

Abstract
Listeria monocytogenes is a gram positive, intracellular, food-borne pathogen that can cause severe illness in humans and animals. Upon infection, it is actively phagocytosed by macrophages1. It then escapes from the phagosome, replicates in the cytosol, and subsequently spreads from cell to cell by a non-lytic mechanism driven by actin polymerization2. Penetration of the phagosomal membrane is initiated by the secreted hemolysin listeriolysin O (LLO), which is essential for vacuolar escape in vitro and for virulence in animal models of infection3. Reduction is required to activate the lytic activity of LLO in vitro 4–6, and we show here that reduction by the enzyme Gamma-interferon Inducible Lysosomal Thiolreductase (GILT) is responsible for the activation of LLO in vivo. GILT is a soluble thiol reductase expressed constitutively within the lysosomes of antigen presenting cells7, 8, and it accumulates in macrophage phagosomes as they mature into phagolysosomes9. The enzyme is delivered by a mannose-6-phosphate receptor-dependent mechanism to the endocytic pathway, where N- and C-terminal pro-peptides are cleaved to generate a 30 kDa mature enzyme7, 8, 10. The active site of GILT contains two cysteine residues in a CXXC motif that catalyzes the reduction of disulfide bonds7, 8. Mice lacking GILT are deficient in generating MHC class II-restricted CD4+ T cell responses to protein antigens that contain disulfide bonds11, 12. Here we show that these mice are resistant to L. monocytogenes infection. Replication of the organism in GILT-negative macrophages, or macrophages expressing an enzymatically inactive GILT mutant, is impaired because of delayed escape from the phagosome. GILT activates LLO within the phagosome by the classical thiol reductase mechanism shared by members of the thioredoxin family. In addition, purified GILT activates recombinant LLO, facilitating membrane permeabilization and red blood cell lysis. The data show GILT is a critical host factor that facilitates L. monocytogenes infection.
GILT is the only known thiol oxidoreductase present in phagosomes and we speculated that it might activate LLO in vivo. Consistent with this idea, when we infected wild type and GILT-deficient mice with L. monocytogenes we observed more rapid bacterial clearance from the spleen and liver of the GILT-deficient mice (Fig. 1a). Initial infection was as effective as in wild type mice, based on the similar bacterial colony forming units (CFUs) observed at day one post infection. No difference in the clearance rates of Salmonella typhimurium was seen between wild type and GILT knockout mice (data not shown). To determine if the difference in clearance was a function of defective intracellular growth, we examined bacterial growth in bone marrow-derived macrophages in vitro. Replication of L. monocytogenes was clearly impaired in macrophages from GILT-deficient mice (Fig. 1b). This was not due to an inherent defect in phagocytosis or bacterial killing by GILT−/− macrophages, which cleared an in vitro infection by non-pathogenic E. coli as efficiently as wild type macrophages (Supplemental Fig. 1). Nor was it due to a problem in phagosomal acidification; acidification of both lysosomes and phagosomes occurred with the same kinetics in GILT −/− and wild type macrophages (Supplemental Fig. 2).
Figure 1.

Growth of L. monocytogenes is decreased in GILT-deficient mice and GILT-deficient macrophages. a, In vivo colony counts of L. monocytogenes (strain 10403s) injected intravenously in wild type B6 or GILT-deficient mice. At each time point three mice per group were sacrificed and spleens and livers were harvested each day for bacterial colony counts to obtain CFU/gram. A representative of three individual experiments is shown. b, In vitro growth of L. monocytogenes in bone marrow-derived macrophages from wild type B6 and GILT-deficient mice. The cells (106 per well) were lysed at each time point shown and plated to obtain the CFU. A representative of three individual experiments is shown. Statistically significant differences with a p < 0.05 are indicated by *, and with a p < 0.01 by **.
Since GILT is active in phagosomes it seemed likely that the growth deficiency was due to a defect in phagosomal escape. Once in the cytosol, L. monocytogenes polymerizes host actin, which allows movement through the cytosol and facilitates intercellular spread2, and actin polymerization provides a convenient assay for cytosolic entry. Indeed, we saw a delay in actin polymerization, detected by phalloidin staining, in infected GILT-deficient macrophages, consistent with impaired escape from the phagosome (Fig. 2a and b). In wild type macrophages actin polymerization was observed as early as 30 min. after infection. This typically results in overwhelming infection and cell death within 36 to 48 h. In GILT-deficient macrophages actin polymerization was not observed until 6–8 h. When wild type or GILT-deficient macrophages were infected with an LLO-negative L. monocytogenes strain the rate of actin polymerization was reduced to a level below that seen with wild type bacteria in the GILT-negative cells (Fig. 2a and b). Consistent with a role for GILT in L. monocytogenes infection, a dramatic increase in cytosolic access was observed upon infection of the GILT-negative human promonocytic cell line THP-1 when human GILT was expressed by retroviral transduction13 (Supplemental Fig. 3). GILT is induced in THP-1 cells upon exposure to bacteria, but more than 24 h is required13.
Figure 2.

Phagosomal escape of L. monocytogenes is delayed in GILT-negative macrophages. a, Actin polymerization in wild type and GILT knockout macrophages infected with wild type L. monocytogenes or ΔLLO L. monocytogenes. Both strains of bacteria express GFP and are green. Phalloidin staining of actin is in red. b, Quantitation of actin polymerization. 1000 cells per sample were counted for each time point. The data derive from the average of three experiments, and statistical significance was assessed by comparing the infection of wild type and GILT −/− macrophages with wild type bacteria. Differences with a p < 0.05 are indicated by *, and those with a p< 0.01 are indicated by **. c, Transmission electron micrographs of wild type or GILT−/− infected macrophages. The arrows indicate intact phagosomal membrane, and the reordering of the cytoplasm in the initial stages of actin polymerization, can be seen around bacteria in the cytosol. The bars represent a length of 1 µm. No arrows are present in the 8 h image for wild type cells because no bacteria are contained within membrane-bound compartments. d, Actin polymerization in infected wild type and GILT-negative macrophages, and GILT-negative macrophages expressing GILT or inactive mutant GILT (C69S, C71S). e, Quantification of actin polymerization in macrophages infected as in (d). The data derive from the average of three experiments and statistical significance was assessed by comparing the wild type and untransduced GILT −/−macrophages and, separately, the GILT−/− macrophages transduced with wild type and mutant GILT retroviruses. Differences with a p < 0.05 are indicated by *, and those with a p < 0.01 by **.
To confirm the escape defect we examined infected cells by electron microscopy (Fig. 2c). After 2 h virtually no bacteria were seen outside of membrane-bound phagosomes in GILT-deficient macrophages, and only limited actin polymerization was observed even 8 h post infection. Furthermore, defective phagosomal escape, detected by the delay in the induction of actin polymerization, was reversed when GILT was expressed in GILT-negative macrophages by retroviral transduction (Fig. 2d and e). However, expression of a double cysteine mutant of GILT (C69S, C71S), lacking both active site cysteine residues and unable to catalyze disulfide bond reduction11, failed to reverse the escape defect.
LLO has a single cysteine residue at position 485 and a mutant with an alanine substitution at that position does not require reduction for activation14. To determine if the mutation also reversed GILT dependence, we repeated the in vitro infection experiments described above with an L. monocytogenes strain expressing this LLO variant. For this organism growth was equivalent in wild type and GILT −/− macrophages (Fig. 3a), as was the rate of escape from the phagosome (Fig. 3b and c). We also examined the capacity of recombinant enzymatically active precursor GILT8, 10, 15 to induce lytic activity in purified recombinant LLO (characterized in Supplemental Fig. 4). GILT clearly was able to activate LLO, determined by lysis of purified bone marrow-derived macrophages (Fig. 3d) and hemolysis of sheep red blood cells (Fig. 3e). Similar lytic curves for macrophages were obtained using trypan blue exclusion (data not shown). Lysis was abrogated if the enzymatic activity of GILT was eliminated by pretreatment with the thiol-reactive reagent, N-ethylmaleimide (NEM).
Figure 3.
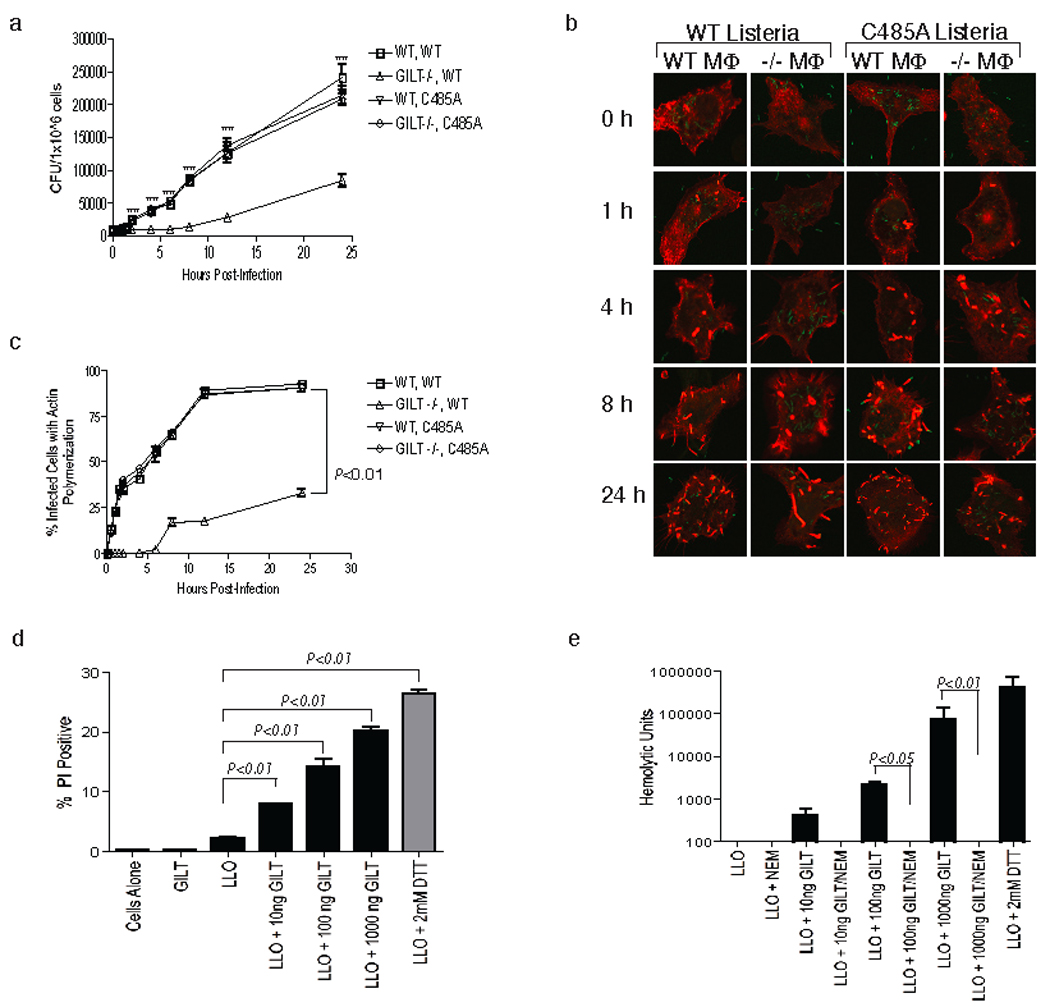
LLO is activated by GILT reduction during infection and in cell-free assays. a, In vitro growth of wild type and C485A LLO mutant L. monocytogenes in wild type and GILT −/− macrophages. The experiment, performed as described in Fig. 1b, was repeated three times and a representative experiment is shown. Differences with a p < 0.05 are indicated by *, and those with a p < 0.01 by **. b, Actin polymerization in wild type and GILT −/−macrophages infected with wild type L. monocytogenes or C485A LLO mutant L. monocytogenes. The bacteria were detected by immunofluorescence (green) and phalloidin staining of actin is in red. c, Quantitation of actin polymerization as described in Fig. 2b. The data represent an average of three independent experiments, and all points after 0.5 h are statistically significant to a p < 0.01. d, Viability of GILT-negative macrophages assessed by propidium iodide staining after incubation of 1×105 cells with 300ng LLO for 30–45 min. Cells were incubated alone, with activated GILT (1µg) and 25µM DTT, with unactivated LLO and 25µM DTT, with DTT-activated LLO, or with LLO and 1µg, 100ng or 10ng activated GILT. The data represent the average of three independent experiments and P values for the samples with significant differences are shown. g, Hemolytic activity of purified LLO pre-incubated with active GILT or GILT inactivated by NEM treatment. The data represent the average of three independent experiments and P values for the samples with significant differences are shown.
GILT shares with thioredoxin a reduction mechanism in which the N-terminal cysteine residue in the CXXC active site reduces a substrate disulfide bond by a nucleophilic attack on one of the involved cysteine residues. This generates a disulfide-linked mixed enzyme-substrate intermediate that is rapidly resolved by an attack of the second active site cysteine residue on the first7, 8. The second step in the reaction, known as the escape pathway16, can be prevented by mutation of the second active site cysteine, generating a ‘trapping mutant’ which allows mixed enzyme-substrate dimers to be isolated. To determine whether GILT uses this mechanism to activate LLO, bone marrow derived macrophages from wild type and GILT −/− mice were again infected with L. monocytogenes, together with GILT −/− macrophages that were retrovirally transduced with a GILT trapping mutant (C71S). Infected cells were lysed in detergent and immunoprecipitated GILT was subjected to SDS-PAGE followed by western blotting to detect associated LLO. LLO co-precipitated with GILT only from infected macrophages expressing the trapping mutant (Fig. 4a). Immunofluorescence microscopy confirmed that in wild type macrophages the organism colocalizes with GILT in LAMP-1-positive phagosomes (data not shown). Thus GILT uses the classical thiol reductase mechanism to activate LLO in phagosomes and initiate the escape of L. monocytogenes to the cytosol.
Figure 4.
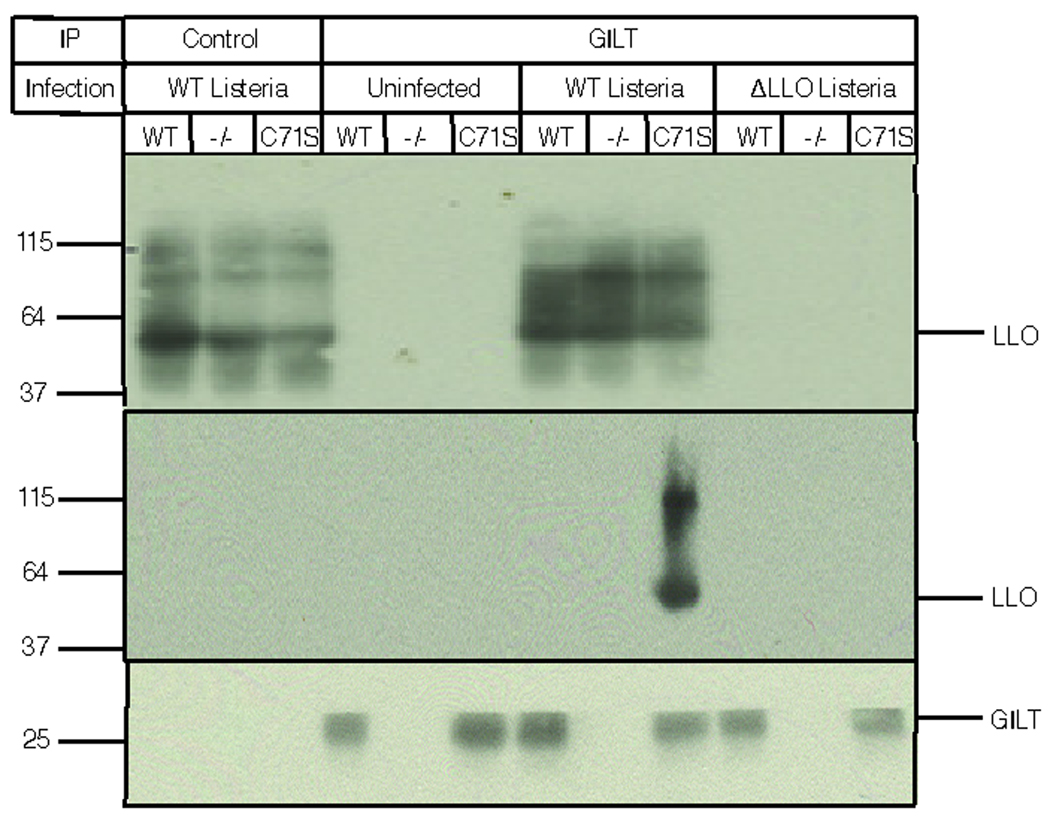
GILT activates LLO by using the classical thioredoxin reduction mechanism. Wild type macrophages, GILT −/− macrophages, or GILT −/− macrophages reconstituted with the C71S GILT trapping mutant were uninfected or infected with either wild type L. monocytogenes or ΔLLO L. monocytogenes for 2 h prior to detergent solubilization, immunoprecipitation, and SDS-PAGE; (top panel), western blot of LLO in the detergent extracts; (middle panel), western blot with an LLO-specific antiserum of immunoprecipitates isolated from the extracts using a control mAb (left three lanes) or an anti GILT mAb (right nine lanes). LLO is only co-precipitated with the C71S trapping mutant; (bottom panel), western blot for GILT in the immunoprecipitates.
In the absence of LLO, L. monocytogenes may eventually escape from the phagosome, just as in the absence of GILT (Fig. 2a). However, when active GILT is present escape of LLO-positive bacteria is extremely rapid. The identity of the disulfide bond targeted by GILT is unclear. The single cysteine present in LLO lies in a short tryptophan-rich sequence that initiates pore formation upon binding to membrane cholesterol. It has been suggested that a small thiol-containing molecule is disulfide linked to the cysteine residue, and that this inhibits activation of the hemolysins17. The equivalent cysteine residue in Perfringolysin O (PFO), a related hemolysin derived from Clostridium perfringens, is not necessary for binding to cholesterol in the membrane, but is necessary for the formation of a pre-pore complex18–20. GILT-mediated exposure of the critical cysteine residue in LLO may facilitate a conformational change that allows the formation of the pre-pore complex and full activation. Notably, the ability of GILT to activate hemolysins is not limited to LLO; GILT can also activate Streptolysin O (SLO), derived from Streptococcus pyogenes, as measured by the hemolysis of sheep red blood cells, but it is not required to activate an SLO mutant that lacks the characteristic single cysteine residue21 (Supplemental Fig. 5).
In most cell types GILT can be induced by IFN-γ, and IFN-γ induction during the early stages of infection may enhance the ability of L. monocytogenes to infect other cells, including hepatocytes where it also replicates in vivo22, 23. The ability of GILT to activate SLO as well as LLO suggests that it may activate other members of this highly conserved family of hemolysins. Phagocytosis of hemolysin-expressing organisms may not be essential for activation of the lytic activity, as we have recently found that Toll-like receptor (TLR)-mediated activation of macrophages by E. coli lipopolysaccharide induces secretion of the enzymatically-active precursor form of GILT13, 24 that activates SLO in vitro (Supplemental Fig. 5). GILT is functional even at neutral pH, retaining approximately 30% of the activity seen at pH4.57, 8, and it has been reported that the local pH can be as low as 5.7 at sites of bacterial infection25. Secretion of GILT by macrophages at such a site could facilitate local hemolysin-mediated tissue damage, perhaps including lysis of inflammatory cells recruited for the purpose of host defense.
METHODS SUMMARY
The L. monocytogenes strains used are listed in the Methods. Infections in C57BL/6 and C57BL/6 GILT knockout mice were performed as described26. Bone marrow derived macrophages were prepared as described24. In vitro infections were done in the presence of gentamicin to prevent extracellular bacterial growth, and at an MOI of 5 (unless otherwise stated in the on-line Methods).
Electron microscopy and immunofluorescence experiments and antibodies used are described in on-line Methods. Colocalization and quantification were performed by direct visualization on a Leica DMIRE2 confocal microscope. Figure assembly was done with Adobe Photoshop and Adobe Illustrator.
LLO protein was purified according to published protocols with modifications described in Methods 27.
The mean +/− standard error is shown in the figures and P values were calculated using a two-tailed two-sample equal variance Student’s t-test. A P value of less than 0.05 was determined to be statistically significant.
METHODS
Bacterial strains
L. monocytogenes were grown in brain heart infusion (BHI) broth (BD). The strains used were: wild type (10403s), L. monocytogenes-GFP (1039), L. monocytogenes ΔLLO (1039ΔLLO) (gifts from Dr. Herve Agaisse at Yale University)28, and DP-L4391 (C485A LLO) (a gift from Dr. D. Portnoy, UC Berkeley). LLO was purified from the DP-E3570 E. coli strain (donated by Dr. D. Portnoy, UC Berkeley) grown in Luria-Bertani (LB) broth (BD) supplemented with kanamycin. Production of LLO was induced by 1mM isopropyl-beta-D-thiogalactopyranoside (IPTG) at 30°C. E. coli used for infection was grown in LB supplemented with ampicillin.
In vivo bacterial infections
Infections of C57BL/6 and C57BL/6 GILT–deficient mice were performed as described previously26. 6–8 week old mice were used for each experiment.
Cell culture
Bone marrow derived macrophages were cultured as described24. Briefly, bone marrow was harvested from the femurs of 8–10 week old mice and cells cultured for 5–6 days in RPMI 1640 containing 20% fetal calf serum (Hyclone), 100 units/ml penicillin (GIBCO), 100 µg/ml streptomycin (GIBCO), 10mM HEPES (GIBCO), 1% non-essential amino acids (GIBCO), 2mM L-glutamine (GIBCO), 1mM sodium pyruvate, and 0.035% beta-mercaptoethanol, supplemented with 10 ng/ml granulocyte-monocyte colony stimulating factor. HEK 293 cells were grown in DMEM with 10% bovine calf serum (Hyclone).
In vitro infections
Infections of macrophages for in vitro growth assays were at an MOI of 5 except for Fig. 1b where an MOI of 0.1 was used and Fig. 3a when an MO1 of 0.5 was used. For infection, 16 h L. monocytogenes cultures were diluted 1:10 in fresh BHI and grown for an additional 2 h at 37°C with shaking. Aliquots of mid-log phase bacteria (∼5×108 CFU/ml) were washed once in PBS and used to infect macrophages in medium without antibiotics. After 30 min. at 37°C gentamicin (50 µg/ml) was added and the cells incubated for another 30 min. at 37°C, washed, and incubated in 1 ml fresh medium at 37°C. Cells were lysed at 0, 2, 4, 6, 8, 10, 12, and 24 h in 1 ml water for 5–10 min. Serial dilutions were plated on BHI plates containing chloramphenicol and colonies were counted the next day to determine CFUs.
Transmission electron microscopy and immunofluorescence
For TEM, cells were fixed in 0.2% glutaraldehyde in 0.1M cacodylate buffer for 1 h at room temperature and processed as described24. For immunofluorescence, macrophages were plated on coverslips 12 h before infection. The cells were fixed in 2% glutaraldehyde for 20 min at room temperature at each time point. For phalloidin staining, the coverslips were washed in PBS and permeabilized with 0.1% Triton X-100 in PBS for 3 min. After washing, methanolic phalloidin stock solution was diluted 40X into 1% BSA in PBS and the coverslips stained for 20 min. at room temperature. Intracellular antibody staining was done after cell permeabilization in 0.1% saponin for 20 min. at room temperature. Primary antibodies were added for 30 min. and secondary antibodies for a further 30 min. The antibodies used were: R.mGILT11, MaP.mGILT6 (mouse anti-mouse GILT mAb)24, phalloidin conjugated to Alexa 546 (Molecular Probes), and rabbit anti-Listeria sp. FITC (Affinity BioReagents). AlexaFluor 546 and 633 conjugated secondary antibodies were used (Molecular Probes). Coverslips were mounted using ProLong Gold mounting solution (Molecular Probes).
Retroviral constructs and spinfection
Wild type mouse GILT cDNA was cloned into the pLPCX vector (Clontech) using BglII and HindIII restriction enzymes. GILT mutants were made by site directed mutagenesis with the following primers: C69S, C71S: 5’ GAGTCCCTGTCCGGAGCTAGCCGCTACTTCCTCCG 3’, and C71S: 5’ CCCTGTGTGGAGCTAGCCGCTACTTCCTC 3’. To produce retrovirus, HEK 293 cells were transfected with 12µg of each pLPCX construct and 12µg pCL-Eco with 60µl Lipofectamine 2000 (Invitrogen). After 12 h the medium was changed to macrophage culture medium and the cells were shifted to 32°C. After 24 h filtered supernatant was added to day 2 macrophage cultures. The cells were spinfected at 32°C, at 2900 rpm for 90 mi. and cultured at 37°C and differentiated as usual.
LLO purification
Purification of LLO was as described except that the β-mercaptoethanol was omitted from all the buffers and 0.5 mM DTT was used for storage27.
Cell lytic assays
Precursor GILT, purified from supernatants of baculovirus infected insect cells as described11, was activated with 25µM DTT at room temperature for 10 min., and for some experiments inactivated with NEM (1.5mM) followed by dialysis against normal saline (pH 5.5). It was then incubated with LLO at 37°C for 30 min. An aliquot of LLO was activated using DTT (2mM) as a positive control. The samples were incubated with macrophages at 37° for 30 min. Viability was assessed by propidium iodide staining and FACs analysis. For hemolysis assays LLO was added to sheep red blood cells (Innovative Research) in PBS, pH 5.5, in 96 well plates on ice. After 30 min. at 37°C lysis was determined spectrophotometrically, with Triton-X100 lysis serving as 100% release. One hemolytic unit (HU) is the amount of toxin that releases half the hemoglobin. SLO (Aalto Bio Reagents Ltd.) was activated by 4mM DTT. Purified SLO with a cysteine to alanine mutation at position 53021 was a gift from Dr. Norma Andrews, Yale University.
LLO co-immunoprecipitation
Macrophages were infected with L. monocytogenes for 2 h and extracted in Tris-buffered saline, pH7.4, containing 1% Triton X-100, protease inhibitors (Roche), and 10mM methyl methanethiolsulfonate for 30 min. on ice. GILT was immunoprecipitated using the mAb MaP.mGILT6, the samples separated by reducing SDS-PAGE and LLO detected by western blot using a rabbit anti-LLO antibody and goat anti-rabbit Ig conjugated with horse radish peroxidase (HRP). An anti-H2-Kb mAb, Y-3, was used as a control. The blot was developed with ECL reagents.
Lysosomal and phagosomal acidification
Lysosomal pH was determined after uptake of Oregon green-labelled dextran (10,000 MW, 25µg/ml) (Molecular Probes) over 45 min. on ice as described29, 30. Cells were extensively washed in PBS at neutral pH and read on a SpectraMax M5 plate reader (Molecular Devices), and analyzed with SoftmaxPro software (Molecular Devices). Fluorescent emission at 520nm was measured with alternating excitation at 450nm and 490nm for 40 min. Conversion of the 450:490 excitation ratio to pH was calculated based on a standard curve generated using excitation ratios of the dextran in standard buffers. Phagosomal pH was determined after the uptake of 3µm carboxy-beads (Polysciences Inc.) covalently labeled with carboxyfluorescein-SE (Molecular Probes) using a similar approach.
Supplementary Material
Acknowledgements
The authors are grateful to Dr. Daniel Portnoy for advice and reagents, and to Nancy Dometios for manuscript preparation. We particularly wish to acknowledge the valuable contribution of the late Dr. Marc Pypaert to the electron microscopy. This work was supported by NIH AI023081 (PC) and the Howard Hughes Medical Institute (PC, RS).
References
- 1.Pizarro-Cerda J, Sousa S, Cossart P. Exploitation of host cell cytoskeleton and signalling during Listeria monocytogenes entry into mammalian cells. C R Biol. 2004;327:523–531. doi: 10.1016/j.crvi.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 2.Tilney LG, Portnoy DA. Actin filaments and the growth, movement, and spread of the intracellular bacterial parasite, Listeria monocytogenes. J. Cell Biol. 1989;109:1597–1608. doi: 10.1083/jcb.109.4.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barry RA, Bouwer HG, Portnoy DA, Hinrichs DJ. Pathogenicity and immunogenicity of Listeria monocytogenes small-plaque mutants defective for intracellular growth and cell-to-cell spread. Infect. Immun. 1992;60:1625–1632. doi: 10.1128/iai.60.4.1625-1632.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Billington SJ, Jost BH, Songer JG. Thiol-activated cytolysins: structure, function and role in pathogenesis. FEMS Microbiol. Lett. 2000;182:197–205. doi: 10.1016/s0378-1097(99)00536-4. [DOI] [PubMed] [Google Scholar]
- 5.Geoffroy C, Gaillard JL, Alouf JE, Berche P. Purification, characterization, and toxicity of the sulfhydryl-activated hemolysin listeriolysin O from Listeria monocytogenes. Infect. Immun. 1987;55:1641–1646. doi: 10.1128/iai.55.7.1641-1646.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Portnoy DA, Chakraborty T, Goebel W, Cossart P. Molecular determinants of Listeria monocytogenes pathogenesis. Infect. Immun. 1992;60:1263–1267. doi: 10.1128/iai.60.4.1263-1267.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arunachalam B, Phan UT, Geuze HJ, Cresswell P. Enzymatic reduction of disulfide bonds in lysosomes: characterization of a gamma-interferon-inducible lysosomal thiol reductase (GILT) Proc. Natl. Acad. Sci. U S A. 2000;97:745–750. doi: 10.1073/pnas.97.2.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Phan UT, Arunachalam B, Cresswell P. Gamma-interferon-inducible lysosomal thiol reductase (GILT). Maturation, activity, and mechanism of action. J. Biol. Chem. 2000;275:25907–25914. doi: 10.1074/jbc.M003459200. [DOI] [PubMed] [Google Scholar]
- 9.Garin J, et al. The phagosome proteome: insight into phagosome functions. J. Cell Biol. 2001;152:165–180. doi: 10.1083/jcb.152.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Phan UT, Lackman RL, Cresswell P. Role of the C-terminal propeptide in the activity and maturation of gamma -interferon-inducible lysosomal thiol reductase (GILT) Proc. Natl. Acad. Sci. U S A. 2002;99:12298–12303. doi: 10.1073/pnas.182430499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maric M, et al. Defective antigen processing in GILT-free mice. Science. 2001;294:1361–1365. doi: 10.1126/science.1065500. [DOI] [PubMed] [Google Scholar]
- 12.Sealy R, et al. Target peptide sequence within infectious human immunodeficiency virus type 1 does not ensure envelope-specific T-helper cell reactivation: influences of cysteine protease and gamma interferon-induced thiol reductase activities. Clin. Vaccine Immunol. 2008;15:713–719. doi: 10.1128/CVI.00412-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lackman RL, Cresswell P. Exposure of the promonocytic cell line THP-1 to Escherichia coli induces IFN-gamma-inducible lysosomal thiol reductase expression by inflammatory cytokines. J. Immunol. 2006;177:4833–4840. doi: 10.4049/jimmunol.177.7.4833. [DOI] [PubMed] [Google Scholar]
- 14.Michel E, Reich KA, Favier R, Berche P, Cossart P. Attenuated mutants of the intracellular bacterium Listeria monocytogenes obtained by single amino acid substitutions in listeriolysin O. Molecular Microbiology. 1990;4:2167–2178. doi: 10.1111/j.1365-2958.1990.tb00578.x. [DOI] [PubMed] [Google Scholar]
- 15.Hastings KT, Lackman RL, Cresswell P. Functional requirements for the lysosomal thiol reductase GILT in MHC class II-restricted antigen processing. J. Immunol. 2006;177:8569–8577. doi: 10.4049/jimmunol.177.12.8569. [DOI] [PubMed] [Google Scholar]
- 16.Walker KW, Gilbert HF. Scanning and escape during protein-disulfide isomerase-assisted protein folding. J. Biol. Chem. 1997;272:8845–8848. doi: 10.1074/jbc.272.14.8845. [DOI] [PubMed] [Google Scholar]
- 17.Alouf JE, Billington SJ, Jost BH. Bacterial Toxins: A Comprehensive Sourcebook. Bacterial Toxins: A Comprehensive Sourcebook. 2005:643–658. [Google Scholar]
- 18.Heuck AP, Tweten RK, Johnson AE. Assembly and topography of the prepore complex in cholesterol-dependent cytolysins. J. Biol. Chem. 2003;278:31218–31225. doi: 10.1074/jbc.M303151200. [DOI] [PubMed] [Google Scholar]
- 19.Soltani CE, Hotze EM, Johnson AE, Tweten RK. Structural elements of the cholesterol-dependent cytolysins that are responsible for their cholesterol-sensitive membrane interactions. Proc. Natl. Acad. Sci. U S A. 2007;104:20226–20231. doi: 10.1073/pnas.0708104105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Soltani CE, Hotze EM, Johnson AE, Tweten RK. Specific protein-membrane contacts are required for prepore and pore assembly by a cholesterol-dependent cytolysin. J. Biol. Chem. 2007;282:15709–15716. doi: 10.1074/jbc.M701173200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pinkney M, Beachey E, Kehoe M. The thiol-activated toxin streptolysin O does not require a thiol group for cytolytic activity. Infect. Immun. 1989;57:2553–2558. doi: 10.1128/iai.57.8.2553-2558.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bouwer HG, et al. Listeria monocytogenes-infected hepatocytes are targets of major histocompatibility complex class Ib-restricted antilisterial cytotoxic T lymphocytes. Infect. Immun. 1998;66:2814–2817. doi: 10.1128/iai.66.6.2814-2817.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haschtmann D, Gerber HJ, Mielke ME. Cytotoxic activity of murine resident peritoneal cells against Listeria monocytogenes-infected hepatocytes in vitro. Microbes Infect. 2005;7:1177–1183. doi: 10.1016/j.micinf.2005.03.031. [DOI] [PubMed] [Google Scholar]
- 24.Lackman RL, Jamieson AM, Griffith JM, Geuze H, Cresswell P. Innate immune recognition triggers secretion of lysosomal enzymes by macrophages. Traffic. 2007;8:1179–1189. doi: 10.1111/j.1600-0854.2007.00600.x. [DOI] [PubMed] [Google Scholar]
- 25.Bryant RE, Rashad AL, Mazza JA, Hammond D. beta-Lactamase activity in human pus. J. Infect. Dis. 1980;142:594–601. doi: 10.1093/infdis/142.4.594. [DOI] [PubMed] [Google Scholar]
- 26.Pamer EG, Wang CR, Flaherty L, Lindahl KF, Bevan MJ. H-2M3 presents a Listeria monocytogenes peptide to cytotoxic T lymphocytes. Cell. 1992;70:215–223. doi: 10.1016/0092-8674(92)90097-v. [DOI] [PubMed] [Google Scholar]
- 27.Gedde MM, Higgins DE, Tilney LG, Portnoy DA. Role of listeriolysin O in cell-to-cell spread of Listeria monocytogenes. Infect. Immun. 2000;68:999–1003. doi: 10.1128/iai.68.2.999-1003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Agaisse H, et al. Genome-wide RNAi screen for host factors required for intracellular bacterial infection. Science. 2005;309:1248–1251. doi: 10.1126/science.1116008. [DOI] [PubMed] [Google Scholar]
- 29.Yates RM, Hermetter A, Russell DG. The kinetics of phagosome maturation as a function of phagosome/lysosome fusion and acquisition of hydrolytic activity. Traffic. 2005;6:413–420. doi: 10.1111/j.1600-0854.2005.00284.x. [DOI] [PubMed] [Google Scholar]
- 30.Yates RM, Hermetter A, Taylor GA, Russell DG. Macrophage activation downregulates the degradative capacity of the phagosome. Traffic. 2007;8:241–250. doi: 10.1111/j.1600-0854.2006.00528.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.