, except peak expression occurs ~7 days earlier. Expression of fibrillin-2, in contrast, is highest in the early embryonic period and then decreases linearly throughout maturation (146, 312). Even when expression of fibrillin-2 is at its highest, it is still appreciably lower in terms of absolute amount than fibrillin-1 or elastin. This suggests that fibrillin-1 is the major fibrillin in the mature aorta, with fibrillin-2 playing a minor role.
Mice lacking the fibrillin-1 gene (Fbn1-/-) have thin, fragmented arterial elastic fibers. The animals die within 2 wk of birth from aortic aneurysms, impaired breathing, and diaphragmatic collapse (27). Mice lacking the fibrillin-2 gene (Fbn2-/-) have no vascular or pulmonary defects, and the arterial elastic fibers look normal. They develop syndactyly, which shows that fibrillin has a much broader developmental role than serving as a scaffold for elastic fibers (7, 27, 29). Fibrillin-1 and -2 probably compensate for each other in these knockout models, because mice lacking both fibrillins (Fbn1-/-; Fbn2-/-) die in utero and show only traces of elastic fibers between SMC layers in the developing aorta. Also, Fbn1+/-; Fbn2-/- mice show a more severe vascular phenotype than Fbn1-/- mice, and half die in utero (27).
Fibrillin-1 mutations lead to the human disease Marfan syndrome (MIM154700), which is characterized by cardiac, skeletal, and ocular abnormalities. The cardiac abnormalities include aortic root dilatation and rupture that can lead to premature death (223). Mice that underexpress fibrillin-1 (mgR/mgR) recapitulate the vascular traits of Marfan syndrome including fragmented elastic fibers and aortic dilation and rupture (213). These mice show increased aortic stiffness and pulse pressure (176).
Mutations in fibrillin-2 are associated with a rare disease in humans called congenital contractural arachnodactyly (Beals Syndrome, MIM121050) with autosomal dominant inheritance and characterized by congenital joint contractures, arachnodactyly, kyphoscoliosis, mal-formed ear helices, and vascular abnormalities (96, 158). Mice deficient in fibrillin-2 are viable and display only a skeletal patterning defect due to altered bone morphogenetic protein (BMP) signaling (7, 27, 29). Mutations in zebrafish fibrillin-2, in contrast, result in notochord and vascular abnormalities (79), most likely through a mechanism that impairs the ability of microfibrils to recruit or correctly position lysyl oxidases at their site of action in the notochord sheath (79, 170). In Xenopus, fibrillin-2 morpholino knockdown results in gastrulation arrest (259), a finding that likely reflects differences in the mechanisms of axial extension used by Xenopus and zebrafish embryos.
Fibrillin is an ancient protein that has been found as far back as jellyfish, whereas elastin is strictly a vertebrate protein. With fibrillin appearing hundreds of million years earlier in evolution, it is unlikely that it evolved with the sole purpose to assemble elastin. In fact, mouse models of fibrillin inactivation have revealed that fibrillin-rich microfibrils function to regulate morphogenetic and tissue homeostatic programs through direct cell-matrix interactions or indirectly by modulating the activity of TGF-β/BMP signals (7, 28, 222, 223, 251, 293). Fibrillins have RGD sequences that interact with integrins (9, 214, 240) and heparin-binding domains that interact with cell surface heparan sulfate proteoglycans (228, 278), suggesting that fibrillins directly signal cells through these receptors.
D. Microfibril-Associated Glycoproteins
The MAGPs are small glycoproteins (~20 kDa for the mature secreted form) with no repeating motifs (88). The two members of the MAGP gene family (MAGP-1 and -2) are related through a 60-amino acid sequence in the middle of the molecule that shares precise alignment of 7 cysteine residues (86). Sequence comparison shows that MAGP-1 is a highly conserved protein; not only among mammals where the similarity level is ~90% overall, but also in lower vertebrates (65% similarity overall, rising to 96% between the cysteine-rich domains of mammals and zebrafish). There is reference in the literature to other proteins that have been given the name microfibril-associated proteins [MFAP-1 (113, 309), MFAP-3 (1), and MAGP-36/MFAP-4 (107, 150, 280, 313)], but these proteins have no structural or sequence similarity to MAGP-1 or -2 and, hence, are not considered members of the same MAGP gene family.
Immunolocalization studies suggest that MAGP-1 and -2 are constitutive components of most microfibrils (84) and together with the fibrillins were predicted to be necessary for elastin assembly (87, 160). However, inactivation of the gene for MAGP-1 (Mfap2) (293) and MAGP-2 (Mfap5) (unpublished results) in mice has no effect on elastin structure or amount and blood vessels, and other elastin-containing tissues are functionally and structurally normal.
The absence of a detectable vascular phenotype in the MAGP-1-null mouse is in contrast to changes that occur when MAGP-1 levels are modified in the zebrafish. Knockdown of MAGP-1 in zebrafish using mfap2 morpholinos results in vascular defects that include dilated vessels in the brain and the eyes, irregular lumens of axial vessels, and a dilated caudal vein with altered venous plexus formation (30). While these studies suggest that MAGP-1 is required for maintaining the structural integrity of the vessel wall and for venous plexus morphogenesis in the fish, it is not clear why similar phenotypes were not found in the mouse. It is also interesting that fibrillin-1 and MAGP-1 morphant embryos exhibit overlapping vascular defects in zebrafish, which is clearly not the case in the mouse.
Rather than the phenotypes expected for loss of an elastic fiber protein, the MAGP-1 phenotypes in mice manifest as bone abnormalities, increased fat deposition, bleeding diathesis (295), and impaired response to injury (293). These traits suggest that MAGP-1 is dispensable for elastic fiber assembly but important for other processes of tissue homeostasis or differentiation. Indeed, MAGP-1 has been shown to bind members of the TGF-β growth factor family, and many of the phenotypes in the MAGP-1 null mouse can be explained by misregulation of TGF-β or BMP signaling (293). The traits that define the MAGP-1 null phenotype are, largely, opposite those described for fibrillin-1-mutant mice and for humans with Marfan syndrome. These differences are particularly interesting in that the fibrillins and MAGP-1 are binding partners and components of the same microfibril. Yet, the phenotypes of the knockout mice suggest contrasting biological functions for the two proteins.
The expression profile of MAGP-1 in the aorta more closely follows that of fibrillin-2 than fibrillin-1, whereas the opposite is true for MAGP-2. Both MAGP-1 and fibrillin-2 are highest in the embryonic period and steadily decrease after birth, although fibrillin-2 decreases to lower levels than does MAGP-1. MAGP-2 and fibrillin-1, in contrast, increase during the embryonic period and remain relatively high after birth. These findings suggest that fibrillin-2 and MAGP-1 are the predominant players in microfibril biology during embryonic and fetal aortic development, whereas fibrillin-1 and both MAGP-1 and -2 are the major components of vascular microfibrils postnatally and into adulthood. While both MAGP-1 and MAGP-2 have been localized to elastin-associated as well as elastin-free microfibrils, MAGP-2 exhibits a pattern of tissue localization and developmental expression that is more restricted than that of MAGP-1 (70, 85).
E. Fibulins
The fibulins are a family of seven ECM proteins that contain EGF-like modules and a distinctive COOH-terminal domain (6, 32, 149). All fibulins except fibulin-6 and fibulin-7 are found in elastic tissues (149), with fibulin-2 and -4 at the interface between the central elastin core and its surrounding microfibrils, fibulin-1 located within the elastin core, and fibulin-5 associated with microfibrils. Mice deficient in fibulin-1 die soon after birth with spontaneous bleeding defects associated with abnormal endothelial integrity in small blood vessels. Depending on genetic background, these animals either show no cardiac phenotype (151) or exhibit cardiac ventricular wall thinning and ventricular septal defects with double outlet right ventricle or overriding aorta (41). Anomalies of the aortic arch arteries are also evident. The spectrum of malformations is consistent with fibulin-1 influencing neural crest cell-dependent development of these tissues.
Inactivation of the fibulin-2 (Fbln2-/-) and -3 (Fbln3-/-) genes in mice has no effect on elastic fiber formation, and the large elastic vessels are normal in both animals (182, 256). Mice lacking either fibulin-4 (Fbln4-/-) or fibulin-5 (Fbln5-/-), however, have highly disrupted and disorganized elastic fibers, leading to defects in skin, arterial blood vessels, and lungs (181, 196, 307). Mice lacking the fibulin-4 gene (Fbln4-/-) die just before birth with severe vascular defects. The aorta is narrowed and tortuous, and the elastic lamina contains irregular elastin aggregates (181). Mice that underexpress fibulin-4 (Fbln4 R/R) live to adulthood but have hypertension, longer tortuous arteries, and dilated aortas with disrupted elastic fibers, thickened walls, and reduced compliance (98). Fibulin-5 knockout (Fbln5-/-) mice show similar, but less severe, phenotypes to Fbln4 R/R mice. Fbln5-/- mice have tortuous, less compliant (260) aortas with disrupted elastic fibers that form abnormal elastin aggregates (196, 307). These results demonstrate that fibulin-4 and fibulin-5 play essential yet nonredundant roles in elastic fiber formation during development. Mutations in the human gene encoding fibulin-5 can cause either autosomal dominant (MIM123700) or autosomal recessive (MIM219100) cutis laxa. Autosomal recessive cutis laxa (MIM219100) with severe systemic connective tissue abnormalities is also caused by mutations in the human fibulin-4 gene (119).
F. EMILIN-1
EMILIN-1 (elastin microfibril interface located protein) is a member of newly defined EMILIN/Multimerin family and is found in elastin-rich tissues such as the skin and arteries. Ultrastructural studies have localized EMILIN-1 (previously known as gp115) to the interface between microfibrils and the amorphous core of elastin (21). Mice lacking the EMILIN-1 gene (Emilin1-/-) live a normal life span but have hypertension and arteries with irregular elastic lamellae, smaller diameters, and thinner walls (310, 311). EMILIN-1 inhibits TGF-β signaling by binding specifically to the pro-TGF-β precursor and preventing its maturation by furin convertases. The result in the knockout is a generalized reduction of blood vessel diameter and increased peripheral resistance. The role of EMILIN-1 in elastic fiber assembly is not yet clear, but its ability to bind both tropoelastin and fibulin-5 suggests it may serve a bridging function between those two molecules (311). EMILIN-1 expression is highest at early stages of aortic development (E14-18) then drops to low levels at birth and during the postnatal period until elastin and collagen production begins to decline at approximately P21-P30. At this point, EMILIN-1 levels increase and persist at high levels into the adult period.
G. Lysyl Oxidase
Lysyl oxidases are a family of five genetically distinct, copper binding proteins with amine oxidase activity that catalyze the formation of lysine and hydroxylysine-derived cross-links in collagens and lysine-derived cross-links in elastin (43, 173, 194). In addition to the canonical lysyl oxidase (LOX), the first member of this family characterized (97, 139, 257, 281), four LOX-like proteins (LOXL1-4) have been identified, although the specific functions of these isoenzymes are unknown (reviewed in Refs. 43, 173). Of the five lysyl oxidases, LOX and LOXL1 are most homologous, with LOXL2-4 forming a second homologous group. The pro-region of LOX and LOXL is important in targeting both enzymes to elastic fibers (275), and the propeptide of LOX plays a role in modulating SMC signaling (127). It is also interesting to note that LOX has been shown to interact with TGF-β and suppress its signaling in bone (8). This raises the interesting possibility that the presence of active LOX may be an important modulator of extracellular growth factor activity. LOX has also been shown to have a direct effect on the collagen type 3 (COL3A) and elastin (ELN) promoters (82, 203), which may provide a pathway for LOX to regulate the production of some of its major protein substrates.
Mice lacking the lysyl oxidase gene, Lox-/-, die just before or soon after birth with large aortic aneurysms (112, 175). The mice have tortuous aortas with thicker walls and smaller lumens. The elastic fibers in the wall are highly fragmented and discontinuous. Interestingly, elastin-associated cross-links are reduced by only 60% in these mice, suggesting that one of the other lysyl oxidase family members contributes to elastin cross-links or partially substitutes for LOX in the knockout. Analysis of lysyl oxidase family expression in the aorta shows that LOX is the most abundant, with an expression pattern similar to that of elastin and the fibrillar collagens. LOX expression increases rapidly during the fetal period to reach its high level at approximately E18. Elevated expression persists through the neonatal period and begins to decline when elastin and collagen expression diminish at approximately P30. Levels then stay low through the adult period. LOXL1 also shows a rapid rise in expression during the fetal period but returns to low levels at birth. Expression remains low during the postnatal phase of collagen and elastin synthesis, then rises to high levels when collagen and elastin expression begin to decrease at approximately P30. Expression remains relatively high into the adult period. The expression pattern for LOXL3 is different from the other LOXs in that it is predominantly expressed during the postnatal period of rapid collagen and elastin expression (P0, P7, P14), with low expression levels before and after. Expression of LOXL2 and LOXL4 was detected in the array, but at extremely low levels.
Mice with a knockout of the LOXL1 gene (Loxl1-/-) have no obvious vascular defects and live a normal life span. However, postpartum Loxl1-/- mice show fragmented elastic fibers, suggesting a role for LOXL1 in elastic fiber maintenance (170). LOXL1 has been shown to interact with fibulin-5 (170) and to interact genetically with fibrillin-2 (79). Mice deficient in LOXL3 have not been reported.
H. Vascular Collagens
Our expression array data identified 17 different collagen types in the mouse aorta, with collagens I, III, IV, V, and VI having the highest expression levels (146, 183). Collagens I, III, and V are fibril-forming collagens, with types I and III being mainly responsible for imparting strength to the vessel wall. The distribution of collagens I and III varies depending on the specific region of the vascular tree being examined (114). In the ascending bovine aorta, collagens I and III colocalize in the media and adventitia. In the descending thoracic aorta and in muscular arteries, type I is distributed mainly in the media and less so in the adventitia, whereas type III is localized predominantly in the adventitia. Human diseases that result from mutations in the collagen I gene have a wide variety of phenotypes, with skin and bone being particularly affected. Osteogenesis imperfecta (MIM166200) is a heritable disorder resulting from mutations in collagen I α-chains and is characterized by bone fragility and low bone mass, with a wide spectrum of clinical expression. Mutations in collagen I can also lead to an autosomal recessive form of Ehlers Danlos syndrome (EDS type VII, MIM225410). Mice homozygous for targeted interruption of the collagen type Iα1 gene die between E12 and E14 from vessel rupture (171).
In humans, mutations in the collagen IIIα1 gene (COL3A1) result in EDS type IV (MIM130050) (216). The vascular phenotype found in this disease includes fragility of blood vessels and a propensity towards rupture in large vessels (248). Mice homozygous for a disrupted Col3A1 allele show 90% perinatal mortality, with survivors dying by 6 mo of age from vascular rupture (169).
Type V collagen plays a critical role in collagen fibril nucleation. Mice deficient in collagen V die in early embryogenesis (about E10) due to a lack of collagen fibril formation (294). Collagen V has been localized to the media of human arteries and has also been seen in the basement membrane. Mutations in collagen type V that cause functional haploinsufficiency are associated with classical EDS in humans (MIM130000) (247). Mice that are heterozygous for a Col5A1 loss-of-function mutation have vessels with decreased stiffness and breaking strength.
Expression of type VI collagen in the mouse aorta is similar to elastin and collagen type I, but while it is a fibril-forming collagen, it does not colocalize in large collagen bundles with collagens I and III. Instead, collagen VI is frequently associated with fibrillin-1 in oxytalan fibers and may serve to connect elastic lamellae to the basement membrane of SMCs, or connect SMCs to other ECM structures (53). In humans, mutations in collagen VI genes result in muscle weakness with progressive muscle wasting (Bethlem myopathy, MIM158810; Ullrich’s disease, MIM254090). Mice with null collagen VI mutations have a phenotype that closely parallels the human Bethlem myopathy (20) with little to no vascular involvement. There may, however, be subtle capillary abnormalities associated with type VI mutations in humans with Ullrich’s disease (197).
I. Proteoglycans
The proteoglycans constitute a number of genetically unrelated families of multidomain proteins that have covalently attached glycosaminoglycan (GAG) chains. Proteoglycans are categorized based on the type of attached GAGs: 1) chondroitin sulfate and dermatan sulfate, consisting of a repeating disaccharide of galactosamine and either glucuronic acid or iduronic acid; 2) heparin and heparan sulfate, consisting of a repeating disaccharide of glucosamine and either glucuronic acid or iduronic acid; and 3) keratan sulfate, consisting of a repeating disaccharide of glucosamine and galactose. GAG chains are usually attached through O-glycosidic linkages to serine residues in the proteoglycan core protein. A characteristic feature of GAG chains is that at physiological pH they contain one to three negative charges per disaccharide due to carboxylate and sulfate groups.
The proteoglycans found in greatest abundance in the vessel wall can be categorized into two classes: large proteoglycans that form large aggregates by interaction with hyaluronan and small leucine-rich proteoglycans. The large proteoglycans interact with hyaluronic acid to form an extensive, interconnected polymeric network in the extracellular space. Hyaluronan is a linear polymer composed of repeating disaccharides of glucuronic acid and N-acetylglucosamine. It is synthesized at the plasma membrane by three different but related hyaluronan synthases: HAS1, HAS2, and HAS3 (273, 292). The growing chain is extruded through the membrane into the pericellular space where it can be anchored to the cell surface via the synthase enzyme or through binding to a cell surface receptor such as CD44 or RHAMM (62).
Versican is the largest proteoglycan in the vessel wall and has been localized to the aortic media and endothelial layers (5, 308). Expression in the mouse aorta shows highest mRNA levels in the fetal period, with peak production at E18-P0. Expression levels drop by ~50% after birth to levels that persist into the adult period. Versican is known to have a wide variety of functions, including induction of cell adhesion, promotion of proliferation, and influencing cell migration (298). Interestingly, in vascular injury models, high versican levels correlate with low elastin content, most likely due to inhibition of elastic fiber assembly by the chondroitin sulfate GAGs on versican (105, 117). Expression of the versican variant V3, which lacks GAG chains, induces elastin synthesis and fiber deposition in vitro, providing further support that chondroitin sulfate GAGs may be biologically relevant in matrix assembly (189). At present, there are no known vascular diseases linked to versican gene mutations, and no versican null mouse has been reported to date.
Aggrecan, a large aggregating proteoglycan most abundant in cartilage, has a smaller core protein than versican but contains nearly threefold more GAG chains. Studies of developing chick aorta show that aggrecan has a different spatial and temporal expression pattern than versican, with aggrecan localized to the outer region of the developing aortic wall and expressed at much lower levels than versican (5). In the mouse aorta, peak expression occurs around E18 with a rapid drop postnatally, reaching low levels at P7 that persist into the adult stage. As in the chick aorta, aggrecan expression in the mouse aorta is substantially lower than versican. The function of aggrecan in the vessel wall is unclear.
The small leucine-rich proteoglycans (SLRP) are a family of secreted proteoglycans that do not interact with hyaluronic acid but, instead, bind ECM molecules such as collagen, tropoelastin, fibronectin, and fibrillin-containing microfibrils, among others (225). The SLRP family includes decorin, biglycan, fibromodulin, osteoglycin, and lumican (129, 242). Both biglycan and decorin bind to and regulate collagen fibrillogenesis (108) and have been shown to bind TGF-β and sequester it to the ECM. Biglycan localizes to all layers of the human aorta by immuno-histochemical staining, whereas decorin is found only in the adventitia (274).
The expression profile of decorin in the mouse aorta closely parallels that of type I collagen in embryonic time points but peaks at P0. Expression decreases somewhat in the postnatal time points but remains constitutively expressed at a moderate level. Biglycan, on the other hand, shows increasing expression over the embryonic time points in a pattern that correlates closely with elastin gene expression. Expression peaks at P7 then falls over the first postnatal month but rises again as the animal enters adulthood (5.5-6 mo). In elastic ligaments, biglycan expression is associated with the elastinogenic phase of elastic fiber formation (226).
Lumican has been localized to the outer layer of medial VSMCs and adventitia of nonatherosclorotic human coronary arteries (205). Mouse aortic expression of lumican occurs from E12 to P6 mo at low to moderate levels. There is a small peak in expression levels at P0. Lumican, like decorin and biglycan, regulates collagen fibrillogenesis.
Studies of mice null for decorin, biglycan, and lumican show phenotypes in bone, tendon, and skin. Specifically, collagen fibril diameter and organization are dys-regulated, with different fibril diameters and organizations dependent on the tissue type examined. No changes in blood vessel structure or stability were reported for decorin- or lumican-deficient animals. There is also no vascular phenotype reported in mice homozygous for a biglycan null mutation in the 129Sv/C57BL6 background (306). However, when bred into the BALB/cA background, male homozygous null mice died suddenly before 2-3 mo of age from aortic rupture (103). Rupture occurs between the media and the adventitia, which is similar to dissections of the aortic wall in Col3A1-deficient mice (103, 169).
VI. EXTRACELLULAR MATRIX AND MECHANICAL DESIGN
A. Microfibril-Based Elasticity
Evolution has provided an interesting glimpse of how vascular cells use different ECM components to achieve the mechanical properties suitable for the unique needs of each organism. As pulse pressure increases with ever-increasing cardiovascular complexity, we see the material properties of large vessels change to provide an appropriate level of elastic recoil. In studies of the mechanical behavior of arteries of several invertebrate and lower vertebrate phyla, investigators (50, 83, 252, 253) have shown that invertebrates with a highly developed, partially open circulatory system have vessels that exhibit elastic behavior with the classical nonlinear mechanics of vertebrate vessels, even though these vessels lack elastin (Fig. 7). Invertebrate arteries, like those of higher vertebrates, are composite structures containing muscle cells and an ECM that changes as the pressure demands increase. The marine whelk (Busycon contrarium), an animal with an open circulatory system that is unlikely to generate significant pulsatile pressure, has a relatively stiff aorta resulting from a vessel wall characterized by an irregular arrangement of SMCs surrounded by collagen fibers (50). In contrast, histological analysis of the aortic wall of the lobster (Homarus americanus) (Fig. 7) and horseshoe crab (Limulus polyphemus), two invertebrates with elastic arteries, shows dense lamellar layers that resemble the elastic lamellae in the vertebrate vessel. Ultrastructural analysis found these dense layers to consist of bundles of fine fibrils (microfibrils) that resemble reticular oxytalan fibers in vertebrate tissues (50, 179). Two primitive vertebrates with a closed circulatory system but relatively low blood pressure, the sea lamprey (Petromyzon marinus) and the Atlantic hagfish (Myxine glutinosa), also have microfibril-based lamellae (50, 133). Characterization of the fibrils in these microfibril-based vessels showed them to be extensible and containing fibrillin, the major structural protein of vertebrate microfibrils (24). Thus the microfibril-based matrix in these species is a compliant material similar to elastin, but employed at lower pressures. Although there are differences between these species in terms of aortic wall architecture, their vessels have mechanical properties similar to vertebrate vessels, characterized by extensibility at low pressures and stiffness at high pressures, and lamprey, hagfish, and lobster vessels have hysteresis properties that show fairly small energy losses per inflation-deflation cycle (Fig. 7) (24, 50).
FIG. 7.

Mechanical properties and wall structure of invertebrate aortae. A: pressure-volume inflation-deflation curves for aortas from lamprey (top) and lobster (bottom). Relative volume is given as V/V0, where V is the instantaneous volume of the vessel and V0 is the volume at the pressure at which the inflation-deflation cycle was started. B: light micrograph showing a transverse section of the abdominal aorta of the lobster, stained with modified Weigert’s technique, showing the positively stained fibrous material forming the internal lamina closest to the lumen (top) dense fibrous matrix within the middle lamina (ML), which contains striated muscle cells (STM) and fibroblasts. No positively staining fibers are observed in the connective tissue of the external layer (EL). Scale bar = 20 μm. C: electron micrograph showing the fibrils within a dense fiber in the middle lamina. Arrowheads indicate the periodicity of the beaded fibrils. Scale bar = 190 nm. [From Davison et al. (50), with permission from the Company of Biologists Ltd.]
B. Elastic Fiber Elasticity
With the transition to a fully closed circulation, a different, more resilient ECM was required to accommodate the altered heart function and overall higher pressures required by this system. It is at this point that elastin emerged as a key vascular protein (Fig. 8). Elastin appeared first in cartilagenous fish and is found in all vertebrate species except for agnathans, the class of jawless vertebrates that includes lamprey (236-238). Elastin-containing vessels in mammals and nonmammals alike function at higher pressures than the microfibril-based invertebrate vessels but show the same nonlinear mechanical properties and low hysteresis. It is interesting that microfibrils seen in invertebrate and lower vertebrate vessels remain an integral part of the vertebrate vessel wall even though their elastomeric function has been superseded by elastin. Mice with no elastin in the vessel wall (Eln-/-) but with normal levels of microfibrils die shortly after birth from aortic obstruction, showing that whatever mechanical properties microfibrils impart, they are clearly not sufficient to maintain vascular function in the high-pressure mammalian system (162).
FIG. 8.

Changes in lamellar ECM composition during transition from an open to a closed circulatory system. The vessel wall in invertebrates with an open circulatory system is exposed to low intraluminal pressure and little or no pulsatile flow. Under these circumstances, the vessel wall does not require elastic recoil for proper function. Ultrastructural analysis of the aorta from the marine whelk, an invertebrate with an open circulatory system, found abundant fibers with the typical appearance of collagen bundles (50). As organisms became more complex and the circulatory system transitions from an open to a closed circuit with increasing pulsatile pressure, the vessel wall developed an extracellular matrix that provides elastic recoil. In invertebrates with a highly developed open circulatory system and in lower vertebrates that have not completed the transition to a fully closed circulatory system (e.g., lamprey and hagfish), the vessel wall contains dense layers of fibrillin-containing microfibrils, which provide elastic recoil not found in the collagen-only vessel of lower invertebrates (50). With the emergence of a fully closed circulation, the major vessels experienced much higher pulse pressure as the entire cardiac output is ejected from the heart during systole. It is at this point in evolution that elastin appears in the wall where it associates with microfibrils to form large elastic structures that provide the elastic recoil required for normal vessel function in vertebrates. [Electron micrographs of whelk and lobster aorta from Davison et al. (50), with permission from The Company of Biologists Ltd., and of bovine aorta from Mecham and Davis (184), copyright Elsevier 1994.]
A comparative study of the mechanical properties of aortas from invertebrates, lower vertebrates, nonmammals, and mammals found that in all species, the physiological pressure range appears to be the region where stresses are transferred from the distensible to the inextensible components of the wall. In vertebrate arteries, the nonlinearity has been attributed to deformation of the elastin component at low strains and collagen at high strains (54). For the microfibril-based materials, McConnell et al. (179) and Bussiere et al. (24) showed that the nonlinearity could be attributed to reorientation of the microfibrils at the low end of the stress-strain curve and to deformation of the microfibrils themselves at the high end of the curve. Collagen could play a role at extremely high strains.
C. Universal Elastic Modulus
Vessels with distensible elasticity have unique relationships between the incremental elastic modulus and the pressure (Fig. 9), which reflects the differing material properties of the vessels themselves. However, when the pressures are normalized to each organism’s mean blood pressure, the incremental elastic modulus of the aorta for all species converges to a value in the range of 0.3-1.0 MPa (Fig. 9) (50, 83, 253). To achieve this functional modulus, each organism must adjust the mix of ECM components in the vessel wall to produce the mechanical properties appropriate for different hemodynamic variables. In this context, the developing vascular SMC must monitor the changing tensional forces within the wall and adjust matrix production accordingly. The finding of a universal vessel wall modulus that applies across species and in vessels with different ECM components is, indeed, surprising. There must be strong evolutionary pressure to ensure that all elastic arteries have similar mechanical properties at each organism’s mean physiological blood pressure. The significance of the physiological modulus being in the range of 0.3-1.0 MPa is not evident, but this appears to be a target mechanical property that is best able to provide capacitance and pulse smoothing in a pulsatile circulatory system. In the section below we will see that changing the mechanical properties of the vessel wall in the mouse by deleting one copy of the elastin gene results in an adaptive response characterized by changes in blood pressure and vessel wall structure, but the physiological elastic modulus of the elastin-insufficient aorta remains in the target range. Thus achieving and maintaining the functional elastic modulus may be a master physiological regulator of ECM gene expression in the vessel wall.
FIG. 9.

Incremental elastic modulus of the aorta in a variety of animal species. Top: plots of incremental elastic modulus (note logarithmic scale) as a function of inflation pressure. Although substantial differences exist among the various animals, in each case the aorta exhibits nonlinear elasticity and the modulus increases dramatically with pressure. Bottom: the same data as above but plotted with pressure normalized to the mean blood pressure for each species. The physiological elastic modulus at P/Pmean = 1 ranged from 0.3 to 1 MPa, with most clustering around 0.4 MPa. The incremental elastic modulus for human (young and old) aorta and wild-type and Eln+/- mouse aorta was determined from data in Learoyd and Taylor (157) and Wagenseil et al. (290), respectively. The modulus for all other animals is from Shadwick (253), with permission from The Company of Biologists Ltd.
VII. MECHANICS OF ELASTIN-INSUFFICIENT ARTERIES: NEW INSIGHT INTO VASCULAR DEVELOPMENT
Constructing a complex, mechanically appropriate matrix requires instructions for assembly, knowledge of the available building materials, and information about the stresses that the final material will have to endure. One way this could occur is through a process where all of the required information is genetically hardwired into the cells participating in the construction project, with no deviation from the blueprint design. Alternatively, the project could be fashioned over time through changing instructive signals from the microenvironment that tell the cells what mix of matrix proteins need to be added at that particular instance. Our studies of elastin-insufficient mice support the latter possibility and show that there is more flexibility in the building plan than previously thought.
A. Eln+/- Mice: Elastin Haploinsufficiency and Supravalvular Aortic Stenosis
Mice with one functional elastin allele (Eln+/-) and half as much elastin live a normal life span and thrive well into adulthood. These animals, however, exhibit remarkable vascular changes that ultimately produce a cardiovascular system that operates at higher blood pressures than wild-type (WT) mice. These traits are similar to conditions associated with supravalvular aortic stenosis (SVAS; MIM185500) in humans, an autosomal dominant disease resulting from elastin haploinsufficiency (63, 164). SVAS can occur sporadically as a result of loss-of-function mutations, or through gene deletion as occurs in Williams-Beuren syndrome (WBS; MIM194050) (206). Thus, in addition to providing information about the role of elastin in vascular development, the Eln+/- mouse is a useful model to study the pathogenesis of an important human disease.
To understand how changes in elastin levels alter cardiovascular function, we assessed vessel mechanics and numerous key hemodynamic parameters in the adult Eln+/- mouse. Blood pressure measurements found that Eln+/- mice have mean arterial pressures 30-40 mmHg higher than WT animals. Surprisingly, left ventricular cardiac hypertrophy, as assessed by heart weight, is minimal at 6 mo of age (~12% increase) considering the high systemic blood pressure. Mechanical testing shows that the abdominal aorta, ascending aorta, and left common carotid artery have smaller inner and outer diameters and thinner vessel walls at all pressures compared with WT (67, 290). The Eln+/- mice also have an increased number of lamellar units in their arteries and a vessel wall with an increased incremental elastic modulus at high pressures (67).
At physiological pressures, the circumferential wall strain and stress are higher in Eln+/- aorta than in WT (67, 290). In typical hypertension, the artery wall would be remodeled to increase the wall thickness and normalize the wall stresses, but Eln+/- arteries have thinner walls, not thicker. The lack of expected remodeling behavior is not due to a defect in the remodeling response of Eln+/- arteries. When pressure is increased in adult Eln+/- and WT mice by clipping of a renal artery, both genotypes show the expected hypertensive remodeling as evidenced by increased heart weight and arterial wall thickness (289). Although circumferential wall stress is higher in the Eln+/- aorta because the vessels have both increased pressure and increased lamellar units, the tension per lamellar unit is the same as WT and is similar to other mammalian species. Therefore, maintaining a constant tension/lamellar unit may be an important stimulus for developmental remodeling. Eln+/- arteries also show altered residual strain compared with WT, with increased circumferential residual strain, decreased longitudinal stretch ratio, and increased residual shear in the longitudinal-circumferential direction. The residual shear causes Eln+/- vessels to loop or curve dramatically upon excision (Fig. 10) (67, 290). Residual and applied stresses and strains in the vessel wall will be discussed in more detail below.
FIG. 10.
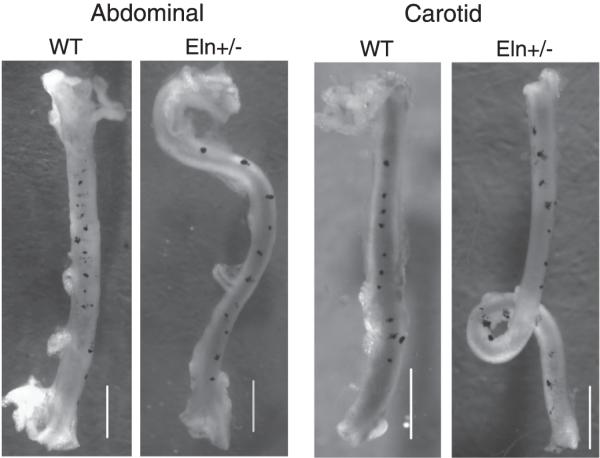
Increased residual shear strain causes the abdominal aorta and left carotid artery of Eln+/- mice to change shape upon excision. Small carbon particles were placed vertically along the length of each vessel in vivo to measure the difference between in vivo and ex vivo length and to document residual shear. Ex vivo, the particles shift to the right along the vessel length, indicating residual shear. The rightward shift is small in wild-type (WT) vessels and becomes more pronounced in Eln+/- vessels. The small residual shear in WT vessels causes only slight curvature in the ex vivo vessel. The increased residual shear in Eln+/- vessels causes either sharp changes in curvature (abdominal aorta) or complete loops (carotid artery) in the ex vivo vessel. Scale bar = 1 mm. [Modified from Wagenseil et al. (290).]
Like the systemic circulation, the pulmonary vasculature is affected by elastin insufficiency. Compared with WT mice, elastin-insufficient mice demonstrate elevated right ventricular (RV) systolic and diastolic pressures (54.6/13.2 mmHg systolic/diastolic for Eln+/- compared with 9.8/3 mmHg for WT) (255). Proximal pulmonary arteries in elastin-insufficient mice have thinner walls and an increased number of elastic lamellae, changes similar to those observed in the systemic circulation. The mice also exhibit RV hypertrophy and peripheral intrapulmonary vascular remodeling, although the changes are much less than expected given the degree of RV pressure elevation.
A major difference between humans and mice with elastin haploinsufficiency is that humans, but not mice, develop severe localized aortic occlusion due to subendothelial SMC proliferation. This may be due to differences in mechanical stimuli such as circumferential stress or strain in the vessel wall, differences in heart rate (~600 beats/min in mouse compared with 60 beats/min in humans), or other differences between the species that are not directly related to the hemodynamics. It is important to note that at their higher physiological pressures, arteries in Eln+/- mice are working close to their maximum strain, suggesting that these animals may be more prone to develop hypertensive cardiovascular pathologies when stressed. At maximal strain, there is a lower potential for distension if the blood pressure increases.
B. Developmental Adaptation Versus Pathological Remodeling
Previous studies have shown that the increase in arterial lamellar number in Eln+/- animals is established at the time of birth (163), suggesting that alterations in vessel wall structure and mechanics occur early in formation of the arterial wall. To better determine when in development the cardiovascular changes take place, we characterized blood pressure, cardiac function, vessel structure, and vessel mechanics in newborn Eln+/- mice (291). As in previous studies with adult animals, we found newborn Eln+/- vessels to have smaller unloaded diameters and longer lengths between branches. Additionally, there is a 25% increase in left ventricular pressure in newborn Eln+/- animals compared with WT, suggesting the existence of a pressure difference during the embryonic period.
Ultrastructural studies show that the change in lamellar number occurs between E16 and birth and that the extra layers are associated with a decrease in adventitial area, indicating that adventitial cells give rise to the additional SMC layers (291). This is interesting in light of numerous studies showing that the adventitia is important in sensing wall stress and that adventitial cells can be activated in response to hypertension or other forms of vascular injury (263). During development, the Eln+/- aorta experiences more stretch with each cardiac cycle than the WT aorta because of its increased pressure, smaller inner diameter, and only slightly thicker wall. The high circumferential stretch could trigger cells in the adventitia to differentiate into SMCs that produce extra layers of elastin and consequently normalize the tension/lamellar unit in the presence of increased pressure. Recent findings have shown that the adventitia contains a progenitor cell population capable of differentiating into SMCs that repopulate the wall (116, 210, 279). It will be interesting to determine if the additional SMC layers in the Eln+/- aorta are derived from this cell population.
The smaller inner diameter of the large arteries in Eln+/- mice coupled with increased arterial stiffness and elevated cardiac output should lead to cardiac hypertrophy, circulatory dysfunction, and possibly death. It is therefore somewhat of a paradox that the Eln+/- animals have a normal life span, exhibit no overt signs of degenerative cardiovascular disease, and show none of the adverse effects observed with other animal models of induced or spontaneous hypertension (17, 94). One explanation for the normal characteristics of the Eln+/- mouse is that the elevated blood pressure is a necessary adaptive response for maintaining cardiovascular function. Figure 11 shows that at the higher systemic physiological pressure, the effective working inner diameter of the Eln+/- ascending aorta is comparable to that of the WT animal. Because the functional demands of the organism require that normal perfusion be preserved, increasing arterial inner diameter through an increase in blood pressure is likely a necessary adaptation to maintain cardiac output and perfusion. Remarkably, despite having half the amount of elastin as WT vessels, the stress-strain relationship of the adult Eln+/- aorta is similar to the WT relationship shown in Figure 1 (290). This provides evidence for adaptation of the ECM component amounts and organization during development to produce a vessel wall with optimized mechanical properties.
FIG. 11.
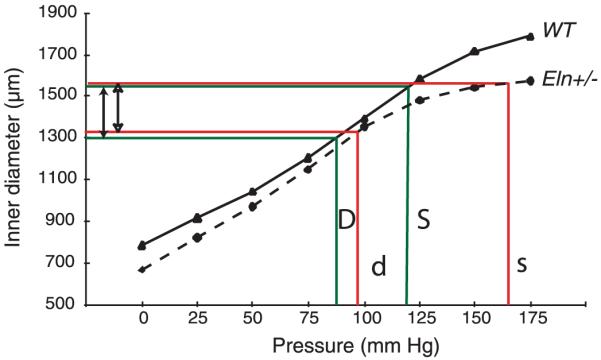
Vessel inner diameters are similar at physiological blood pressures of WT and Eln+/- genotypes. Comparison of ascending aorta inner diameters of adult WT and Eln+/- mice at their respective physiological pressures shows that the vessel in Eln+/- animals is smaller at every intravascular pressure. Because the basal blood pressure in the Eln+/- genotype is higher, however, its effective working diameter (open arrows) is comparable to that of the WT animal (solid arrows). Green lines show the working diameter of the WT artery at its physiological systolic (S) and diastolic (D) pressures. Red lines show that the systolic (s) and diastolic (d) dimensions of the Eln+/- artery at its higher pressures are similar to the WT values.
Despite the similar stress-strain relationship, Eln+/- arteries operate at higher physiological stresses and strains than WT because of the increased physiological pressure. As discussed above, when adult vessels are exposed to high pressure, such as in adult-onset hypertension, arterial geometry is subsequently remodeled to increase the wall thickness and reduce the circumferential wall stress back to normal values. Vessels also produce a scarlike ECM (mostly collagen) that leads to stiffer vessels, further increases in blood pressure, and, eventually, cardiac hypertrophy. Contrast those changes to vessel wall changes in the Eln+/- mouse that has extremely high blood pressure. In these animals, there is no significant increase in collagen or elastin concentration in the vessel wall, cardiac hypertrophy is minimal, life span is not appreciably compromised, vessel walls are thinner not thicker, and the thickness-to-lumen ratio is approximately the same as in WT vessels.
These different responses to increased blood pressure reflect what vascular cells are appropriately programmed to do at two different stages of vascular development. Changes in Eln+/- vessels occur during the fetal period when mechanical stress associated with changing blood pressure provides normal and required informational signals to help guide SMCs in building a mechanically appropriate ECM. This is a period when the proliferative and synthetic properties of SMCs are highest and the cardiovascular system is able to adapt to working at different physiological set points. During the adult period, however, SMCs enter a state where ECM synthesis is minimal, they undergo little, if any, proliferation, and are programmed to recognize increased hemodynamic stress as an injury and initiate a wound response characterized by deposition of a scarlike ECM. The uniqueness of these two phases is superbly illustrated by vascular changes in Eln+/- mice before and after renal artery clipping. When exposed to high pressure during the embryonic/fetal period, the cardiovascular system of the Eln+/- mouse undergoes adaptive remodeling (described above) to work at the higher pressure. When the blood pressure is raised even higher as adults through renal artery clipping, the vessels show pathological remodeling characterized by thicker, stiffer, collagen-rich vessels that lead to cardiac hypertrophy (289).
Despite the changes in vessel wall structure and ECM composition, the Eln+/- incremental elastic modulus at mean physiological pressure remains in the range observed for numerous invertebrate and vertebrate animals (Fig. 9) (253). The unique cardiovascular remodeling seen in elastin-insufficient mice indicates that the developing vascular cell can adapt its building process to accommodate environmental changes and produce a different overall wall structure that operates at the desired physiological setpoint. However, this can only be done during a short period of development when the matrix is being formed and appropriate gene sets are being expressed in a spatially and temporally appropriate manner.
C. Eln-/- Mice: The Absence of the Windkessel Effect
Mice with a null mutation in the elastin gene (Eln-/-) die within a few days of birth due to SMC overproliferation that eventually occludes the vessel lumen (162). Overproliferation begins around E17.5, and all Eln-/- mice are dead by P4.5. Because these animals are viable through fetal development when many of the critical cardiovascular structural and hemodynamic changes are occurring, they provide a glimpse of how a closed circulatory system develops when vertebrate vessels cannot provide the elastic recoil (i.e., windkessel effect) required for normal heart function.
At birth, mice with no elastin (Eln-/-) have tortuous, stenotic, stiff vessels that show little diameter change between systole and diastole (291). As a consequence, the animals have elevated left ventricular pressures and develop larger hearts with impaired function characterized by reduced ejection fraction and cardiac output. Without elastic vessels to act as an elastic reservoir, left ventricular pressure increases in an attempt to maintain cardiac output and perfusion pressure through the smaller, stiffer aorta. The combination of high blood pressure and increased afterload is most likely responsible for the observed cardiac hypertrophy. While hypertrophic cardiac remodeling is expected with noncompliant, stenotic vessels, it is remarkable that cardiac development proceeds to birth with vessels as stiff and morphologically abnormal as those seen in Eln-/- mice. If we look earlier in development, we find that most of the adverse changes in heart function and vessel structure occur during the last 3-4 days of gestation. At E15.5, the aortae in Eln-/- and WT animals look similar by histology, and both have about eight circumferential cell layers (162). There is little elastin or collagen in the wall at this stage (146, 183), so the wall stresses are borne mostly by the cells. Because wall structure is similar in the two genotypes, hemodynamic forces are most likely similar as well. As shown in Figure 4, there is a dramatic increase in elastin and collagen expression beginning around E14, which suggests that the wall is strengthening in response to increased hemodynamic stimuli. When elastin cannot be produced at appropriate levels to accommodate increased stress, as in the Eln-/- mouse, SMCs on the luminal side of the wall proliferate to lower wall stresses by decreasing the inner diameter and increasing wall thickness. By doing so, the physiological circumferential stresses in the thicker Eln-/- aorta at P1 are almost equal to WT, despite the fact that left ventricular pressure is almost twofold higher in the knockout animal. For reasons that are not yet understood, SMC proliferation continues in Eln-/- aorta until the lumen becomes occluded.
Overproliferation occurs in all arteries of the Eln-/- mouse and was demonstrated in aortic sections grown ex vivo. These results suggest that elastin participates in regulating the proliferation and organization of SMCs during development (162). Further studies with isolated SMCs from Eln-/- aorta show that elastin inhibits proliferation, induces a contractile phenotype, controls migration, and signals through a G protein-coupled signaling pathway (106, 141, 142, 185). In its polymerized form, elastin might also create a physical barrier and/or landmark that serves to spatially compartmentalize SMCs in the aortic wall. Throughout the last trimester of mouse development, vascular SMCs produce abundant amounts of elastin and collagen such that by P1, the elastic lamellae are almost continuous and each SMC is in contact with the lamella above and below it in the wall. In a sense, the SMCs are spatially “trapped” between the two continuous lamellar structures (48, 146, 183). The loss of this physical restraint in vessels without elastin may contribute to the changes in SMC organization and phenotype observed in the elastin-null mouse. In in vitro assays, mechanical strain induces proliferation in SMCs plated on collagen, fibronectin, and vitronectin, but not on elastin or laminin, showing that ECM-specific signals and mechanical strain and/or stress may work coordinately to alter SMC phenotype (299).
The elastic recoil imparted by the extracellular elastic fibers is a required property for all large conducting vessels in higher vertebrates. Without elastin, the organism does not survive. With only half as much elastin (at least in mice), the organism adapts. It is this adaptation that offers a powerful tool to begin understanding how the developing vascular cell recognizes and calibrates tensional forces, and how mechanically induced signals translate into gene regulation and vessel wall formation.
VIII. VASCULAR MECHANICS
Building or adjusting the vessel wall structure to withstand applied forces requires SMCs to recognize and translate a variety of mechanical signals. These mechanical signals include forces and deformations from blood flow and pressure, as well as those applied by surrounding tissue and during vessel growth. Stresses can be calculated from the forces to account for differences in vessel geometry and make comparisons between different vessels and across species. Stress is the force applied to an object divided by the surface area over which that force is applied. The stresses imparted on the vessel wall include shear stresses from the flow of blood across the vessel lumen, longitudinal stresses from surrounding tissue, and circumferential stresses from the blood pressure. The relationships between the vessel dimensions, wall stresses, and physiological parameters are shown in Figure 12. Additionally, residual stresses develop during formation of the vessel wall from nonuniform growth. The forces that generate stresses also induce deformations in the vessel wall that can be converted into strains. Strain relates the deformed dimensions of an object to the undeformed dimensions and quantifies deformation regardless of the original geometry. For materials like rubber or steel, stress is linearly related to the measured strain and can be calculated from the Young’s elastic modulus or stiffness. For materials like the vessel wall, the relationship between stress and strain is nonlinear, and empirically derived equations are necessary.
FIG. 12.

Relationships between the vessel wall dimensions, stresses, and physiological parameters. The dimensions include inner radius (ri), length (l), and thickness (t). The stresses include wall shear stress (τw), longitudinal stress (σz), and average circumferential stress (σθ). The physiological parameters include volumetric blood flow (Q), longitudinal tethering forces [F(l)], and blood pressure (P). The viscosity of blood (μ) is also included in the wall shear stress equation. The equations show how changes in the vessel wall dimensions and/or physiological parameters can alter the wall stresses.
In simple geometries, such as a cylindrical blood vessel, the stresses and strains can be calculated from in vitro experiments where the pressure, longitudinal force, and unloaded and loaded dimensions are measured. These calculations require several assumptions, including incompressibility and uniform strain across the vessel wall. Incompressibility requires that the total wall volume remains constant during deformation and has been shown to be a reasonable assumption for most blood vessels (66, 121). As the inner and outer diameters expand with increased pressure, the wall thins radially, maintaining a constant volume. Assuming incompressibility allows the deformed inner diameter to be calculated from the undeformed dimensions and the deformed outer diameter and length. This is necessary because it is often difficult to detect the inner diameter during in vitro testing of large, thick-walled vessels. Uniform strain, however, is not a valid assumption if it is based solely on the boundary conditions at the inner and outer wall. The inner wall is exposed to physiological blood pressure, while the outer wall is exposed to atmospheric pressure, leading to a strain gradient across the wall. The calculated strain differential is normalized by the circumferential residual strain, which consists of compression in the inner wall and tension in the outer wall (167). Residual strain can be measured by cutting open a vessel section and measuring the “opening angle” formed between the two ends and the midpoint of the cut section. Residual strain can be caused by differential growth at the outer and inner wall boundaries and serves to normalize the strains experienced by cells throughout the wall thickness (77, 269). Like strain, circumferential stress will vary with position in the wall, but the residual strain causes a residual stress, which normalizes the transmural wall stress (77, 218). Therefore, the average circumferential stress is a good estimate for the stress at most points in the vessel wall.
The circumferential stress-strain relationship in blood vessels varies with longitudinal stress or strain. For physiologically relevant relationships, it is important to test the vessel at its in vivo length. In vivo longitudinal strain varies from 10 to 70% depending on location in the arterial tree, with distal vessels generally being more stretched than proximal vessels (95). The proximal ascending aorta has an in vivo strain starting at ~10%, but this increases to 15-20% at points in the cardiac cycle due to motion of the heart (55). In vivo longitudinal strain manifests during development as the overall length or height of the animal increases more than the length of the blood vessels. Because the circumferential stress-strain relationship depends on the longitudinal strain, controlling the vessel length may be another way to regulate the circumferential stress experienced by SMCs (60).
A. Developmental Growth and Remodeling
All of the parameters discussed above (circumferential and longitudinal stress and strain, shear stress, and residual stress and strain) can be important stimuli for growth and remodeling in arteries. It is difficult to determine which parameter is directly stimulating cellular responses because many of the parameters are correlated. For example, increased strain will cause increased stress and vice versa. Rates of change of these parameters may also be important stimuli. Thoma first observed the relationships between mechanical stimuli and vessel form and function in 1893. Thoma’s conclusions about the developing vasculature in the chick embryo include the following: 1) vessel lumen size depends on blood flow, 2) vessel length depends on longitudinal force exerted by connective tissues, and 3) vessel wall thickness depends on blood pressure (36). Another way to state Thoma’s conclusions is that the geometric properties of the vessel are optimized for the applied forces and serve to normalize the mechanical stresses. As shown by the equations in Figure 12, the relationship between vessel lumen and blood flow provides constant shear stress, the relationship between vessel length and longitudinal force provides constant longitudinal stress, and the relationship between wall thickness and blood pressure provides constant circumferential wall stress. More recent studies have followed up on Thoma’s original observations both in developing and adult animals.
In 1918, Clark investigated Thoma’s laws by observing the growth of blood vessels in the tail of frog larvae (36). He found support for Thoma’s first law, although he argued that the vessel diameter was dependent on blood volume, not blood flow. It must be noted that his measurements of blood volume and flow rate were purely qualitative and not quantitative. Also, the shear stress on the artery wall depends on the volumetric flow rate and so depends on both factors. Clark observed that capillaries that showed a decrease in blood volume would decrease in diameter and eventually disappear, while capillaries that showed an increase in blood volume would increase in diameter and eventually become arterioles or venules. In mammals, abrupt changes in blood flow occur soon after birth due to redistribution of blood between the placental and adult circulation. Before birth in sheep, the thoracic and abdominal aortas experience similar volumetric blood flows and have similar diameters. After birth, the loss of placental circulation reduces the abdominal aortic blood flow by more than 90%, while flow is maintained in the thoracic aorta. Consistent with Thoma’s observations, the abdominal aorta shows a marked decrease in diameter, while the diameter of the thoracic aorta increases slightly (154).
Clark also found support for Thoma’s second law that the length of blood vessels depends on the tension exerted by surrounding tissue (36). He found that the increase in capillary length in each direction was the same as the growth of the frog larvae tail in each direction. Berry et al. (15) also showed a linear relationship between vessel length and crown-rump length in developing rats.
Several investigators have addressed Thoma’s assertion that vessel wall thickness depends on blood pressure. Gerrity and Cliff (81) found that blood pressure and medial wall thickness increase simultaneously in developing rats. In mammals, large changes in pressure occur soon after birth with closure of the ductus arteriosus and the separation of the pulmonary and arterial vascular systems. After birth, the aorta and pulmonary artery carry similar volumetric blood flows, but experience very different blood pressures. Consistent with Thoma’s observations, the diameters of these two arteries in rabbits remain similar, but the wall thickness increases in the aorta and decreases in the pulmonary artery to accommodate the different pressure in each vessel (161).
Wolinsky and Glagov (304) extended Thoma’s third law to hypothesize that the number of lamellar units in the artery depends on wall tension. Wall tension is the blood pressure times the inner radius and is similar to wall stress, but is not normalized by the wall thickness. They found that the tension/lamellar unit was 1-3 MPa in 10 mammalian species ranging from mice to sheep. The lamellar units are formed during the later half of embryonic development, and the number of units does not change after birth (40, 211), even in the presence of increased hemodynamic stress (302). Wolinsky and Glagov’s observations make the increased lamellar units in Eln+/- aorta even more remarkable. However, these mice do not break the rule of constant tension/lamellar unit. Because both the pressure and lamellar units are increased in Eln+/- aorta, the tension/lamellar unit is similar to WT mice and to other mammalian species.
Gibbons and Shadwick (83) calculated the tension/lamellar unit in nonmammalian species and found that it was lower than mammals and in the range of 0.3-0.5 MPa. They suggest that the lamellar unit is structurally and mechanically different in lower vertebrates than in mammals. As demonstrated by the lobster aorta in Figure 7, the vessel wall in lower vertebrates and some invertebrates has a lamellar organization, but the lamellar structure itself is made up of fibrillin-containing microfibrils lacking elastin (50). Gibbons and Shadwick (83) argue that the physiological elastic modulus is a more important evolutionary parameter than tension/lamellar unit. Shadwick found that the physiological elastic modulus is ~0.4 MPa in animals varying from squid to snake to rat (253) and concluded that all species are designed to be relatively compliant at physiological pressure. The physiological modulus of human thoracic aorta is 0.6-1.0 MPa (157), WT mouse aorta is 0.4 MPa, and Eln+/- mouse aorta is 0.9 MPa (Fig. 9) (290). While the Eln+/- modulus is not completely normalized to WT values, it is within the same range, and it is possible that maintaining this modulus within the narrow range found in all vertebrate and invertebrate species is the most important variable controlling remodeling in the Eln+/- aorta.
B. Growth and Remodeling in Adult Vessels
Thoma’s laws apply to remodeling of adult arteries as well. When flow is altered, the first step is for SMCs to contract or relax to change the diameter of the artery wall. This may occur through signaling from endothelial cells at the artery lumen that sense changes in shear stress (52, 201, 204). Over the long term, SMCs remodel themselves and the ECM to increase or decrease the artery diameter to accommodate the new volumetric flow rate and maintain a constant shear stress. In dog carotid arteries, for example, the internal diameter changes to normalize shear stresses to within 15% of control values after 6-8 mo of altered flow (140). In rat arteries, diameter increases in response to increased flow are caused by growth at both the inner and outer wall, with slightly more growth at the outer wall, which leads to normalization of the shear stresses and a decrease in the circumferential residual strain (172). The growth occurs through increases in elastin and collagen content and SMC hypertrophy (11). In rabbits, as aortic diameter increases with increased flow, the internal elastic lamina is remodeled and develops large gaps that are subsequently filled with endothelial cells (177). Rabbit arteries also show reduced diameter with decreased flow to normalize reduced shear stress, and the change is specific to mean flow velocity, not flow amplitude (155). In this study, weanling rabbits had a larger diameter change than adult rabbits, and the remodeling was manifested by different methods, providing evidence that developing animals adapt in different ways than adult animals.
Longitudinal stretch applied to arteries in vivo causes growth in the longitudinal direction to normalize the longitudinal stress. When rabbit carotid arteries were stretched 22% over their original stretch ratio, the arteries grew longitudinally until the resultant strain was completely normalized within 7 days. The longitudinal growth resulted from increases in endothelial cell and SMC replication rates and increases in total DNA, elastin, and collagen contents (135). Porcine carotid arteries stretched in vitro at least 43% over their in vivo strecth ratios for 7 days also show longitudinal growth due to SMC proliferation in an attempt to normalize the longitudinal stress (49). Growth in the longitudinal direction after an increase in stress or strain is much faster (on the order of days) than growth in the circumferential direction (on the order of months) after an equivalent stimulus. This implies that maintaining homeostatic values of longitudinal stress and/or strain is critical for proper vessel function. Also, because increased longitudinal strain affects the circumferential stress-strain relationship, two stresses are normalized by growth in the longitudinal direction (123).
Curiously, a decrease in longitudinal strain is not normalized. When an artery is decreased in length, it does not shorten to normalize the longitudinal stress and strain, but actually lengthens, exacerbating the problem and leading to arterial tortuosity. The lengthening is accomplished in a similar manner (increased cell proliferation rates, increased elastin and collagen amounts) to the lengthening that follows an increase in longitudinal strain (134, 135). This behavior demonstrates that, unlike shear stress where arteries can adapt to increased or decreased stress, arteries can only adapt to increased longitudinal stress and/or strain.
Increased pressure stretches SMCs in the artery wall and increases the circumferential stress experienced by the cells. In adult animals, this increased pressure is directly related to changes in arterial wall thickness that serve to normalize the circumferential stress. In vitro, isolated SMCs respond to stretch by stimulating signaling pathways such as the small GTPase Rac (144), protein kinase A, protein kinase C (192), and p38 mitogen-activated protein kinase (31, 165) and by upregulating collagen (59), elastin (265), and proteoglycan synthesis (159). In vivo, these signaling and synthetic SMC responses translate into geometric and morphological changes in the artery wall that normalize the circumferential stresses. Like shear stress, but unlike longitudinal stress, increased or decreased circumferential stress can be normalized. Arterial wall thickness is decreased in response to a step decrease in blood pressure in rats, with the medial layer showing a greater decrease than the adventitial layer (166). In response to a step increase in mean blood pressure of 67%, the carotid artery wall thickness in rats increases ~7% in 2 days and 57% over 60 days. This is accomplished through increased collagen, elastin, and SMC content and by thickening the individual lamellar units (76). Liu and Fung (168) found that the change in thickness for an equal step change in pressure was identical in the rat ileac and splenic arteries and that the lumen diameter did not change with pressure in either artery. Increasing the thickness normalizes the circumferential stress after the pressure increase and maintaining the diameter keeps the circumferential shear constant because flow is unchanged. Wolinsky (302) increased pressure in adult rats and found that the medial wall thickness increased ~63% after a 67% increase in systolic blood pressure for 18 wk. This was accomplished through increases in total elastin and collagen content. The circumferential stress was still ~60% higher in the hypertensive compared with normotensive rats, but without the increased thickness, it would have been more than double the normotensive value. Arterial remodeling following increases in shear and longitudinal stresses seems to be able to return those stresses close to the normotensive values, but remodeling for circumferential stress may be incomplete, at least in adult animals.
It is important to remember the differences between adult and developmental remodeling. During development, the stresses influence the structure and patterning of the arterial system. Vessel remodeling can be viewed as advantageous and optimized for the ultimate stresses experienced by the adult animal. SMCs are expressing numerous ECM molecules and can adapt the ECM mixture to accommodate developmental hemodynamic changes. In adulthood, SMCs are no longer producing all the ECM molecules that were necessary during development to build the optimal vessel wall. When adult stresses are altered through changes in blood flow, longitudinal strain, or blood pressure, SMCs undergo what often becomes pathological remodeling. The cells remodel the vessel wall to change the inner diameter, length, or thickness and bring stresses back to physiological levels, but this is done by changing the ECM content of the wall. Collagen is the ECM molecule that contributes to most of the geometric changes in the vessel wall. Collagen has a higher incremental elastic modulus than elastin, and more collagen increases the vessel stiffness. This increases the load on the heart and can start a negative-feedback cycle that ultimately leads to heart failure.
IX. MECHANICAL MODELS
The unique, two-phase composition of the vessel wall provides nonlinear stress-strain relationships (Fig. 1). At low strains, the incremental elastic modulus of elastin dominates the mechanical behavior and the wall is relatively extensible. At high strains, the elastic modulus of collagen dominates and the wall is relatively inextensible. The overlap at physiological pressure provides optimal behavior for expansion and contraction of the vessel wall during the cardiac cycle and limits distension of the wall when exposed to extreme pressures. The nonlinear behavior also complicates mathematical descriptions of the stress-strain relationship. The stress at a given strain cannot be calculated from a simple constant and a linear equation, but must be calculated from a set of constants and an exponential, logarithmic or polynomial equation. Mathematical descriptions are necessary to extrapolate experimental results to more relevant in vivo loading conditions, as it is not possible to duplicate the complete range of in vivo loading conditions in the laboratory. Mathematical descriptions are necessary to compare stress-strain relationships between different animal species, genotypes, and disease states. Also, if the stress-strain relationship is known, then strains can be measured noninvasively with various imaging techniques, and the stresses can be calculated. Calculated stresses can be used to predict vessel wall stability (i.e., has the vessel wall reached a homeostatic state, will it continue to remodel, or will it fail?) and determine when treatment is necessary. For example, it has been shown that the stress-strain relationship changes with age in dilated human ascending aorta. The same strains in the vessel wall of an older patient will cause higher stresses and may increase the chance of dissection and rupture (202).
Mathematical descriptions of the stress-strain relationship are called constitutive equations, and the constants in the equations are called material properties. Early constitutive equations were phenomenological; that is, the equations fit the stress-strain data for intact arteries, but were not directly related to the vessel microstructure. Most of these models assume that the artery wall can be modeled as a continuum material. Phenomenological models allow quantification of changes in the vessel wall material properties, but do not account for the changes with any physical explanation, such as changes in the ECM composition. More recent models have been developed that are based on the vessel microstructure, namely, the arrangement of elastin, collagen, and SMCs. These models assign different mechanical behavior to each component with constraints on how they deform relative to each other and to the composite artery. By relating the mechanics to the microstructure, specific interventions can be designed and tested. For example, the drug ramipril decreases collagen and increases elastin and fibrillin-1 production in SMC cultures (2). These changes in ECM composition alter the material properties of the composite artery and lead to improved cardiovascular outcome in patients with peripheral artery disease that are treated with ramipril.
Both phenomenological and microstructural models have been adapted for growth and development and can be used to predict the effects of increasing forces on the artery wall. These models are critical because many disease states (i.e., hypertension and atherosclerosis) involve changes in the static and hemodynamic forces and consequent stresses on the artery wall. Using models to better understand and predict the remodeling process will help us design strategies to reverse it in disease and to reproduce it in tissue engineering. The application of stresses and strains during the culture of artificial blood vessels has been shown to significantly improve mechanical properties through optimization of the ECM component amounts and organization (130, 198, 249). Most models of growth and development have been applied to remodeling of adult arteries because there is considerable data available to validate the model predictions. There is a pressing need to extend these models to developmental remodeling because the process is fundamentally different in development and adulthood. Developmental remodeling is more applicable than adult remodeling to the design of tissue-engineered blood vessels because embryonic or neonatal cells are often used and the initial matrices are either mechanically weak or degradable and are designed to be significantly remodeled by the cells and supplemented by new cell-produced matrices. It has been shown that incremental increases in the cyclic strain applied to fibrin-based tissue constructs results in increased collagen production per cell and increased stiffness and tensile strength compared with constant cyclic strain conditioning (266). The incremental increases are similar to what may be experienced in vivo during development as the pressures and flows increase faster than the diameter and thickness of the vessel wall.
A. Continuum Models
Vito and Dixon (286) reviewed a wide variety of blood vessel constitutive equations, a few of which will be briefly summarized here. Although blood vessels are viscoelastic, for modeling purposes they are usually considered to be pseudoelastic, which means that the loading and unloading behavior is treated separately and models are applied to the loading behavior after suitable preconditioning cycles (78). The vessels are considered to be nonlinear, anisotropic (different properties in different directions), and incompressible. Some models include the circumferential residual stress (35, 178, 284). Most continuum models define a strain energy density function (W), which requires that the stress depends only on the current strain state and not on the path between initial and final strain states. Strain energy density functions relate the Green strain tensor (which is a function of the deformation tensor) to the second Piola-Kirchoff stress or to the Cauchy stress (which are two different forms of the stress tensor). While arteries are anisotropic, they show some material symmetry and are usually considered transversely isotropic (symmetric about one plane of isotropy, such as about cells and fibers arranged in the circumferential direction) or orthotropic (different properties in different orthogonal directions, such as circumferential, longitudinal, and radial in a cylindrical artery). The functional form of W for these two types of materials is well known (286).
Continuum models vary mostly in the specific forms chosen for W and the number of constants included in each equation. A good model will fit the stress-strain behavior of the vessel, provide unique material constants, and give realistic predictions for all possible physiological deformation states. Seven-parameter polynomial (285), four-parameter logarithmic (271), and four- or seven-parameter exponential (34) equations can all reasonably describe the stress-strain behavior of mammalian arteries. Increased numbers of parameters improve the fit of the equation to the experimental data but have a tendency to become over-parameterized and produce nonunique material parameters. Fung et al. (78) compared their four-parameter exponential to the seven-parameter polynomial of Vaishnav et al. (285) and found that their exponential equation provided a better fit to the stress-strain data and more stable material constants for rabbit arteries than the polynomial equation. Humphrey (120) compared the predictive capability of three commonly used forms for W including the four- and seven-parameter exponential and the logarithmic equations. He found that the four-parameter exponential and the logarithmic equations have limited capability to describe the anisotropic behavior observed in mammalian carotid arteries and that the logarithmic equation yields physically unrealistic predictions of the deformation tensor within the experimental range. Humphrey (120) concluded that the seven-parameter exponential equation is the best available form for W to describe the stress-strain behavior of elastic arteries. Many investigators still use the four-parameter exponential equation despite its limited anisotropy because it significantly reduces the number of parameters to fit and eliminates the tendency for overparameterization with the seven-parameter exponential equation. Many of the microstructural models discussed below use one of these forms of W to describe the behavior of individual arterial components.
B. Microstructural Models
Microstructural models incorporate the composition of the vessel wall into the constitutive relationships. These models are beneficial because changes in mechanical behavior can be directly related to measurable changes in the amount or organization of the wall components. Recognizing the different microstructure of the media and adventitia, the earliest attempts at relating mechanical behavior to wall structure used separate forms of W for each wall layer (287). Knowing that elastin and collagen have different mechanical properties, other investigators used separate forms of W for the contribution of each protein, with elastin behaving like a linear, isotropic material and collagen behaving like a nonlinear, anisotropic material (109). Some investigators have incorporated the fibrous nature of ECM proteins into their mechanical models. Wuyts et al. (305) developed a model where collagen was represented by a distribution of wavy, linearly elastic fibers and elastin and SMCs were considered interconnected sheets of linearly elastic material. The collagen shows nonlinear behavior as more fibers are recruited from the wavy to the stretched state and begin to contribute to the total tissue stress. Zulliger and Stergiopulos (314) expanded the model of Wuyts et al. (305) to include longitudinal stress, residual strain, and local wall stress. They used it to predict mechanical changes in aging arteries based solely on measurable alterations in the waviness of the collagen fibers. Rachev and Hayashi (219) used a logarithmic strain energy density function for the arterial wall and added an active stress component due to SMC activation. They used the model to predict changes in residual strain with SMC tone and hypertension. Combining many of the above studies, Humphrey and Rajagopal (122) presented a constrained mixture model, which includes elastin with a linear stress response function, collagen with an exponential form of W, and SMCs with active and passive stress components. They also allowed for different natural configurations, which translates to different residual strains, for each component. Their model was applied to growth and remodeling of arteries in response to increased flow. All of these models can predict, to some degree, changes in the vessel wall mechanical behavior based on changes in the wall components, and vice versa. Describing and predicting the structure-mechanical function relationship of the vessel wall will help to better design treatment strategies for human diseases and to recreate the ideal vessel wall in tissue engineering.
C. Growth and Remodeling
In 1892, Wolff (300) proposed that trabecular bone grows in orientations that correspond to the principal mechanical stresses acting on the bone and that mathematical laws can explain the growth patterns. Wolff’s law, as it has come to be known, is a formalization of the observations that Thoma had on blood vessel growth a year later (36). These ideas have been generalized to explain growth of any biologic tissue in response to applied stresses or strains (115, 258). Several mechanical models have been developed specifically to describe and predict this behavior. Rodriguez et al. (230) introduced a continuum model for finite growth in soft tissues that depends on the tissue stress and leads to natural residual stresses as each differential element grows. The residual stresses are consistent with those observed by the opening angle in blood vessels. To describe stress-dependent growth, the growth law must relate the growth deformation tensor (or growth rate deformation tensor) to the stress tensor. This requires a constitutive equation relating the growth-induced strains to the stresses. For blood vessels, investigators often use one of the forms of W discussed above.
Taber and Eggers (268) applied the theory of Rodriguez et al. (230) to describe growth of the rat aorta from the first heartbeat through maturity and during experimentally induced hypertension. The authors modeled the aorta with a linear, local stress-dependent growth law with no predefined equilibrium stress. The aorta has two layers (media and adventitia) that may have different growth constants, but follow the same constitutive equation with an exponential form for W. Longitudinal stress from surrounding tissue and shear stress from blood flow were neglected, but changes in blood pressure through development or hypertension were included. The authors were able to reproduce most of the geometric changes in the artery wall and in the residual strain observed in developing and hypertensive arteries. Rachev (217) also used a two-layer model to predict arterial growth in hypertension. He used an exponential form of W for the constitutive equation and allowed each layer to have different material coefficients and different residual stresses. He restricted the inner radius of the artery to remain unchanged so that shear stresses were not altered and assumed that the arterial geometry and residual stress configuration are remodeled in a way to maintain stress and strain configurations that are identical under normo- and hypertensive conditions. His model was also able to recreate experimental changes in geometry and residual strain observed for hypertensive arteries. Fridez et al. (75) expanded Rachev’s model (217) to include active SMC contributions to the remodeling process.
Microstructural models of growth and remodeling extend the above ideas to account for adaptation and growth of individual arterial wall components. Tsamis and Stergiopulos altered models of Rachev et al. (217, 220, 221) by replacing the continuum based constitutive equation with the microstructural one introduced by Zulliger and Stergiopulos (314). They predict hypertensive remodeling based on growth-induced changes in the collagen waviness profile. Humphrey and Rajagopal’s (122) constrained mixture model was developed specifically to include growth of individual wall components by including the turnover kinetics for SMCs, elastin, and collagen. This model has been applied to flow- (91) and pressure-induced changes (90) in carotid arteries and the effects of spatial variations in wall constituents (4). Most of these models are designed to apply to growth and remodeling in adulthood and development, but have been verified only with adult data because there are little data for the developmental period. To extend the models to developing vessels, additional data are needed on the hemodynamics, mechanics, geometry, ultrastructure, and component amounts of arteries at various developmental stages. The data will help verify current models and determine necessary changes to describe the unique process of growth and remodeling in developing vessels. In genetic diseases, such as SVAS and Marfan’s syndrome, the ECM composition is altered embryonically. This induces changes in the vessel mechanical properties, the blood flow, and pressure and consequently the stresses and strains in the vessel wall. Being able to predict these changes and design early intervention strategies to compensate for the changes in ECM composition or to reverse some of the mechanical effects may significantly improve the life expectancy in these patients.
X. CONCLUSIONS
We have focused on the unique composition of mammalian elastic arteries and the interplay between mechanical signals, SMC behavior, ECM components, and vessel wall structure during remodeling and development. The artery wall is designed for optimal performance given the mechanical demands of the closed, high pressure, pulsatile, and vertebrate cardiovascular system. When these demands are changed, either through genetic alterations or adult-onset disease, the vessel wall adjusts to meet the new demands and to normalize the stresses experienced by the SMCs. For genetic alterations, the mechanical demands are changed during development and the embryonic vascular cells are able to sense and adapt to the changing stimuli. For adult-onset diseases, the mechanical demands are placed on differentiated SMCs that are no longer capable of producing the optimal ECM component mix to remodel the vessel wall. Hence, the wall is remodeled to shield SMCs from increased stress, but the remodeling is often detrimental to heart function and starts a negative-feedback cycle that progressively worsens the disease. Embryonic vascular cells are only able to remodel advantageously if the mechanical changes are below a certain threshold. In Eln+/- mice, this remodeling leads to an unexpected vessel wall structure and a completely normal life span, despite hypertension that would usually cause cardiac hypertrophy and eventual cardiac failure. In humans with elastin insufficiency, the mechanical and morphological changes are similar to Eln+/- mice, but humans still develop vascular disease characterized by focal aortic stenosis. In Eln-/- mice, the mechanical changes include the elimination of vessel elasticity, and the animal cannot survive beyond birth. By characterizing the process of embryonic adaptation including the mechanical stimuli, genetic signaling pathways, ECM production, and SMC differentiation, we will be able to design strategies to encourage advantageous remodeling in developmental diseases and to recreate this process in tissue engineering of artificial blood vessels. Developing models that describe and predict the developmental remodeling process will assess the efficacy of intervention strategies before their application in clinical practice.
ACKNOWLEDGMENTS
We thank Gilles Faury, Sean McLean Adrian Shifren, Russ Knutsen, and Christopher Ciliberto for their contribution to data used in this report. We also thank Sean McLean for providing the photomicrograph in Figure 2 and Brigham Mecham for assistance with the gene array analysis. We acknowledge a long-standing collaboration with Dean Li at the University of Utah who produced the elastin knockout mice.
GRANTS
This review was made possible through research funding from National Heart, Lung, and Blood Institute Grants HL-53325, HL-71960, and HL-74138 (to R. P. Mecham) and HL-87563 (to J. E. Wagenseil) and by a grant from the National Marfan Foundation (to R. P. Mecham).
REFERENCES
- 1.Abrams WR, Ma RI, Kucich U, Bashir MM, Decker S, Tsipouras P, McPherson JD, Wasmuth JJ, Rosenbloom J. Molecular cloning of the microfibrillar protein MFAP3 and assignment to the gene to human chromosome 5q32-q33.2. Genomics. 1995;26:47–54. doi: 10.1016/0888-7543(95)80081-v. [DOI] [PubMed] [Google Scholar]
- 2.Ahimastos AA, Natoli AK, Lawler A, Blombery PA, Kingwell BA. Ramipril reduces large-artery stiffness in peripheral arterial disease and promotes elastogenic remodeling in cell culture. Hypertension. 2005;45:1194–1199. doi: 10.1161/01.HYP.0000168945.44069.aa. [DOI] [PubMed] [Google Scholar]
- 3.Albert EN. Developing elastic tissue. An electron microscopic study. Am J Pathol. 1972;69:89–102. [PMC free article] [PubMed] [Google Scholar]
- 4.Alford PW, Humphrey JD, Taber LA. Growth and remodeling in a thick-walled artery model: effects of spatial variations in wall constituents. Biomech Model Mechanobiol. 2008;7:245–262. doi: 10.1007/s10237-007-0101-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arciniegas E, Neves CY, Candelle D, Parada D. Differential versican isoforms and aggrecan expression in the chicken embryo aorta. Anat Rec A Discov Mol Cell Evol Biol. 2004;279:592–600. doi: 10.1002/ar.a.20042. [DOI] [PubMed] [Google Scholar]
- 6.Argraves WS, Greene LM, Cooley MA, Gallagher WM. Fibulins: physiological and disease perspectives. EMBO Rep. 2003;4:1127–1131. doi: 10.1038/sj.embor.7400033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arteaga-Solis E, Gayraud B, Lee SY, Shum L, Sakai L, Ramirez F. Regulation of limb patterning by extracellular microfibrils. J Cell Biol. 2001;154:275–281. doi: 10.1083/jcb.200105046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Atsawasuwan P, Mochida Y, Katafuchi M, Kaku M, Fong KS, Csiszar K, Yamauchi M. Lysyl oxidase binds transforming growth factor-beta and regulates its signaling via amine oxidase activity. J Biol Chem. 2008;283:34229–34240. doi: 10.1074/jbc.M803142200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bax DV, Bernard SE, Lomas A, Morgan A, Humphries J, Shuttleworth CA, Humphries MJ, Kielty CM. Cell adhesion to fibrillin-1 molecules and microfibrils is mediated by alpha 5 beta 1 and alpha v beta 3 integrins. J Biol Chem. 2003;278:34605–34616. doi: 10.1074/jbc.M303159200. [DOI] [PubMed] [Google Scholar]
- 10.Beck LJ, D’Amore PA. Vascular development: cellular and molecular regulation. FASEB J. 1997;11:365–373. [PubMed] [Google Scholar]
- 11.Ben Driss A, Benessiano J, Poitevin P, Levy BI, Michel JB. Arterial expansive remodeling induced by high flow rates. Am J Physiol Heart Circ Physiol. 1997;272:H851–H858. doi: 10.1152/ajpheart.1997.272.2.H851. [DOI] [PubMed] [Google Scholar]
- 12.Bendeck MP, Keeley FW, Langille BL. Perinatal accumulation of arterial wall constituents: relation to hemodynamic changes at birth. Am J Physiol Heart Circ Physiol. 1994;267:H2268–H2279. doi: 10.1152/ajpheart.1994.267.6.H2268. [DOI] [PubMed] [Google Scholar]
- 13.Bendeck MP, Langille BL. Rapid accumulation of elastin and collagen in the aortas of sheep in the immediate perinatal period. Circ Res. 1991;69:1165–1169. doi: 10.1161/01.res.69.4.1165. [DOI] [PubMed] [Google Scholar]
- 14.Bergwerff M, DeRuiter MC, Poelmann RE, Gittenberger-deGroot AC. Onset of elastogenesis and downregulation of smooth muscle actin as distinguishing phenomena in artery differentiation in the chick embryo. Anat Embryol. 1996;194:545–557. doi: 10.1007/BF00187468. [DOI] [PubMed] [Google Scholar]
- 15.Berry CL, Looker T, Germain J. The growth and development of the rat aorta. I. Morphological aspects. J Anat. 1972;113:1–16. [PMC free article] [PubMed] [Google Scholar]
- 16.Berry CL, Sosa-Melgarejo JA, Greenwald SE. The relationship between wall tension, lamellar thickness, and intercellular junctions in the fetal and adult aorta: its relevance to the pathology of dissecting aneurysm. J Pathol. 1993;169:15–20. doi: 10.1002/path.1711690104. [DOI] [PubMed] [Google Scholar]
- 17.Berry CL, Greenwald SE. Effects of hypertension on the static mechanical properties and chemical composition of the rat aorta. Cardiovasc Res. 1976;10:437–451. doi: 10.1093/cvr/10.4.437. [DOI] [PubMed] [Google Scholar]
- 18.Berry CL, Greenwald SE, Rivett JF. Static mechanical properties of the developing and mature rat aorta. Cardiovasc Res. 1975;9:669–678. doi: 10.1093/cvr/9.5.669. [DOI] [PubMed] [Google Scholar]
- 19.Berry CL, Looker T. Nucleic acid and scleroprotein content of the developing human aorta. J Pathol. 1972;108:265–274. doi: 10.1002/path.1711080402. [DOI] [PubMed] [Google Scholar]
- 20.Bonaldo P, Braghetta P, Zanetti M, Piccolo S, Volpin D, Bressan GM. Collagen VI deficiency induces early onset myopathy in the mouse: an animal model for Bethlem myopathy. Hum Mol Genet. 1998;7:2135–2140. doi: 10.1093/hmg/7.13.2135. [DOI] [PubMed] [Google Scholar]
- 21.Bressan GM, Daga-Gordini D, Colombatti A, Castellani I, Marigo V, Volpin D. Emilin, a component of elastic fibers preferentially located at the elastin-microfibrils interface. J Cell Biol. 1993;121:201–212. doi: 10.1083/jcb.121.1.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bunton TE, Biery NJ, Myers L, Gayraud B, Ramirez F, Dietz HC. Phenotypic alteration of vascular smooth muscle cells precedes elastolysis in a mouse model of Marfan syndrome. Circ Res. 2001;88:37–43. doi: 10.1161/01.res.88.1.37. [DOI] [PubMed] [Google Scholar]
- 23.Burton AC. Relation of structure to function of the tissues of the wall of blood vessels. Physiol Rev. 1954;34:619–642. doi: 10.1152/physrev.1954.34.4.619. [DOI] [PubMed] [Google Scholar]
- 24.Bussiere CT, Wright GM, DeMont ME. The mechanical function and structure of aortic microfibrils in the lobster Homarus americanus. Comp Biochem Physiol A Mol Integr Physiol. 2006;143:417–428. doi: 10.1016/j.cbpa.2005.09.020. [DOI] [PubMed] [Google Scholar]
- 25.Cantor JO, Keller S, Parshley MS, Darnule TV, Darnule AT, Cerreta JM, Turino GM, Mandl I. Synthesis of crosslinked elastin by an endothelial cell culture. Biochem Biophys Res Commun. 1980;95:1381–1386. doi: 10.1016/s0006-291x(80)80050-7. [DOI] [PubMed] [Google Scholar]
- 26.Carnes WH, Abraham PA, Buonassisi V. Biosynthesis of elastin by an endothelial cell culture. Biochem Biophys Res Commun. 1979;90:1393–1399. doi: 10.1016/0006-291x(79)91190-2. [DOI] [PubMed] [Google Scholar]
- 27.Carta L, Pereira L, Arteaga-Soli E, Lee-Arteaga SY, Lenart B, Starcher B, Merkel CA, Sukoyan M, Kerkis A, Hazeki N, Keene DR, Sakai LY, Ramirez F. Fibrillins 1 and 2 perform partially overlapping functions during aortic development. J Biol Chem. 2006;281:8016–8023. doi: 10.1074/jbc.M511599200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Charbonneau NL, Ono RN, Corson GM, Keene DR, Sakai LY. Fine tuning of growth factor signals depends on fibrillin microfibril networks. Birth Defects Res C Embryo Today. 2004;72:37–50. doi: 10.1002/bdrc.20000. [DOI] [PubMed] [Google Scholar]
- 29.Chaudhry SS, Gazzard J, Baldock C, Dixon J, Rock MJ, Skinner GC, Steel KP, Kielty CM, Dixon MJ. Mutation of the gene encoding fibrillin-2 results in syndactyly in mice. Hum Mol Genet. 2001;10:835–843. doi: 10.1093/hmg/10.8.835. [DOI] [PubMed] [Google Scholar]
- 30.Chen E, Larson JD, Ekker SC. Functional analysis of zebrafish microfibril-associated glycoprotein-1 (Magp1) in vivo reveals roles for microfibrils in vascular development and function. Blood. 2006;107:4364–4374. doi: 10.1182/blood-2005-02-0789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen Q, Li W, Quan Z, Sumpio BE. Modulation of vascular smooth muscle cell alignment by cyclic strain is dependent on reactive oxygen species and P38 mitogen-activated protein kinase. J Vasc Surg. 2003;37:660–668. doi: 10.1067/mva.2003.95. [DOI] [PubMed] [Google Scholar]
- 32.Chu ML, Tsuda T. Fibulins in development and heritable disease. Birth Defects Res C Embryo Today. 2004;72:25–36. doi: 10.1002/bdrc.20003. [DOI] [PubMed] [Google Scholar]
- 33.Chung MI, Ming M, Stahl RJ, Chan E, Parkinson J, Keeley FW. Sequences and domain structures of mammalian, avian, amphibian and teleost tropoelastins: clues to the evolutionary history of elastins. Matrix Biol. 2006;25:492–504. doi: 10.1016/j.matbio.2006.08.258. [DOI] [PubMed] [Google Scholar]
- 34.Chuong CJ, Fung YC. Three-dimensional stress distribution in arteries. J Biomech Eng. 1983;105:268–274. doi: 10.1115/1.3138417. [DOI] [PubMed] [Google Scholar]
- 35.Chuong CJ, Fung YC. On residual stresses in arteries. J Biomech Eng. 1986;108:189–192. doi: 10.1115/1.3138600. [DOI] [PubMed] [Google Scholar]
- 36.Clark ER. Studies on the growth of blood-vessels in the tail of the frog larva: by observation and experiment on the living animal. Am J Anat. 1918;23:37–88. [Google Scholar]
- 37.Clark JM, Glagov S. Transmural organization of the arterial media. The lamellar unit revisited. Arteriosclerosis. 1985;5:19–34. doi: 10.1161/01.atv.5.1.19. [DOI] [PubMed] [Google Scholar]
- 38.Cleary EG, Gibson MA. Elastin-associated microfibrils and microfibrillar proteins. Int Rev Connect Tiss Res. 1983;10:97–209. doi: 10.1016/b978-0-12-363710-9.50009-5. [DOI] [PubMed] [Google Scholar]
- 39.Cleary EG, Sandberg LB, Jackson DS. The changes in chemical composition during development of the bovine nuchal ligament. J Cell Biol. 1983;33:469–479. doi: 10.1083/jcb.33.3.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cliff WJ. The aortic tunica media in growing rats studied with the electron microscope. Lab Invest. 1967;17:599–615. [PubMed] [Google Scholar]
- 41.Cooley MA, Kern CB, Fresco VM, Wessels A, Thompson RP, McQuinn TC, Twal WO, Mjaatvedt CH, Drake CJ, Argraves WS. Fibulin-1 is required for morphogenesis of neural crest-derived structures. Dev Biol. 2008;319:336–345. doi: 10.1016/j.ydbio.2008.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Corson GM, Charbonneau NL, Keene DR, Sakai LY. Differential expression of fibrillin-3 adds to microfibril variety in human and avian, but not rodent, connective tissues. Genomics. 2004;83:461–472. doi: 10.1016/j.ygeno.2003.08.023. [DOI] [PubMed] [Google Scholar]
- 43.Csiszar K. Lysyl oxidases: a novel multifunctional amine oxidase family. Prog Nucleic Acid Res Mol Biol. 2001;70:1–32. doi: 10.1016/s0079-6603(01)70012-8. [DOI] [PubMed] [Google Scholar]
- 44.Czirok A, Zach J, Kozel BA, Mecham RP, Davis EC, Rongish BJ. Elastic fiber macro-assembly is a hierarchical, cell motion-mediated process. J Cell Physiol. 2006;207:97–106. doi: 10.1002/jcp.20573. [DOI] [PubMed] [Google Scholar]
- 45.Daga-Gordini D, Bressan GM, Castellani I, Volpin D. Fine mapping of tropoelastin-drived components in the aorta of developing chick. Histochem J. 1987;19:623–632. doi: 10.1007/BF01676168. [DOI] [PubMed] [Google Scholar]
- 46.Damiano V, Tsang A, Weinbaum G, Christner P, Rosenbloom J. Secretion of elastin in the embryonic chick aorta as visualized by immunoelectron microscopy. Coll Relat Res. 1984;4:153–164. doi: 10.1016/s0174-173x(84)80022-9. [DOI] [PubMed] [Google Scholar]
- 47.Davis EC. Smooth muscle cell to elastic lamina connections in developing mouse aorta. Role in aortic medial organization. Lab Invest. 1993;68:89–99. [PubMed] [Google Scholar]
- 48.Davis EC. Endothelial cell connecting filaments anchor endothelial cells to the subjacent elastic lamina in the developing aortic intima of the mouse. Cell Tiss Res. 1993;272:211–219. doi: 10.1007/BF00302726. [DOI] [PubMed] [Google Scholar]
- 49.Davis NP, Han HC, Wayman B, Vito R. Sustained axial loading lengthens arteries in organ culture. Ann Biomed Eng. 2005;33:867–877. doi: 10.1007/s10439-005-3488-x. [DOI] [PubMed] [Google Scholar]
- 50.Davison IG, Wright GM, DeMont ME. The structure and physical properties of invertebrate and primitive vertebrate arteries. J Exp Biol. 1995;198:2185–2196. doi: 10.1242/jeb.198.10.2185. [DOI] [PubMed] [Google Scholar]
- 51.DeRuiter MC, Poelmann RE, VanMunsteren JC, Mironov V, Markwald RR, Gittenberger-de Groot AC. Embryonic endothelial cells transdifferentiate into mesenchymal cells expressing smooth muscle actins in vivo and in vitro. Circ Res. 1997;80:444–451. doi: 10.1161/01.res.80.4.444. [DOI] [PubMed] [Google Scholar]
- 52.Diamond SL, Sharefkin JB, Dieffenbach C, Frasier-Scott K, McIntire LV, Eskin SG. Tissue plasminogen activator messenger RNA levels increase in cultured human endothelial cells exposed to laminar shear stress. J Cell Physiol. 1990;143:364–371. doi: 10.1002/jcp.1041430222. [DOI] [PubMed] [Google Scholar]
- 53.Dingemans KP, Teeling P, Lagendijk JH, Becker AE. Extracellular matrix of the human aortic media: an ultrastructural histochemical and immunohistochemical study of the adult aortic media. Anat Rec. 2000;258:1–14. doi: 10.1002/(SICI)1097-0185(20000101)258:1<1::AID-AR1>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 54.Dobrin PB. Mechanical properties of arteries. Physiol Rev. 1978;58:397–460. doi: 10.1152/physrev.1978.58.2.397. [DOI] [PubMed] [Google Scholar]
- 55.Dobrin PB. Physiology and pathophysiology of blood vessels. In: Sidawy A, Sbe N, DePalma RG, editors. The Basic Science of Vascular Disease. Futura; New York: 1997. pp. 69–105. chapt. 3. [Google Scholar]
- 56.Drake CJ, Hungerford JE, Little CD. Morphogenesis of the first blood vessels. Ann NY Acad Sci. 1998;857:155–179. doi: 10.1111/j.1749-6632.1998.tb10115.x. [DOI] [PubMed] [Google Scholar]
- 57.Dubick MA, Rucker RB, Cross CE, Last JA. Elastin metabolism in rodent lung. Biochim Biophys Acta. 1981;672:303–306. doi: 10.1016/0304-4165(81)90297-x. [DOI] [PubMed] [Google Scholar]
- 58.Dumont DJ, Fong GH, Puri MC, Gradwohl G, Alitalo K, Breitman ML. Vascularization of the mouse embryo: a study of flk-1, tek, tie, and vascular endothelial growth factor expression during development. Dev Dyn. 1995;203:80–92. doi: 10.1002/aja.1002030109. [DOI] [PubMed] [Google Scholar]
- 59.Durante W, Liao L, Reyna SV, Peyton KJ, Schafer AI. Physiological cyclic stretch directs l-arginine transport and metabolism to collagen synthesis in vascular smooth muscle. FASEB J. 2000;14:1775–1783. doi: 10.1096/fj.99-0960com. [DOI] [PubMed] [Google Scholar]
- 60.Dye WW, Gleason RL, Wilson E, Humphrey JD. Altered biomechanical properties of carotid arteries in two mouse models of muscular dystrophy. J Appl Physiol. 2007;103:664–672. doi: 10.1152/japplphysiol.00118.2007. [DOI] [PubMed] [Google Scholar]
- 61.Effmann EL, Whitman SA, Smith BR. Aortic arch development. Radiographics. 1986;6:1065–1089. doi: 10.1148/radiographics.6.6.3685519. [DOI] [PubMed] [Google Scholar]
- 62.Evanko SP, Tammi MI, Tammi RH, Wight TN. Hyaluronan-dependent pericellular matrix. Adv Drug Deliv Rev. 2007;59:1351–1365. doi: 10.1016/j.addr.2007.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ewart AK, Jin WS, Atkinson D, Morris CA, Keating MT. Supravalvular aortic stenosis associated with a deletion disrupting the elastin gene. J Clin Invest. 1994;93:1071–1077. doi: 10.1172/JCI117057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Eyre ER, Paz MA, Gallop PM. Cross-linking in collagen and elastin. Annu Rev Biochem. 1984;53:717–748. doi: 10.1146/annurev.bi.53.070184.003441. [DOI] [PubMed] [Google Scholar]
- 65.Fahrenbach WH, Sandberg LB, Cleary EG. Ultrastructural studies on early elastogenesis. Anat Rec. 1966;155:563–576. [Google Scholar]
- 66.Faury G, Maher GM, Li DY, Keating MT, Mecham RP, Boyle WA. Relation between outer and luminal diameter in cannulated arteries. Am J Physiol Heart Circ Physiol. 1999;277:H1745–H1753. doi: 10.1152/ajpheart.1999.277.5.H1745. [DOI] [PubMed] [Google Scholar]
- 67.Faury G, Pezet M, Knutsen RH, Boyle WA, Heximer SP, McLean SE, Minkes RK, Blumer KJ, Kovacs A, Kelly DP, Li DY, Starcher B, Mecham RP. Developmental adaptation of the mouse cardiovascular system to elastin haploinsufficiency. J Clin Invest. 2003;112:1419–1428. doi: 10.1172/JCI19028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fazio MJ, Olsen DR, Kauh EA, Baldwin CT, Indik Z, Ornstein GN, Yeh H, Rosenbloom J. Cloning of full-length elastin cDNAs from a human skin fibroblast recombinant cDNA library: further elucidation of alternative splicing utilizing exon-specific oligonucleotides. J Invest Dermatol. 1988;91:458–464. doi: 10.1111/1523-1747.ep12476591. JU. [DOI] [PubMed] [Google Scholar]
- 69.Francis G, John R, Thomas J. Biosynthetic pathway of desmosines in elastin. Biochem J. 1973;136:45–55. doi: 10.1042/bj1360045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Frankfater C, Maus E, Gaal K, Segade F, Copeland NG, Gilbert DJ, Jenkins NA, Shipley JM. Organization of the mouse microfibril-associated glycoprotein-2 (MAGP-2) gene. Mam Genom. 2000;11:191–195. doi: 10.1007/s003350010036. [DOI] [PubMed] [Google Scholar]
- 71.Franzblau C. Elastin. Comp Biochem Physiol C Pharmacol. 1971;26:659–712. [Google Scholar]
- 72.Franzblau C, Baris B, Lent RW, Salcedo LL, Smith B, Jaffe R, Crombie G. Chemistry and biosynthesis of crosslinks in elastin. In: Balazs EA, editor. Chemistry and Molecular Biology of the Intracellular Matrix. Vol. 1. Academic; New York: 1969. pp. 617–639. [Google Scholar]
- 73.Frid MG, Kale VA, Stenmark KR. Mature vascular endothelium can give rise to smooth muscle cells via endothelial-mesenchymal transdifferentiation: in vitro analysis. Circ Res. 2002;90:1189–1196. doi: 10.1161/01.res.0000021432.70309.28. [DOI] [PubMed] [Google Scholar]
- 74.Frid MG, Moiseeva EP, Stenmark KR. Multiple phenotypically distinct smooth muscle cell populations exist in the adult and developing bovine pulmonary arterial media in vivo. Circ Res. 1994;75:669–681. doi: 10.1161/01.res.75.4.669. [DOI] [PubMed] [Google Scholar]
- 75.Fridez P, Rachev A, Meister JJ, Hayashi K, Stergiopulos N. Model of geometrical and smooth muscle tone adaptation of carotid artery subject to step change in pressure. Am J Physiol Heart Circ Physiol. 2001;280:H2752–H2760. doi: 10.1152/ajpheart.2001.280.6.H2752. [DOI] [PubMed] [Google Scholar]
- 76.Fridez P, Zulliger M, Bobard F, Montorzi G, Miyazaki H, Hayashi K, Stergiopulos N. Geometrical, functional, and histomorphometric adaptation of rat carotid artery in induced hypertension. J Biomech. 2003;36:671–680. doi: 10.1016/s0021-9290(02)00445-1. [DOI] [PubMed] [Google Scholar]
- 77.Fung YC. What are the residual stresses doing in our blood vessels? Ann Biomed Eng. 1991;19:237–249. doi: 10.1007/BF02584301. [DOI] [PubMed] [Google Scholar]
- 78.Fung YC, Fronek K, Patitucci P. Pseudoelasticity of arteries and the choice of its mathematical expression. Am J Physiol Heart Circ Physiol. 1979;237:H620–H631. doi: 10.1152/ajpheart.1979.237.5.H620. [DOI] [PubMed] [Google Scholar]
- 79.Gansner JM, Madsen EC, Mecham RP, Gitlin JD. Essential role for fibrillin-2 in zebrafish notochord and vascular morphogenesis. Dev Dyn. 2008;237:2844–2861. doi: 10.1002/dvdy.21705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gerrity RG, Cliff WJ. The aortic tunica intima in young and aging rats. Exp Mol Pathol. 1972;16:382–402. doi: 10.1016/0014-4800(72)90012-3. [DOI] [PubMed] [Google Scholar]
- 81.Gerrity RG, Cliff WJ. The aortic tunica media of the developing rat. I. Quantitative stereologic and biochemical analysis. Lab Invest. 1975;32:585–600. [PubMed] [Google Scholar]
- 82.Giampuzzi M, Botti G, Di Duca M, Arata L, Ghiggeri G, Gusmano R, Ravazzolo R, Di Donato A. Lysyl oxidase activates the transcription activity of human collagene III promoter. Possible involvement of Ku antigen. J Biol Chem. 2000;275:36341–36349. doi: 10.1074/jbc.M003362200. [DOI] [PubMed] [Google Scholar]
- 83.Gibbons CA, Shadwick RE. Functional similarities in the mechanical design of the aorta in lower vertebrates and mammals. Experientia. 1989;45:1083–1088. doi: 10.1007/BF01950164. [DOI] [PubMed] [Google Scholar]
- 84.Gibson MA, Hughes JL, Fanning JC, Cleary EG. The major antigen of elastin-associated microfibrils is a 31-kDa glycoprotein. J Biol Chem. 1986;261:11429–11436. [PubMed] [Google Scholar]
- 85.Gibson MA, Finnis ML, Kumaratilake JS, Cleary EG. Microfibril-associated glycoprotein-2 (MAGP-2) is specifically associated with fibrillin-containing microfibrils but exhibits more restricted patterns of tissue localization and developmental expression than its structural relative MAGP-1. J Histochm Cytochem. 1998;46:871–885. doi: 10.1177/002215549804600802. [DOI] [PubMed] [Google Scholar]
- 86.Gibson MA, Hatzinikolas G, Kumaratilake JS, Sandberg LB, Nicholl JK, Sutherland GR, Cleary EG. Further characterization of proteins associated with elastic fiber microfibrils including the molecular cloning of MAGP-2 (MP25) J Biol Chem. 1996;271:1096–1103. doi: 10.1074/jbc.271.2.1096. [DOI] [PubMed] [Google Scholar]
- 87.Gibson MA, Kumaratilake JS, Cleary EG. The protein components of the 12-nanometer microfibrils of elastic and non-elastic tissues. J Biol Chem. 1989;264:4590–4598. [PubMed] [Google Scholar]
- 88.Gibson MA, Sandberg LB, Grosso LE, Cleary EG. Complementary DNA cloning establishes microfibril-associated glycoprotein (MAGP) to be a discrete component of the elastin-associated microfibrils. J Biol Chem. 1991;266:7596–7601. [PubMed] [Google Scholar]
- 89.Gittenberger-de Groot AC, DeRuiter MC, Bergwerff M, Poelmann RE. Smooth muscle cell origin and its relation to heterogeneity in development and disease. Arterioscler Thromb Vasc Biol. 1999;19:1589–1594. doi: 10.1161/01.atv.19.7.1589. [DOI] [PubMed] [Google Scholar]
- 90.Gleason RL, Humphrey JD. A mixture model of arterial growth and remodeling in hypertension: altered muscle tone and tissue turnover. J Vasc Res. 2004;41:352–363. doi: 10.1159/000080699. [DOI] [PubMed] [Google Scholar]
- 91.Gleason RL, Taber LA, Humphrey JD. A 2-D model of flow-induced alterations in the geometry, structure and properties of carotid arteries. J Biomech Eng. 2004;126:371–381. doi: 10.1115/1.1762899. [DOI] [PubMed] [Google Scholar]
- 92.Glukhova MA, Koteliansky VE. Integrins, cytoskeletal and extracellular matrix proteins in developing smooth muscle cells of human aorta. In: Schwartz SM, Mecham RP, editors. The Vascular Smooth Muscle Cell: Molecular and Biological Responses to the Extracellular Matrix. Academic; San Diego, CA: 1995. pp. 37–79. [Google Scholar]
- 93.Greenwald SE, Moore JE, Jr, Rachev A, Kane TP, Meister JJ. Experimental investigation of the distribution of residual strains in the artery wall. J Biomech Eng. 1997;119:438–444. doi: 10.1115/1.2798291. [DOI] [PubMed] [Google Scholar]
- 94.Greenwald SE, Berry CL. Static mechanical properties and chemical composition of the aorta of spontaneously hypertensive rats: a comparison with the effects of induced hypertension. Cardiovas Res. 1978;12:364–372. doi: 10.1093/cvr/12.6.364. [DOI] [PubMed] [Google Scholar]
- 95.Guo X, Kono Y, Mattrey R, Kassab GS. Morphometry and strain distribution of the C57BL/6 mouse aorta. Am J Physiol Heart Circ Physiol. 2002;283:H1829–H1837. doi: 10.1152/ajpheart.00224.2002. [DOI] [PubMed] [Google Scholar]
- 96.Gupta PA, Putnam EA, Carmical SG, Kaitila I, Steinmann B, Child A, Danesino C, Metcalf K, Berry SA, Chen E, Delorme CV, Thong MT, Adés LC, Milewicz DM. Ten novel FBN2 mutations in congenital contractural arachnodactyly: delineation of the molecular pathogenesis and clinical phenotype. Hum Mutat. 2002;19:39–48. doi: 10.1002/humu.10017. [DOI] [PubMed] [Google Scholar]
- 97.Hamalainen ER, Jones TA, Sheer D, Taskinen K, Pihlajaniemi T, Kivirikko KI. Molecular cloning of human lysyl oxidase and assignment of the gene to chromosome 5q23.3-312. Genomics. 1991;11:508–516. doi: 10.1016/0888-7543(91)90057-l. [DOI] [PubMed] [Google Scholar]
- 98.Hanada K, Vermeij M, Garinis GA, de Waard MC, Kunen MG, Myers L, Maas A, Duncker DJ, Meijers C, Dietz HC, Kanaar R, Essers J. Perturbations of vascular homeostasis and aortic valve abnormalities in fibulin-4 deficient mice. Circ Res. 2007;100:738–746. doi: 10.1161/01.RES.0000260181.19449.95. [DOI] [PubMed] [Google Scholar]
- 99.Handford PA, Downing AK, Reinhardt DP, Sakai LY. Fibrillin: from domain structure to supramolecular assembly. Matrix Biol. 2000;19:457–470. doi: 10.1016/s0945-053x(00)00100-1. [DOI] [PubMed] [Google Scholar]
- 100.Harkness ML, Harkness RD, McDonald DA. The collagen and elastin content of the arterial wall in the dog. Proc R Soc Lond B Biol Sci. 1957;146:541–551. doi: 10.1098/rspb.1957.0029. [DOI] [PubMed] [Google Scholar]
- 101.Hartenstein V, Mandal L. The blood/vascular system in a phylogenetic perspective. Bioessays. 2006;28:1203–1210. doi: 10.1002/bies.20497. [DOI] [PubMed] [Google Scholar]
- 102.Haust MD, More RH, Benscome SA, Balis JU. Elastogenesis in human aorta: an electron microscopic study. Exp Mol Pathol. 1965;4:508–524. doi: 10.1016/0014-4800(65)90015-8. [DOI] [PubMed] [Google Scholar]
- 103.Heegaard AM, Corsi A, Danielsen CC, Nielsen KL, Jorgensen HL, Riminucci M, Young MF, Bianco P. Biglycan deficiency causes spontaneous aortic dissection and rupture in mice. Circulation. 2007;115:2731–2738. doi: 10.1161/CIRCULATIONAHA.106.653980. [DOI] [PubMed] [Google Scholar]
- 104.Heuser JE. Quick-freeze, deep etch preparation of samples for 3-D electron microscopy. Trends Biochem Sci. 1981:64–68. [Google Scholar]
- 105.Hinek A, Wrenn DS, Mecham RP, Barondes SH. The elastin receptor: a galactoside-binding protein. Science. 1988;239:1539–1541. doi: 10.1126/science.2832941. [DOI] [PubMed] [Google Scholar]
- 106.Hinek A, Rabinovitch M, Keeley F, Okamura-Oho Y, Callahan J. The 67-kD elastin/laminin-binding protein is related to an enzymatically inactive, alternatively spliced form of beta-galactosidase. J Clin Invest. 1993;91:1198–1205. doi: 10.1172/JCI116280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hirose H, Ozsvath KJ, Xia S, Tilson MD. Molecular cloning of the complementary DNA for an additional member of the family of aortic aneurysm antigenic proteins. J Vasc Surg. 1997;26:313–318. doi: 10.1016/s0741-5214(97)70194-0. [DOI] [PubMed] [Google Scholar]
- 108.Hocking AM, Shinomura T, McQuillan DJ. Leucine-rich repeat glycoproteins of the extracellular matrix. Matrix Biol. 1998;17:1–19. doi: 10.1016/s0945-053x(98)90121-4. [DOI] [PubMed] [Google Scholar]
- 109.Holzapfel GA, Weizsacker HW. Biomechanical behavior of the arterial wall and its numerical characterization. Comput Biol Med. 1998;28:377–392. doi: 10.1016/s0010-4825(98)00022-5. [DOI] [PubMed] [Google Scholar]
- 110.Holzenberger M, Lievre CA, Robert L. Tropoelastin gene expression in the developing vascular system of the chicken: an in situ hybridization study. Anat Embryol. 1993;188:481–492. doi: 10.1007/BF00190142. [DOI] [PubMed] [Google Scholar]
- 111.Hoofnagle MH, Wamhoff BR, Owens GK. Lost in transdifferentiation. J Clin Invest. 2004;113:1249–1251. doi: 10.1172/JCI21761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hornstra IK, Birge S, Starcher B, Bailey AJ, Mecham RP, Shapiro SD. Lysyl oxidase is required for vascular and diaphragmatic development in mice. J Biol Chem. 2003;278:14387–14393. doi: 10.1074/jbc.M210144200. [DOI] [PubMed] [Google Scholar]
- 113.Horrigan SK, Rich CB, Streeten BW, Li ZY, Foster JA. Characterization of an associated microfibril protein through recombinant DNA techniques. J Biol Chem. 1992;267:10087–10095. [PubMed] [Google Scholar]
- 114.Howard PS, Macarak EJ. Localization of collagen types in regional segments of the fetal bovine aorta. Lab Invest. 1989;61:548–555. [PubMed] [Google Scholar]
- 115.Hsu FH. The influences of mechanical loads on the form of a growing elastic body. J Biomech. 1968;1:303–311. doi: 10.1016/0021-9290(68)90024-9. [DOI] [PubMed] [Google Scholar]
- 116.Hu Y, Zhang Z, Torsney E, Afzal AR, Davison F, Metzler B, Xu Q. Abundant progenitor cells in the adventitia contribute to atherosclerosis of vein grafts in ApoE-deficient mice. J Clin Invest. 2004;113:1258–1265. doi: 10.1172/JCI19628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Huang R, Merrilees MJ, Braun K, Beaumont B, Lemire J, Clowes AW, Hinek A, Wight TN. Inhibition of versican synthesis by antisense alters smooth muscle cell phenotype and induces elastic fiber formation in vitro and in neointima after vessel injury. Circ Res. 2006;98:370–377. doi: 10.1161/01.RES.0000202051.28319.c8. [DOI] [PubMed] [Google Scholar]
- 118.Huang Y, Guo X, Kassab GS. Axial nonuniformity of geometric and mechanical properties of mouse aorta is increased during postnatal growth. Am J Physiol Heart Circ Physiol. 2006;290:H657–H664. doi: 10.1152/ajpheart.00803.2005. [DOI] [PubMed] [Google Scholar]
- 119.Hucthagowder V, Sausgruber N, Kim KH, Angle B, Marmorstein LY, Urban Z. Fibulin-4: a novel gene for an autosomal recessive cutis laxa syndrome. Am J Hum Genet. 2006;78:1075–1080. doi: 10.1086/504304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Humphrey JD. An evaluation of pseudoelastic descriptors used in arterial mechanics. J Biomech Eng. 1999;121:259–262. doi: 10.1115/1.2835113. [DOI] [PubMed] [Google Scholar]
- 121.Humphrey JD. Cardiovascular Solid Mechanics. Springer-Verlag; New York: 2002. [Google Scholar]
- 122.Humphrey JD, Rajagopal KR. A constrained mixture model for arterial adaptations to a sustained step change in blood flow. Biomech Model Mechanobiol. 2003;2:109–126. doi: 10.1007/s10237-003-0033-4. [DOI] [PubMed] [Google Scholar]
- 123.Humphrey JD, Eberth JF, Dye WW, Gleason RL. Fundamental role of axial stress in compensatory adaptations by arteries. J Biomech. 2009;42:1–8. doi: 10.1016/j.jbiomech.2008.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hungerford JE, Little CD. Developmental biology of the vascular smooth muscle cell: building a multilayered vessel wall. J Vasc Res. 1999;36:2–27. doi: 10.1159/000025622. [DOI] [PubMed] [Google Scholar]
- 125.Hungerford JE, Hoeffler JP, Bowers CW, Dahm LM, Flachetto R, Shabanowitz J, Hunt DF, Little CD. Identification of a novel marker for primordial smooth muscle and its differential expression pattern in contractile vs. nonontractile cells. J Cell Biol. 1997;137:925–937. doi: 10.1083/jcb.137.4.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hungerford JE, Owens GK, Argraves WS, Little CD. Development of the aortic vessel wall as defined by vascular smooth muscle and extracellular matrix markers. Dev Biol. 1996;178:375–392. doi: 10.1006/dbio.1996.0225. [DOI] [PubMed] [Google Scholar]
- 127.Hurtado PA, Vora S, Sume SS, Yang D, St Hilaire C, Guo Y, Palamakumbura AH, Schreiber BM, Ravid K, Trackman PC. Lysyl oxidase propeptide inhibits smooth muscle cell signaling and proliferation. Biochem Biophys Res Commun. 2008;366:156–161. doi: 10.1016/j.bbrc.2007.11.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Indik Z, Yoon K, Morrow SD, Cicila B, Rosenbloom JC, Rosenbloom J, Ornstein-Goldstein N. Structure of the 3′ region of the human elastin gene: great abundance of Alu repetitive sequences and few coding sequences. Connect Tiss Res. 1987;16:197–211. doi: 10.3109/03008208709006976. [DOI] [PubMed] [Google Scholar]
- 129.Iozzo RV. The biology of the small leucine-rich proteoglycans. Functional network of interactive proteins. J Biol Chem. 1999;274:18843–18846. doi: 10.1074/jbc.274.27.18843. [DOI] [PubMed] [Google Scholar]
- 130.Isenberg BC, Tranquillo RT. Long-term cyclic distention enhances the mechanical properties of collagen-based media-equivalents. Ann Biomed Eng. 2003;31:937–949. doi: 10.1114/1.1590662. [DOI] [PubMed] [Google Scholar]
- 131.Ishii T, Kuwaki T, Masuda Y, Fukuda Y. Postnatal development of blood pressure and baroreflex in mice. Auton Neurosci. 2001;94:34–41. doi: 10.1016/S1566-0702(01)00339-3. [DOI] [PubMed] [Google Scholar]
- 132.Ishiwata T, Nakazawa M, Pu WT, Tevosian SG, Izumo S. Developmental changes in ventricular diastolic function correlate with changes in ventricular myoarchitecture in normal mouse embryos. Circ Res. 2003;93:857–865. doi: 10.1161/01.RES.0000100389.57520.1A. [DOI] [PubMed] [Google Scholar]
- 133.Isokawa K, Takagi M, Toda Y. Ultrastructural cytochemistry of aortic microfibrils in the Arctic lamprey, Lampetra japonica. Anat Rec. 1989;223:158–164. doi: 10.1002/ar.1092230207. [DOI] [PubMed] [Google Scholar]
- 134.Jackson ZS, Dajnowiec D, Gotlieb AI, Langille BL. Partial off-loading of longitudinal tension induces arterial tortuosity. Arterioscler Thromb Vasc Biol. 2005;25:957–962. doi: 10.1161/01.ATV.0000161277.46464.11. [DOI] [PubMed] [Google Scholar]
- 135.Jackson ZS, Gotlieb AI, Langille BL. Wall tissue remodeling regulates longitudinal tension in arteries. Circ Res. 2002;90:918–925. doi: 10.1161/01.res.0000016481.87703.cc. [DOI] [PubMed] [Google Scholar]
- 136.Jevon M, Dorling A, Hornick PI. Progenitor cells and vascular disease. Cell Prolif. 2008;41(Suppl 1):146–164. doi: 10.1111/j.1365-2184.2008.00488.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ji RP, Phoon CK, Aristizabal O, McGrath KE, Palis J, Turnbull DH. Onset of cardiac function during early mouse embryogenesis coincides with entry of primitive erythroblasts into the embryo proper. Circ Res. 2003;92:133–135. doi: 10.1161/01.res.0000056532.18710.c0. [DOI] [PubMed] [Google Scholar]
- 138.Kagan HM, Li W. Lysyl oxidase: properties, specificity, and biological roles inside and outside of the cell. J Cell Biochem. 2003;88:660–672. doi: 10.1002/jcb.10413. [DOI] [PubMed] [Google Scholar]
- 139.Kagan HM. Characterization and regulation of lysyl oxidase. In: Mecham RP, editor. Regulation of Matrix Accumulation. Academic; New York: 1986. pp. 321–398. [Google Scholar]
- 140.Kamiya A, Togawa T. Adaptive regulation of wall shear stress to flow change in the canine carotid artery. Am J Physiol Heart Circ Physiol. 1980;239:H14–H21. doi: 10.1152/ajpheart.1980.239.1.H14. [DOI] [PubMed] [Google Scholar]
- 141.Karnik SK, Brooke BS, Bayes-Genis A, Sorensen L, Wythe JD, Schwartz RS, Keating MT, Li DY. A critical role for elastin signaling in vascular morphogenesis and disease. Development. 2003;130:411–423. doi: 10.1242/dev.00223. [DOI] [PubMed] [Google Scholar]
- 142.Karnik SK, Wythe JD, Sorensen L, Brooke BS, Urness LD, Li DY. Elastin induces myofibrillogenesis via a specific domain, VGVAPG. Matrix Biol. 2003;22:409–425. doi: 10.1016/s0945-053x(03)00076-3. [DOI] [PubMed] [Google Scholar]
- 143.Karrer HE. An electron microscope study of the aorta in young and aging mice. J Ultrastruct Res. 1961;5:1–27. doi: 10.1016/s0022-5320(61)80002-6. [DOI] [PubMed] [Google Scholar]
- 144.Katsumi A, Milanini J, Kiosses WB, del Pozo MA, Kaunas R, Chien S, Hahn KM, Schwartz MA. Effects of cell tension on the small GTPase Rac. J Cell Biol. 2002;158:153–164. doi: 10.1083/jcb.200201105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Keeley FW. The synthesis of soluble and insoluble elastin in chicken aorta as a function of development and age. Effect of a high cholesterol diet. Can J Biochem. 1979;57:1273–1280. doi: 10.1139/o79-169. [DOI] [PubMed] [Google Scholar]
- 146.Kelleher CM, McLean SE, Mecham RP. Vascular extracellular matrix and aortic development. Curr Top Dev Biol. 2004;62:153–188. doi: 10.1016/S0070-2153(04)62006-0. [DOI] [PubMed] [Google Scholar]
- 147.Keller BB, MacLennan MJ, Tinney JP, Yoshigi M. In vivo assessment of embryonic cardiovascular dimensions and function in day-10.5 to -14.5 mouse embryos. Circ Res. 1996;79:247–255. doi: 10.1161/01.res.79.2.247. [DOI] [PubMed] [Google Scholar]
- 148.Kielty CM, Wess TJ, Haston L, Ashworth JL, Sherratt MJ, Shuttleworth CA. Fibrillin-rich microfibrils: elastic biopolymers of the extracellular matrix. J Muscle Res Cell Motil. 2002;23:581–596. [PubMed] [Google Scholar]
- 149.Kobayashi N, Kostka G, Garbe JH, Keene DR, Bachinger HP, Hanisch FG, Markova D, Tsuda T, Timpl R, Chu ML, Sasaki T. A comparative analysis of the fibulin protein family. Biochemical characterization, binding interactions, and tissue localization. J Biol Chem. 2007;282:11805–11816. doi: 10.1074/jbc.M611029200. [DOI] [PubMed] [Google Scholar]
- 150.Kobayashi R, Tashima R, Masuda H, Shozawa T, Numata Y, Miyauchi K, Hayakawa T. Isolation and characterization of a new 36-kDa microfibril associated glycoprotein from porcine aorta. J Biol Chem. 1989;264:17437–17444. [PubMed] [Google Scholar]
- 151.Kostka G, Giltay R, Bloch W, Addicks K, Timpl R, Fassler R, Chu ML. Perinatal lethality and endothelial cell abnormalities in several vessel compartments of fibulin-1-deficient mice. Mol Cell Biol. 2001;21:7025–7034. doi: 10.1128/MCB.21.20.7025-7034.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Kozel BA, Rongish BJ, Czirok A, Zach J, Little CD, Davis EC, Knutsen RH, Wagenseil JE, Levy MA, Mecham RP. Elastic fiber formation: a dynamic view of extracellular matrix assembly using timer reporters. J Cell Physiol. 2006;207:87–96. doi: 10.1002/jcp.20546. [DOI] [PubMed] [Google Scholar]
- 153.Kucera T, Strilic B, Regener K, Schubert M, Laudet V, Lammert E. Ancestral vascular lumen formation via basal cell surfaces. PLoS ONE. 2009;4:e4132. doi: 10.1371/journal.pone.0004132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Langille BL. Remodeling of developing and mature arteries: endothelium, smooth muscle, and matrix. J Cardiovasc Pharmacol. 1993;21(Suppl 1):S11–S17. doi: 10.1097/00005344-199321001-00003. [DOI] [PubMed] [Google Scholar]
- 155.Langille BL, Bendeck MP, Keeley FW. Adaptations of carotid arteries of young and mature rabbits to reduced carotid blood flow. Am J Physiol Heart Circ Physiol. 1989;256:H931–H939. doi: 10.1152/ajpheart.1989.256.4.H931. [DOI] [PubMed] [Google Scholar]
- 156.Le Lievre CS, Le Douarin NM. Mesenchymal derivatives of the neural crest: analysis of chimaeric quail and chick embryos. J Embryol Exp Morphol. 1975;34:125–154. [PubMed] [Google Scholar]
- 157.Learoyd BM, Taylor MG. Alterations with age in the viscoelastic properties of human arterial walls. Circ Res. 1966;18:278–292. doi: 10.1161/01.res.18.3.278. [DOI] [PubMed] [Google Scholar]
- 158.Lee B, Godfrey M, Vitale E, Hori H, Mattei MG, Sarfarazi M, Tsipouras P, Ramirez F, Hollister DW. Linkage of Marfan syndrome and a phenotypically related disorder to two different fibrillin genes. Nature. 1991;352:330–334. doi: 10.1038/352330a0. [DOI] [PubMed] [Google Scholar]
- 159.Lee RT, Yamamoto C, Feng Y, Potter-Perigo S, Briggs WH, Landschulz KT, Turi TG, Thompson JF, Libby P, Wight TN. Mechanical strain induces specific changes in the synthesis and organization of proteoglycans by vascular smooth muscle cells. J Biol Chem. 2001;276:13847–13851. doi: 10.1074/jbc.M010556200. [DOI] [PubMed] [Google Scholar]
- 160.Lemaire R, Bayle J, Mecham RP, Lafyatis R. Microfibril-associated MAGP-2 stimulates elastic fiber assembly. J Biol Chem. 2007;282:800–888. doi: 10.1074/jbc.M609692200. [DOI] [PubMed] [Google Scholar]
- 161.Leung DY, Glagov S, Mathews MB. Elastin and collagen accumulation in rabbit ascending aorta and pulmonary trunk during postnatal growth. Correlation of cellular synthetic response with medial tension. Circ Res. 1977;41:316–323. doi: 10.1161/01.res.41.3.316. [DOI] [PubMed] [Google Scholar]
- 162.Li DY, Brooke B, Davis EC, Mecham RP, Sorensen LK, Boak BB, Eichwald E, Keating MT. Elastin is an essential determinant of arterial morphogenesis. Nature. 1998;393:276–280. doi: 10.1038/30522. [DOI] [PubMed] [Google Scholar]
- 163.Li DY, Faury G, Taylor DG, Davis EC, Boyle WA, Mecham RP, Stenzel P, Boak B, Keating MT. Novel arterial pathology in mice and humans hemizygous for elastin. J Clin Invest. 1998;102:1783–1787. doi: 10.1172/JCI4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Li DY, Toland AE, Boak BB, Atkinson DL, Ensing GJ, Morris CA, Keating MR. Elastin point mutations cause an obstructive vascular disease, supravalvular aortic stenosis. Hum Mol Genet. 1997;6:1021–1028. doi: 10.1093/hmg/6.7.1021. [DOI] [PubMed] [Google Scholar]
- 165.Li W, Chen Q, Mills I, Sumpio BE. Involvement of S6 kinase and p38 mitogen activated protein kinase pathways in strain-induced alignment and proliferation of bovine aortic smooth muscle cells. J Cell Physiol. 2003;195:202–209. doi: 10.1002/jcp.10230. [DOI] [PubMed] [Google Scholar]
- 166.Li ZJ, Huang W, Fung YC. Changes of zero-bending-moment states and structures of rat arteries in response to a step lowering of the blood pressure. Ann Biomed Eng. 2002;30:379–391. doi: 10.1114/1.1470181. [DOI] [PubMed] [Google Scholar]
- 167.Liu SQ, Fung YC. Zero-stress states of arteries. J Biomech Eng. 1988;110:82–84. doi: 10.1115/1.3108410. [DOI] [PubMed] [Google Scholar]
- 168.Liu SQ, Fung YC. Indicial functions of arterial remodeling in response to locally altered blood pressure. Am J Physiol Heart Circ Physiol. 1996;270:H1323–H1333. doi: 10.1152/ajpheart.1996.270.4.H1323. [DOI] [PubMed] [Google Scholar]
- 169.Liu X, Wu H, Byrne M, Krane S, Jaenisch R. Type III collagen is crucial for collagen I fibrillogenesis and for normal cardiovascular development. Proc Natl Acad Sci USA. 1997;94:1852–1856. doi: 10.1073/pnas.94.5.1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Liu X, Zhao Y, Gao J, Pawlyk B, Starcher B, Spencer JA, Yanagisawa H, Zuo J, Li T. Elastic fiber homeostasis requires lysyl oxidase-like 1 protein. Nat Genet. 2004;36:178–182. doi: 10.1038/ng1297. [DOI] [PubMed] [Google Scholar]
- 171.Lohler J, Timpl R, Jaenisch R. Embryonic lethal mutation in mouse collagen I gene causes rupture of blood vessels and is associated with erythropoietic and mesenchymal cell death. Cell. 1984;38:597–607. doi: 10.1016/0092-8674(84)90514-2. [DOI] [PubMed] [Google Scholar]
- 172.Lu X, Zhao JB, Wang GR, Gregersen H, Kassab GS. Remodeling of the zero-stress state of femoral arteries in response to flow overload. Am J Physiol Heart Circ Physiol. 2001;280:H1547–H1559. doi: 10.1152/ajpheart.2001.280.4.H1547. [DOI] [PubMed] [Google Scholar]
- 173.Lucero HA, Kagan HM. Lysyl oxidase: an oxidative enzyme and effector of cell function. Cell Mol Life Sci. 2006;63:2304–2316. doi: 10.1007/s00018-006-6149-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Majesky MW. Developmental basis of vascular smooth muscle diversity. Arterioscler Thromb Vasc Biol. 2007;27:1248–1258. doi: 10.1161/ATVBAHA.107.141069. [DOI] [PubMed] [Google Scholar]
- 175.Maki JM, Rasanen J, Tikkanen H, Sormunen R, Makikallio K, Kivirikko KI, Soininen R. Inactivation of the lysyl oxidase gene Lox leads to aortic aneurysms, cardiovascular dysfunction, and perinatal death in mice. Circulation. 2002;106:2503–2509. doi: 10.1161/01.cir.0000038109.84500.1e. [DOI] [PubMed] [Google Scholar]
- 176.Marque V, Kieffer P, Gayraud B, Lartaud-Idjouadiene I, Ramirez F, Atkinson J. Aortic wall mechanics and composition in a transgenic mouse model of Marfan syndrome. Arterioscler Thromb Vasc Biol. 2001;21:1184–1189. doi: 10.1161/hq0701.092136. [DOI] [PubMed] [Google Scholar]
- 177.Masuda H, Zhuang YJ, Singh TM, Kawamura K, Murakami M, Zarins CK, Glagov S. Adaptive remodeling of internal elastic lamina and endothelial lining during flow-induced arterial enlargement. Arterioscler Thromb Vasc Biol. 1999;19:2298–2307. doi: 10.1161/01.atv.19.10.2298. [DOI] [PubMed] [Google Scholar]
- 178.Matsumoto T, Hayashi K. Stress and strain distribution in hypertensive and normotensive rat aorta considering residual strain. J Biomech Eng. 1996;118:62–73. doi: 10.1115/1.2795947. [DOI] [PubMed] [Google Scholar]
- 179.McConnell CJ, DeMont ME, Wright GM. Microfibrils provide non-linear elastic behaviour in the abdominal artery of the lobster Homarus americanus. J Physiol. 1997;499:513–526. doi: 10.1113/jphysiol.1997.sp021945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.McGrath KE, Koniski AD, Malik J, Palis J. Circulation is established in a stepwise pattern in the mammalian embryo. Blood. 2003;101:1669–1676. doi: 10.1182/blood-2002-08-2531. [DOI] [PubMed] [Google Scholar]
- 181.McLaughlin PJ, Chen Q, Horiguchi M, Starcher BC, Stanton JB, Broekelmann TJ, Marmorstein AD, McKay B, Mecham R, Nakamura T, Marmorstein LY. Targeted disruption of fibulin-4 abolishes elastogenesis and causes perinatal lethality in mice. Mol Cell Biol. 2006;26:1700–1709. doi: 10.1128/MCB.26.5.1700-1709.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.McLaughlin PJ, Bakall B, Choi J, Liu Z, Sasaki T, Davis EC, Marmorstein AD, Marmorstein LY. Lack of fibulin-3 causes early aging and herniation, but not macular degeneration in mice. Hum Mol Genet. 2007;16:3059–3070. doi: 10.1093/hmg/ddm264. [DOI] [PubMed] [Google Scholar]
- 183.McLean SE, Mecham BH, Kelleher CM, Mariani TJ, Mecham RP. Extracellular matrix gene expression in developing mouse aorta. In: Miner JH, editor. Extracellular Matrices and Development. Elsevier; New York: 2005. pp. 82–128. [Google Scholar]
- 184.Mecham RP, Davis EC. Elastic fiber structure and assembly. In: Yurchenko PD, Birk DE, Mecham RP, editors. Extracellular Matrix Assembly and Structure. Academic; San Diego, CA: 1994. pp. 281–314. [Google Scholar]
- 185.Mecham RP, Hinek A, Entwistle R, Wrenn DS, Griffin GL, Senior RM. Elastin binds to a multifunctional 67-kilodalton peripheral membrane protein. Biochemistry. 1989;28:3716–3722. doi: 10.1021/bi00435a014. [DOI] [PubMed] [Google Scholar]
- 186.Mecham RP, Madaras J, McDonald JA, Ryan U. Elastin production by cultured calf pulmonary artery endothelial cells. J Cell Physiol. 1983;116:282–288. doi: 10.1002/jcp.1041160304. [DOI] [PubMed] [Google Scholar]
- 187.Mecham RP, Heuser J. Three-dimensional organization of extracellular matrix in elastic cartilage as viewed by quick freeze, deep etch electron microscopy. Connect Tissue Res. 1990;24:83–93. doi: 10.3109/03008209009152425. [DOI] [PubMed] [Google Scholar]
- 188.Megill WM, Gosline JM, Blake RW. The modulus of elasticity of fibrillin-containing elastic fibres in the mesoglea of the hydromedusa Polyorchis penicillatus. J Exp Biol. 2005;208:3819–3834. doi: 10.1242/jeb.01765. [DOI] [PubMed] [Google Scholar]
- 189.Merrilees MJ, Lemire JM, Fischer JW, Kinsella MG, Braun KR, Clowes AW, Wight TN. Retrovirally mediated overexpression of versican v3 by arterial smooth muscle cells induces tropoelastin synthesis and elastic fiber formation in vitro and in neointima after vascular injury. Circ Res. 2002;90:481–487. doi: 10.1161/hh0402.105791. [DOI] [PubMed] [Google Scholar]
- 190.Miano JM, Cserjesi P, Ligon KL, Periasamy M, Olson EN. Smooth muscle myosin heavy chain exclusively marks the smooth muscle lineage during mouse embryogenesis. Circ Res. 1994;75:803–812. doi: 10.1161/01.res.75.5.803. [DOI] [PubMed] [Google Scholar]
- 191.Miao M, Bruce AE, Bhanji T, Davis EC, Keeley FW. Differential expression of two tropoelastin genes in zebrafish. Matrix Biol. 2007;26:115–124. doi: 10.1016/j.matbio.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 192.Mills I, Cohen CR, Kamal K, Li G, Shin T, Du W, Sumpio BE. Strain activation of bovine aortic smooth muscle cell proliferation and alignment: study of strain dependency and the role of protein kinase A and C signaling pathways. J Cell Physiol. 1997;170:228–234. doi: 10.1002/(SICI)1097-4652(199703)170:3<228::AID-JCP2>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 193.Mithieux SM, Weiss AS. Elastin. Adv Protein Chem. 2005;70:437–461. doi: 10.1016/S0065-3233(05)70013-9. [DOI] [PubMed] [Google Scholar]
- 194.Molnar J, Fong KS, He QP, Hayashi K, Kim Y, Fong SF, Fogelgren B, Szauter KM, Mink M, Csiszar K. Structural and functional diversity of lysyl oxidase and the LOX-like proteins. Biochim Biophys Acta. 2003;1647:220–224. doi: 10.1016/s1570-9639(03)00053-0. [DOI] [PubMed] [Google Scholar]
- 195.Nakamura H. Electron microscopic study of the prenatal development of the thoracic aorta in the rat. Am J Anat. 1988;181:406–418. doi: 10.1002/aja.1001810409. [DOI] [PubMed] [Google Scholar]
- 196.Nakamura T, Lozano PR, Ikeda Y, Iwanaga Y, Hinek A, Minamisawa S, Cheng CF, Kobuke K, Dalton N, Takada Y, Tashiro K, Ross J, Jr, Honjo T, Chien KR. Fibulin-5/DANCE is essential for elastogenesis in vivo. Nature. 2002;415:171–175. doi: 10.1038/415171a. [DOI] [PubMed] [Google Scholar]
- 197.Niiyama T, Higuchi I, Hashiguchi T, Suehara M, Uchida Y, Horikiri T, Shiraishi T, Saitou A, Hu J, Nakagawa M, Arimura K, Osame M. Capillary changes in skeletal muscle of patients with Ullrich’s disease with collagen VI deficiency. Acta Neuropathol. 2003;106:137–142. doi: 10.1007/s00401-003-0714-1. [DOI] [PubMed] [Google Scholar]
- 198.Niklason LE, Gao J, Abbott WM, Hirschi KK, Houser S, Marini R, Langer R. Functional arteries grown in vitro. Science. 1999;284:489–493. doi: 10.1126/science.284.5413.489. [DOI] [PubMed] [Google Scholar]
- 199.Ninomiya K, Takahashi A, Fujioka Y, Ishikawa Y, Yokoyama M. Transforming growth factor-beta signaling enhances transdifferentiation of macrophages into smooth muscle-like cells. Hypertens Res. 2006;29:269–276. doi: 10.1291/hypres.29.269. [DOI] [PubMed] [Google Scholar]
- 200.O’Connell MK, Murthy S, Phan S, Xu C, Buchanan J, Spilker R, Dalman RL, Zarins CK, Denk W, Taylor CA. The three-dimensional micro- and nanostructure of the aortic medial lamellar unit measured using 3D confocal and electron microscopy imaging. Matrix Biol. 2008;27:171–181. doi: 10.1016/j.matbio.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Ohno M, Gibbons GH, Dzau VJ, Cooke JP. Shear stress elevates endothelial cGMP. Role of a potassium channel and G protein coupling. Circulation. 1993;88:193–197. doi: 10.1161/01.cir.88.1.193. [DOI] [PubMed] [Google Scholar]
- 202.Okamoto RJ, Wagenseil JE, DeLong WR, Peterson SJ, Kouchoukos NT, Sundt TM., 3rd. Mechanical properties of dilated human ascending aorta. Ann Biomed Eng. 2002;30:624–635. doi: 10.1114/1.1484220. [DOI] [PubMed] [Google Scholar]
- 203.Oleggini R, Gastaldo N, Di Donato A. Regulation of elastin promoter by lysyl oxidase and growth factors: cross control of lysyl oxidase on TGF-beta1 effects. Matrix Biol. 2007;26:494–505. doi: 10.1016/j.matbio.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 204.Olesen SP, Clapham DE, Davies PF. Haemodynamic shear stress activates a K+ current in vascular endothelial cells. Nature. 1988;331:168–170. doi: 10.1038/331168a0. [DOI] [PubMed] [Google Scholar]
- 205.Onda M, Ishiwata T, Kawahara K, Wang R, Naito Z, Sugisaki Y. Expression of lumican in thickened intima and smooth muscle cells in human coronary atherosclerosis. Exp Mol Pathol. 2002;72:142–149. doi: 10.1006/exmp.2002.2425. [DOI] [PubMed] [Google Scholar]
- 206.Osborne LR, Marindale D, Scherer SW, Shi XM, Huizenga J, Costa T, Pober B, Lew L, Brinkman J, Rommens J, Koop B, Tsui LC. Identification of genes from a 500-kb region at 7q11.23 that is commonly deleted in Williams syndrome patients. Genomics. 1996;36:328–336. doi: 10.1006/geno.1996.0469. HHQH. [DOI] [PubMed] [Google Scholar]
- 207.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 208.Owens GK. Regulation of differentiation of vascular smooth muscle cells. Physiol Rev. 1995;75:487–517. doi: 10.1152/physrev.1995.75.3.487. [DOI] [PubMed] [Google Scholar]
- 209.Partridge SM. Elastin. Adv Prot Chem. 1962;17:227–302. [Google Scholar]
- 210.Passman JN, Dong XR, Wu SP, Maguire CT, Hogan KA, Bautch VL, Majesky MW. A sonic hedgehog signaling domain in the arterial adventitia supports resident Sca1+ smooth muscle progenitor cells. Proc Natl Acad Sci USA. 2008;105:9349–9354. doi: 10.1073/pnas.0711382105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Paule WJ. Electron microscopy of the newborn rat aorta. J Ultrastruct Res. 1963;8:219–235. doi: 10.1016/s0022-5320(63)90004-2. [DOI] [PubMed] [Google Scholar]
- 212.Pease DC, Paule WJ. Electron microscopy of elastic arteries; the thoracic aorta of the rat. J Ultrastruct Res. 1960;3:469–483. doi: 10.1016/s0022-5320(60)90023-x. [DOI] [PubMed] [Google Scholar]
- 213.Pereira L, Andrikopoulos K, Tian J, Lee SY, Keene DR, Ono R, Reinhardt DP, Sakai LY, Biery NJ, Bunton T, Dietz HC, Ramirez F. Targetting of the gene encoding fibrillin-1 recapitulates the vascular aspect of Marfan syndrome. Nat Genet. 1997;17:218–222. doi: 10.1038/ng1097-218. [DOI] [PubMed] [Google Scholar]
- 214.Pfaff M, Reinhardt DP, Sakai LY, Timpl R. Cell adhesion and integrin binding to recombinant human fibrillin-1. FEBS Lett. 1996;384:247–250. doi: 10.1016/0014-5793(96)00325-0. [DOI] [PubMed] [Google Scholar]
- 215.Piontkivska H, Zhang Y, Green ED, Elnitski L. Multi-species sequence comparison reveals dynamic evolution of the elastin gene that has involved purifying selection and lineage-specific insertions/deletions. BMC Genomics. 2004;5:31. doi: 10.1186/1471-2164-5-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Pope FM, Martin GR, Lichtenstein JR, Penttinen R, Gerson B, Rowe DW, McKusick VA. Patients with Ehlers-Danlos syndrome type IV lack type III collagen. Proc Natl Acad Sci USA. 1975;72:1314–1316. doi: 10.1073/pnas.72.4.1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Rachev A. Theoretical study of the effect of stress-dependent remodeling on arterial geometry under hypertensive conditions. J Biomech. 1997;30:819–827. doi: 10.1016/s0021-9290(97)00032-8. [DOI] [PubMed] [Google Scholar]
- 218.Rachev A, Greenwald SE. Residual strains in conduit arteries. J Biomech. 2003;36:661–670. doi: 10.1016/s0021-9290(02)00444-x. [DOI] [PubMed] [Google Scholar]
- 219.Rachev A, Hayashi K. Theoretical study of the effects of vascular smooth muscle contraction on strain and stress distributions in arteries. Ann Biomed Eng. 1999;27:459–468. doi: 10.1114/1.191. [DOI] [PubMed] [Google Scholar]
- 220.Rachev A, Stergiopulos N, Meister JJ. Theoretical study of dynamics of arterial wall remodeling in response to changes in blood pressure. J Biomech. 1996;29:635–642. doi: 10.1016/0021-9290(95)00108-5. [DOI] [PubMed] [Google Scholar]
- 221.Rachev A, Stergiopulos N, Meister JJ. A model for geometric and mechanical adaptation of arteries to sustained hypertension. J Biomech Eng. 1998;120:9–17. doi: 10.1115/1.2834313. [DOI] [PubMed] [Google Scholar]
- 222.Ramirez F, Dietz HC. Fibrillin-rich microfibrils: structural determinants of morphogenetic and homeostatic events. J Cell Physiol. 2007;213:326–330. doi: 10.1002/jcp.21189. [DOI] [PubMed] [Google Scholar]
- 223.Ramirez F, Sakai LY, Rifkin DB, Dietz HC. Extracellular microfibrils in development and disease. Cell Mol Life Sci. 2007;64:2437–2446. doi: 10.1007/s00018-007-7166-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Reber-Muller S, Spissinger T, Schuchert P, Spring J, Schmid V. An extracellular matrix protein of jellyfish homologous to mammalian fibrillins forms different fibrils depending on the life stage of the animal. Dev Biol. 1995;169:662–672. doi: 10.1006/dbio.1995.1177. [DOI] [PubMed] [Google Scholar]
- 225.Reinboth B, Hanssen E, Cleary EG, Gibson MA. Molecular interactions of biglycan and decorin with elastic fiber components: biglycan forms a ternary complex with tropoelastin and microfibril-associated glycoprotein 1. J Biol Chem. 2002;277:3950–3957. doi: 10.1074/jbc.M109540200. [DOI] [PubMed] [Google Scholar]
- 226.Reinboth BJ, Finnis ML, Gibson MA, Sandberg LB, Cleary EG. Developmental expression of dermatan sulfate proteoglycans in the elastic bovine nuchal ligament. Matrix Biol. 2000;19:149–162. doi: 10.1016/s0945-053x(00)00060-3. [DOI] [PubMed] [Google Scholar]
- 227.Risau W, Flamme I. Vasculogenesis. Annu Rev Cell Dev Biol. 1995;11:73–91. doi: 10.1146/annurev.cb.11.110195.000445. [DOI] [PubMed] [Google Scholar]
- 228.Ritty TM, Broekelmann T, Werneck CC, Mecham RP. Fibrillin-1 and -2 contain heparin-binding sites important for matrix deposition and that support cell attachment. Biochem J. 2003;375:425–432. doi: 10.1042/BJ20030649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Roach MR, Burton AC. The reason for the shape of the distensibility curves of arteries. Can J Biochem Physiol. 1957;35:681–690. [PubMed] [Google Scholar]
- 230.Rodriguez EK, Hoger A, McCulloch AD. Stress-dependent finite growth in soft elastic tissues. J Biomech. 1994;27:455–467. doi: 10.1016/0021-9290(94)90021-3. [DOI] [PubMed] [Google Scholar]
- 231.Rosenquist TH, Beall AC. Elastogenic cells in the developing cardiovascular system. Smooth muscle, nonmuscle, and cardiac neural crest. Ann NY Acad Sci. 1990;588:106–119. doi: 10.1111/j.1749-6632.1990.tb13201.x. [DOI] [PubMed] [Google Scholar]
- 232.Rosenquist TH, McCoy JR, Waldo KL, Kirby ML. Origin and propagation of elastogenesis in the developing cardiovascular system. Anat Rec. 1988;221:860–871. doi: 10.1002/ar.1092210411. [DOI] [PubMed] [Google Scholar]
- 233.Ross JJ, Hong Z, Willenbring B, Zeng L, Isenberg B, Lee EH, Reyes M, Keirstead SA, Weir EK, Tranquillo RT, Verfaillie CM. Cytokine-induced differentiation of multipotent adult progenitor cells into functional smooth muscle cells. J Clin Invest. 2006;116:3139–3349. doi: 10.1172/JCI28184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Ross R. The elastic fiber. J Histochem Cytochem. 1973;21:199–208. doi: 10.1177/21.3.199. [DOI] [PubMed] [Google Scholar]
- 235.Ross R, Bornstein P. The elastic fiber. I. The separation and partial characterization of its macromolecular components. J Cell Biol. 1969;40:366–381. doi: 10.1083/jcb.40.2.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Sage H. The evolution of elastin: correlation of functional properties with protein structure and phylogenetic distribution. Comp Biochem Physiol B Comp Biochem. 1983;74:373–380. doi: 10.1016/0305-0491(83)90197-9. [DOI] [PubMed] [Google Scholar]
- 237.Sage H, Gray WR. Studies on the evolution of elastin. I. Phylogenetic distribution. Comp Biochem Physiol B Comp Biochem. 1979;64:313–327. doi: 10.1016/0305-0491(79)90277-3. [DOI] [PubMed] [Google Scholar]
- 238.Sage H, Gray WR. Studies on the evolution of elastin. III. The ancestral protein. Comp Biochem Physiol B Comp Biochem. 1981;68:473–480. doi: 10.1016/0305-0491(79)90277-3. [DOI] [PubMed] [Google Scholar]
- 239.Sakai LY, Keene DR, Engvall E. Fibrillin, a new 350-kD glycoprotein, is a component of extracellular microfibrils. J Cell Biol. 1986;103:2499–2509. doi: 10.1083/jcb.103.6.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Sakamoto H, Broekelmann T, Cheresh DA, Ramirez F, Rosenbloom J, Mecham RP. Cell-type specific recognition of RGD- and non-RGD-containing cell binding domains in fibrillin-1. J Biol Chem. 1996;271:4916–4922. [PubMed] [Google Scholar]
- 241.Sato S, Burdett I, Hughes RC. Secretion of the baby hamster kidney 30-kDa galactose-binding lectin from polarized and nonpolarized cells: a pathway independent of the endoplasmic reticulum-Golgi complex. Exp Cell Res. 1993;207:8–18. doi: 10.1006/excr.1993.1157. [DOI] [PubMed] [Google Scholar]
- 242.Schaefer L, Iozzo RV. Biological functions of the small leucine-rich proteoglycans: from genetics to signal transduction. J Biol Chem. 2008;283:21305–21309. doi: 10.1074/jbc.R800020200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Schulze-Bauer CA, Regitnig P, Holzapfel GA. Mechanics of the human femoral adventitia including the high-pressure response. Am J Physiol Heart Circ Physiol. 2002;282:H2427–H2440. doi: 10.1152/ajpheart.00397.2001. [DOI] [PubMed] [Google Scholar]
- 244.Schwartz SM, Benditt EP. Studies on aortic intima. I. Structure and permeability of rat thoracic aortic intima. Am J Pathol. 1972;66:241–264. [PMC free article] [PubMed] [Google Scholar]
- 245.Schwartz SM, Majesky MW, Murry CE. The intima: development and monoclonal responses to injury. Atherosclerosis. 1995;118(Suppl):S125–S140. [PubMed] [Google Scholar]
- 246.Schwartz SM, Mecham RP. The Vascular Smooth Muscle Cell: Molecular, and Biological Responses to the Extracellular Matrix. Academic; San Diego, CA: 1995. [Google Scholar]
- 247.Schwarze U, Atkinson M, Hoffman GG, Greenspan DS, Byers PH. Null alleles of the COL5A1 gene of type V collagen are a cause of the classical forms of Ehlers-Danlos syndrome (types I and II) Am J Hum Genet. 2000;66:1757–1765. doi: 10.1086/302933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Schwarze U, Schievink WI, Petty E, Jaff MR, Babovic-Vuksanovic D, Cherry KJ, Pepin M, Byers PH. Haploinsufficiency for one COL3A1 allele of type III procollagen results in a phenotype similar to the vascular form of Ehlers-Danlos syndrome, Ehlers-Danlos syndrome type IV. Am J Hum Genet. 2001;69:989–1001. doi: 10.1086/324123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Seliktar D, Black RA, Vito RP, Nerem RM. Dynamic mechanical conditioning of collagen-gel blood vessel constructs induces remodeling in vitro. Ann Biomed Eng. 2000;28:351–362. doi: 10.1114/1.275. [DOI] [PubMed] [Google Scholar]
- 250.Selmin O, Volpin D, Bressan GM. Changes of cellular expression of mRNA for tropelastin in the intraembryonic arterial vessels of developing chick revealed by in situ hybridization. Matrix. 1991;11:347–358. doi: 10.1016/s0934-8832(11)80206-4. [DOI] [PubMed] [Google Scholar]
- 251.Sengle G, Charbonneau NL, Ono RN, Sasaki T, Alvarez J, Keene DR, Bachinger HP, Sakai LY. Targeting of bone morphogenetic protein growth factor complexes to fibrillin. J Biol Chem. 2008;283:13874–13888. doi: 10.1074/jbc.M707820200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Shadwick RE, Gosline JM. Elastic arteries in invertebrates: mechanics of the octopus aorta. Science. 1981;213:759–761. doi: 10.1126/science.7256277. [DOI] [PubMed] [Google Scholar]
- 253.Shadwick RE. Mechanical design in arteries. J Exp Biol. 1999;202:3305–3313. doi: 10.1242/jeb.202.23.3305. [DOI] [PubMed] [Google Scholar]
- 254.Shapiro SD, Endicott SK, Province MA, Pierce JA, Campbell EJ. Marked longevity of human lung parenchymal elastic fibers deduced from prevalence of d-aspartate and nuclear weapons-related radiocarbon. J Clin Invest. 1991;87:1828–1834. doi: 10.1172/JCI115204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Shifren A, Durmowicz AG, Knutsen RH, Faury G, Mecham RP. Elastin insufficiency predisposes to elevated pulmonary circulatory pressures through changes in elastic artery structure. J Appl Physiol. 2008;105:1610–1619. doi: 10.1152/japplphysiol.90563.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Sicot FX, Tsuda T, Markova D, Klement JF, Arita M, Zhang RZ, Pan TC, Mecham RP, Birk DE, Chu ML. Fibulin-2 is dispensable for mouse development and elastic fiber formation. Mol Cell Biol. 2008;28:1061–1067. doi: 10.1128/MCB.01876-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Siegel RC, Pinnell SR, Martin GR. Cross-linking of collagen and elastin. Properties of lysyl oxidase. Biochemistry. 1970;9:4486–4492. doi: 10.1021/bi00825a004. [DOI] [PubMed] [Google Scholar]
- 258.Skalak R, Dasgupta G, Moss M, Otten E, Dullumeijer P, Vilmann H. Analytical description of growth. J Theor Biol. 1982;94:555–577. doi: 10.1016/0022-5193(82)90301-0. [DOI] [PubMed] [Google Scholar]
- 259.Skoglund P, Keller R. Xenopus fibrillin regulates directed convergence and extension. Dev Biol. 2007;301:404–416. doi: 10.1016/j.ydbio.2006.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Spencer JA, Hacker SL, Davis EC, Mecham RP, Knutsen RH, Li DY, Gerard RD, Richardson JA, Olson EN, Yanagisawa H. Altered vascular remodeling in fibulin-5-deficient mice reveals a role of fibulin-5 in smooth muscle cell proliferation and migration. Proc Natl Acad Sci USA. 2005;102:2946–2951. doi: 10.1073/pnas.0500058102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Starcher BC. Elastin and the lung. Thorax. 1986;41:577–585. doi: 10.1136/thx.41.8.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Starcher BC, Partidge SM, Elsden DF. Isolation and partial characterization of a new amino acid from reduced elastin. Biochemistry. 1967;6:2425–2432. doi: 10.1021/bi00860a019. [DOI] [PubMed] [Google Scholar]
- 263.Stenmark KR, Davie N, Frid M, Gerasimovskaya E, Das M. Role of the adventitia in pulmonary vascular remodeling. Physiology. 2006;21:134–145. doi: 10.1152/physiol.00053.2005. [DOI] [PubMed] [Google Scholar]
- 264.Suri C, Jones PF, Patan S, Bartunkova S, Maisonpierre PC, Davis S, Sato TN, Yancopoulos GD. Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell. 1996;87:1171–1180. doi: 10.1016/s0092-8674(00)81813-9. [DOI] [PubMed] [Google Scholar]
- 265.Sutcliffe MC, Davidson JM. Effect of static stretching on elastin production by porcine aortic smooth muscle cells. Matrix. 1990;10:148–153. doi: 10.1016/s0934-8832(11)80163-0. [DOI] [PubMed] [Google Scholar]
- 266.Syedain ZH, Weinberg JS, Tranquillo RT. Cyclic distension of fibrin-based tissue constructs: evidence of adaptation during growth of engineered connective tissue. Proc Natl Acad Sci USA. 2008;105:6537–6542. doi: 10.1073/pnas.0711217105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Szabo Z, Levi-Minzi SA, Christiano AM, Struminger C, Stoneking M, Batzer MA, Boyd CD. Sequential loss of two neighboring exons of the tropoelastin gene during primate evolution. J Mol Evol. 1999;49:664–671. doi: 10.1007/pl00006587. [DOI] [PubMed] [Google Scholar]
- 268.Taber LA, Eggers DW. Theoretical study of stress-modulated growth in the aorta. J Theor Biol. 1996;180:343–357. doi: 10.1006/jtbi.1996.0107. [DOI] [PubMed] [Google Scholar]
- 269.Taber LA, Humphrey JD. Stress-modulated growth, residual stress, and vascular heterogeneity. J Biomech Eng. 2001;123:528–535. doi: 10.1115/1.1412451. [DOI] [PubMed] [Google Scholar]
- 270.Tada T, Kishimoto H. Ultrastructural and histological studies on closure of the mouse ductus arteriosus. Acta Anat. 1990;139:326–334. doi: 10.1159/000147020. [DOI] [PubMed] [Google Scholar]
- 271.Takamizawa K, Hayashi K. Strain energy density function and uniform strain hypothesis for arterial mechanics. J Biomech. 1987;20:7–17. doi: 10.1016/0021-9290(87)90262-4. [DOI] [PubMed] [Google Scholar]
- 272.Tamburro AM, Bochicchio B, Pepe A. The dissection of human tropoelastin: from the molecular structure to the self-assembly to the elasticity mechanism. Pathol Biol. 2005;53:383–389. doi: 10.1016/j.patbio.2004.12.014. [DOI] [PubMed] [Google Scholar]
- 273.Tammi MI, Day AJ, Turley EA. Hyaluronan and homeostasis: a balancing act. J Biol Chem. 2002;277:4581–4584. doi: 10.1074/jbc.R100037200. [DOI] [PubMed] [Google Scholar]
- 274.Theocharis AD, Karamanos NK. Decreased biglycan expression and differential decorin localization in human abdominal aortic aneurysms. Atherosclerosis. 2002;165:221–230. doi: 10.1016/s0021-9150(02)00231-9. [DOI] [PubMed] [Google Scholar]
- 275.Thomassin L, Werneck CC, Broekelmann TJ, Gleyzal C, Hornstra IK, Mecham RP, Sommer P. The pro-regions of lysyl oxidase and lysyl oxidase-like 1 are required for deposition onto elastic fibers. J Biol Chem. 2005;280:42848–42855. doi: 10.1074/jbc.M506832200. [DOI] [PubMed] [Google Scholar]
- 276.Thompson RP, Fitzharris TP. Morphogenesis of the truncus arteriosus of the chick embryo heart: tissue reorganization during septation. Am J Anat. 1979;156:251–264. doi: 10.1002/aja.1001560206. [DOI] [PubMed] [Google Scholar]
- 277.Thyberg J, Hinek A, Nilsson J, Friberg U. Electron microscopic and cytochemical studies of rat aorta. Intracellular vesicles containing elastin- and collagen-like material. Histochem J. 1979;11:1–17. doi: 10.1007/BF01041263. [DOI] [PubMed] [Google Scholar]
- 278.Tiedemann K, Batge B, Muller PK, Reinhardt DP. Interactions of fibrillin-1 with heparin/heparan sulfate, implications for microfibrillar assembly. J Biol Chem. 2001;276:36035–36042. doi: 10.1074/jbc.M104985200. [DOI] [PubMed] [Google Scholar]
- 279.Torsney E, Hu Y, Xu Q. Adventitial progenitor cells contribute to arteriosclerosis. Trends Cardiovasc Med. 2005;15:64–68. doi: 10.1016/j.tcm.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 280.Toyoshima T, Ishida T, Nishi N, Kobayashi R, Nakamura T, Itano T. Differential gene expression of 36-kDa microfibril-associated glycoprotein (MAGP-36/MFAP4) in rat organs. Cell Tissue Res. 2008;332:271–278. doi: 10.1007/s00441-008-0587-7. [DOI] [PubMed] [Google Scholar]
- 281.Trackman PC, Pratt AM, Wolanski A, Tang SS, Offner GD, Troxler RF, Kagan HM. Cloning of rat aorta lysyl oxidase cDNA: complete codons and predicted amino acid sequence. Biochemistry. 1990;29:4863–4870. doi: 10.1021/bi00472a016. [DOI] [PubMed] [Google Scholar]
- 282.Umeda H, Nakamura F, Suyama K. Oxodesmosine and isooxodesmosine, candidates of oxidative metabolic intermediates of pyridinium cross-links in elastin. Arch Biochem Biophys. 2001;385:209–219. doi: 10.1006/abbi.2000.2145. [DOI] [PubMed] [Google Scholar]
- 283.Umeda H, Takeuchi M, Suyama K. Two new elastin cross-links having pyridine skeleton. Implication of ammonia in elastin cross-linking in vivo. J Biol Chem. 2001;276:12579–12587. doi: 10.1074/jbc.M009744200. [DOI] [PubMed] [Google Scholar]
- 284.Vaishnav RN, Vossoughi J. Residual stress and strain in aortic segments. J Biomech. 1987;20:235–239. doi: 10.1016/0021-9290(87)90290-9. [DOI] [PubMed] [Google Scholar]
- 285.Vaishnav RN, Young JT, Patel DJ. Distribution of stresses and of strain-energy density through the wall thickness in a canine aortic segment. Circ Res. 1973;32:577–583. doi: 10.1161/01.res.32.5.577. [DOI] [PubMed] [Google Scholar]
- 286.Vito RP, Dixon SA. Blood vessel constitutive models-1995-2002. Annu Rev Biomed Eng. 2003;5:413–439. doi: 10.1146/annurev.bioeng.5.011303.120719. [DOI] [PubMed] [Google Scholar]
- 287.Von Maltzahn WW, Besdo D, Wiemer W. Elastic properties of arteries: a nonlinear two-layer cylindrical model. J Biomech. 1981;14:389–397. doi: 10.1016/0021-9290(81)90056-7. [DOI] [PubMed] [Google Scholar]
- 288.Vuillemin M, Pexieder T. Normal stages of cardiac organogenesis in the mouse. II. Development of the internal relief of the heart. Am J Anat. 1989;184:114–128. doi: 10.1002/aja.1001840203. [DOI] [PubMed] [Google Scholar]
- 289.Wagenseil JE, Knutsen RH, Li D, Mecham RP. Elastin insufficient mice show normal cardiovascular remodeling in 2K1C hypertension, despite higher baseline pressure and unique cardiovascular architecture. Am J Physiol Heart Circ Physiol. 2007;293:H574–H582. doi: 10.1152/ajpheart.00205.2007. [DOI] [PubMed] [Google Scholar]
- 290.Wagenseil JE, Nerurkar NL, Knutsen RH, Okamoto RJ, Li DY, Mecham RP. Effects of elastin haploinsufficiency on the mechanical behavior of mouse arteries. Am J Physiol Heart Circ Physiol. 2005;289:H1209–H1217. doi: 10.1152/ajpheart.00046.2005. [DOI] [PubMed] [Google Scholar]
- 291.Wagenseil JE, Ciliberto CH, Knutsen RH, Levy MA, Kovacs A, Mecham RP. Reduced vessel elasticity alters cardiovascular structure and function in developing mice. Circ Res. 2009;104:1217–1224. doi: 10.1161/CIRCRESAHA.108.192054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Weigel PH, Hascall VC, Tammi M. Hyaluronan synthases. J Biol Chem. 1997;272:13997–14000. doi: 10.1074/jbc.272.22.13997. [DOI] [PubMed] [Google Scholar]
- 293.Weinbaum JS, Broekelmann TJ, Pierce RA, Werneck CC, Segade F, Craft CS, Knutsen RH, Mecham RP. Deficiency in microfibril-associated glycoprotein-1 leads to complex phenotypes in multiple organ systems. J Biol Chem. 2008;283:25533–25543. doi: 10.1074/jbc.M709962200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Wenstrup RJ, Florer JB, Brunskill EW, Bell SM, Chervoneva I, Birk DE. Type V collagen controls the initiation of collagen fibril assembly. J Biol Chem. 2004;279:53331–53337. doi: 10.1074/jbc.M409622200. [DOI] [PubMed] [Google Scholar]
- 295.Werneck CC, Vicente CP, Weinberg JS, Shifren A, Pierce RA, Broekelmann TJ, Tollefsen DM, Mecham RP. Mice lacking the extracellular matrix protein MAGP1 display delayed thrombotic occlusion following vessel injury. Blood. 2008;111:4137–4144. doi: 10.1182/blood-2007-07-101733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Wessels A, Markwald R. Cardiac morphogenesis and dysmorphogenesis. I. Normal development. Methods Mol Biol. 2000;136:239–259. doi: 10.1385/1-59259-065-9:239. [DOI] [PubMed] [Google Scholar]
- 297.Wiesmann F, Ruff J, Hiller KH, Rommel E, Haase A, Neubauer S. Developmental changes of cardiac function and mass assessed with MRI in neonatal, juvenile, and adult mice. Am J Physiol Heart Circ Physiol. 2000;278:H652–H657. doi: 10.1152/ajpheart.2000.278.2.H652. [DOI] [PubMed] [Google Scholar]
- 298.Wight TN. Versican: a versatile extracellular matrix proteoglycan in cell biology. Curr Opin Cell Biol. 2002;14:617–623. doi: 10.1016/s0955-0674(02)00375-7. [DOI] [PubMed] [Google Scholar]
- 299.Wilson E, Mai Q, Sudhir K, Weiss RH, Ives HE. Mechanical strain induces growth of vascular smooth muscle cells via autocrine action of PDGF. J Cell Biol. 1993;123:741–747. doi: 10.1083/jcb.123.3.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Wolff J. The Law of Bone Remodeling. Springer; Berlin: 1986. [Google Scholar]
- 301.Wolinsky H, Glagov S. Nature of species differences in the medial distribution of aortic vasa vasorum in mammals. Circ Res. 1967;20:409–421. doi: 10.1161/01.res.20.4.409. [DOI] [PubMed] [Google Scholar]
- 302.Wolinsky H. Response of the rat aortic media to hypertension. Morphological and chemical studies. Circ Res. 1970;26:507–522. doi: 10.1161/01.res.26.4.507. [DOI] [PubMed] [Google Scholar]
- 303.Wolinsky H, Glagov S. Structural basis for the static mechanical properties of the aortic media. Circ Res. 1964;14:400–413. doi: 10.1161/01.res.14.5.400. [DOI] [PubMed] [Google Scholar]
- 304.Wolinsky H, Glagov S. A lamellar unit of aortic medial structure and function in mammals. Circ Res. 1967;20:99–111. doi: 10.1161/01.res.20.1.99. [DOI] [PubMed] [Google Scholar]
- 305.Wuyts FL, Vanhuyse VJ, Langewouters GJ, Decraemer WF, Raman ER, Buyle S. Elastic properties of human aortas in relation to age and atherosclerosis: a structural model. Phys Med Biol. 1995;40:1577–1597. doi: 10.1088/0031-9155/40/10/002. [DOI] [PubMed] [Google Scholar]
- 306.Xu T, Bianco P, Fisher LW, Longenecker G, Smith E, Goldstein S, Bonadio J, Boskey A, Heegaard AM, Sommer B, Satomura K, Dominguez P, Zhao C, Kulkarni AB, Robey PG, Young MF. Targeted disruption of the biglycan gene leads to an osteoporosis-like phenotype in mice. Nat Genet. 1998;20:78–82. doi: 10.1038/1746. [DOI] [PubMed] [Google Scholar]
- 307.Yanagisawa H, Davis EC, Starcher BC, Ouchi T, Yanagisawa M, Richardson JA, Olson EN. Fibulin-5 is an elastin-binding protein essential for elastic fibre development in vivo. Nature. 2002;415:168–171. doi: 10.1038/415168a. [DOI] [PubMed] [Google Scholar]
- 308.Yao LY, Moody C, Schonherr E, Wight TN, Sandell LJ. Identification of the proteoglycan versican in aorta and smooth muscle cells by DNA sequence analysis, in situ hybridization and immunohistochemistry. Matrix Biol. 1994;14:213–225. doi: 10.1016/0945-053x(94)90185-6. [DOI] [PubMed] [Google Scholar]
- 309.Yeh H, Chow M, Abrams WR, Fan J, Foster JA, Mitchell H, Muenke M, Rosenbloom J. Structure of the human gene encoding the associated microfibrillar protein (MFAP1) and localization to chromosome 15Q15-Q21. Genomics. 1994;23:443–449. doi: 10.1006/geno.1994.1521. [DOI] [PubMed] [Google Scholar]
- 310.Zacchigna L, Vecchione C, Notte A, Cordenonsi M, Dupont S, Maretto S, Cifelli G, Ferrari A, Maffei A, Fabbro C, Braghetta P, Marino G, Selvetella G, Aretini A, Colonnese C, Bettarini U, Russo G, Soligo S, Adorno M, Bonaldo P, Volpin D, Piccolo S, Lembo G, Bressan GM. Emilin1 links TGF-beta maturation to blood pressure homeostasis. Cell. 2006;124:929–942. doi: 10.1016/j.cell.2005.12.035. [DOI] [PubMed] [Google Scholar]
- 311.Zanetti M, Braghetta P, Sabatelli P, Mura I, Doliana R, Colombatti A, Volpin D, Bonaldo P, Bressan GM. EMILIN-1 deficiency induces elastogenesis and vascular cell defects. Mol Cell Biol. 2004;24:638–650. doi: 10.1128/MCB.24.2.638-650.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Zhang H, Hu W, Ramirez F. Developmental expression of fibrillin genes suggests heterogeneity of extracellular microfibrils. J Cell Biol. 1995;129:1165–1176. doi: 10.1083/jcb.129.4.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Zhao A, Lee CC, Hiralerspong S, Juyal RC, Lu F, Baldini A, Greenberg F, Caskey CT, Patel PI. The gene for human microfibril-associated glycoprotein is commonly deleted in Smith-Magenis syndrome patients. Hum Mol Genet. 1995;4:589–597. doi: 10.1093/hmg/4.4.589. [DOI] [PubMed] [Google Scholar]
- 314.Zulliger MA, Stergiopulos N. Structural strain energy function applied to the ageing of the human aorta. J Biomech. 2007;40:3061–3069. doi: 10.1016/j.jbiomech.2007.03.011. [DOI] [PubMed] [Google Scholar]