

Summary
The MVB pathway plays essential roles in several eukaryotic cellular processes. Proper function of the MVB pathway requires reversible membrane association of the ESCRTs, a process catalyzed by Vps4 ATPase. Vta1 regulates the Vps4 activity but its mechanism of action was poorly understood. We report the high-resolution crystal structures of the Did2- and Vps60-binding N-terminal domain and the Vps4-binding C-terminal domain of S. cerevisiae Vta1. The C-terminal domain also mediates Vta1 dimerization and both subunits are required for its function as a Vps4 regulator. Emerging from our analysis is a mechanism of regulation by Vta1 in which the C-terminal domain stabilizes the ATP-dependent double ring assembly of Vps4. In addition, the MIT motif containing N-terminal domain, projected by a long disordered linker, allows contact between the Vps4 disassembly machinery and the accessory ESCRT-III proteins. This provides an additional level of regulation and coordination for ESCRT-III assembly and disassembly.
Introduction
The multivesicular body (MVB) is a special form of the late endosome, generated when the limiting membrane of the endosome invaginates and buds into its own lumen. During this process, proteins residing in the endosomal membrane are sorted into the forming vesicles. Subsequent fusion of MVBs with the lysosome results in the delivery of cargo-containing intralumenal vesicles to the hydrolytic environment of the lysosome (for reviews see (Babst, 2005; Gruenberg and Stenmark, 2004; Katzmann et al., 2002; Piper and Katzmann, 2007)). The MVB pathway plays key roles in eukaryotic cellular processes such as cell surface growth factor receptor downregulation (for reviews see (Katzmann et al., 2002; Piper and Katzmann, 2007)), budding of retroviruses from the host cell (for a review see (Morita and Sundquist, 2004)) and cytokinesis (Carlton and Martin-Serrano, 2007; Morita et al., 2007). Entry into the MVB pathway is a tightly regulated process and depends on the action of class E Vps (Vacuolar Protein Sorting) proteins, a majority of which are subunits of three distinct protein complexes called endosomal sorting complexes required for transport (ESCRT-I, -II, and -III, for reviews see (Hurley and Emr, 2006; Katzmann et al., 2002; Piper and Katzmann, 2007; Williams and Urbe, 2007)). These complexes transiently assemble on the endosomal membrane during the cargo sorting process. For sustained protein trafficking through the MVB pathway, the ESCRTs need to be dissociated and disassembled from the membrane and recycled back into the cytoplasm. Vps4, together with its regulator Vta1, catalyzes the process of ESCRT disassembly in an ATP-dependent reaction (Babst et al., 1997).
Vps4 is a member of the AAA-protein (ATPase associated with a variety of activities) family (Frickey and Lupas, 2004; Vale, 2000). It cycles between alternate molecular assembly states in vitro depending on the state of ATP binding and hydrolysis (Babst et al., 1998). The protein exists as a 90 kDa molecular species that upon ATP binding oligomerizes into a large molecular complex of 440 kDa, which also has a higher affinity for the substrate ESCRT-III subunits (Xiao et al., 2007). ATP hydrolysis by Vps4 in turn results in the disassembly of the oligomer, coincident with the release of the ESCRTs from the endosomal membrane. Since ATP hydrolysis represents an irreversible step in the MVB pathway, regulation of the Vps4 ATPase activity is critical for the proper function of the MVB sorting reaction. Vta1 has been recently shown to act as a positive regulator of Vps4 by stimulating its ATPase activity (Azmi et al., 2006; Lottridge et al., 2006). Furthermore, this mechanism of regulation appears to be conserved during evolution, since Vta1 orthologs have been identified from yeast to humans and contain highly homologous sequence motifs that are thought to mediate interactions with Vps4 (Fujita et al., 2004; Haas et al., 2007; Yeo et al., 2003). In addition, Vta1 also interacts with other proteins acting in the late stage of the MVB pathway, such as the accessory ESCRT-III proteins Vps60 and Vps46/Did2 (Azmi et al., 2006; Lottridge et al., 2006; Shiflett et al., 2004). These interactions are also important for the MVB sorting and the retrovirus release processes (Ward et al., 2005). However, the structural basis for the function of Vta1 remains elusive.
Here we report biochemical and high-resolution structural characterization of S. cerevisiae Vta1. We show that Vta1 has a modular structure with two well-folded terminal domains bridged by a region dominated by random-coil structure. Crystal structures of the Vps60- and Did2-binding N-terminal domain and the Vps4-binding C-terminal domain reveal the structural details for the respective binding sites. The C-terminal domain also mediates Vta1 dimerization, and mutagenesis studies demonstrate that both subunits in the dimer are necessary for the function of Vta1 as a Vps4 regulator. Our study suggests a mechanism of regulation by Vta1 in which the C-terminal domain promotes the ATP-dependent double ring assembly of Vps4, while the N-terminal domain, projected by the disordered middle linker region, allows contact between the Vps4 disassembly machinery and accessory ESCRT-III proteins to coordinate ESCRT-III assembly and disassembly.
Results
Structure determination
Initial attempt to crystallize the intact S. cerevisiae Vta1 was not successful. Limited proteolysis of Vta1 by chymotrypsin cleaved the protein after Tyr279 and resulted in two major fragments; of which the C-terminal fragment Vta1280-330 was readily crystallized. Secondary structure prediction for Vta1 suggested that the region between residues 168 and 279 contains little regular structure. Therefore, an N-terminal fragment Vta11-167 was generated and crystals were subsequently obtained. In the following sections, we will refer Vta11-167 as Vta1NTD and Vta1280-330 as Vta1CTD.
The structure of Vta1NTD was determined by single isomorphous replacement with anomalous scattering (SIRAS) from a native crystal and a selenomethionyl crystal. The protein structure was refined against the native data to a working R factor of 21.5% and a free R factor of 26.0% at 2.9 Å (Table 1). The entire structure can be visualized except for residues 65-75, which are presumed disordered. There are two polypeptide chains, related by a non-crystallographic 2-fold symmetry, in the asymmetric unit. However, gel filtration and sedimentation equilibrium analytical ultracentrifugation experiments suggested that Vta1NTD is a monomer in solution. The structure of Vta1CTD was determined by multi-wavelength anomalous dispersion (MAD) from a selenomethionyl crystal at 1.8 Å and later refined against a native data set to a working R factor of 22.0% and a free R factor of 24.9% at 1.5 Å (Table 1). There are a total of six polypeptide chains that form three dimer pairs in the asymmetric unit. While the C-termini of the six molecules are all well defined, the N-termini show great structural variability among the six molecules and the region between residues 280 and 289 must be modeled into several alternative positions.
Table 1.
Data collection and refinement statistics
| Vta1-NTD | Vta1-CTD | |||||
|---|---|---|---|---|---|---|
| Data collection | ||||||
| Native | SeMet | Native | SeMet | |||
| Wavelength | 0.9787 Å | 0.9787 Å | 0.9793 Å | 0.9794Å | 0.9566Å | |
| Space group | I4 | I4 | P21212 | P21212 | ||
| Unit cell (Å) | a=b=126.169 Å, c=70.252 Å | a=b=126.623 Å, c=69.046 Å | a=102.420 Å b=50.707 c=74.353 Å |
a=102.429 Å, b=50.695, c=74.317 Å | ||
| Resolution (Å) | 2.9 | 3.0 | 1.5 | 1.8 | 1.8 | 1.8 |
| Completeness (%) | 99.5 (96.7) | 95.2 (73.0) | 94.2 (91.3) | 99.9 (100.0) | 99.9 (100.0) | 99.7 (100.0) |
| I / σ | 37.6 (3.3) | 34.5 (2.4) | 23.7 (5.2) | 38.8 (9.9) | 39.6 (11.0) | 41.2 (11.6) |
| Rmerge (%) | 6.2 (41.8) | 8.1 (48.7) | 6.2 (20.2) | 6.5 (21.2) | 5.8 (19.4) | 5.4 (17.9) |
| Redundancy | 7.4 | 9.7 | 4.0 | 8.1 | 8.1 | 8.2 |
| Unique reflections | 12,169 | 10,527 | 59,057 | 36,525 | 36,514 | 36,570 |
| Refinement | ||||||
| Rwork/ Rfree | 21.5/26.0 (CNS) | 22.0/24.9 (REFMAC) | ||||
| Rmsd, bonds (Å) | 0.009 | 0.014 | ||||
| Rmsd, angles (°) | 1.108 | 1.240 | ||||
| Mean B (Å2) | 81.8 | 19.8 (residue B after TLS refinement) | ||||
| Ramachandran | 92.5% core 7.5% allow | 96.7% core 3.3% allow | ||||
The crystal structure of Vta1NTD
Vta1NTD contains seven α-helices (α1-α7) arranged into two anti-parallel three-helix bundle modules (Figure 1A). Helices α1 (1-18), α2 (22-36) and α3 (42-64) form the first module; and helices α5 (86-110), α6 (115-135) and α7 (141-163) form the second module (Supplemental Figure 1). A short helix α4 (76-84) and a disordered loop between α3 and α4 connect α3 and α5 at one end of the molecule. The two modules have an overall very similar structure and are related by a pseudo-dyad with α1/α5, α2/α6, and α3/α7 forming the corresponding pairs. They stack together via α1-α2 and α5-α6 with their helical axes perpendicular to each other. Extensive hydrophobic interactions exist between module 1, α4 and module 2 such that a total of 1809 Å2 is buried at the interface of the three structural elements. Therefore the entire assembly should be considered as a single domain. This explains the fact that attempts to express the two modules separately lead to either no or aggregated protein expression (data not shown).
Figure 1. The crystal structure of Vta1NTD.
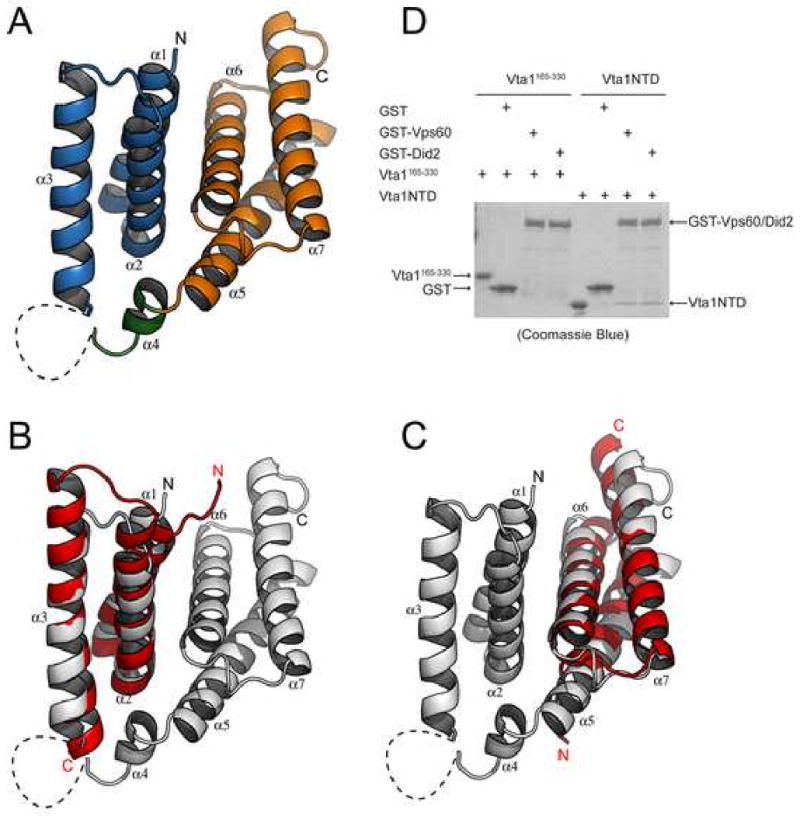
(A) A ribbon representation of the Vta1NTD structure. Helices α1, α2, and α3 (MIT1) are colored blue; helix α4 is colored green; and helices α5, α6, and α7 (MIT2) are colored orange. The disordered loop region connecting helices α3 and α4 is drawn as a dash line. The N- and C-termini of the molecule are indicated. (B) Structural overlay of Vta1NTD-MIT1 with the MIT domain of human Vps4A (PDB ID: 1YXR). Vta1NTD is colored gray. Vps4 MIT is colored red. The N- and C-termini for both molecules are indicated. (C) Structural Overlay of Vta1NTD-MIT2 with the MIT domain of human Vps4A. (D) In vitro analysis of Vta1-Vps60/Did2 interaction. GST or GST-tagged Vps60/Did2 was used to pull down purified Vta1NTD or Vta1165-330 as indicated. Proteins retained on the beads were analyzed by SDS-PAGE and visualized by Coomassie blue staining. Figures 1A, 1B and 1C are prepared with Pymol (DeLano Scientific LLC).
Vta1NTD consists of two MIT motifs
When the two structural modules within Vta1NTD were used to search for proteins of similar structure in the protein database, the N-terminal ESCRT-III binding domains of Vps4 were among the best matches (PDB ID: 1YXR and 2CPT, for human Vps4A and Vps4B, respectively). This domain contains a fold of three helices also known as the microtubule interacting and transport (MIT) motif (Scott et al., 2005b; Takasu et al., 2005). When the three MIT domain helices of Vps4 were aligned with those in the two modules of Vta1NTD (MIT1 and MIT2), root mean square deviations for the Cα atoms are 1.3Å and 1.7Å over 52 and 54 residues, respectively, for human Vps4A (Figure 1B, 1C).
The MIT domain of Vps4 mediates its interaction with the ESCRT-III subunits. The site of interaction involves a surface groove formed by helix 2 and helix 3 of the MIT domain (Scott et al., 2005b; Obita et al., 2007; Stuchell-Brereton et al., 2007). The ESCRT-III subunits interact with Vps4 by providing a fourth helix to complete a non-canonical tetratricopeptide repeat (TPR) fold. Although Vta1 does not bind to the ESCRT-III subunits, it does interact with two ESCRT-III like proteins, Vps60 and Vps46/Did2 (Lottridge et al., 2006; Shiflett et al., 2004). Given the structural similarity between Vta1NTD and Vps4 MIT domain, it is likely that Vta1 interacts with Vps60 and Vps46/Did2 via Vta1NTD in a manner similar to that between Vps4 and ESCRT-III. Indeed, purified Vta1NTD alone is sufficient to bind to GST-Vps60 and GST-Did2, while purified Vta1 lacking the NTD fails to bind to either (Figure 1D). This in vitro result is also consistent with an earlier finding that deletion of the N-terminal 68 residues of Vta1 abolishes its in vivo interaction with Vps60 (Azmi et al., 2006).
Potential Vps60 and Did2 binding surface on Vta1NTD
Since the site of protein-protein interaction in the Vps4 MIT domain involves a surface groove formed by helix 2 and helix 3 of the motif, we examined the two corresponding surface areas in Vta1NTD. Both surfaces contain a hydrophobic center surrounded by polar/charged regions. Surface 1 in MIT1 is formed by conserved hydrophobic residues Leu-29, Val-32, Leu-36, Leu-53, and Ile-56, and conserved charged/polar residues Glu-33, Arg-41, Thr-46, Thr-50, Asp-54, Glu-57, Lys-60, and Lys-61 (Figure 2A, 2B). In fact, this region on MIT1 represents the largest conserved area on the surface of Vta1NTD. The corresponding residues for Leu-29, Val-32, Leu-36, Leu-53, Ile-56, Glu-33, Thr-46, Asp-54, Glu-57 and Lys-60 in the Vps4 MIT domain (Y34, L37, M41, L64, A67, D38, R57, N65, E68 and K71) were shown to be involved in its interaction with the ESCRT-III subunit Vps2 (Obita et al., 2007; Stuchell-Brereton et al., 2007). Compared with surface 1, surface 2 on MIT2 is much less conserved (Figure 2A, 2C). Residues that form the surface include hydrophobic residues Trp-122, Ile-125, Leu-132 and Ile151, and charged/polar residues Lys-118, Ser-129, His-133, Lys-136, Glu-137, Glu-141, Thr-144, Gln-148, Lys-152 and Lys155. Of these, only Trp-122, Leu-132, Glu-137, Glu-141 and Lys-152 appear to be conserved in other Vta1 proteins.
Figure 2. Potential Vps60 binding sites on Vta1NTD.
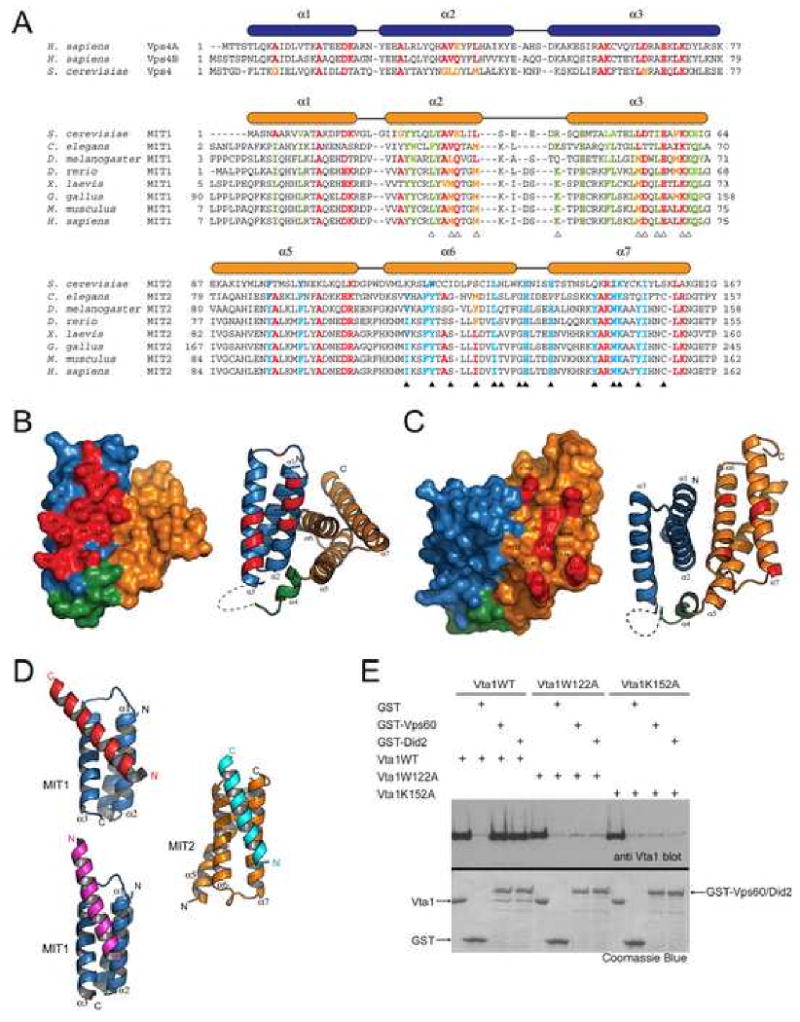
(A) Structure-based sequence alignment for the MIT domains in Vps4 and the two MIT motifs in Vta1. Secondary structure elements are shown above the sequences. Residues conserved in all MIT domains are colored red and yellow. Residues conserved only in MIT1 are colored green; and residues conserved only in MIT2 are colored blue. Residues involved in the potential functional surfaces of MIT1 and MIT2 are marked by white and black triangles, respectively. (B, C) Conserved molecular surfaces on MIT1 (B) and MIT2 (C). Surface and ribbon representations of molecules in the same orientation are shown on the left and right panels, respectively. MIT1 is colored blue; helix α4 is colored green; and MIT2 is colored orange. Conserved molecular surfaces are colored red and underlying residues are labeled. (D) The two conserved surfaces are involved in interacting with an α-helix from molecules in the crystal lattice. MIT1 is colored blue and MIT2 is colored orange. The helices they interact with in the crystal lattice are colored red, magenta, and cyan, respectively. (E) Residues in MIT2 are important for Vps60 and Did2 binding. GST or GST-tagged Vps60/Did2 was used to pull down purified wild-type Vta1 or mutants as indicated. Proteins retained on the beads were analyzed by SDS-PAGE and visualized by Coomassie blue staining or western blotting with anti-Vta1 antibody (see the accompanying study (Azmi et al., 2007) for anti-Vta1 polyclonal antibody production). Figures 2B, 2C and 2D are prepared with Pymol (DeLano Scientific LLC).
In the lattice of Vta1NTD crystals, each Vta1NTD non-crystallographic “dimer” interacts with four neighboring molecules. Three of these interactions involve a helix from a neighboring molecule binding to the potential Vps60 binding surface in either MIT1 or MIT2 (Figure 2D). In all three cases, a majority of the above-described conserved residues are buried at the interface. These findings suggest that surface grooves formed by helix 2 and helix 3 of the MIT motifs in Vta1NTD are capable of participating in protein-protein interactions and therefore are excellent candidates for binding to Vps60 and Vps46/Did2.
To address this possibility, mutational analysis of the putative Vps60 and Did2 binding surface(s) of Vta1 was performed. Conserved surface residues within MIT1 (Leu-29, Leu-36, Leu-53, Ile-56, Glu-57) and MIT2 (Trp-122, Leu-132, Glu-137, Lys-149, Lys-152) were mutated to alanines, the mutant proteins were expressed in bacteria, and the purified proteins were examined for binding to GST-Did2 and GST-Vps60. While most mutations did not dramatically reduce binding (data not shown), this analysis identified 2 residues, Trp-122 and Lys-152, as important for Vta1 interaction with both Did2 and Vps60 (Figure 2E). This result implicated the surface groove formed by helix 2 (α6) and helix 3 (α7) of MIT2 as the putative binding site for both Vps60 and Did2. Functional analysis of this interaction is presented in the accompanying study (Azmi et al., 2007).
The crystal structure of Vta1CTD
Vta1CTD is organized into a pair of anti-parallel α-helices (α8-α9) (Figure 3A). Helix α8 (281-310) is significantly longer than helix α9 (312-330) such that inter-helical interactions only exist between α9 and the C-terminal half of α8. As will be discussed in more detail, the structure where inter-helical interaction occurs is also involved in protein dimerization. Therefore, it should be considered as the structural core of Vta1CTD. Interestingly, regions outside the core display great conformational variability. Of the six independently determined Vta1CTD molecules in the asymmetric unit, helix α8 displays different degrees of bending at Arg-290, ranging from -30° to 14° (Figure 3B). These results suggest that the N-terminal part of helix α8 is highly flexible relative to the core of Vta1CTD.
Figure 3. The crystal structure of Vta1CTD.
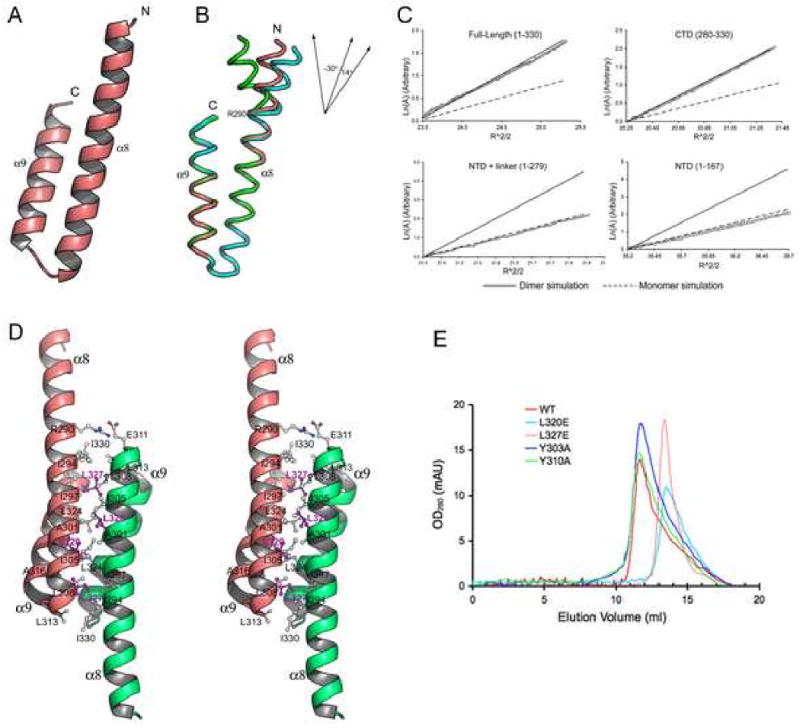
(A) A ribbon representation of the monomer structure of Vta1CTD. The N- and C-termini of the molecule are indicated. (B) Structure flexibility within Vta1CTD. Three Vta1CTD molecules that represent the largest differences among the six molecules in the asymmetric unit are shown as ribbons and colored green, pink, and cyan, respectively. Top right insert: a schematic diagram illustrating different degrees of helix α8 bending at Arg-290. (C) LnA vs R2 plot of sedimentation equilibrium analytical ultracentrifugation data. The slope of the plot is proportional to the molecular weight of the protein. Data is plotted alongside simulations using theoretical values for the molecular weight of a dimer (solid line) or a monomer (dash line) species, with an arbitrary Y-axis for direct comparison. (D) Stereo view of the dimer structure of Vta1CTD. One molecule is colored pink, the other colored green. Residues located at the dimer interface are labeled. Leu-320 and Leu-327 are highlighted in magenta. (E) Gel filtration analysis. Gel filtration experiments were performed on a Supedex™ 200 column (Amersham). Compared with the wild-type protein (red), L320E (cyan) and L327E (pink) eluted at a monomer position; while Y303A (blue) and Y310A (green) maintained their dimeric structures. Figures 2A, 2B and 2D are prepared with Pymol (DeLano Scientific LLC).
Oligomerization state of Vta1
The crystal structure of Vta1CTD shows that the molecule forms a dimeric structure in the crystal lattice where three independent dimer pairs were observed. This is consistent with previous findings that Vta1 is molecular dimer in solution and the C-terminal region is required for dimerization (Azmi et al., 2006). To determine whether Vta1CTD alone is sufficient for dimerization, the oligomerization state of Vta1CTD was analyzed using sedimentation equilibrium analytical ultracentrifugation (Figure 3C). The results showed that, like Vta1, Vta1CTD is a dimer in solution. In contrast, only a monomer species was observed for Vta1 lacking the C-terminal domain (Vta11-279), consistent with results from previous studies using an in vivo assay (Azmi et al., 2006). Therefore, Vta1CTD is necessary and sufficient for protein dimerization.
In forming the dimer, the anti-parallel α-helices within each Vta1CTD subunit interacts with their helical axes aligned to create a four-helix bundle dimer structure (Figure 3D). In the four-helix bundle, symmetry-related helices are packed against each other and the molecular dyad is perpendicular to the helical axes. As a result, the N-terminal ends of the two subunits point way from the four-helix bundle and in opposite directions. The dimer interface covers 21% of the solvent accessible surface for each subunit. The interface is predominantly hydrophobic with many highly conserved residues involved, including Ala-304, Leu-308, Leu-313, Ala-316, Leu-320, Leu-324 and Leu-327. At the periphery of the interface, a few polar interactions are also observed including a pair of salt bridges between Arg-290 and a highly conserved Glu-311.
To assess the contribution of the conserved residues at the Vta1CTD dimer interface to the stability of Vta1 dimer structure, two single point mutants, L320E and L327E, were generated. Both residues are in the central region of the hydrophobic dimer interface, and we predicted that substitution of them with charged glutamic acids would disrupt the packing within the dimer structure. Mutant proteins were purified and their oligomerization states were analyzed using gel-filtration chromatography. Indeed, compared with the wild type Vta1 dimer, the elution profile of the L320E and L327E mutants were shifted towards the monomeric species on gel filtration (Figure 3E). These biochemical results confirmed that the conserved interface observed in Vta1CTD structure is responsible for Vta1 dimerization.
Vps4 binding surface on Vta1CTD
The C-terminal region of Vta1 has been previously shown to interact with Vps4 (Azmi et al., 2006). With purified Vps4E233Q (an ATPase deficient mutant), we further demonstrated that Vta1 interacts with Vps4 in an adenine nucleotide dependent manner (Figure 4A). Interestingly, the N-terminal region of Vta1 appears to have an effect on this interaction as Vta1CTD displays a stronger ADP-dependent binding to Vps4.
Figure 4. Vps4 binding sites on Vta1CTD.
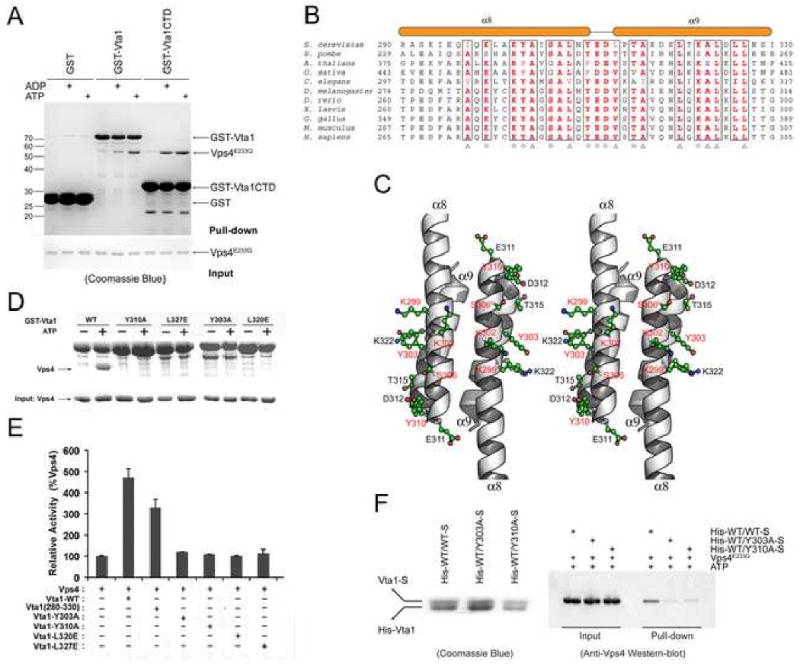
(A) In vitro analysis of Vta1-Vps4 interaction. Glutathione agarose beads pre-loaded with GST, GST-Vta1 or GST-Vta1CTD were incubated with purified Vps4E233Q under indicated conditions. Proteins retained on the beads were analyzed by SDS-PAGE and visualized by Coomassie blue staining. (B) Sequence alignment of Vta1CTD. Triangles indicate conserved residues involved in the dimer formation. Stars indicate conserved surface residues. Secondary structure elements are shown above the sequences. (C) Stereo view of the conserved surface residues on Vta1CTD. The residues demonstrated to be important for Vps4 interaction are highlighted in red. (D) Interactions between Vps4 and mutant Vta1 proteins. Purified Vps4E233Q were incubated with GST-tagged wild-type or point mutant Vta1 proteins in the presence of 2 mM ATP. Materials bound to glutathione agarose beads were analyzed by SDS PAGE and visualized by Coomassie blue staining. (E) ATPase activity of Vps4 in the presence of Vta1 proteins. The activity was normalized to Vps4 alone to compare the stimulation effects of different Vta1 proteins (error bars represent standard deviation of results from three independent measurements). (F) Interactions between Vps4 and Vta1 heterodimers. Vta1 heterodimers were co-expressed in pETDuet-1 vector such that the N-terminal His-tagged wild-type protein is in the first expression cassette and the C-terminal S-tagged wild-type or mutant protein is in the second expression cassette. Heterodimer proteins were isolated by tandem purification with Ni++-NTA resin and S-protein resin (left panel). Heterodimers retained on the S-protein resin were then used to pull down Vps4E233Q in the presence of 2mM ATP. Bound Vps4 proteins were analyzed on SDS-PAGE and detected by Western blotting with anti-Vps4 antibody (right panel). Figure 4C is prepared with Pymol (DeLano Scientific LLC).
Sequence alignment of Vta1 proteins revealed a high degree of overall conservation for Vta1CTD among representative members from yeast, plants, and mammals (Figure 4B). Many of the conserved residues are located at the dimer interface and are important for supporting the dimer structure. There are, however, a few conserved surface exposed residues including Lys-299, Lys-302, Tyr-303, Ser-306, Tyr-310, Glu-311, Asp-312, Thr-315 and Lys-322 (Figure 4C). Previous studies showed that mutations in Lys-299 and Lys-302 completely abolished the ability of Vta1 to stimulate the ATPase activity of Vps4 while mutation in Lys-322 had no effect (Azmi et al., 2006). Since Lys-299 and Lys-302 are located on helix α8 and Lys-322 is located on helix α9, these results suggest that α8 is the site of Vps4 binding.
Four more conserved residues on α8 and the short linker between α8 and α9, residues Tyr-303, Tyr-310, Glu-311, and Asp-312, were mutated into alanines to assess their roles in mediating Vps4 interaction. Of the four, Y303A and Y310A were deficient in interaction with Vps4 as demonstrated in a GST pull-down assay while maintaining their dimeric structures (Figure 4D, 3E). In addition, when the mutant proteins were purified and used in the ATPase assay, they failed to stimulate the ATPase activity of Vps4, in contrast to the wild type protein (Figure 4E). Therefore, it is likely that these two residues are directly involved in Vps4 interaction as well. The other two mutants, E311A and D312A, also did not bind to Vps4 but they were monomers in solution (data not shown). Hence it is not clear whether they directly participate in binding to the ATPase. Taken together, these results showed that the site of Vps4 binding on Vta1 is located on the solvent-exposed side of helix α8 and the binding characteristic appears to be largely hydrophilic in nature.
The dimer structure of Vta1 is required for Vps4 interaction
The fact that monomer mutants E311A and D312A failed to interact with Vps4 prompted us to examine the role of Vta1 dimer in regulating the function of Vps4. Indeed, the monomer mutants L320E and L327E also failed either to pull down Vps4 in our GST pull-down assay or to stimulate the ATPase activity of Vps4 suggesting that a stable association between Vta1 and Vps4 requires the presence of both Vps4 binding sites on Vta1 (Figure 4D, 4E). To further characterize this structural requirement, His-tagged wild-type Vta1 was co-expressed with S-tagged S-tagged wild-type Vta1, Y303A, or Y310A mutant in E. coli. Vta1 heterodimers were isolated by tandem purification with Ni++-NTA resin and S-protein resin (Figure 4F). Heterodimers retained on the S-protein resin were then used to pull down Vps4E233Q in the presence of ATP. Although these heterodimers behaved similarly during the process of expression and purification, only the wild-type Vta1 heterodimer could interact with Vps4. Neither WT/Y303A nor WT/Y310A heterodimer, which contains one of the two functional Vps4 binding sites, could form a stable complex with Vps4E233Q. These results strongly suggested that both Vps4 binding sites on the Vta1 dimer are necessary for its stable association with Vps4.
To further examine the effect of Vta1 dimerization and Vps4 binding on the function of Vta1 in the MVB pathway, we examined the sorting of carboxypeptidase S (CPS) in yeast cells expressing wild-type VTA1 versus mutant vta1 alleles. Sorting of the MVB cargo GFP-CPS was analyzed by fluorescence microscopy in vta1Δ cells transformed with mutant forms of vta1 (Figure 5). Four mutants were used in the study: two Vps4-binding deficient mutants Y303A and Y310A and two dimerization-deficient mutants L320E and L327E. Cells transformed with empty plasmid display a dramatic mis-localization of GFP-CPS to the limiting membrane of the vacuole, indicative of the MVB sorting defect observed in vta1Δ cells. As expected, transformation with wild type VTA1 restores delivery of GFP-CPS into the MVB pathway as indicated by fluorescence within the vacuole lumen. However, none of the mutants is capable of complementing the MVB sorting defect observed in vta1Δ cells. GFP-CPS was mis-localized in cells expressing these mutants as in vta1Δ cells. These results highlight the importance of both Vta1 dimerization and Vps4 binding for the in vivo function of Vta1.
Figure 5. Residues involved in dimerization and Vps4 binding are required for cargo sorting in the MVB pathway.
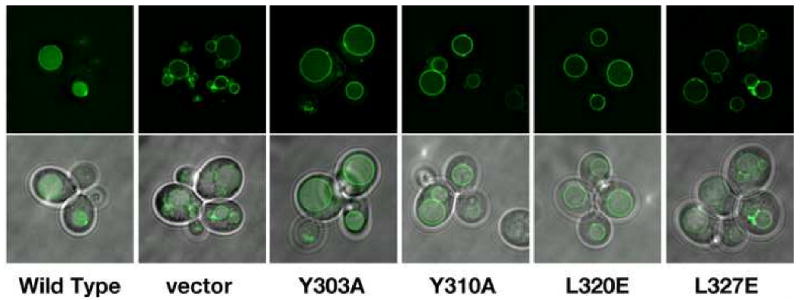
vta1 cells (BY4742 genetic background) were transformed with both cen-copy plasmids expressing the indicated form of Vta1 and GFP-CPS. Live cells were visualized using fluorescence and brightfield microscopy and fluorescent images were processed using Deltavision deconvolution software.
The structure of intact Vta1
The region between Vta1NTD and Vta1CTD (residues 168-279) was not defined crystallographically in the current study. Although this linker is present in all Vta1 proteins, there appears to be no strong sequence conservation. In addition, the length of the linker varies greatly among different Vta1 orthologs. While structure prediction for the linker suggests that it contains little regular ordered secondary structure, we sought to confirm the prediction experimentally by measuring the circular dichroism (CD) spectrum in the far UV region for the linker. Since the linker alone cannot be expressed, we obtained its spectrum indirectly by measuring the spectra for the intact and various truncation forms of Vta1. The CD spectrum of the intact Vta1 is dominated by contributions from α-helices (Figure 6A). The CD spectrum of the intact protein is nearly superimposable to a linear combination of the spectra of Vta11-279 and Vta1CTD suggesting that these isolated fragments retain their native structures as in the intact protein (Figure 6B). The CD spectrum for the linker can be obtained by subtracting the spectrum of Vta1NTD from that of Vta11-279, or, alternatively, by subtracting the spectra of Vta1NTD and Vta1CTD from that of the intact protein. These two calculated CD spectra agree with each other very well and both have a strong minimum near 195nm (Figure 6C). This feature is typical for random coil structures, suggesting that the linker region is mostly devoid of ordered secondary structure and likely adopts a random coil conformation.
Figure 6. The structure of intact Vta1.
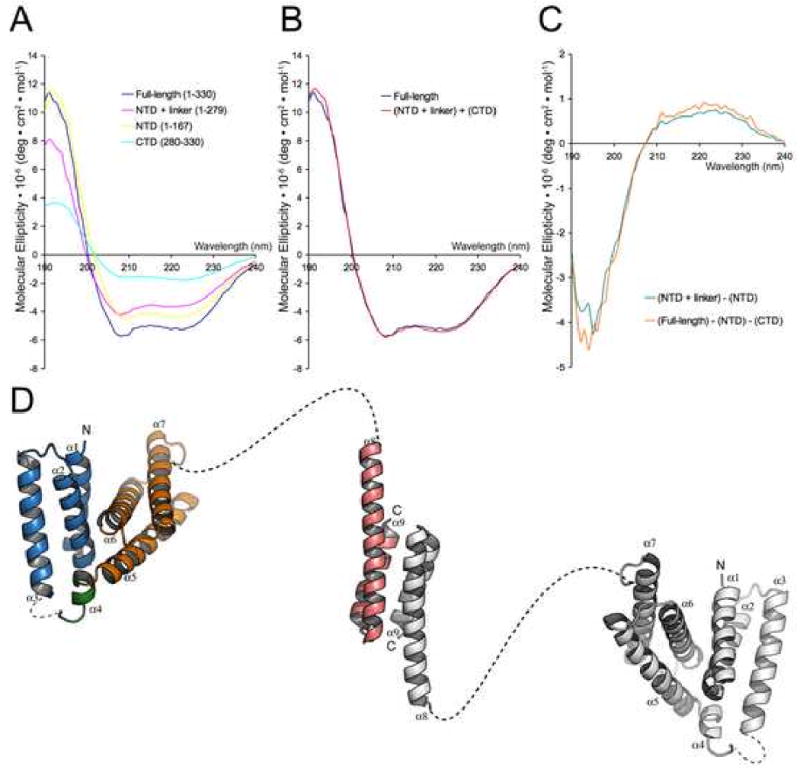
(A) Experimental far-UV CD spectra of Vta1, Vta11-279, Vta11-167 (Vta1NTD) and Vta1280-330 (Vta1CTD) show α-helix-containing structures. (B) The experimental CD spectrum of Vta1 is superimposable with the reconstructed CD spectrum calculated from the experimental spectra of Vta11-279 and Vta1280-330. (C) Calculated CD spectra of the linker region (Vta1168-279) show random coil structure. (D) A model of the full-length Vta1 dimer. One molecule is colored as in Figures 1A and 3A and the other is colored gray. The linkers between helices α3 and α4 and between helices α7 and α8 are drawn as dash lines.
Based on the structural information from the crystallographic and CD spectral analysis, we propose that the structure of intact Vta1 likely adopts an extremely elongated shape with Vta1CTD at the center and Vta1NTD pointing to the opposite ends, connected by the largely unstructured middle linker (Figure 6D). The Stokes radii of Vta1, Vta1CTD, and Vta1NTD are 5.9nm, 2.2nm and 1.9nm respectively, as determined by dynamic light scattering. These numbers are consistent either with the model (Vta1) or with experimentally determined structures (Vta1NTD and Vta1CTD). Interestingly, the N-terminal portion of Vta1CTD can bend to different degrees depending on its environment, strongly suggesting that Vta1 is a highly flexible molecule with the Vps60 binding N-terminal domain acting like tentacles relative to its dimeric Vps4 binding core.
Discussion
Vta1 functions in the MVB pathway by interacting with at least three other class E Vps proteins, Vps4, Vps60 and Vps46/Did2. Consistent with its biological function, the structure of Vta1 is organized into three distinct regions: an N-terminal Vps60- and Did2-binding domain, a random-coiled linker region and a C-terminal Vps4 binding domain. In addition, the C-terminal domain also mediates protein homo-dimerization and the dimer structure is critical to its in vitro and in vivo function as a Vps4 regulator.
The structure of Vta1 suggests a possible mechanism of interaction between Vta1 and Vps4. Vps4 contains a unique three-stranded “β domain” structure that has been shown to mediate its interaction with Vta1 (Scott et al., 2005a). When Vps4 forms a high molecular weight ring-shaped oligomer, the β domain is situated at the periphery of the ring based on structural analogy to other AAA-ATPases whose oligomeric structures are known. Although it is still possible that the two symmetry-related Vps4 binding surfaces on Vta1CTD constitute a single Vps4 binding site, the discontinuous nature of the two surfaces would argue that they each bind to one Vps4 subunit. Given the symmetry within Vta1, the two Vps4 subunits they bind to must also obey the same matching two-fold symmetry. This in turn suggests that the nucleotide-dependent Vps4 oligomer is a molecule with two-fold symmetries, i.e., a double-ring structure.
One could therefore envision a model where Vta1 promotes the nucleotide-dependent assembly of a double-ring structure of Vps4 by binding to one subunit of Vps4 from each ring (Figure 7). The activity of Vta1 would then lie in its ability to strengthen the inter-ring subunit interaction rather than the intra-ring subunit interaction, which is likely to be solely due to nucleotide binding. In fact, nucleotide-dependent oligomerization must precede the action of Vta1 as only very weak interaction is observed in the absence of nucleotide. On the other hand, while a stable Vps4 oligomer only exists in the presence of ATP, addition of Vta1 can stabilize the oligomeric structure even with ADP as the affinity between Vps4 and Vta1 increases significantly with the addition of ADP in our in vitro pull-down assay (Figure 4A). Assuming each ring of Vps4 oligomer consists of six subunits, simultaneous binding of six Vta1 molecules will significantly stabilize the structure of Vps4 oligomer.
Figure 7. A model of Vps4-Vta1 complex.
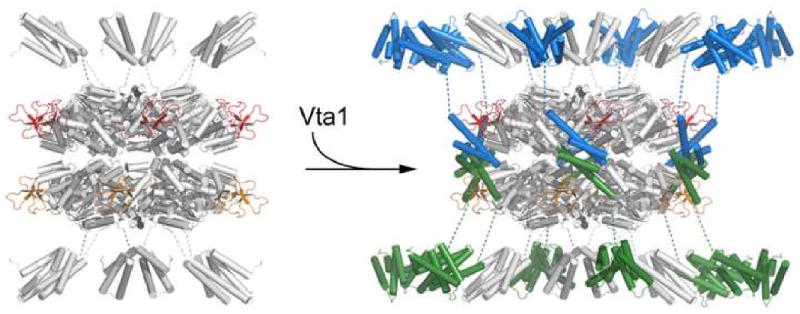
Vps4 dodecameric ring structure (colored white) is modeled using coordinates of S. cerevisiae Vps4 ATPase domain (Xiao et al., 2007) based on its homology to p97 (1S3S) and FtsK (2IUU). ESCRT-III binding MIT domains (colored white, 1YXR) of Vps4 are placed at both ends of the double-ring structure. One Vta1CTD dimer (colored blue and green) is modeled between two Vta1-binding β-domains of Vps4 (one from each ring, colored red and orange) so that contacts between the known interacting structural elements from the two proteins are maximized. The long linker between Vta1CTD and Vta1NTD likely projects Vta1NTD near the MIT domain of Vps4.
While the biological role of Vps4-Vta1 interaction is now clearly defined, we are less certain about the function of the interaction between Vta1 and accessory ESCRT-III proteins partly due to the lack of a well-defined biochemical role for these proteins. Like other class E Vps proteins, Vps60 and Vps46/Did2 are required for proper MVB sorting and have a structural organization similar to that of the ESCRT-III subunits (Williams and Urbe, 2007). In an accompanying study, Vta1 membrane recruitment is demonstrated to be dependent on Did2 while Vps60 membrane recruitment is dependent on Vta1 (Azmi et al., 2007). In addition, Vps60- and Did2-binding to Vta1 is demonstrated to enhance stimulation of Vps4 in vitro. These results suggest that interactions between Vta1 and the accessory ESCRT-III proteins direct membrane recruitment as well as stimulation of Vps4. Our structural analysis of Vta1NTD suggests that the overall mode of interaction between Vta1 and the accessory ESCRT-III proteins likely mimics that between Vps4 and the ESCRT-III subunits and differences may lie in the critical residues involved. It would be important to illustrate these structural features in future work.
Although the middle linker region of Vta1 contains little ordered structure, it may nevertheless be functionally important. In a simple picture, the sizable linker can project the N-terminal region of Vta1 significantly beyond its dimeric C-terminal core, allowing Vta1NTD and its binding partner to interact with other proteins in the MVB pathway. For example, Vta1NTD might be close to the N-terminal domain of Vps4, thereby bringing the ESCRT-III and accessory ESCRT-III proteins together. Interestingly, we have noticed that the linker also appears to have certain effects on the affinities of Vta1 for Vps4 and the accessory ESCRT-III proteins. Both Vta1NTD and Vta1CTD show higher affinities for their respective binding partners than the full-length protein in our pull-down assays (Data not shown and Figure 4A). These observations suggested that the linker may even function as a communicator between the two terminal domains more than just as a simple connector.
In summary, we have determined the crystal structures of the two functionally important domains of Vta1 and showed that Vta1 is an elongated molecule with a Vps4-binding dimeric core in the middle and two Did2- and Vps60-binding domains pointing in opposite direction and connected to the core by long, flexible regions. The structural information, coupled with biochemical and cell biology experiments, has provided important insights to the molecular mechanism of Vta1 function. Emerging from this study is a model where six Vta1 molecules bind to a double-ring shaped Vps4 near its mid-section and twelve Did2- and Vps60-binding domains project to both ends of the Vps4 double-ring. In this picture, these MIT motif-containing domains will be close to the N-terminal ESCRT-III binding domain of Vps4 suggesting that the ESCRT-III like proteins might be spatially close to the ESCRT-III proteins and participate in regulating the assembly and disassembly of the ESCRT-III complex. Therefore, we predict that Vta1 plays a much more complicated role in regulating the MVB pathway than previously envisioned.
Methods
Cloning, expression and purification
DNA fragments encoding Vta1 and Vps4 were amplified from the Saccharomyces cerevisiae genomic DNA. Vta1NTD was expressed in E. coli BL21(DE3) using a modified pET28b vector with a SUMO protein inserted between a His6-tag and the Vta1NTD coding region. The His6-SUMO-Vta1NTD protein was purified by Ni2+-NTA affinity chromatography following standard procedures. ULP1 protease was then added to remove the His6-SUMO tag, and the protein mixture was passed over a second Ni2+-NTA column. Vta1NTD was further purified by anion exchange chromatography on a Resource-Q column (Amersham Pharmacia). Vta1CTD was expressed in BL21(DE3) using a modified pET21a vector with an N-terminal His8-tag followed by a TEV protease cleavage site. The His8-Vta1CTD was first purified by Ni2+-NTA affinity chromatography. Overnight TEV protease digestion cleaved the N-terminal His8-tag, and a second Ni2+-NTA step removed the tag, uncleaved protein and TEV protease. Vta1CTD was further purified using an anion exchange Source-Q column (Amersham Pharmacia). Selenomethionyl protein derivatives were expressed in E. coli B834(DE3) using a minimal medium where methionines were replaced with selenomethionines. Derivative proteins were purified in the same way as native proteins.
Crystallization and data collection
Native crystals of Vta1NTD were grown by the sitting drop method at 20°C. Proteins at 25 mg/ml were mixed in a 1:1 ratio with a reservoir solution of 3.5 M sodium formate in a final volume of 4 μl and equilibrated against the reservoir solution. Crystals grew to full size in several days and were transferred into 4 M sodium formate, 10% glucose before flash-frozen under liquid nitrogen. Selenomethionyl protein crystals of Vta1NTD were grown from 3.6-3.8 M sodium formate, 0.1 M HEPES (pH 7.5) and harvested in the same way. Native and selenomethinyl crystals diffracted to 2.9 Å and 3.0 Å, respectively. Diffraction data were collected at the Advanced Photon Source beam line 21-ID. Native crystals of Vta1CTD were also grown by the sitting drop method at 20°C. Proteins at 20 mg/ml were mixed in a 1:1 ratio with a reservoir solution of 30% MPD, 0.1 M Na-Acetate (pH 4.5), and 20 mM CaCl2 in a final volume of 4 μl and equilibrated against the reservoir solution. Crystals were transferred into the reservoir solution before flash-frozen under liquid nitrogen. Selenomethionyl protein crystals of Vta1CTD were obtained in a similar way. Native and selenomethinyl crystals diffracted to 1.5 Å and 1.8 Å, respectively. Diffraction data were collected at the Advanced Photon Source beam line 23-ID. All data were integrated and scaled by using the program HKL2000 (HKL Research).
Structure determination and refinement
The structure of Vta1NTD was determined by the SIRAS method using a native data set and a data set collected at the peak wavelength of the selenium atom (λ = 0.9787 Å). Crystals belong to the I4 space group with a unit cell dimension of a = b = 126.17 Å, c = 70.25 Å. There are two molecules in the asymmetric unit. Six of the twelve expected selenium sites were found and cross-confirmed with SnB (Weeks and Miller, 1999) and CNS (Brunger et al., 1998). Four additional sites were identified using anomalous difference Fourier method as implemented in SHARP (Vonrhein et al., 2006). Parameters for these ten sites were refined and SIRAS phases were calculated and improved by solvent flattening with SHARP. The resulting experimental map is of high quality to allow unambiguous tracing of the model. The model was built manually with O (Jones et al., 1991) and refined against the native data with CNS using 5% randomly selected reflections for cross-validation. Initial refinement was carried out with torsion angle dynamics simulated annealing using the maximum likelihood target function with the experimental phases as a prior phase distribution (MLHL) followed by rebuilding in O. At later stages of the refinement, maximum likelihood target function using amplitudes (MLF) was used and individual restrained atom B-factor was refined. Although no non-crystallographic symmetry (NCS) restraint was utilized during the refinement process, the two protomers in the asymmetric unit appear to be almost identical, with a root mean square deviation of 0.551 Å for the Cα atoms. The final model contains residues 1-64 and 76-152 and an N-terminal alanine derived from the vector for each protomer and 11 water molecules.
The structure of Vta1CTD was determined by the MAD method using three data sets collected at the peak, the edge and a remote wavelength of the selenium atom. Crystals belong to the P21212 space group with a unit cell dimension of a = 102.42 Å, b = 50.71 Å, c = 74.35 Å. There are six molecules in the asymmetric unit. Heavy atom search, phase calculation and refinement, and density modification were carried out with autoSHARP (Vonrhein et al., 2006). An initial model was automatically built using ARP/WARP (Morris et al., 2003). The model was further refined against the native data set using CNS with intersperse manual rebuilding with O. Later rounds of refinement were performed with REFMAC5 in the CCP4 (CCP4, 1994) suite with TLS parameters incorporated (Winn et al., 2003). Each Vta1CTD dimer in the asymmetric unit was defined as one TLS group. The regions for 281-289 in protomer C and 280-289 in protomer F were modeled with alternative conformations with 50% occupancy for each conformation (Supplemental Figure 2). The final structure contains residues 280-330 for chain A, 281-330 for chain B, 280-330 for chain C, 289-330 for chain D, 286-329 for chain E, and 280-330 for chain F and a total of 295 water molecules. Three protomers (A, C, and F) contain extra residues derived from the vector: Gly-Ser for A and C, and a single serine for F.
In vitro binding experiments
GST pull-down experiments were performed following standard procedures in PBS buffer supplemented with 1mM DTT and 0.1% Triton X-100. Purified proteins were incubated with either GST alone or GST-tagged proteins immobilized on glutathione agarose beads for 40min at 4°C. The beads were then washed extensively with above buffer, and bound proteins were separated on SDS-PAGE and visualized by Coomassie staining.
The heterodimer of Vta1 were expressed using pETDuet-1 vector (Novagen) with the wild-type protein cloned in the first cassette and either wild-type or mutant proteins cloned in the second cassette. To purify the heterodimer, cells were lysed in 50mM Tris•HCl (pH 8.0), 300 mM NaCl, 15 mM imidazole, 5 mM β- mercaptoethanol, 0.1% Triton X-100 and 10 μg/ml PMSF. Cell lysate was centrifuged and the supernatant was applied to Ni++-NTA resin. After extensive wash, bound proteins were eluted with the above buffer containing 500 mM imidazole. The eluted protein sample was subsequently diluted into the GST pull-down buffer (see above) and incubated with S-protein agarose (Novagen) for 1 hr at 4°C. Beads with heterodimer protein bound were harvested by centrifugation, washed extensively, and either analyzed by SDS PAGE or used for pull-down experiments as described above. Due to the similar migration positions of S-tagged Vta1 and Vps4 on SDS-PAGE, bound Vps4 proteins were detected by Western blotting with anti-Vps4 antibody (Santa Cruz, sc-21821).
Analytical ultracentrifugation
Protein samples were prepared in 25 mM Tris•HCl (pH 7.5), 100 mM NaCl, 1 mM EDTA, and 2 mM DTT. Three protein concentrations at OD280 of 0.7, 0.5, and 0.3 were used for each sample. All sedimentation equilibrium experiments were carried out at 4°C by using An-50 Ti rotor in a Beckman ProteomeLab XL-1 analytical ultracentrifuge (Beckman Instruments, Fullerton, CA). Data were collected at three different speeds (32,000, 36,000, and 39,600 rpm) and represented an average of 50 scans using a scan step size of 0.001 cm. Data were analyzed using the UltraScan II program from B. Demeler (University of Texas Health Science Center, San Antonio, TX) and were converted to a plot of Ln(absorbance) versus radius square. The slope of the plot is proportional to the molecular weight of the protein. Data is plotted alongside simulations using theoretical values for molecular weight of a dimer and a monomer species, and plotted with an arbitrary Y-axis for direct comparison.
Circular dichroism (CD) spectral analysis
Far-UV CD spectra were measured with a Jasco J-810 CD spectropolarimeter at 25°C using a 1-mm cuvette. The protein of 0.3 mg/ml was in a buffer containing 20 mM KH2PO4 (pH 7.5). The samples were centrifuged and degassed before measurement. Spectra represent the average of five runs with buffer blanks subtracted, and the resulting spectra were normalized to molar ellipticities against the known protein concentrations with Jasco Series 810 software.
ATPase assay
The ATPase activity of Vps4 was measured using an assay based on the conversion of phosphoenolpyruvate (PEP) to pyruvate by pyruvate kinase (PK) coupled to the conversion of pyruvate to lactate by lactate dehydrogenase (LDH) (Huang and Hackney, 1994). Briefly, 1.25 μM Vps4 and 1.5 μM GST-Vta1 variants were first incubated in 800 μl of reaction buffer (50 mM Tris-acetate, pH7.5, 1 mM MgCl2, 1 mM DTT, 3 mM phosphoenolpyruvate, 17.5 units/ml of pyruvate kinase, 12.3 units/ml lactate dehydrogenase, 0.21 mM NADH) at room temperature. After 1.5 mM ATP was added to initiate the reaction, absorbance was monitored at 340 nm with 10 seconds interval from 300 seconds using a HP 8453E Spectroscopy system and the kinetic data were analyzed as described. Protein concentrations were measured using Bradford method.
Fluorescence Microscopy
Yeast plasmids containing mutant forms of vta1 were generated by subcloning the mutated open reading frame out of the bacterial expression vectors into the plasmid pRS416-PnTAP with BamHI/SalI (Azmi et al., 2006). This resulted in TAP-tagged forms of Vta1 with the desired mutation that were subsequently subcloned into pRS415 as a Sac1/Xho1 fragment. Complementation analyses were performed by transformation of the pRS415 constructs together with GFP-tagged CPS into vta1Δ cells (BY4742 genetic background, Open Biosystems). Fluorescence microscopy was performed on live cells using a Nikon microscope fitted with an eGFP filter set and digital camera (Coolsnap HQ, Photometrix), and images were processed using Delta Vision deconvolution software (Applied Precision Inc.).
Acknowledgments
We thank staffs of the Center for Structural Biology at the University of Michigan for maintaining the X-ray facility and staffs at the Advanced Photon Source beam lines 21-ID and 23-ID of Argonne National Laboratory for access and help with data collection; K. Yoshino-Koh for helps at the early stage of this project; X. Chen for technical assistance; M. Amenta-Ilbert and U. Jakob for access and help with CD spectral analysis; R. Matthews, J. Tesmer and L. Weisman for critically reading the manuscript. This work was supported in part by NIH grants to Z.X. (R01-DK65980) and D.J.K. (R01-GM 73024). IA is supported by a predoctoral fellowship from the American Heart Association (AHA 07-155882).
Footnotes
Protein Data Bank accession number: Atomic coordinates and structure factors for yeast Vta1NTD and Vta1CTD have been deposited in the RCSB Protein Data Bank with accession code 2RKK and 2RKL, respectively.
Author Contributions: JX, HX, JZ and ZX conceived and designed the experiments. JX produced Vta1NTD crystal and determined both Vta1NTD and Vta1CTD structures. HX produced Vta1CTD crystal and characterized the interaction between Vta1CTD and Vps4. JX, IA, BAD, and DJK characterized the interaction between Vta1NTD and Vps60/Did2. IA, BAD, and DJK carried out yeast cell biology experiments. JX conducted the circular dichroism measurements. JX and ZX wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Azmi I, Davies B, Dimaano C, Payne J, Eckert D, Babst M, Katzmann DJ. Recycling of ESCRTs by the AAA-ATPase Vps4 is regulated by a conserved VSL region in Vta1. J Cell Biol. 2006;172:705–717. doi: 10.1083/jcb.200508166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azmi J, Davies B, Xiao J, Babst M, Xu Z, Katzmann DJ. ESCRT-III family members stimulate Vps4 ATPase activity directly or via Vta1. Dev Cell. 2007 doi: 10.1016/j.devcel.2007.10.021. [DOI] [PubMed] [Google Scholar]
- Babst M. A protein's final ESCRT. Traffic. 2005;6:2–9. doi: 10.1111/j.1600-0854.2004.00246.x. [DOI] [PubMed] [Google Scholar]
- Babst M, Sato TK, Banta LM, Emr SD. Endosomal transport function in yeast requires a novel AAA-type ATPase, Vps4p. Embo J. 1997;16:1820–1831. doi: 10.1093/emboj/16.8.1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babst M, Wendland B, Estepa EJ, Emr SD. The Vps4p AAA ATPase regulates membrane association of a Vps protein complex required for normal endosome function. Embo J. 1998;17:2982–2993. doi: 10.1093/emboj/17.11.2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Carlton JG, Martin-Serrano J. Parallels between cytokinesis and retroviral budding: a role for the ESCRT machinery. Science. 2007;316:1908–1912. doi: 10.1126/science.1143422. [DOI] [PubMed] [Google Scholar]
- CCP4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. Collaborative Computational Project, Number 4. [DOI] [PubMed] [Google Scholar]
- Frickey T, Lupas AN. Phylogenetic analysis of AAA proteins. J Struct Biol. 2004;146:2–10. doi: 10.1016/j.jsb.2003.11.020. [DOI] [PubMed] [Google Scholar]
- Fujita H, Umezuki Y, Imamura K, Ishikawa D, Uchimura S, Nara A, Yoshimori T, Hayashizaki Y, Kawai J, Ishidoh K, et al. Mammalian class E Vps proteins, SBP1 and mVps2/CHMP2A, interact with and regulate the function of an AAA-ATPase SKD1/Vps4B. J Cell Sci. 2004;117:2997–3009. doi: 10.1242/jcs.01170. [DOI] [PubMed] [Google Scholar]
- Gruenberg J, Stenmark H. The biogenesis of multivesicular endosomes. Nat Rev Mol Cell Biol. 2004;5:317–323. doi: 10.1038/nrm1360. [DOI] [PubMed] [Google Scholar]
- Haas TJ, Sliwinski MK, Martinez DE, Preuss M, Ebine K, Ueda T, Nielsen E, Odorizzi G, Otegui MS. The Arabidopsis AAA ATPase SKD1 is involved in multivesicular endosome function and interacts with its positive regulator LYST-INTERACTING PROTEIN5. Plant Cell. 2007;19:1295–1312. doi: 10.1105/tpc.106.049346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang TG, Hackney DD. Drosophila kinesin minimal motor domain expressed in Escherichia coli. Purification and kinetic characterization. J Biol Chem. 1994;269:16493–16501. [PubMed] [Google Scholar]
- Hurley JH, Emr SD. The ESCRT Complexes: Structure and Mechanism of a Membrane-Trafficking Network. Annu Rev Biophys Biomol Struct. 2006;35:277–298. doi: 10.1146/annurev.biophys.35.040405.102126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47(Pt 2):110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Katzmann DJ, Odorizzi G, Emr SD. Receptor downregulation and multivesicular-body sorting. Nat Rev Mol Cell Biol. 2002;3:893–905. doi: 10.1038/nrm973. [DOI] [PubMed] [Google Scholar]
- Lottridge JM, Flannery AR, Vincelli JL, Stevens TH. Vta1p and Vps46p regulate the membrane association and ATPase activity of Vps4p at the yeast multivesicular body. Proc Natl Acad Sci U S A. 2006;103:6202–6207. doi: 10.1073/pnas.0601712103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita E, Sandrin V, Chung HY, Morham SG, Gygi SP, Rodesch CK, Sundquist WI. Human ESCRT and ALIX proteins interact with proteins of the midbody and function in cytokinesis. Embo J. 2007;26:4215–4227. doi: 10.1038/sj.emboj.7601850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita E, Sundquist WI. Retrovirus budding. Annu Rev Cell Dev Biol. 2004;20:395–425. doi: 10.1146/annurev.cellbio.20.010403.102350. [DOI] [PubMed] [Google Scholar]
- Morris RJ, Perrakis A, Lamzin VS. ARP/wARP and automatic interpretation of protein electron density maps. Methods Enzymol. 2003;374:229–244. doi: 10.1016/S0076-6879(03)74011-7. [DOI] [PubMed] [Google Scholar]
- Obita T, Saksena S, Ghazi-Tabatabai S, Gill DJ, Perisic O, Emr SD, Williams RL. Structural basis for selectiv recognition of ESCRT-III by the AAA ATPase Vps4. Nature. 2007;449:735–739. doi: 10.1038/nature06171. [DOI] [PubMed] [Google Scholar]
- Piper RC, Katzmann DJ. Biogenesis and Function of Multivesicular Bodies. Annu Rev Cell Dev Biol. 2007;23:519–547. doi: 10.1146/annurev.cellbio.23.090506.123319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott A, Chung HY, Gonciarz-Swiatek M, Hill GC, Whitby FG, Gaspar J, Holton JM, Viswanathan R, Ghaffarian S, Hill CP, et al. Structural and mechanistic studies of VPS4 proteins. Embo J. 2005a;24:3658–3669. doi: 10.1038/sj.emboj.7600818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott A, Gaspar J, Stuchell-Brereton MD, Alam SL, Skalicky JJ, Sundquist WI. Structure and ESCRT-III protein interactions of the MIT domain of human VPS4A. Proc Natl Acad Sci U S A. 2005b;102:13813–12818. doi: 10.1073/pnas.0502165102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiflett SL, Ward DM, Huynh D, Vaughn MB, Simmons JC, Kaplan J. Characterization of Vta1p, a class E Vps protein in Saccharomyces cerevisiae. J Biol Chem. 2004;279:10982–10990. doi: 10.1074/jbc.M312669200. [DOI] [PubMed] [Google Scholar]
- Stuchell-Brereton MD, Skalicky JJ, Kieffer C, Karren MA, Ghaffarian S, Sundquist WI. ESCRT-III recognition by Vps4 ATPases. Nature. 2007;449:740–744. doi: 10.1038/nature06172. [DOI] [PubMed] [Google Scholar]
- Takasu H, Jee JG, Ohno A, Goda N, Fujiwara K, Tochio H, Shirakawa M, Hiroaki H. Structural characterization of the MIT domain from human Vps4b. Biochem Biophys Res Commun. 2005;334:460–465. doi: 10.1016/j.bbrc.2005.06.110. [DOI] [PubMed] [Google Scholar]
- Vale RD. AAA proteins. Lords of the ring. J Cell Biol. 2000;150:F13–19. doi: 10.1083/jcb.150.1.f13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonrhein C, Blanc E, Roversi P, Bricogne G. Automated Structure Solution With autoSHARP. Methods Mol Biol. 2006;364:215–230. doi: 10.1385/1-59745-266-1:215. [DOI] [PubMed] [Google Scholar]
- Ward DM, Vaughn MB, Shiflett SL, White PL, Pollock AL, Hill J, Schnegelberger R, Sundquist WI, Kaplan J. The role of LIP5 and CHMP5 in multivesicular body formation and HIV-1 budding in mammalian cells. J Biol Chem. 2005;280:10548–10555. doi: 10.1074/jbc.M413734200. [DOI] [PubMed] [Google Scholar]
- Weeks CM, Miller R. The design and implementation of SnB v2.0. J Appl Cryst. 1999;32:120–124. [Google Scholar]
- Williams RL, Urbe S. The emerging shape of the ESCRT machinery. Nat Rev Mol Cell Biol. 2007;8:355–368. doi: 10.1038/nrm2162. [DOI] [PubMed] [Google Scholar]
- Winn MD, Murshudov GN, Papiz MZ. Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 2003;374:300–321. doi: 10.1016/S0076-6879(03)74014-2. [DOI] [PubMed] [Google Scholar]
- Xiao J, Xia H, Yoshino-Koh K, Zhou J, Xu Z. Structural chracterization of the ATPase reaction cycle of endosomal AAA protein Vps4. J Mol Biol. 2007 doi: 10.1016/j.jmb.2007.09.067. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo SC, Xu L, Ren J, Boulton VJ, Wagle MD, Liu C, Ren G, Wong P, Zahn R, Sasajala P, et al. Vps20p and Vta1p interact with Vps4p and function in multivesicular body sorting and endosomal transport in Saccharomyces cerevisiae. J Cell Sci. 2003;116:3957–3970. doi: 10.1242/jcs.00751. [DOI] [PubMed] [Google Scholar]