

Abstract
While epidemiologic studies indicate that combined exposures to cigarette smoke and alcohol increase risk and severity of liver diseases, the molecular mechanisms responsible for hepatotoxicity are unknown. Similarly, emerging evidence indicates a linkage among hepatic steatosis and cardiovascular disease. Herein, we hypothesize that combined exposure to alcohol and environmental tobacco smoke (ETS) on a hypercholesterolemic background increase liver injury through oxidative/nitrative stress, hypoxia, and mitochondrial damage. To test this, male apoE-/- mice were exposed to an ethanol-containing diet, ETS alone, or a combination, and histology and functional endpoints were compared to filtered air, ethanol-naïve controls. While ethanol consumption induced a mild steatosis, combined exposure to ethanol + ETS resulted in increased hepatic steatosis, inflammation, alpha smooth muscle actin, and collagen. Exposure to ethanol + ETS induced the largest increase on CYP2E1 and iNOS protein, as well as increased 3-nitrotyrosine, mtDNA damage, and decreased cytochrome c oxidase protein compared to all other groups. Similarly, the largest increase in HIF1α expression was observed in the ethanol + ETS group indicating enhanced hypoxia. These studies demonstrate that ETS increases alcohol-dependent steatosis and hypoxic stress. Therefore, ETS may be a key environmental “hit” that accelerates and exacerbates alcoholic liver disease in hypercholesterolemic apoE-/- mice.
Keywords: liver, alcohol, steatosis, mitochondria, cigarette smoke, hypercholesterolemia, oxidative stress, hypoxia, cytochrome c oxidase, blue native gel electrophoresis
Introduction
Excessive consumption of alcohol (i.e. ethanol) causes liver disease by a combination of pathologic mechanisms including oxidative/nitrative stress, inflammation, redox alterations, and mitochondrial damage [1, 2]. During the early stages of alcoholic liver disease gut-derived endotoxin activates Kupffer cells, the resident hepatic macrophages, which release cytokines and reactive oxygen/nitrogen species (ROS/RNS) that negatively affect hepatocyte function [3, 4] particularly at the level of the mitochondrion. This alcohol-dependent depression in mitochondrial function is proposed to contribute to disease progression and severity via increased oxidative stress and bioenergetic defects leading to hepatocyte cell death [5]. However, it has been proposed that additional exposures or stressors are necessary for severe liver pathology to occur in the chronic alcohol consumer [6]. This concept is reinforced by statistics showing that only 25% of heavy drinkers develop alcoholic steatohepatitis with even fewer progressing to cirrhosis [7]. These observations have lead to a new hypothesis in the hepatology field that genetic, viral, metabolic, and/or environmental stressors or “hits” accelerate the progression from simple fatty liver (steatosis) to more severe liver pathologies including steatohepatitis, fibrosis, cirrhosis, and hepatocellular carcinoma [8].
While the direct hepatotoxic effects of alcohol have been extensively studied, the impacts of metabolic and/or environmental stressors or “hits” on those processes that lead to alcohol-induced hepatotoxicity remain undefined. Examples of these “hits” include metabolic stressors such as hyperglycemia, hypertriglyceridemia, and hypercholesterolemia that are associated with cardiovascular diseases and diabetes, whereas environmental stressors may include exposures to dietary factors, industrial pollutants, and tobacco smoke. It is estimated that alcohol and cigarette smoke exposures are the underlying causes in 35% and 50% of all deaths, respectively [9]. Moreover, while “second-hand” or environmental tobacco smoke (ETS), a widespread environmental toxicant, is a known risk factor for heart disease [10], the hepatotoxic potential of direct and indirect cigarette smoke exposure is only now being recognized [11]. Seminal work by Klatsky and colleagues showed that cigarette smoke exposure promotes alcoholic cirrhosis, increases certain cancers, and possibly increases mortality in heavy alcohol drinkers [12, 13]. Recent epidemiologic studies also suggest that cigarette smoke exposure accelerates a number of chronic liver diseases including hepatitis C and primary biliary cirrhosis, and increases the risk for hepatocellular carcinoma [14-18]. Importantly, obesity has been identified as a risk factor for alcoholic liver disease [19, 20] and alcohol, tobacco smoke, and obesity have been shown to be synergistic risk factors for hepatocellular carcinoma [21]. Taken together, these studies clearly support the hypothesis that tobacco smoke and other environmental “hits” may worsen the pathologic effects of alcohol in the liver, especially in obese individuals and those suffering from pre-existing illnesses like cardiovascular disease.
Although findings from epidemiologic studies demonstrate a correlative association between ethanol and ETS to worsen liver disease, the molecular mechanisms responsible for the interactive effects of ETS and ethanol to aggravate liver pathology are not known. Emerging evidence from the cardiovascular field strongly implicates oxidative stress and mitochondrial dysfunction with altered nitric oxide (NO) bioavailability in the disease process. For example, cigarette smoke damages the microvasculature via endothelial cell dysfunction and smooth muscle cell proliferation leading to impaired NO signaling and tissue hypoxia [22]. Rats exposed to ETS from only two cigarettes per day for two months have severely damaged myocardial oxidative phosphorylation function following ischemia-reperfusion [23]. Furthermore, ETS exposure in hypercholesterolemic mice causes significant aortic mitochondrial DNA (mtDNA) damage, increased tyrosine nitration, and inactivation of manganese superoxide dismutase when compared to hypercholesterolemia alone [24, 25]. These studies also demonstrate that when combined with hypercholesterolemia, ETS accelerates both mitochondrial damage and atherosclerosis [25]. Importantly, our laboratories have demonstrated that combined exposure to ethanol and ETS exacerbates atherosclerosis, which is accompanied by increased mtDNA damage and oxidant stress in heart and aorta when compared to hypercholesterolemic mice exposed to ethanol or ETS alone [26]. Due to the large body of literature advancing the cardio-protective benefits provided by mild-to-moderate alcohol consumption [27, 28], the study by Cakir and colleagues highlights the importance of considering the potential negative impacts of alcohol on the cardiovascular system when combined with known cardiovascular risk factors like hypercholesterolemia and ETS [26]. Indeed, the cardio-protective effects of alcohol have recently been questioned in several excellent review articles [29-31].
Because exposure to ETS is widespread and often occurs concurrently with alcohol consumption, investigations of their combined toxicities on the progression and severity of alcoholic liver disease are warranted and in the interest of public health. The importance of this is further supported because many of the responses elicited by cardiovascular risk factors like ETS and hypercholesterolemia are identical to those found in liver following chronic alcohol consumption. Indeed, there are overlapping impacts of chronic ethanol consumption, ETS, and hypercholesterolemia on mitochondrial function, which involve increased production of ROS/RNS and accumulation of mtDNA damage. Given this, and the likelihood that the chronic alcohol consumer is also likely to suffer from conditions of the cardiometabolic syndrome, it is crucial to begin to define the molecular mechanisms through which ETS and cardiovascular risk factors may accelerate alcoholic liver disease. To this end, several laboratories are using dyslipidemic mice models traditionally reserved for cardiovascular disease investigations to inform studies on the pathobiologic mechanisms underlying fatty liver disease. One approach has been to use hypercholesterolemic mouse models like the apoE-/- especially because it is known that apoE lipoprotein may play a key role in hepatic lipid metabolism and β-oxidation [32]. Specifically, studies show that triglyceride-very low density lipoprotein (VLDL) secretion is impaired in livers of ldlr-/- and apoE-/- mice and leads to development of fatty liver when mice are fed a Western diet [33-35]. These findings suggest that the apoE-/- mouse might be a novel model to study mechanisms of fatty liver disease from both alcohol and non-alcohol mediated sources, e.g. from high fat or high carbohydrate diets. Therefore, the current study builds upon our previous work demonstrating increased cardiovascular damage from combined exposures to ethanol and ETS [26] and was conducted to test the hypothesis that concurrent ethanol and ETS exposure on a dyslipidemic background significantly increase liver steatosis and injury through increased oxidative/nitrative stress, hypoxia, and mitochondrial damage.
Materials and methods
Animals
Male apoE-/- mice (C57BL/6 background, 6 weeks of age) were purchased from Jackson Laboratories (Bar Harbor, ME) and allowed to acclimate for 1 wk before beginning liquid diets. The apoE-/- mouse lacks apolipoprotein E, which is required for receptor mediated low density lipoprotein (LDL) cholesterol uptake from the blood. Consequently, mice are dyslipidemic with elevated levels of serum VLDL, LDL, cholesterol, and triglycerides, which predispose mice to develop atherosclerotic lesions similar to humans [36]. Mice were handled in accordance with guidelines outlined in the “Guide for the Care and Use of Laboratory Animals” (NIH publication 86-23 revised 1985) and the research protocol for these studies was approved by the institutional animal care and use committee.
Chronic ethanol consumption and ETS exposures
Animal exposures for these studies were performed at the Institute of Toxicology and Environmental Health inhalation facilities (University of California, Davis) as described previously [26]. Mice were fed nutritionally adequate control and ethanol-containing liquid diets formulated by Bio-Serv (Frenchtown, NJ). The ethanol-containing diet provided 28.8% of the total daily caloric intake as ethanol, 35% as fat, 18% as protein, and 18.2% as carbohydrate (maltose-dextrin). Control animals were pair-fed identical diets except that ethanol calories were replaced isocalorically with carbohydrate [37]. The ethanol concentration in the diet was increased from 0 to 28.8% of total daily caloric intake over a 9 day period and then maintained at the highest dose, 4% (w/v) ethanol, for the duration of the study to achieve blood ethanol concentrations of approximately 10 mM, which are in agreement with previously reported values [37]. ETS exposures and filtered air exposures began on day 14 after mice were acclimated to the 4% (w/v) ethanol-containing diet. Mice were exposed to ETS (10 mg/m3 total suspended particulate, TSP) or filtered air 5 hr per day, 5 days a week (Mon-Fri) for 4 additional weeks [26]. The concentration of carbon monoxide and nicotine at this ETS dose averaged 32.6 ± 5.6 ppm and 1.41 ± 0.2 mg/m3, respectively, throughout the 4 week exposure period. As noted previously [26] this dose of ETS is well below that reported in similar studies in rodents [38] and mimics that of physiologically relevant doses experienced by humans exposed to passive and/or active cigarette smoking [39]. In summary, there were 4 treatment groups for this study: 1) control diet + filtered air; 2) ethanol diet + filtered air; 3) control diet + ETS; and 4) ethanol diet + ETS.
Liver histopathology and Sirius red staining
Formalin-fixed liver samples were processed for hematoxylin-eosin staining and evaluated for steatosis, hepatocyte ballooning, and Mallory bodies by two hepatologists that were blinded to the experimental conditions [40]. The degree of steatosis (% of hepatocytes containing fat), the presence of intra-acinar (i.e. lobular) inflammation, and hepatocyte ballooning were used to define the severity of pathology. Steatosis was assessed by determining the percentage of hepatocytes containing fat droplets as 0 (< 5%); 1+ (6-25%); 2+ (26-50%); and 3+ (51-75%). Intra-acinar inflammation was scored as the presence of inflammatory cell aggregates: absence of inflammation was scored 0, 1 foci was scored 1, and 2-4 foci were scored 2 per 10X objective.
Collagen fibers were visualized in liver using Sirius red staining as described in [41]. Briefly, formalin-fixed liver sections were de-paraffinized and rehydrated by standard methods and then stained with 0.1% (w/v) Sirius red (Direct Red 80, Sigma-Aldrich, St. Louis, MO) in a saturated aqueous solution of picric acid for 60 min. The red-stained collagen fibers were quantified using the ImageJ software package (National Institutes of Health, Bethesda, MD) and expressed as percent area [41].
Immunoblot analyses for alpha smooth muscle actin, cytochrome P450 2E1 (CYP2E1), and inducible nitric oxide synthase (iNOS)
Immunoblots were performed by loading 20-100 μg of whole liver homogenate onto SDS-PAGE gels with proteins electroblotted onto nitrocellulose membranes for incubations with primary antibodies. Levels of iNOS protein were detected using a 1:5,000 dilution of antibody (Chemicon International, Temucula, CA). CYP2E1 protein was detected using a 1:1,000 dilution (Chemicon International) and alpha smooth muscle actin protein was detected using a 1:1,000 dilution of antibody (Dako, Carpinteria, CA). After incubation of membranes with appropriate HRP-conjugated secondary antibodies (Sigma Chemical Company, St. Louis), proteins were visualized using chemiluminescence detection methods and the intensity of immunoreactive protein bands were quantified using NIH Image (Bethesda, MD). The protein concentration loaded onto gels was within the linear range of the chemiluminescence system used for all proteins.
Immunostaining for hepatic 3-nitrotyrosine (3-NT), hypoxia inducible factor 1alpha (HIF1α), and alpha smooth muscle actin
A hallmark feature of RNS generation is nitration of tyrosine residues forming 3-NT residues, which can be detected by immunohistochemistry as described in [37]. Formalin-fixed liver sections were de-pariffinized in xylene and rehydrated through incubations in graded ethanol concentrations. Tissue sections were subjected to antigen retrieval in 10 mM citrate buffer, pH 6.0, endogenous peroxidase activity was quenched by incubation with 3% H2O2 for 10 min, and blocking was performed for 1 hr at room temperature with 10% goat serum diluted in a 1% (w/v) BSA-containing Tris-buffered saline with Tween-20 (TBS-T) solution. Sections were incubated for 2 hr at room temperature in a humidified chamber with rabbit polyclonal 3-NT antiserum at a 1:150 dilution. Following incubation, sections were washed, incubated with secondary antibody at a 1:200 dilution, and bound antibody was visualized with diaminobenzidine.
Hypoxia was verified by assessing the response of the hypoxia-sensitive protein HIF1α essentially as described above with minor modifications. Liver sections were subjected to antigen retrieval and blocking, followed by an overnight incubation at 4°C in a humidified chamber with anti-HIF1α antiserum (Lab Vision Corp., CA) at a 1:50 dilution. Following incubation, sections were washed in a 0.025% (w/v) Triton X-100, TBS solution and incubated with the secondary antibody, Alexa Fluor® 350 conjugated goat anti-mouse (Invitrogen, Eugene, OR) at a 1:50 dilution for 1 hr. Sections were washed with TBS and counterstained with DAPI (8 μg/mL, Sigma, St. Louis, MO) for 10 min to visualize nuclei.
Immunostaining for alpha smooth muscle actin was performed by standard methods [42]. After liver sections were de-parrifinized, rehydrated, incubated with 3% H2O2, and washed, they were blocked with 10% (w/v) BSA/TBS-T. Liver sections were incubated with alpha smooth muscle actin antiserum at a 1:800 dilution. After washings, liver sections were incubated with biotinylated anti-mouse IgG (Vector Laboratories, Burlingame, CA) at a 1:200 dilution followed by diaminobenzidine to visualize alpha smooth muscle actin. Smooth muscle actin and 3-NT were visualized as brown staining with brown area intensity quantified using either ImageJ software package (National Institutes of Health, Bethesda, MD) or Simple PCI software (Hamamatsu Corporation, Sewickley, PA), respectively. For HIF1α, images were taken and analyzed using a Leica fluorescent microscope with IPLAB Spectrum (BD Biosciences Bioimaging, Scanalytics, Rockville, MD) and the intensity of fluorescence quantified using Simple PCI software.
Blue native gel electrophoresis (BN-PAGE)
Mitochondria were isolated by differential centrifugation and BN-PAGE was performed as described in [40, 43]. For BN-PAGE, mitochondrial protein (1 mg) was suspended in extraction buffer (0.75 M aminocaproic acid, 50 mM Bis-Tris, and 1% (w/v) N-docecyl-β-D maltoside) and incubated on ice to release the intact respiratory complexes from the inner membrane. After incubation, Coomassie Blue G-250 was added and mitochondrial extracts (250 μg) were loaded on to non-denaturing gels to separate the individual protein complexes [44]. Gels were stained and complex densities were quantified using Scion Image Beta 4.01 (Scion Image Corp.) as described in [43]. There was no difference in mitochondrial protein yield and citrate synthase activities among groups [control + filtered air, 0.506 ± 0.021; ethanol + filtered air, 0.474 ± 0.042; control + ETS, 0.525 ± 0.027; ethanol + ETS, 0.545 ± 0.022 μmol/min/mg protein, p=0.41]. This is important because it demonstrates that the high levels of fat in liver of the ethanol + ETS group had no effect on isolation, overall content, and purity of mitochondria used for BN-PAGE. Cytochrome c oxidase activity was measured using a standard spectrophotometric method that monitors the oxidation of ferrocytochrome c at 550 nm [45] and oxidase activities were normalized to citrate synthase activities [46] for each individual mitochondrial sample.
Quantitative polymerase chain reaction (QPCR) for mtDNA damage
Genomic DNA was extracted from flash-frozen liver tissue, quantified using PicoGreen (Molecular Probes, Eugene, OR), and QPCR performed as described previously [40, 43]. The principle of this method is that DNA lesions block the rTth polymerase, which results in decreased amplification of the gene target of interest, i.e. mitochondrial genome. Sensitivity of the assay is increased via amplification of large targets thereby increasing the probability of encountering a DNA lesion. A 16,059 bp QPCR product that includes all but 236 bp of the NADH5/6 genes in the mouse mtDNA genome was amplified using a Gene AMP PCR system 2400 with Accuprime polymerase and a specified primer set [25]. Copy number differences in mtDNA were normalized using a short QPCR reaction (base pairs 12281-13361 of the mtDNA), which yields products directly related to gene copy numbers. Mitochondrial DNA damage was quantified by comparing the relative efficiency of amplification of large (>15 kb) fragments of DNA and normalizing this to gene copy numbers by the amplification of smaller (<250 bp) fragments, which have a statistically negligible likelihood of containing damaged bases. DNA lesion frequencies were calculated as described in [47].
Statistical Analyses
Data represent the mean ± SEM for 3-6 pairs of animals in each group. Significant differences between treatments were obtained using paired and unpaired t-tests and two-factor ANOVA, p<0.05. The Mann-Whitney rank sum test was used for pathology analyses.
Results
Male apoE-/- mice were pair-fed either a control or ethanol-containing liquid diet for 6 weeks with ETS or filtered air exposure during the last 4 weeks of the experimental study as described previously [26]. Mice were exposed to ETS at 10 mg/m3 total suspended particulate 5 hr per day/5 days per week; an exposure that is well below those in published reports on ETS exposure in rodents [48]. Ethanol and ETS exposure had no effect on body weight [control + filtered air, 24.3 ± 0.7; ethanol + filtered air, 23.7 ± 1.3; control + ETS, 24.0 ± 1.73; and ethanol + ETS, 27.3 ± 1.5 g, p=0.18]. Similarly, there was no difference in ethanol consumption between the ETS and filtered air exposure groups throughout the feeding period [ethanol + filtered air, 12.3 ± 0.3 and ethanol + ETS, 13.6 ± 0.3 mL of diet/day, p=0.6]. These results of ethanol consumption are similar to those previously seen in our laboratories when using mice [37] and demonstrate that ETS exposure has no effect on ethanol consumption.
Histologic examination of livers showed a large accumulation of macrovesicular, as well as microvesicular fat in the livers of apoE-/- mice exposed to ETS and fed an ethanol-containing diet when compared to all other experimental groups (Figure 1A, panel d). Livers from the ethanol alone group show only a mild steatosis (Figure 1A, panel b), which agrees with our previous studies using wild-type and iNOS-/- mice fed the same ethanol-containing diet [37]. Quantification of steatosis demonstrates that while only 5% of hepatocytes contained fat droplets in the ethanol alone group, 50% of hepatocytes across all zones of the liver lobule contained fat in the ethanol + ETS group (Figure 1B). Moreover, livers from both control groups (ETS alone or filtered air) show no signs of fat accumulation and exhibited no pathological changes (Figure 1A, panels a and c), consistent with previous studies using this same corn oil based-high fat diet in mice [37]. Hepatocyte ballooning was observed in 1 of 3 ethanol exposed mice and in 1 of 3 ethanol + ETS treated mice, whereas hepatocyte ballooning was absent in both control groups.
Fig. 1. ETS increases ethanol dependent steatosis and inflammation.
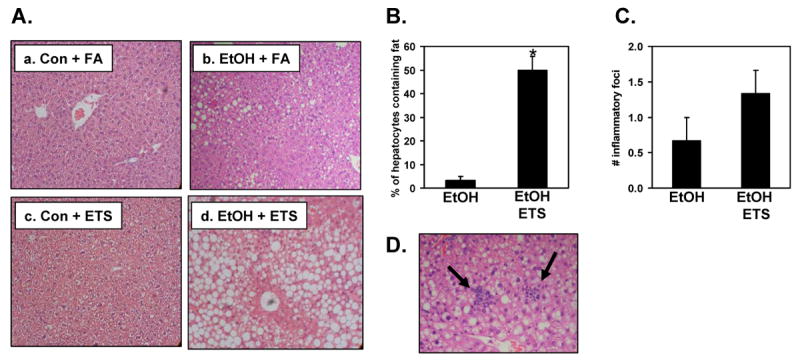
Liver was fixed in 10% buffered formalin, embedded in paraffin, sectioned, and stained with hematoxylin-eosin. Panel A, Livers from mice fed control diets (Con) exposed to filtered air (FA) (a) or ETS (c) show no pathological changes or fat deposition in hepatocytes. Mice fed an ethanol-containing diet (EtOH) and exposed to filtered air (b) show mild steatosis, whereas ethanol-fed mice exposed to ETS (d) have severe steatosis. Magnification is 100X. Panel B, Quantification of steatosis. Panel C, Quantification of inflammatory foci. Panel D, Photomicrograph showing aggregates of neutrophils and lymphocytes within liver parenchyma of ethanol + ETS group (arrows). Magnification is 200X.
This large increase in steatosis in the ethanol + ETS group was accompanied by increased inflammatory foci and necrosis. Figure 1D shows increased neutrophils and lymphocytes within the liver parenchyma from animals exposed to both ethanol and ETS (see arrows). While there was increased inflammation in the ethanol + ETS group as compared to ethanol alone this difference was not statistically significant (Figure 1C). Accompanying the histologic presence of steatosis, co-exposure to ethanol + ETS also resulted in a significant increase in the liver weight and liver/body weight ratio as compared to ethanol and ETS only groups (Figure 2) indicating liver injury. ETS exposure alone had no effect on these two parameters when compared to the control diet only group.
Fig. 2. ETS significantly enhances the ethanol dependent increase in liver weight and liver/bodyweight ratio.

Mice were pair-fed control (Con) and ethanol (EtOH)-containing diets for 6 weeks while being exposed to filtered air (FA) or ETS (10 mg TSP/m3) for the last 4 weeks of the study. ETS exposure significantly increased the liver weight and liver/body weight ratio in ethanol-fed mice. Data represent the mean ± SEM for 6 mice per group. *p<0.05, compared to corresponding control; **p<0.05, compared to ethanol + filtered air. For liver weight; two-factor ANOVA, ethanol, p=0.0008; ETS, p=0.0023; ethanol × ETS, p=0.017. For liver/body weight ratio; two-factor ANOVA, ethanol, p=0.016; ETS, p=0.042; ethanol × ETS, 0.27.
Previous studies have shown that hypoxia is induced in liver by both acute and chronic ethanol consumption and contributes to hepatotoxicity [49, 50]. To determine whether ETS enhanced ethanol-mediated hypoxia, HIF1α expression was measured by immunofluorescence. Figure 3 shows a significant increase in HIF1α staining in liver from ethanol + filtered air mice (Figure 3A, panel b) when compared to their corresponding control (Figure 3A, panel a). There was no difference in HIF1α expression in mice fed control diets and those exposed to ETS only (Figure 3A, compare panels a and c). Interestingly, there was a significant increase in HIF1α expression in liver of mice co-exposed to ethanol and ETS (Figure 3A, panel d) when compared to the ethanol only group (Figure 3A, panel b). Quantification of HIF1α expression is presented in Figure 3B.
Fig. 3. ETS enhances ethanol-dependent hypoxia in liver.
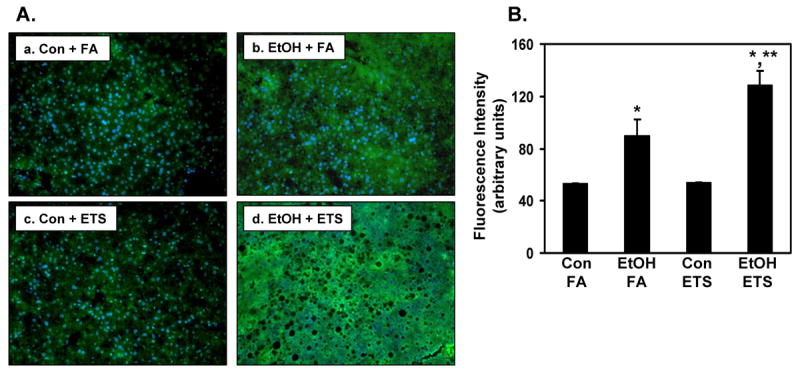
Immunofluorescence was performed on formalin fixed liver tissue using HIF1α antiserum (green) and nuclei were counterstained with DAPI (blue). No statistical difference in HIF1α levels was observed between control groups (Con + FA, panel a and Con + ETS, panel c), whereas there was a significant increase in HIF1α staining in the ethanol + filtered air group (EtOH + FA, panel b) compared to corresponding control. This increase in HIF1α was amplified in the ethanol + ETS group (panel d) with widespread HIF1α expression in liver. Panel B shows quantification with *p<0.05, compared to corresponding controls, and **p<0.05, compared to ethanol + filtered air. Two-factor ANOVA, ethanol, p=0.00013; ETS, p=0.043; ethanol × ETS, p=0.048. Data represent the mean ± SEM for n=3 pairs per group. Magnification is 100X.
Because these findings suggested acceleration in liver injury by combined exposures to ethanol and ETS, the levels of alpha smooth muscle actin were determined as an early marker of fibrogenesis. Immunoblot analysis demonstrates that combined exposure to ethanol + ETS increased the level of alpha smooth muscle actin in liver by 65% when compared to the ETS alone group and 40% when compared to the ethanol alone group (Figure 4A). Ethanol consumption alone increased the levels of alpha smooth muscle actin by 50% compared to the control + filtered air; however, this increase was not statistically significant (p=0.078). ETS had no effect on the levels of alpha smooth muscle actin in control fed animals. The ethanol and ETS-associated effects on alpha smooth muscle actin were also confirmed and visualized with immunohistochemistry (Figure 4B) with increased staining around veins and in tissue (Figure 4B, panel d). In addition, livers from ethanol + ETS group showed increased staining for collagen compared to all treatment groups (Figure 5). Increased collagen staining (i.e. faint, red fibers) was observed radiating out from the central vein regions in livers from the ethanol + ETS group (see arrows, Figure 5A, panel d) whereas red staining is only seen around vessels in the other treatment groups. Quantification of Sirius red staining is presented in Figure 5B.
Fig. 4. Induction of alpha smooth muscle actin by ethanol and ETS in liver.
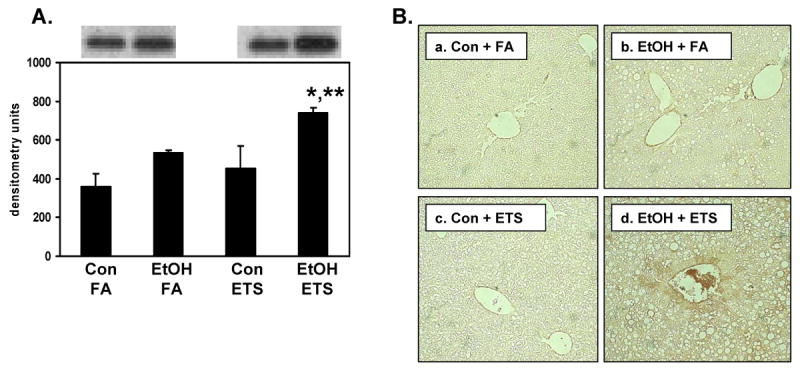
Panel A, Liver homogenates were subjected to SDS-PAGE and immunoblotting for quantification of alpha smooth muscle actin protein. The top panel in 4A shows representative immunoblots from one pair of control (Con) and ethanol (EtOH)-fed mice exposed to either filtered air (FA) or ETS. The bottom panel in 4A represents the quantification of alpha smooth muscle actin protein in immunoblots. Panel B, Immunohistochemistry was also performed on formalin fixed tissue and representative photomicrographs from each treatment group are shown: a, Con + FA; b, EtOH + FA; c, Con + ETS; and d, EtOH + ETS. Increased brown staining for alpha smooth muscle actin was observed in the EtOH + ETS group (d) compared to other treatment groups. Data represent the mean ± SEM for n=3 pairs per group. Immunoblot statistics - *p<0.05, compared to corresponding control; **p<0.05, compared to ethanol + filtered air. Two-factor ANOVA; ethanol, p=0.011; ETS, p=0.067; ethanol × ETS, p=0.46. Immunohistochemistry data and statistics – Con + FA, 5.4 ± 0.15; EtOH + FA, 11.4 ± 0.9; Con + ETS, 5.7 ± 0.4; and EtOH + ETS, 16.0 ± 1.9 arbitrary intensity units. Two factor ANOVA; ethanol, p<0.001; ETS, p=0.049; ethanol × ETS, p=0.08. Magnification is 200X.
Fig. 5. Increased collagen fibers by ethanol and ETS in liver.
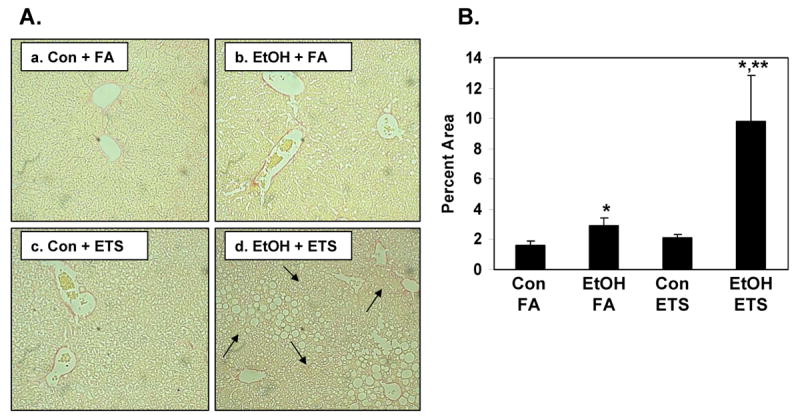
Liver sections were stained with Sirius red and analyzed to detect and quantify collagen. Panel A, There was increased Sirius red staining in EtOH + FA group (b) compared to control diet alone (a). Increased Sirius red staining was observed in the EtOH + ETS group (d) with increased collagen detected around the central veins that radiated out into the parenchyma (arrows, panel d). Panel B, Image analysis (percent area) demonstrated increased Sirius red staining in liver from EtOH + ETS group compared to all other groups. Data represent the mean ± SEM for 3 pairs of mice. Magnification is 200X. *p<0.05, compared to each corresponding control; **p<0.05, compared to ethanol + filtered air. Two-factor ANOVA; ethanol, p=0.019; ETS, p=0.044; ethanol × ETS, 0.074.
There are several potential sources of ROS/RNS in hepatocytes, which are induced or altered by chronic ethanol consumption resulting in increased production of oxidants. The data shown in Figure 6 confirms results demonstrating induction of hepatic CYP2E1 protein in response to chronic ethanol consumption [40]. More important, is the observation that the combination of ethanol and ETS resulted in a much larger increase in CYP2E1 protein levels when compared to the ethanol alone group (Figure 6).
Fig. 6. Combined exposure to ethanol and ETS significantly increases hepatic levels of CYP2E1 protein.
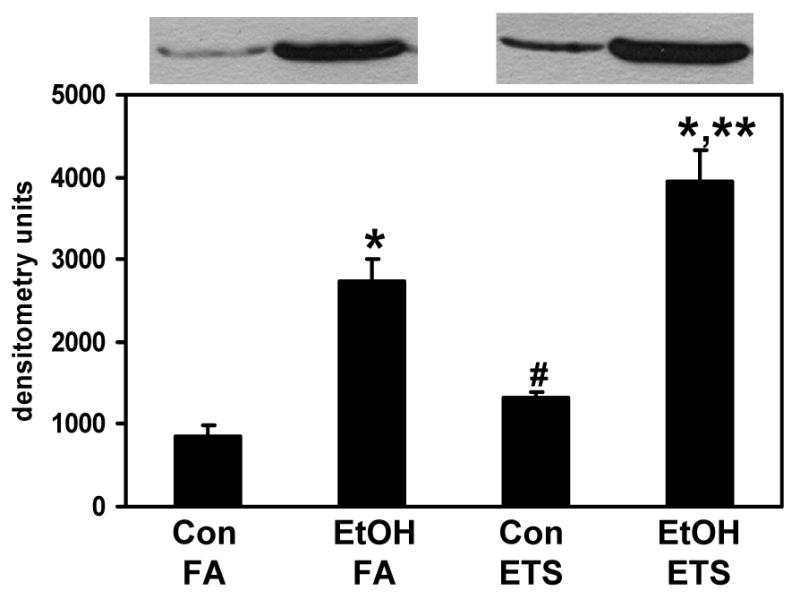
Liver homogenates were subjected to SDS-PAGE and immunoblotting for quantification of CYP2E1 protein. Top panel shows representative immunoblots from one pair of control (Con) and ethanol (EtOH)-fed mice exposed to either filtered air (FA) or ETS. Bottom panel represents the quantification of CYP2E1 protein. Data represent the mean ± SEM for n=3 pairs per group. *p<0.05, compared to corresponding control; **p<0.05, compared to ethanol + filtered air; #p<0.05, compared to control + filtered air. Two-factor ANOVA; ethanol, p<0.0001; ETS, p=0.008; ethanol × ETS, 0.17.
Increased expression of iNOS has also been associated with chronic alcohol-dependent oxidative/nitrative stress and inflammation [37, 51]; however, the impact of ETS on hepatic iNOS expression is not known. While there was a modest ethanol associated increase in iNOS protein, the combination of ethanol and ETS significantly increased hepatic iNOS protein levels when compared to either ethanol or ETS alone (Figure 7). These results are in agreement with those showing increased heart iNOS transcripts and protein following ethanol and ETS exposure [26]. Similarly, we assessed the effect of ethanol and ETS on protein nitration by measuring the levels of 3-NT in liver tissue as described previously [37]. As shown in Figure 8, little to no 3-NT was observed in the liver of mice exposed to the control diet only (Figure 8A, panel a), with increased 3-NT in the ETS alone (panel c) and ethanol alone (panel b) groups. In contrast, there was a large increase in 3-NT staining in the ethanol + ETS group (panel d), which was significantly increased over all treatments (Figure 8B). These results, in combination with those presented for iNOS, suggest that simultaneous exposure to ethanol and ETS causes a significant increase in RNS production and potential nitrative stress in liver.
Fig. 7. Exposure to ethanol and ETS significantly increases hepatic levels of iNOS.
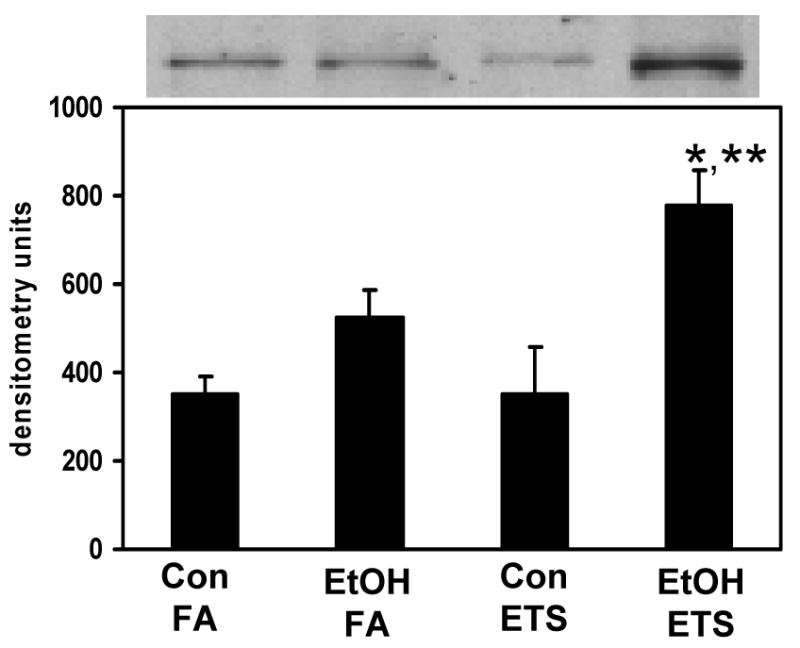
Liver homogenates were subjected to SDS-PAGE and immunoblotting for quantification of iNOS protein. Top panel shows representative immunoblots from one pair of control (Con) and ethanol (EtOH)-fed mice exposed to either filtered air (FA) or ETS. Bottom panel represents the quantification of iNOS. Data represent the mean ± SEM for n=3 pairs per group. *p<0.05, compared to corresponding control; **p<0.05, compared to ethanol + filtered air. Two-factor ANOVA; ethanol, p=0.002; ETS, p=0.13; ethanol × ETS, p=0.12.
Fig. 8. Hepatic levels of 3-NT are increased by exposure to ethanol and ETS.
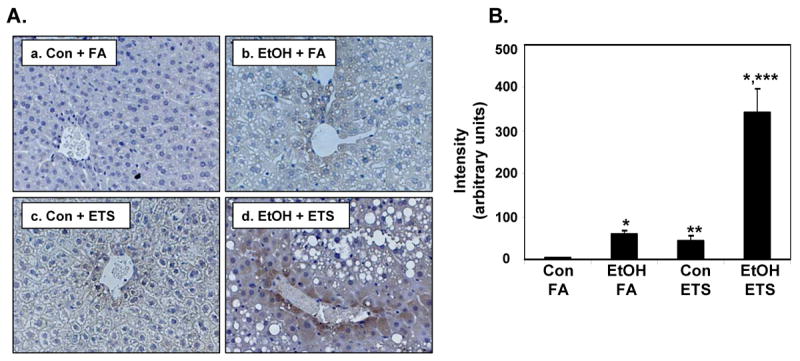
Immunohistochemistry was performed on formalin fixed tissues using 3-NT antiserum (brown) against a hemtoxylin nuclear counterstain (blue) in liver sections of control (Con) and ethanol (EtOH)-fed mice exposed to either filtered air (FA) or ETS. Panel A, There was increased 3-NT in control + ETS group (c) and the EtOH + FA group (b) compared to control diet alone (a). Increased 3-NT was observed in the EtOH + ETS group (d) with intense staining for 3-NT around the central veins. Panel B, Image analysis demonstrated increased intensity of 3-NT in liver from EtOH + ETS group compared to all other groups. Data represent the mean ± SEM for 3 pairs of mice. Magnification is 200X. *p<0.05, compared to each corresponding control; **p<0.05, compared to control + filtered air; ***p<0.05, compared to ethanol + filtered air. Two-factor ANOVA; ethanol, ETS, and ethanol × ETS, p<0.0001, ethanol, ETS.
Based on results indicating increased oxidative and nitrative stress in liver of mice exposed to ethanol and/or ETS, it was predicted that there would be increased mitochondrial damage in response to both toxicant exposures. To examine the effect of ethanol and ETS on mitochondria, mtDNA damage was assessed using QPCR. As shown in Figure 9, mtDNA damage progressively increased from low levels of mtDNA damage in the ethanol alone group to a substantially higher level in the ETS and ethanol + ETS exposure groups. Thus, through damaging the mtDNA it is hypothesized that combined exposure to ethanol and ETS will increase mitochondrial dysfunction and worsen liver injury.
Fig. 9. Liver mitochondrial DNA damage is increased by ethanol and ETS.

Genomic DNA was prepared from livers and mtDNA damage was assessed using QPCR. Data presented in the bar graph indicate the relative amount of mtDNA damage (lesions per 16 kb) in liver from ethanol-fed (EtOH), ETS exposed, and EtOH + ETS animals compared with their corresponding controls. Controls are set at zero lesions per 16kb. Data represent the mean ± SEM for n=3 pairs animals per group, *p<0.05, compared to corresponding control.
To assess the effect of ethanol, ETS, and ethanol + ETS on the mitochondrial oxidative phosphorylation system, BN-PAGE was used. BN-PAGE is a specialized form of gel electrophoresis that is particularly well-suited for the separation and analysis of the large, hydrophobic respiratory complexes that reside in the mitochondrial inner membrane and comprise the oxidative phosphorylation system [52]. Figure 10A shows a representative BN-PAGE separation of the five oxidative phosphorylation system complexes from each treatment group. The complexes are separated intact on a non-denaturing gel, arranged vertically based on molecular weight, and are visualized as large protein bands (Figure 10A, arrows). Quantification of the density of each complex revealed modest decreases in protein content in response to ethanol or ethanol + ETS for complexes I, II, III, and V; however, none of these differences were statistically significant (Figure 10B). However, there was a significant decrease in the content of cytochrome c oxidase (i.e. complex IV) in the ethanol + ETS group when compared to the ethanol only group (p=0.022). Similarly, two-factor ANOVA analyses showed a statistically significant effect of ETS on complex IV content compared to filtered air groups (p=0.015). Cytochrome c oxidase activity was also decreased in the ethanol + ETS group compared to the ethanol only group; however, this difference was not statistically significant [control + filtered air, 0.093 ± 0.02; ethanol + filtered air, 0.086 ± 0.02; control + ETS, 0.091 ± 0.03; ethanol + ETS, 0.071 ± 0.015 U/units citrate synthase, p=0.19, ethanol only compared to ethanol + ETS].
Fig. 10. Ethanol and ETS decrease cytochrome c oxidase in liver.
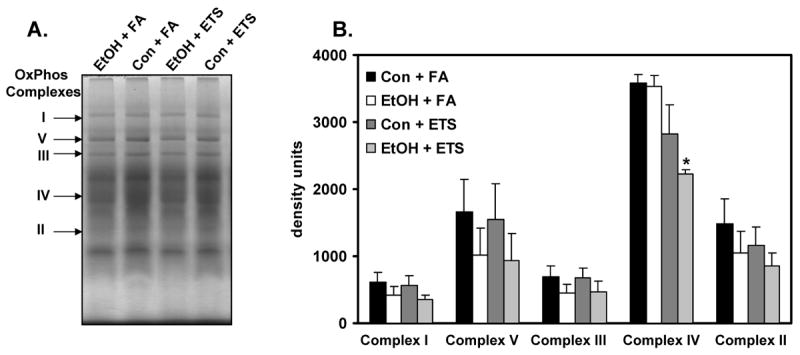
Representative BN-PAGE gels of mitochondria from control + filtered (Con + FA), ethanol + filtered air (EtOH + FA), control + ETS (Con + ETS), and ethanol + ETS (EtOH + ETS) are shown in panel A. For these gels, 250 μg of mitochondrial protein was loaded onto non-denaturing 5-15% gradient gels to separate the oxidative phosphorylation complexes intact and in native form. The complexes are labeled by the arrows. Panel B presents a comparison of the relative densities (i.e. quantities) of complexes I, V, III, IV, and II from each of the 4 treatment groups. Data represent the mean ± SEM for 3 pairs per group. *p=0.022, compared to ethanol + filtered air group. Two-factor ANOVA, ethanol, p=0.55; ETS, p=0.015; ethanol × ETS, p=0.66.
Discussion
Studies designed to investigate the combined toxicities of ethanol and cigarette smoke on liver pathogenesis have largely relied on ethanol exposure in combination with nicotine injections, which have shown decreased antioxidants, increased lipid peroxidation, and increased CYP2E1 levels [53-55]. Despite these efforts, it has been difficult to investigate the physiologic and mechanistic mechanisms through which cigarette smoke and especially “second-hand” exposure (i.e. ETS), worsen liver disease in the ethanol consumer. To address this issue, Gentry-Nielsen and colleagues made significant progress by developing a physiologically relevant rat model of combined ethanol and cigarette smoke exposure [38]. Using this model, they observed alcohol-dependent defects in liver function, decreased asialoglycoprotein receptor binding, and alterations in blood cholesterol levels with no additional metabolic disturbances observed when rats were co-exposed to cigarette smoke [38]. Based on this [38] and the knowledge that fatty liver disease is now associated with cardiovascular disease risk [56, 57], we propose that experimental models of dyslipidemia and atherosclerosis, like the apoE-/- hypercholesterolemic mouse, may be important tools to better understand mechanisms of liver disease from combined exposures to ethanol and other environmental toxicants. This concept is strengthened by reports that hypercholesterolemic ldlr-/- and apoE-/-mice develop steatohepatitis when fed a high fat, Western diet supplemented with cholesterol [33-35]. While the mechanism for increased steatosis in these hypercholesterolemic mouse models is not clear, it has been suggested that in addition to its role in lipoprotein clearance, apoE protein may function in VLDL assembly and secretion [33]. As a consequence, apoE deficiency may contribute, in part, to the development of steatosis due to reduced VLDL-triglyceride secretion, which is not related to defects in triglyceride turnover, diacylglycerol acyltransferase activity, or microsomal triglyceride transfer protein levels [58].
Additionally, impaired VLDL secretion may not be the sole cause for steatosis in the apoE-/- mouse. For example, changes in the expression of β-oxidation and lipid metabolism proteins may underlie fat accumulation as shown in mice expressing the mutant APOE*3 Leiden isoform that were fed a high fat/cholesterol-containing diet [32]. In humans, the mutant APOE*Leiden leads to dysbetalipoproteinemia [59] and mice expressing this transgene develop hyperlipidemia due to impaired binding of mutant-remnant lipoproteins to the LDL receptor [60]. To date, it is unclear whether there is a link between hypercholesterolemia (i.e. APOE genotype) and the incidence and severity of hepatic steatosis associated with the cardiometabolic syndrome or excessive alcohol intake. However, two recent studies indicate an association between APOE polymorphisms in alcohol and nonalcoholic steatohepatitis. Specifically, Panduro and colleagues found an association between hypertriglyceridemia and the APOE*2 allele with early onset alcoholic cirrhosis [61], whereas the APOE*3 allele was overrepresented in a group of nonalcoholic steatohepatitis patients [62]. This, taken together with data presented herein demonstrating a large increase in steatosis in apoE-/- mice exposed to ethanol and ETS, supports the concept that genetically determined stressors like hypercholesterolemia may be important additional metabolic “hits” that should be considered in studies of the molecular events involved in liver disease pathogenesis. To clarify this, future studies using wild-type mice exposed to ethanol and ETS are warranted to shed light on the precise role of hypercholesterolemia in this “multi-hit” model of fatty liver disease. These studies are critical to determining whether or not a dyslipidemic genetic background is required for the steatosis-enhancing properties of ETS in a model of alcoholic liver disease. It is possible that ETS may not enhance alcohol-dependent steatosis in the absence of dyslipidemia.
Using well-established models of ETS exposure and chronic ethanol consumption, we observed that steatosis was significantly increased by simultaneous exposures to ETS and ethanol, which was accompanied by increased liver weight and liver/body weight ratio; additional measures of liver injury. Combined exposure to ETS and ethanol also increased the number of inflammatory foci in liver, as well as alpha smooth muscle actin levels and collagen; early indicators of fibrogenesis. As discussed earlier, the metabolic defects associated with hypercholesterolemia may increase fat accumulation in liver through alterations in lipoprotein and triglyceride processing [58], thereby predisposing liver to increased damage when exposed to “hits” like ethanol and ETS. It should be pointed that previous studies showing increased fat in untreated apoE-/- mice were conducted using Western diets high in butter fat or palm oil with varying concentrations of added cholesterol [33, 34]. In contrast, the lack of overt steatosis in the untreated control group in this current study may be due to differences in dietary fat type. For example, the diet used herein was a corn oil based (largely linoleate) and contained no added cholesterol. It is predicted that the inclusion of cholesterol in this diet may have enhanced steatosis in all groups. Previously, we demonstrated that chronic ethanol consumption increased total plasma cholesterol in apoE-/- mice, which when combined with ETS exposure was elevated to much higher levels [26]. Important to the current study, is the report from Mari et al [63] demonstrating that cholesterol loading of liver and mitochondria increases tumor necrosis factor α-dependent liver injury due to mitochondrial glutathione depletion, thereby sensitizing the liver to oxidative and nitrative stress. Consequently, increased cholesterol is proposed to accelerate the progression from simple steatosis to more severe liver pathologies like steatohepatitis. Taken together, these findings suggest that an additional metabolic “hit” like hypercholesterolemia may contribute, in part, to increase the severity of fatty liver disease in populations consuming alcohol and exposed to environmental toxicants like cigarette smoke.
Increased expression of iNOS and production of RNS are associated with inflammation observed in alcohol-induced fatty liver disease [37, 51]. NO and its metabolites have been linked to mechanisms of hepatotoxicity through mtDNA damage, as well as through post-translational modification of cellular and mitochondrial proteins via oxidation, nitrosation, and nitration [64-66]. Specifically, excess NO reacts with superoxide (O2•−) to produce ONOO−, a highly reactive species, which has been implicated as a key mediator in NO-dependent cellular and mitochondrial dysfunction via protein nitration [67]. Whereas the levels of iNOS and CYP2E1, a source of O2•−, were elevated over control values by ethanol consumption alone, greater increases were observed in the ethanol + ETS group. Similarly, nitration and mtDNA damage were increased in the ethanol + ETS group compared to other groups. These results are in agreement with those observed in heart tissue of apoE-/- mice exposed to ethanol + ETS, which showed increased iNOS, oxidant damage, and atherosclerosis [26]. Taken together, this suggests a common mechanism involving oxidative and/or nitrative stress in the pathobiology of atherosclerosis and fatty liver disease; two conditions of the cardiometabolic syndrome.
Hypoxia also has been shown to contribute to hepatotoxicity from a number of toxicants including alcohol and with other pathologies linked to steatosis. Studies show that alcohol consumption increases hypoxia in the pericentral region of the liver (i.e. zone 3) due to increased O2 consumption in response to ethanol oxidation [49]. Similarly, chronic hypoxia associated with obstructive sleep apnea has been implicated as a contributing factor to the development of fatty liver disease [68] and atherosclerosis [69] in obese individuals. It is proposed that the interaction of hypoxia with other stressors or “hits”, either metabolic or environmental, may participate in the progression from simple steatosis to steatohepatitis. What hasn't been studied to date is whether combined exposure to ETS exacerbates ethanol-induced hypoxia.
It is well established that a critical adaptation to reduced oxygen availability is upregulation of the transcription factor HIF1α and adaptive pathways to prevent hypoxic injury. Consistent with the histopathology presented herein, is the observation that liver from the ethanol + ETS group showed the highest level of HIF1α expression indicating enhanced hypoxia from this combined toxicant exposure. ETS may enhance liver hypoxia due to the presence of low amounts of carbon monoxide in tobacco smoke, which could have the effect of reducing the amount of oxygen delivery to hepatocytes through formation of carboxyhemoglobin [70]. Similarly, the presence of these multiple “hits” could increase oxygen demand in liver due to increased metabolic activity thereby enhancing tissue hypoxia. This may be particularly relevant given that increased mtDNA damage and cytochrome c oxidase defects were also observed in the ethanol + ETS group. It is possible that one precarious consequence of these damaged mitochondria may be a disturbance in the normal O2 gradient within the liver lobule resulting in increased hypoxia. In particular, it is proposed that defective mitochondria in the ethanol + ETS group will have increased O2 uptake to attempt to maintain sufficient ATP levels in hepatocytes for use in detoxification and/or repair pathways. This would result in a “steepening” of the liver O2 gradient due to increased mitochondrial O2 consumption in the upstream O2-rich periportal hepatocytes, which would place the downstream O2-poor pericentral hepatocytes under increased hypoxia.
Interestingly, these results contribute to an ongoing debate in the literature focused on whether or not small decreases, e.g. 15-30% in the levels or activities of key respiratory chain enzymes like cytochrome c oxidase negatively impact energy conservation and contribute to tissue pathology. While it is evident that impaired ATP synthesis can not be linked to a specific defect in cytochrome c oxidase per se, it is possible that a defect in oxidase or one of the other respiratory complexes could alter the respiratory threshold or reserve capacity needed to maintain bioenergetic homeostasis. It is expected that under conditions of pathological stress (i.e. chronic ethanol, ETS, and dyslipidemia) the bioenergetic demands on hepatocytes are likely increased due to increased repair and detoxification while the threshold capacity may be limited due to a loss in cytochrome c oxidase function. In support of this hypothesis we and others have shown that similar decreases in state 3 respiration following chronic alcohol consumption is associated with the inability of the mitochondrion to meet the energetic demands of the tissue. Specifically, decreased activity in respiratory chain complexes results in depressed ATP synthesis in liver [71]. Importantly, this can be related directly to cell viability because the inability to maintain adequate ATP levels increases hepatocyte death when steatotic hepatocytes are placed under low oxygen tensions [72]. Whether this occurs in this “multi-hit” model of alcoholic steatosis is not known; however, it is possible because in the current study we observed amplified hepatic hypoxia in vivo in the ethanol + ETS group. Therefore, when ATP synthesis is impaired this could enhance hepatocyte death particularly if ATP needs are increased acutely. Moreover, this may be critical for pathology as a loss in the capacity for maintenance of hepatic ATP levels may lead to more serious or permanent damage due to a depression in anabolic processes responsible for replacing damaged and/or lost cellular components.
The interaction of NO with the mitochondrion has been implicated as a key mediator in how cells respond to hypoxic stress. NO modulates mitochondrial function through control of mitochondrial biogenesis [73] and respiration by binding to the heme center in cytochrome c oxidase [74, 75]. While the interaction of NO and cytochrome c oxidase has been proposed to extend O2 gradients and increase O2 tensions in healthy tissue via inhibition of respiration [76], recent studies show that under pathologic conditions the inability to adapt through use of this signaling pathway occurs when mitochondria are damaged in cardiac hypertrophy, alcoholic fatty liver disease, and high fat diet hepatotoxicity resulting in ROS/RNS production and bioenergetic defects [77-80]. Previously, we reported that mitochondrial respiration becomes more sensitive to inhibition by exogenous NO exposure in response to chronic alcohol exposure [79] and that this response is iNOS dependent because increased sensitivity is not observed in mitochondria isolated from livers of iNOS-/- mice fed ethanol [37]. Important to this, is the observation that chronic alcohol-mediated steatosis is attenuated in iNOS-/- mice fed ethanol suggesting a link between NO, mitochondria, and pathology [37, 51]. Therefore, via iNOS induction and continual NO exposure, chronic alcohol consumption may change how endogenous NO controls mitochondrial respiration leading to enhanced hypoxia, increased ROS/RNS production, and compromised bioenergetics. Similarly, ETS may further exacerbate this alcohol dependent change in NO-control of respiration limiting the capacity of mitochondria to respond to cellular stress. Even though the interactive mechanisms responsible for the effects of hypercholesterolemia, ETS, and/or ethanol on NO biology and adaptation to hypoxia are poorly understood, we propose that toxicant exposure on a dyslipidemic background increases mitochondrial damage and susceptibility to hypoxic injury, which may be a critical and currently unrecognized factor or “hit” in the development of serious liver disease like steatohepatitis.
Summary/conclusions
The findings reported in this study provide strong support for the emerging paradigm that environmental and metabolic stressors, also referred to as “hits”, influence the progression from simple fatty liver (i.e. steatosis) to more serious liver diseases observed in the chronic alcohol consumer [6]. This study indicates that pro-inflammatory and pro-oxidant conditions induced by these multiple stressors or “hits” may disrupt the physiologic role of NO signaling and in doing so may contribute, in part, to mitochondrial damage and hypoxic liver injury. Furthermore, we postulate that it is under these conditions that increased post-translational modification of proteins, particularly mitochondrial proteins, will occur rendering proteins of key metabolic pathways catalytically inactive amplifying bioenergetic defects and increasing liver toxicity. Because the chronic alcohol consumer is likely to suffer from combinations of metabolic disorders and be exposed to multiple environmental toxicants, it will be critical for future investigations to identify the molecular targets and mechanisms through which these potential stressors or “hits” interact to accelerate and worsen alcoholic liver disease.
Acknowledgments
The authors would like to acknowledge the technical assistance of Dr. Rui-Ming Liu, Dr. Soniya Pawar, and Ms. Gloria Robinson at the University of Alabama at Birmingham. We thank Dr. Richard A. Rippe, University of North Carolina - Chapel Hill for technical assistance with alpha smooth muscle actin immunohistochemistry. Similarly, we thank Dr. Alvaro G. Estevez, Weill Medical College of Cornell University, for kindly providing the 3-nitrotyrosine antiserum and Dr. Victor M. Darley-Usmar, University of Alabama at Birmingham, for his critical comments on the results of this study. This work was supported by NIH grants AA15172 (SMB) and HL77419 and ES11172 (SWB).
List of abbreviations
- ATP
adenosine triphosphate
- BN-PAGE
blue native gel electrophoresis
- CYP2E1
cytochrome P450 2E1
- ETS
environmental tobacco smoke
- FA
filtered air
- HIF1α
hypoxia inducible factor 1α
- iNOS
inducible nitric oxide synthase
- LDL
low density lipoprotein
- mtDNA
mitochondrial DNA
- NO
nitric oxide
- 3-NT
3-nitrotyrosine
- O2
oxygen
- ONOO−
peroxynitrite
- QPCR
quantitative polymerase chain reaction
- ROS
reactive oxygen species
- RNS
reactive nitrogen species
- TBS-T
Tris-buffered saline with Tween-20
- VLDL
very low density lipoprotein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mantena SK, King AL, Andringa KK, Landar A, Darley-Usmar V, Bailey SM. Novel interactions of mitochondria and reactive oxygen/nitrogen species in alcohol mediated liver diseases. World J Gastroenterol. 2007;13:4967–4973. doi: 10.3748/wjg.v13.i37.4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thakur V, McMullen MR, Pritchard MT, Nagy LE. Regulation of macrophage activation in alcoholic liver disease. J Gastroenterol Hepatol. 2007;22 1:S53–56. doi: 10.1111/j.1440-1746.2006.04650.x. [DOI] [PubMed] [Google Scholar]
- 3.Han DW. Intestinal endotoxemia as a pathogenetic mechanism in liver failure. World J Gastroenterol. 2002;8:961–965. doi: 10.3748/wjg.v8.i6.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hines IN, Wheeler MD. Recent advances in alcoholic liver disease III. Role of the innate immune response in alcoholic hepatitis. Am J Physiol Gastrointest Liver Physiol. 2004;287:G310–314. doi: 10.1152/ajpgi.00094.2004. [DOI] [PubMed] [Google Scholar]
- 5.Bailey SM. A review of the role of reactive oxygen and nitrogen species in alcohol-induced mitochondrial dysfunction. Free Radic Res. 2003;37:585–596. doi: 10.1080/1071576031000091711. [DOI] [PubMed] [Google Scholar]
- 6.Day CP. Genes or environment to determine alcoholic liver disease and non-alcoholic fatty liver disease. Liver Int. 2006;26:1021–1028. doi: 10.1111/j.1478-3231.2006.01323.x. [DOI] [PubMed] [Google Scholar]
- 7.Bellentani S, Saccoccio G, Costa G, Tiribelli C, Manenti F, Sodde M, Saveria Croce L, Sasso F, Pozzato G, Cristianini G, Brandi G. Drinking habits as cofactors of risk for alcohol induced liver damage. The Dionysos Study Group. Gut. 1997;41:845–850. doi: 10.1136/gut.41.6.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cave M, Deaciuc I, Mendez C, Song Z, Joshi-Barve S, Barve S, McClain C. Nonalcoholic fatty liver disease: predisposing factors and the role of nutrition. J Nutr Biochem. 2007;18:184–195. doi: 10.1016/j.jnutbio.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 9.Hurt RD, Offord KP, Croghan IT, Gomez-Dahl L, Kottke TE, Morse RM, Melton LJ., 3rd Mortality following inpatient addictions treatment. Role of tobacco use in a community-based cohort. JAMA. 1996;275:1097–1103. doi: 10.1001/jama.275.14.1097. [DOI] [PubMed] [Google Scholar]
- 10.Steenland K, Thun M, Lally C, Heath C., Jr Environmental tobacco smoke and coronary heart disease in the American Cancer Society CPS-II cohort. Circulation. 1996;94:622–628. doi: 10.1161/01.cir.94.4.622. [DOI] [PubMed] [Google Scholar]
- 11.Bataller R. Time to ban smoking in patients with chronic liver diseases. Hepatology. 2006;44:1394–1396. doi: 10.1002/hep.21484. [DOI] [PubMed] [Google Scholar]
- 12.Klatsky AL, Armstrong MA. Alcohol, smoking, coffee, and cirrhosis. Am J Epidemiol. 1992;136:1248–1257. doi: 10.1093/oxfordjournals.aje.a116433. [DOI] [PubMed] [Google Scholar]
- 13.Klatsky AL, Friedman GD, Siegelaub AB. Alcohol and mortality. A ten-year Kaiser-Permanente experience. Ann Intern Med. 1981;95:139–145. doi: 10.7326/0003-4819-95-2-139. [DOI] [PubMed] [Google Scholar]
- 14.El-Zayadi AR. Heavy smoking and liver. World J Gastroenterol. 2006;12:6098–6101. doi: 10.3748/wjg.v12.i38.6098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hezode C, Lonjon I, Roudot-Thoraval F, Mavier JP, Pawlotsky JM, Zafrani ES, Dhumeaux D. Impact of smoking on histological liver lesions in chronic hepatitis C. Gut. 2003;52:126–129. doi: 10.1136/gut.52.1.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pessione F, Ramond MJ, Njapoum C, Duchatelle V, Degott C, Erlinger S, Rueff B, Valla DC, Degos F. Cigarette smoking and hepatic lesions in patients with chronic hepatitis C. Hepatology. 2001;34:121–125. doi: 10.1053/jhep.2001.25385. [DOI] [PubMed] [Google Scholar]
- 17.Zein CO, Beatty K, Post AB, Logan L, Debanne S, McCullough AJ. Smoking and increased severity of hepatic fibrosis in primary biliary cirrhosis: A cross validated retrospective assessment. Hepatology. 2006;44:1564–1571. doi: 10.1002/hep.21423. [DOI] [PubMed] [Google Scholar]
- 18.Zhu K, Moriarty C, Caplan LS, Levine RS. Cigarette smoking and primary liver cancer: a population-based case-control study in US men. Cancer Causes Control. 2007;18:315–321. doi: 10.1007/s10552-006-0105-8. [DOI] [PubMed] [Google Scholar]
- 19.Naveau S, Giraud V, Borotto E, Aubert A, Capron F, Chaput JC. Excess weight risk factor for alcoholic liver disease. Hepatology. 1997;25:108–111. doi: 10.1002/hep.510250120. [DOI] [PubMed] [Google Scholar]
- 20.Raynard B, Balian A, Fallik D, Capron F, Bedossa P, Chaput JC, Naveau S. Risk factors of fibrosis in alcohol-induced liver disease. Hepatology. 2002;35:635–638. doi: 10.1053/jhep.2002.31782. [DOI] [PubMed] [Google Scholar]
- 21.Marrero JA, Fontana RJ, Fu S, Conjeevaram HS, Su GL, Lok AS. Alcohol, tobacco and obesity are synergistic risk factors for hepatocellular carcinoma. J Hepatol. 2005;42:218–224. doi: 10.1016/j.jhep.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 22.Zhang WZ, Venardos K, Chin-Dusting J, Kaye DM. Adverse effects of cigarette smoke on NO bioavailability: role of arginine metabolism and oxidative stress. Hypertension. 2006;48:278–285. doi: 10.1161/01.HYP.0000231509.27406.42. [DOI] [PubMed] [Google Scholar]
- 23.van Jaarsveld H, Kuyl JM, Alberts DW. Exposure of rats to low concentration of cigarette smoke increases myocardial sensitivity to ischaemia/reperfusion. Basic Res Cardiol. 1992;87:393–399. doi: 10.1007/BF00796524. [DOI] [PubMed] [Google Scholar]
- 24.Ballinger SW, Bouder TG, Davis GS, Judice SA, Nicklas JA, Albertini RJ. Mitochondrial genome damage associated with cigarette smoking. Cancer Res. 1996;56:5692–5697. [PubMed] [Google Scholar]
- 25.Knight-Lozano CA, Young CG, Burow DL, Hu ZY, Uyeminami D, Pinkerton KE, Ischiropoulos H, Ballinger SW. Cigarette smoke exposure and hypercholesterolemia increase mitochondrial damage in cardiovascular tissues. Circulation. 2002;105:849–854. doi: 10.1161/hc0702.103977. [DOI] [PubMed] [Google Scholar]
- 26.Cakir Y, Yang Z, Knight CA, Pompilius M, Westbrook D, Bailey SM, Pinkerton KE, Ballinger SW. Effect of alcohol and tobacco smoke on mtDNA damage and atherogenesis. Free Radic Biol Med. 2007;43:1279–1288. doi: 10.1016/j.freeradbiomed.2007.07.015. [DOI] [PubMed] [Google Scholar]
- 27.de Gaetano G, Di Castelnuovo A, Costanzo S, Donati MB, Iacoviello L. Alcohol, cardiovascular risk, and health: there is a window for benefits. J Thromb Haemost. 2006;4:1156–1157. 1157–1158. doi: 10.1111/j.1538-7836.2006.01872.x. author reply. [DOI] [PubMed] [Google Scholar]
- 28.Di Castelnuovo A, Costanzo S, Bagnardi V, Donati MB, Iacoviello L, de Gaetano G. Alcohol dosing and total mortality in men and women: an updated meta-analysis of 34 prospective studies. Arch Intern Med. 2006;166:2437–2445. doi: 10.1001/archinte.166.22.2437. [DOI] [PubMed] [Google Scholar]
- 29.Fuchs FD, Chambless LE. Is the cardioprotective effect of alcohol real? Alcohol. 2007;41:399–402. doi: 10.1016/j.alcohol.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 30.Kloner RA, Rezkalla SH. To drink or not to drink? That is the question. Circulation. 2007;116:1306–1317. doi: 10.1161/CIRCULATIONAHA.106.678375. [DOI] [PubMed] [Google Scholar]
- 31.O'Keefe JH, Bybee KA, Lavie CJ. Alcohol and cardiovascular health: the razor-sharp double-edged sword. J Am Coll Cardiol. 2007;50:1009–1014. doi: 10.1016/j.jacc.2007.04.089. [DOI] [PubMed] [Google Scholar]
- 32.de Roos B, Duivenvoorden I, Rucklidge G, Reid M, Ross K, Lamers RJ, Voshol PJ, Havekes LM, Teusink B. Response of apolipoprotein E*3-Leiden transgenic mice to dietary fatty acids: combining liver proteomics with physiological data. FASEB J. 2005;19:813–815. doi: 10.1096/fj.04-2974fje. [DOI] [PubMed] [Google Scholar]
- 33.Mensenkamp AR, Van Luyn MJ, Havinga R, Teusink B, Waterman IJ, Mann CJ, Elzinga BM, Verkade HJ, Zammit VA, Havekes LM, Shoulders CC, Kuipers F. The transport of triglycerides through the secretory pathway of hepatocytes is impaired in apolipoprotein E deficient mice. J Hepatol. 2004;40:599–606. doi: 10.1016/j.jhep.2003.12.011. [DOI] [PubMed] [Google Scholar]
- 34.Tous M, Ferre N, Camps J, Riu F, Joven J. Feeding apolipoprotein E-knockout mice with cholesterol and fat enriched diets may be a model of non-alcoholic steatohepatitis. Mol Cell Biochem. 2005;268:53–58. doi: 10.1007/s11010-005-2997-0. [DOI] [PubMed] [Google Scholar]
- 35.Yoshimatsu M, Terasaki Y, Sakashita N, Kiyota E, Sato H, van der Laan LJ, Takeya M. Induction of macrophage scavenger receptor MARCO in nonalcoholic steatohepatitis indicates possible involvement of endotoxin in its pathogenic process. Int J Exp Pathol. 2004;85:335–343. doi: 10.1111/j.0959-9673.2004.00401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reddick RL, Zhang SH, Maeda N. Atherosclerosis in mice lacking apo E. Evaluation of lesional development and progression. Arterioscler Thromb. 1994;14:141–147. doi: 10.1161/01.atv.14.1.141. [DOI] [PubMed] [Google Scholar]
- 37.Venkatraman A, Shiva S, Wigley A, Ulasova E, Chhieng D, Bailey SM, Darley-Usmar V. The role of iNOS in alcohol-dependent hepatoxicity and mitochondrial dysfunction. Hepatology. 2004;40:565–573. doi: 10.1002/hep.20326. [DOI] [PubMed] [Google Scholar]
- 38.Gentry-Nielsen MJ, Top EV, Snitily MU, Casey CA, Preheim LC. A rat model to determine the biomedical consequences of concurrent ethanol ingestion and cigarette smoke exposure. Alcohol Clin Exp Res. 2004;28:1120–1128. doi: 10.1097/01.alc.0000136383.45378.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hoffman D, Wtnder EL. Active and passive smoking. In: Marquart K, editor. Toxicology. New York: Academic Press; 1999. pp. 878–898. [Google Scholar]
- 40.Bailey SM, Robinson G, Pinner A, Chamlee L, Ulasova E, Pompilius M, Page GP, Chhieng D, Jhala N, Landar A, Kharbanda KK, Ballinger S, Darley-Usmar V. S-adenosylmethionine prevents chronic alcohol-induced mitochondrial dysfunction in the rat liver. Am J Physiol Gastrointest Liver Physiol. 2006;291:G857–867. doi: 10.1152/ajpgi.00044.2006. [DOI] [PubMed] [Google Scholar]
- 41.Tipoe GL, Liong EC, Casey CA, Donohue TM, Jr, Eagon PK, So H, Leung TM, Fogt F, Nanji AA. A voluntary oral ethanol-feeding rat model associated with necroinflammatory liver injury. Alcohol Clin Exp Res. 2008;32:669–682. doi: 10.1111/j.1530-0277.2008.00623.x. [DOI] [PubMed] [Google Scholar]
- 42.Gabele E, Froh M, Arteel GE, Uesugi T, Hellerbrand C, Scholmerich J, Brenner DA, Thurman RG, Rippe RA. TNFalpha is required for cholestasis-induced liver fibrosis in the mouse. Biochem Biophys Res Commun. 2008 doi: 10.1016/j.bbrc.2008.10.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Venkatraman A, Landar A, Davis AJ, Chamlee L, Sanderson T, Kim H, Page G, Pompilius M, Ballinger S, Darley-Usmar V, Bailey SM. Modification of the mitochondrial proteome in response to the stress of ethanol-dependent hepatotoxicity. J Biol Chem. 2004;279:22092–22101. doi: 10.1074/jbc.M402245200. [DOI] [PubMed] [Google Scholar]
- 44.Brookes PS, Pinner A, Ramachandran A, Coward L, Barnes S, Kim H, Darley-Usmar VM. High throughput two-dimensional blue-native electrophoresis: a tool for functional proteomics of mitochondria and signaling complexes. Proteomics. 2002;2:969–977. doi: 10.1002/1615-9861(200208)2:8<969::AID-PROT969>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 45.Wharton DC, Tzagoloff A. Cytochrome oxidase from beef heart mitochondria. Methods Enzymol. 1967;10:245–250. [Google Scholar]
- 46.Shepherd JA, Garland GP. Citrate synthase from rat liver. Methods Enzymol. 1969;13:11–19. [Google Scholar]
- 47.Ballinger SW, Van Houten B, Jin GF, Conklin CA, Godley BF. Hydrogen peroxide causes significant mitochondrial DNA damage in human RPE cells. Exp Eye Res. 1999;68:765–772. doi: 10.1006/exer.1998.0661. [DOI] [PubMed] [Google Scholar]
- 48.Witschi H, Joad JP, Pinkerton KE. The toxicology of environmental tobacco smoke. Annu Rev Pharmacol Toxicol. 1997;37:29–52. doi: 10.1146/annurev.pharmtox.37.1.29. [DOI] [PubMed] [Google Scholar]
- 49.Arteel GE, Iimuro Y, Yin M, Raleigh JA, Thurman RG. Chronic enteral ethanol treatment causes hypoxia in rat liver tissue in vivo. Hepatology. 1997;25:920–926. doi: 10.1002/hep.510250422. [DOI] [PubMed] [Google Scholar]
- 50.Arteel GE, Raleigh JA, Bradford BU, Thurman RG. Acute alcohol produces hypoxia directly in rat liver tissue in vivo: role of Kupffer cells. Am J Physiol Gastrointest Liver Physiol. 1996;271:G494–500. doi: 10.1152/ajpgi.1996.271.3.G494. [DOI] [PubMed] [Google Scholar]
- 51.McKim SE, Gabele E, Isayama F, Lambert JC, Tucker LM, Wheeler MD, Connor HD, Mason RP, Doll MA, Hein DW, Arteel GE. Inducible nitric oxide synthase is required in alcohol-induced liver injury: studies with knockout mice. Gastroenterology. 2003;125:1834–1844. doi: 10.1053/j.gastro.2003.08.030. [DOI] [PubMed] [Google Scholar]
- 52.Schagger H, von Jagow G. Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal Biochem. 1991;199:223–231. doi: 10.1016/0003-2697(91)90094-a. [DOI] [PubMed] [Google Scholar]
- 53.Husain K, Scott BR, Reddy SK, Somani SM. Chronic ethanol and nicotine interaction on rat tissue antioxidant defense system. Alcohol. 2001;25:89–97. doi: 10.1016/s0741-8329(01)00176-8. [DOI] [PubMed] [Google Scholar]
- 54.Micu AL, Miksys S, Sellers EM, Koop DR, Tyndale RF. Rat hepatic CYP2E1 is induced by very low nicotine doses: an investigation of induction, time course, dose response, and mechanism. J Pharmacol Exp Ther. 2003;306:941–947. doi: 10.1124/jpet.103.052183. [DOI] [PubMed] [Google Scholar]
- 55.Schoedel KA, Tyndale RF. Induction of nicotine-metabolizing CYP2B1 by ethanol and ethanol-metabolizing CYP2E1 by nicotine: summary and implications. Biochim Biophys Acta. 2003;1619:283–290. doi: 10.1016/s0304-4165(02)00487-7. [DOI] [PubMed] [Google Scholar]
- 56.Kotronen A, Yki-Jarvinen H. Fatty liver: a novel component of the metabolic syndrome. Arterioscler Thromb Vasc Biol. 2008;28:27–38. doi: 10.1161/ATVBAHA.107.147538. [DOI] [PubMed] [Google Scholar]
- 57.Lizardi-Cervera J, Chavez-Tapia NC, Perez-Bautista O, Ramos MH, Uribe M. Association among C-reactive protein, Fatty liver disease, and cardiovascular risk. Dig Dis Sci. 2007;52:2375–2379. doi: 10.1007/s10620-006-9262-6. [DOI] [PubMed] [Google Scholar]
- 58.Mensenkamp AR, Havekes LM, Romijn JA, Kuipers F. Hepatic steatosis and very low density lipoprotein secretion: the involvement of apolipoprotein E. J Hepatol. 2001;35:816–822. doi: 10.1016/s0168-8278(01)00249-5. [DOI] [PubMed] [Google Scholar]
- 59.de Knijff P, van den Maagdenberg AM, Stalenhoef AF, Leuven JA, Demacker PN, Kuyt LP, Frants RR, Havekes LM. Familial dysbetalipoproteinemia associated with apolipoprotein E3-Leiden in an extended multigeneration pedigree. J Clin Invest. 1991;88:643–655. doi: 10.1172/JCI115349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.van Vlijmen BJ, van den Maagdenberg AM, Gijbels MJ, van der Boom H, HogenEsch H, Frants RR, Hofker MH, Havekes LM. Diet-induced hyperlipoproteinemia and atherosclerosis in apolipoprotein E3-Leiden transgenic mice. J Clin Invest. 1994;93:1403–1410. doi: 10.1172/JCI117117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hernandez-Nazara ZH, Ruiz-Madrigal B, Martinez-Lopez E, Roman S, Panduro A. Association of the epsilon 2 allele of APOE gene to hypertriglyceridemia and to early-onset alcoholic cirrhosis. Alcohol Clin Exp Res. 2008;32:559–566. doi: 10.1111/j.1530-0277.2007.00607.x. [DOI] [PubMed] [Google Scholar]
- 62.Sazci A, Akpinar G, Aygun C, Ergul E, Senturk O, Hulagu S. Association of apolipoprotein e polymorphisms in patients with non-alcoholic steatohepatitis. Dig Dis Sci. 2008;53:3218–3224. doi: 10.1007/s10620-008-0271-5. [DOI] [PubMed] [Google Scholar]
- 63.Mari M, Caballero F, Colell A, Morales A, Caballeria J, Fernandez A, Enrich C, Fernandez-Checa JC, Garcia-Ruiz C. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 2006;4:185–198. doi: 10.1016/j.cmet.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 64.Cooper CE, Patel RP, Brookes PS, Darley-Usmar VM. Nanotransducers in cellular redox signaling: modification of thiols by reactive oxygen and nitrogen species. Trends Biochem Sci. 2002;27:489–492. doi: 10.1016/s0968-0004(02)02191-6. [DOI] [PubMed] [Google Scholar]
- 65.Costa NJ, Dahm CC, Hurrell F, Taylor ER, Murphy MP. Interactions of mitochondrial thiols with nitric oxide. Antioxid Redox Signal. 2003;5:291–305. doi: 10.1089/152308603322110878. [DOI] [PubMed] [Google Scholar]
- 66.Radi R. Nitric oxide, oxidants, and protein tyrosine nitration. Proc Natl Acad Sci U S A. 2004;101:4003–4008. doi: 10.1073/pnas.0307446101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Savransky V, Nanayakkara A, Vivero A, Li J, Bevans S, Smith PL, Torbenson MS, Polotsky VY. Chronic intermittent hypoxia predisposes to liver injury. Hepatology. 2007;45:1007–1013. doi: 10.1002/hep.21593. [DOI] [PubMed] [Google Scholar]
- 69.Savransky V, Nanayakkara A, Li J, Bevans S, Smith PL, Rodriguez A, Polotsky VY. Chronic intermittent hypoxia induces atherosclerosis. Am J Respir Crit Care Med. 2007;175:1290–1297. doi: 10.1164/rccm.200612-1771OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Couch NP. On the arterial consequences of smoking. J Vasc Surg. 1986;3:807–812. doi: 10.1067/mva.1986.avs0030807. [DOI] [PubMed] [Google Scholar]
- 71.Cunningham CC, Coleman WB, Spach PI. The effects of chronic ethanol consumption on hepatic mitochondrial energy metabolism. Alcohol Alcohol. 1990;25:127–136. doi: 10.1093/oxfordjournals.alcalc.a044987. [DOI] [PubMed] [Google Scholar]
- 72.Bailey SM, Cunningham CC. Effect of dietary fat on chronic ethanol-induced oxidative stress in hepatocytes. Alcohol Clin Exp Res. 1999;23:1210–1218. [PubMed] [Google Scholar]
- 73.Nisoli E, Clementi E, Moncada S, Carruba MO. Mitochondrial biogenesis as a cellular signaling framework. Biochem Pharmacol. 2004;67:1–15. doi: 10.1016/j.bcp.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 74.Cleeter MW, Cooper JM, Darley-Usmar VM, Moncada S, Schapira AH. Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial respiratory chain, by nitric oxide. Implications for neurodegenerative diseases. FEBS Lett. 1994;345:50–54. doi: 10.1016/0014-5793(94)00424-2. [DOI] [PubMed] [Google Scholar]
- 75.Cooper CE, Giulivi C. Nitric oxide regulation of mitochondrial oxygen consumption II: molecular mechanism and tissue physiology. Am J Physiol Cell Physiol. 2007 doi: 10.1152/ajpcell.00310.2006. [DOI] [PubMed] [Google Scholar]
- 76.Thomas DD, Liu X, Kantrow SP, Lancaster JR., Jr The biological lifetime of nitric oxide: implications for the perivascular dynamics of NO and O2. Proc Natl Acad Sci U S A. 2001;98:355–360. doi: 10.1073/pnas.011379598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Brookes PS, Zhang J, Dai L, Zhou F, Parks DA, Darley-Usmar VM, Anderson PG. Increased sensitivity of mitochondrial respiration to inhibition by nitric oxide in cardiac hypertrophy. J Mol Cell Cardiol. 2001;33:69–82. doi: 10.1006/jmcc.2000.1276. [DOI] [PubMed] [Google Scholar]
- 78.Shiva S, Oh JY, Landar AL, Ulasova E, Venkatraman A, Bailey SM, Darley-Usmar VM. Nitroxia: The pathological consequence of dysfunction in the nitric oxide-cytochrome c oxidase signaling pathway. Free Radic Biol Med. 2005;38:297–306. doi: 10.1016/j.freeradbiomed.2004.10.037. [DOI] [PubMed] [Google Scholar]
- 79.Venkatraman A, Shiva S, Davis AJ, Bailey SM, Brookes PS, Darley-Usmar V. Chronic alcohol consumption increases the senstivity of rat liver mitochondrial respiration to inhibition by nitric oxide. Hepatology. 2003;38:141–147. doi: 10.1053/jhep.2003.50293. [DOI] [PubMed] [Google Scholar]
- 80.Mantena SK, Vaughn DP, Jr, Andringa KK, Eccleston HB, King AL, Abrams GA, Doeller JE, Kraus DW, Darley-Usmar V, Bailey SM. High fat diet induces dysregulation of hepatic oxygen gradients and mitochondrial function in vivo. Biochem J. 2009;417:183–193. doi: 10.1042/BJ20080868. [DOI] [PMC free article] [PubMed] [Google Scholar]