

Abstract
Apoptosis-inducing factor (AIF)-deficient harlequin (Hq) mice undergo neurodegeneration associated with a 40–50% reduction in complex I level and activity. We tested the hypothesis that AIF and complex I regulate reactive oxygen species (ROS) production by brain mitochondria. Isolated Hq brain mitochondria oxidizing complex I substrates displayed no difference compared to wild type (WT) in basal ROS production, H2O2 removal, or ROS production stimulated by complex I inhibitors rotenone or 1-methyl-4-phenylpyridinium. In contrast, ROS production caused by reverse electron transfer to complex I was attenuated by ~50% in Hq mitochondria oxidizing the complex II substrate succinate. Basal and rotenone-stimulated rates of H2O2 release from in situ mitochondria did not differ between Hq and WT synaptosomes metabolizing glucose, nor did the level of in vivo oxidative protein carbonyl modifications detected in synaptosomes, brain mitochondria, or homogenates. Our results suggest that AIF does not directly modulate ROS release from brain mitochondria. In addition, they demonstrate that in contrast to ROS produced by mitochondria oxidizing succinate, ROS release from in situ synaptosomal mitochondria or from isolated brain mitochondria oxidizing complex I substrates is not proportional to the amount of complex I. These findings raise the important possibility that complex I contributes less to physiological ROS production by brain mitochondria than previously suggested.
Keywords: apoptosis-inducing factor, electron transport chain, neurodegeneration, oxidative stress, protein carbonyl, synaptosome
Introduction
Whether oxidative stress is a cause or consequence of neurodegeneration is heavily debated and likely depends on the mechanism of injury [1]. Although animal models that display age onset-dependent chronic neurodegeneration are rare, one example is the harlequin (Hq) mutant mouse that harbors a retroviral insertion in intron 1 of the pcd8 gene encoding apoptosis-inducing factor (AIF). This mutation results in an AIF hypomorph, with an 80–90% global impairment in AIF expression [23]. These AIF deficient mice undergo progressive degeneration in several brain regions including the cerebellum, cortex, striatum, and thalamus [16,23].
Oxidative stress associated with AIF deficiency led to the suggestion that the flavin-binding AIF protein functions as a mitochondrial anti-oxidant [23,24,33,57,58,63]. The crystal structure of AIF, however, does not resemble an anti-oxidant enzyme [34,62]. Furthermore, recombinant AIF functions as a superoxide-generating NAD(P)H oxidase in vitro [35] or forms a relatively stable NAD(P)H-bound dimer [12], depending on whether it is purified using an N-terminal or C-terminal tag, respectively. Genetic and RNA interference knockdown models of AIF deficiency have failed to provide a consistent picture of the role of AIF in regulating mitochondrial and/or cellular ROS, with reports suggesting that ROS increases [3], decreases [55], or does not change [41,56] when mitochondrial AIF is reduced. AIF is released from mitochondria in a calpain-dependent fashion in neural cells treated with the complex I inhibitors rotenone or 1-methyl-4-phenylpyridinium (MPP+) [11,31,32,40]. Whether this event influences net mitochondrial ROS generation is unknown.
Recent evidence that AIF deficiency results in a decrease in assembled complex I [3,5,8,22,41,55,56] raises the possibility that changes in ROS attributed to AIF are instead a downstream consequence of alterations in electron transport chain function. Complex I is generally considered to be the primary physiological source of mitochondrial ROS [2,4,7,19,28,47,59], although support for this hypothesis comes predominantly from pharmacological rather than genetic studies. Inhibition of complex I oxidation by rotenone or MPP+ exacerbates ROS production by brain mitochondria incubated with complex I substrates [2,17,27,38,59], while rotenone inhibits ROS production by brain mitochondria oxidizing succinate [13,27,51,54,59]. Systemic administration of rotenone or MPTP recapitulates the neuropathology of Parkinson’s disease in rodents and primates [6,9], with arguments both for and against oxidative stress as the chief cause of neuronal demise [45,60].
In this study we employed the harlequin genetic model of AIF and complex I deficiency to assess whether AIF and complex I depletion influence ROS production by isolated or in situ brain mitochondria from mice prior to the onset of neurodegeneration. The use of isolated mitochondria enabled us to directly test the contribution of AIF and complex I to ROS production in the presence of defined substrates and compare results to the behavior of in situ mitochondria within glucose-metabolizing intact nerve terminals (synaptosomes). An increase in rotenone or MPP+-stimulated ROS production from AIF-depleted Hq mitochondria or synaptosomes was predicted if AIF serves an anti-oxidant role while an attenuation was expected if the primary influence of AIF on ROS was via down-regulation of assembled complex I. Our surprising results suggest that brain mitochondria oxidizing complex I substrates under physiologically relevant conditions are able to tolerate large decreases in both AIF (~90%) and complex I (>40%) without overt effects on mitochondrial ROS production.
Experimental Procedures
Materials
Amplex® UltraRed was obtained from Invitrogen (Carlsbad, CA). Antimycin A was purchased from MP Biochemicals (Solon, OH). Percoll™ was from GE Healthcare (Piscataway, NJ). The complex I immunocapture monoclonal antibody (MS101c) was from MitoSciences (Eugene, OR). The OxyBlot Protein Oxidation Detection Kit was obtained from Chemicon (Temecula, CA). Mouse monoclonal antibody to AIF (clone E-1) was from Santa Cruz Biotechnology (Santa Cruz, CA). Mouse monoclonal antibody to cytochrome c (clone 7H8.2C12) was from BD Biosciences (San Jose, CA). Rabbit polyclonal antibody to MnSOD was from Novus Biologicals (Littleton, CO). Mouse monoclonal antibody to catalase (clone CAT-505) or β-actin (clone AC-74) was from Sigma-Aldrich (St. Louis, MO). Rabbit polyclonal antibody to MWFE was generated as described [61]. All other reagents were purchased from Sigma unless otherwise noted.
Animals
Male mice hemizygous for the harlequin (Hq) mutation in the pcd8 gene encoding AIF (Strain name B6CBACa Aw-J/A-Aifm1Hq/J) and age-matched male wild type (WT) controls on the same background were obtained from The Jackson Laboratory (Bar Harbor, ME). Mice were housed according to standard animal care protocols, fed ad libitum, and kept on a 12 hour light/dark cycle until sacrifice. All protocols were approved by the Institutional Animal Care and Use Committee (IACUC) and were in accordance with the NIH Guide for the Care and Use of Laboratory Animals.
Preparation of Isolated Mitochondria and Intact Synaptosomes
Non-synaptosomal brain mitochondria were isolated from 2–3 Hq male mice and from 2–3 male wild type littermates in parallel and purified on a Percoll™ density gradient as previously described [25,46]. Synaptosomes were isolated from the same preparations at the interface of 15% and 23% Percoll™, washed in ‘HBM medium’ consisting of 140 mM NaCl, 5 mM KCl, 5 mM NaHCO3, 1 mM MgCl2, 1.2 mM Na2HPO4, 10 mM glucose, 20 mM HEPES, pH 7.4, centrifuged at 16,600 × g for 10 min at 4° C, and finally resuspended to a concentration of 10–20 mg/ml in HBM medium. Liver mitochondria were isolated by standard differential centrifugation [30]. Mitochondrial and synaptosomal protein concentrations were determined by the Bio-Rad (Hercules, CA) Bradford method. Mitochondria and synaptosomes were stored on ice and utilized for oxygen consumption and H2O2 measurements on the day of isolation. Aliquots of samples were frozen at −80° C for the later determination of citrate synthase activity or protein levels by immunoblot.
Citrate Synthase Assay
Citrate synthase activity was measured in brain mitochondria according to published procedure [44,48]. Briefly, 100 μg of mitochondrial protein dissolved in 10 μl of 0.1% Triton X-100 in 0.1 M Tris-Hcl, pH 8.0 was added to assay medium consisting of 0.1 M Tris-Hcl, pH 8.0, 0.1 mM 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB), 0.36 mM acetyl-CoA, and 0.5 mM oxalacetate at 25° C. The conversion of DTNB to 5-thio-2-nitrobenzoic acid was monitored in a Varian Cary 100 Bio spectrophotometer (Walnut Creek, CA) by measuring absorbance at 412 nm.
Oxygen Consumption Measurements
Mitochondrial oxygen consumption was measured using a MIP-730 Dip-type O2 microelectrode (Microelectrodes, Inc., Bedford, NH) in a microcuvette (Hellma Cells, Inc. Plainview, NY) with a custom-built gastight lid at 30° C. WT or Hq mitochondria (0.25 mg protein/ml) were incubated in ‘KCl medium’ consisting of 125 mM KCl, 2 mM K2HPO4, 20 mM HEPES, pH 7.0. The medium was supplemented with 5 mM malate, 5 mM glutamate, 4 mM MgCl2, 3 mM ATP, 10 μM CaCl2, 1 μM ruthenium red, and 10 μM tetraphenylboron anion (TPB−) to mimic the conditions chosen for investigation of ROS (see below). Respiratory control ratios were calculated as the ratio of the rate of oxygen consumption in the presence of 1 mM ADP (state 3) to the rate of O2 consumption following the addition of 3 μg/ml oligomycin (state 4).
H2O2 detection using Amplex® UltraRed
H2O2 release from WT and Hq brain mitochondria was measured in parallel using two Hellma quartz microcuvettes in a Perkin-Elmer LS50 spectrofluorometer. Amplex® UltraRed was excited at 560 nm, and emission was collected at 585 nm using 10 nm excitation and emission slit widths and a cuvette cycle time of 5 sec. Most experiments were conducted in KCl medium additionally containing 5 mM malate, 5 mM glutamate, 4 mM MgCl2, 3 mM ATP, 10 μM CaCl2, 1 μM ruthenium red, 10 μM TPB−, 40 U/ml bovine liver Cu/Zn superoxide dismutase (SOD1), 5 μM Amplex® UltraRed, and 10 U/ml horseradish peroxidase. In experiments examining ROS production mediated by reverse electron flow, 5 mM malate and 5 mM glutamate were replaced by 5 mM succinate. H2O2 release from synaptosomes (0.5 mg/ml) was also measured by coupling 5 μM Amplex® UltraRed and 10 U/ml horseradish peroxidase, but in HBM medium additionally containing 1.3 mM CaCl2. Amplex® UltraRed signals were sensitive to added catalase (Sigma, 15,000 U/ml), confirming that the fluorescent resorufin product measured was due to the oxidation of Amplex® UltraRed by H2O2. In control experiments, it was confirmed that ruthenium red does not interfere with the Amplex® UltraRed assay. Traces were calibrated in pmol H2O2 for each experiment as described using freshly prepared H2O2 stocks serially diluted from a stabilized 30% H2O2 solution [59].
We chose to investigate ROS in State 4 with a physiologically realistic concentration of ATP for two reasons. First, ROS production is membrane potential sensitive [49,59] and ROS produced by ADP-stimulated mitochondria in state 3 is below the threshold for reliable detection. Second, the presence of ATP is known to stabilize brain mitochondria against artifactual induction of the permeability transition (38, 39) TPB− was included because it is known to potentiate the uptake and consequent toxicity of MPP+ [36]. Results without TPB-were qualitatively similar but required a 25-fold increase in the concentration of MPP+ (data not shown).
Immunoblotting
Mitochondria (0.1 mg/ml in 0.75 ml) were pelleted by centrifugation at 13,400 × g for 5 min at 4° C at the conclusion of Amplex® UltraRed assays for H2O2 detection. Supernatants were saved for analysis of released proteins and pellets were resuspended to 0.1 or 0.2 mg/ml in KCl medium containing 1% Triton X-100 and 1x Protease Inhibitor Cocktail III (EMD Biosciences). Twenty to 30μl of mitochondrial supernatant or pellet fractions or 20 μg of synaptosomal protein were subjected to SDS-PAGE on 4–20% Tris-HEPES gradient gels and protein was transferred to polyvinylidene difluoride membranes. Immunodetection was carried out by standard procedures using the following dilutions of primary antibodies: AIF, 1:500, β-actin, 1:5000, catalase, 1:1000, cytochrome c, 1:1000, MnSOD, 1:2000, MWFE, 1:1000, NDUFA9, 1:1000. Protein quantification was performed from linear exposures of X-ray film using the Bio-Rad GS-710 Calibrated Imaging Densitometer (Hercules, CA) and Quantity-One software.
Immunocapture and Detection of Complex I Subunits
WT or Hq mitochondria (0.5 mg protein) were washed with 20 mM Tris-HCl, 1 mM EDTA, pH 7.5, and resuspended in the same buffer containing protease inhibitor cocktail. This suspension was solubilized by adding n-dodecyl-β-D-maltoside to a final concentration of 1% at a 5 mg/ml protein concentration and incubated for 30 min on ice. A very minor fraction of insoluble material from this suspension that did not differ between WT and Hq was removed by centrifugation at 55,000 × g for 30 min at 4°C and soluble supernatant was taken for semi-quantative immunoprecipitation of mitochondrial complex I using the MS101 Complex I Immunocapture kit from MitoSciences (Eugene, OR) [42]. Briefly, 10μl of MitoSciences immunocapture antibody coupled to agarose beads was incubated with the solubilized mitochondrial supernatant for at least 3 hours at room temperature. Following incubation, the beads were collected by gentle centrifugation at 3,200 × g in a microcentrifuge and washed six times with PBS supplemented with 0.05% n-dodecyl-β-D-maltoside. Immunocaptured NADH dehydrogenase was eluted with 10 μl of 0.1 M glycine, pH 2.5, supplemented with 0.05% n-dodecyl-β-D-maltoside. The samples were run on 10% SDS-PAGE gels following elution and then stained with SYPRO® Ruby protein gel stain (Bio-Rad) to visualize complex I subunits.
Detection of Protein Carbonyl Modifications in Mitochondria, Synaptosomes, or Brain Homogenates
Oxidative protein carbonyl modifications were detected in mitochondria, synaptosomes, or brain homogenates (20 μg protein) using The OxyBlot Protein Oxidation Detection Kit (Chemicon) according to the instructions of the manufacturer. The brain homogenates were prepared by sonicating cerebellum, cortex, or striatum in RIPA lysis buffer (150 mM NaCl, 50 mM Tris, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% sodium dodecylsulfate, and Protease Inhibitor Cocktail Set III, pH 7.4) in three 5 sec bursts using a 550 Sonic dismembrator (Fisher Scientific, Pittsburgh, PA) set at 3. Densiometric DNP quantification was performed from entire lanes using ImageJ software (National Institutes of Health, Bethesda, MD).
Statistics
Protein levels, DNP staining, respiratory control ratios, citrate synthase activity, and H2O2 rates were compared by paired T-test using SigmaStat 3.5 and values were expressed as mean ± standard error. P<0.05 was considered significant.
Results
Non-synaptosomal forebrain mitochondria were isolated in parallel from male AIF-deficient Hq mice and age-matched WT littermates at 4–14 weeks of age. Hq mice at this stage are considered pre-symptomatic and do not display overt signs of neurodegeneration in the forebrain [16,23]. The amount of citrate synthase activity per milligram protein did not differ significantly between WT and Hq brain mitochondria (Fig. 1A, p=0.50, n=3). Additionally, the cytosolic and peroxisomal enzyme catalase could not be detected in Percoll™ gradient-purified WT or Hq brain mitochondria in contrast to liver mitochondria isolated by standard differential centrifugation (Fig. 1B). Thus differences in mitochondrial purity or contamination by non-mitochondrial anti-oxidant enzymes were unlikely to confound the results of subsequent assays.
Fig. 1.

The purity and respiratory characteristics of WT and AIF-deficient Hq brain mitochondria. (A) Citrate synthase (CS) activity was measured as described in Experimental Procedures. Mean ± standard error are expressed in international units (IU)/mg mitochondrial protein where one IU is equal to the formation of one μmol of citrate/min (n=3). (B) Immunodetection for catalase (~60 kD) and cytochrome c (cyt c, ~13 kD) in brain and liver mitochondria was performed using identical film exposure times for 20 μg of protein loaded on the same gel. (C) Oxygen consumption was monitored for WT and Hq brain mitochondria (0.25 mg/ml) incubated in KCl medium containing 5 mM malate, 5 mM glutamate, 4 mM MgCl2, 3 mM ATP, 10 μM CaCl2, 1 μM ruthenium red, and 10 μM TPB− at 30° C. ADP (1 mM) and oligomycin (3 μg/ml) were added when indicated by arrows. Numbers represent oxygen consumption rates in nmol O2/min/mg mitochondrial protein. (D) Oxygen consumption for WT and Hq mitochondria (0.25 mg/ml) was monitored as described in (C) in the presence of 1 mM ADP. Rotenone additions of 2 nM are indicated by filled arrowheads. Numbers following each rotenone addition represent percentage of the initial O2 consumption rate (0 nM rotenone). Results are representative of 3 independent experiments.
WT and Hq rates of ADP-stimulated state 3 respiration or resting state 4 respiration were not significantly different for mitochondria oxidizing the complex I-linked substrates malate and glutamate under our experimental conditions (Fig. 1C, Table 1). Respiratory control ratios (RCR) calculated as the state 3 ADP-stimulated rate divided by the state 4 resting rate (measured in the presence of the ATP synthase inhibitor oligomycin) were also equivalent between the two types of mitochondria. However Hq mitochondria respiring on complex I-linked substrates displayed increased sensitivity to respiratory inhibition with rotenone (Fig. 1D), consistent with the reported deficit in assembled complex I level and activity [5,56]. This deficit was confirmed by immunodetection for complex I subunits NDUFA9 and MWFE (Fig. 2A–C) and by semi-quantitative complex I immunocapture and protein stain (Fig. 2D). The level of NDUFA9 reduction was consistent with the amount of complex I depletion detected in Hq brain mitochondria by blue native polyacrylamide gel electrophoresis or biochemical activity [56].
Table 1.
Mean oxygen consumption rates (nmol O2/min/mg mitochondrial protein) and respiratory control ratios (RCR) of WT and Hq brain mitochondria isolated in parallel (n=3).
| Rate | Mitochondria | Mean | Std Error |
|---|---|---|---|
| State 3a | WT | 148.3 | ± 23.7 |
| Hq | 153.0 | ± 19.1 | |
| Hq (% WT)b | 104.4% | ± 5.3% | |
|
| |||
| State 4a | WT | 27.5 | ± 4.8 |
| Hq | 27.8 | ± 3.3 | |
| Hq (% WT) | 103.6% | ± 10.9% | |
|
| |||
| RCRa | WT | 5.4 | ± 0.4 |
| Hq | 5.5 | ± 0.07 | |
| Hq (% WT) | 101.9% | ± 5.9% | |
No significant difference between WT and Hq (p>0.05).
Numbers in shaded rows represent the mean ± standard error when Hq data are expressed as a percentage of WT values for each isolation.
Fig. 2.
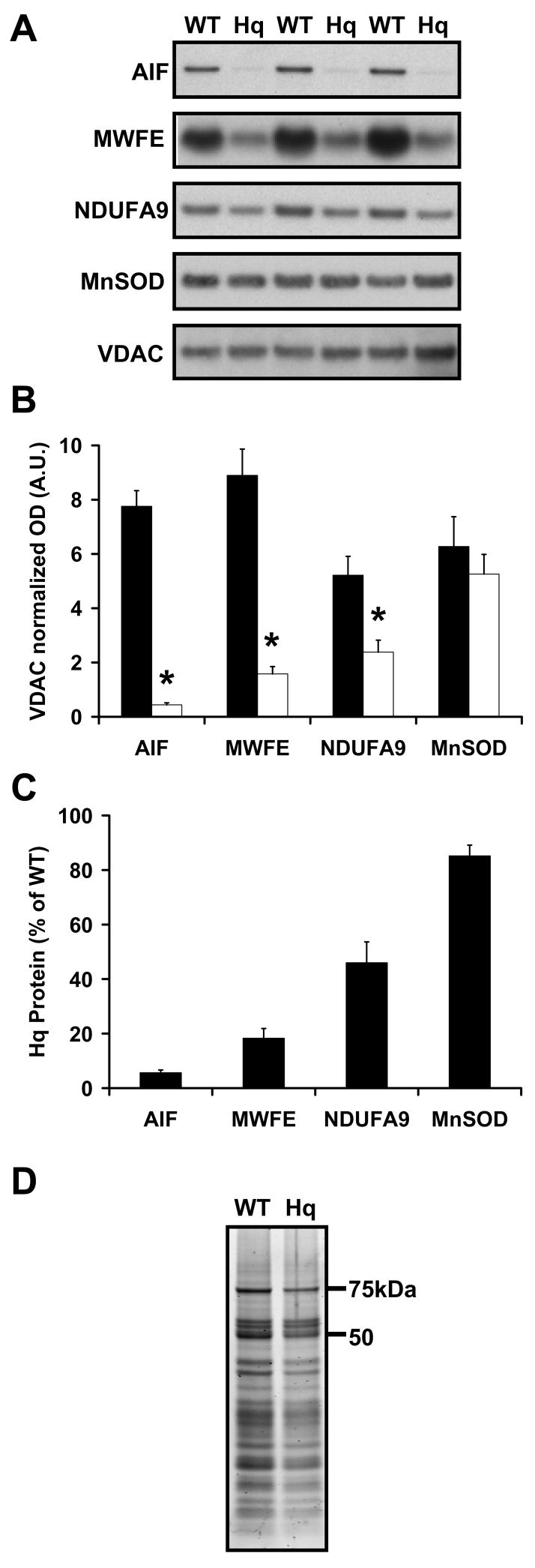
AIF-deficient Hq brain mitochondria exhibit a significant reduction in complex I subunits but not MnSOD. AIF, MnSOD, VDAC, and the MWFE and NDUFA9 subunits of complex I were detected by immunoblot in WT vs. Hq brain mitochondria in A, then quantified by densitometry in B (WT, closed bars, Hq, open bars, n=3). Results in B are normalized to VDAC to control for equal loading and optical density (OD) arbitrary units (A.U.) are adjusted to scale. Asterisks indicate a significant difference from WT (p<0.05). (C) Proteins in Hq mitochondria expressed as a percentage of WT levels after values were normalized to VDAC as a loading control. (D) Complex I was isolated from WT or Hq brain mitochondria by semi-quantitative immunoprecipitation and subjected to SDS-PAGE. Complex I subunits were visualized using SYPRO® Ruby protein gel stain.
We next investigated the influence of the AIF and complex I deficiencies on ROS regulation by brain mitochondria using the Amplex® UltraRed-horseradish peroxidase assay. Superoxide produced by the mitochondrial electron transport chain in the matrix is converted to membrane-permeable hydrogen peroxide by endogenous MnSOD (SOD2). The addition of exogenous bovine liver Cu/Zn SOD (SOD1) converts any superoxide generated in the intermembrane space to H2O2. Therefore the oxidation of the Amplex® UltraRed dye by hydrogen peroxide is expected to reveal any difference in net superoxide or H2O2 generation in the matrix or intermembrane space by Hq mitochondria compared to WT.
We found that the low basal rate of ROS release captured by our assay was unaltered in AIF- and complex I-deficient brain mitochondria compared to WT (Fig. 3A, p=0.92, n=4). However it is known that mitochondria have a very efficient endogenous system for ROS removal [2,64]. To determine whether Hq mitochondria compensate for a defect in ROS generation with a change in the rate of ROS detoxification, we employed the enzymatic xanthine/xanthine oxidase (X/XO) superoxide generating system coupled to Cu/Zn SOD to assess rates of H2O2 removal by the mitochondria. Xanthine oxidase (1.33 milliunits/ml) generated a linear rate of H2O2 production in the presence of 100 μM xanthine and 40 units/ml SOD1 that was impaired by the addition of 0.2 mg/ml isolated brain mitochondria (Fig. 3B). This finding is consistent with the reported ability of brain mitochondria to scavenge hydrogen peroxide [64]. The capacity of Hq mitochondria to inhibit the rate of X/XO/SOD1-mediated H2O2 production was the same as that of WT (54.1 ± 2.4% inhibition for Hq compared to 51.0 ± 4.5% for WT, p=0.38, n=3), arguing against the possibility that an alteration in H2O2 removal compensated for a difference in basal ROS release by Hq mitochondria. MnSOD protein was also equivalent in Hq and WT brain mitochondria (Fig 2A–C), consistent with previous reports [5,23]. Therefore it is unlikely that we failed to detect a difference in superoxide production in the two populations of mitochondria due to impaired conversion to H2O2.
Fig. 3.

WT and AIF-deficient Hq brain mitochondria do not differ in basal ROS release or H2O2 removal. (A) WT and Hq mitochondria (0.1 mg/ml) were incubated in parallel as described in Experimental Procedures and basal H2O2 release with malate and glutamate as substrates was measured using Amplex® UltraRed. Numbers represent H2O2 release rates in pmol H2O2/min/mg mitochondrial protein. Results are representative of 4 independent experiments. (B) Amplex® UltraRed oxidation was measured under the conditions described in Experimental Procedures with the added presence of xanthine oxidase (1.33 milliunits/ml), xanthine (100 μM) and SOD1 (40 units/ml) to generate a linear rate of H2O2 production in the absence of mitochondria. WT or Hq brain mitochondria (0.2 mg/ml) were added at the arrow. Numbers represent rates in pmol H2O2/min and numbers in parenthesis indicate percent inhibition by mitochondria of the initial rate [100−(rate initial/rate final*100)]. Results are representative of 3 independent experiments.
Mitochondrial complex I inhibitors recapitulate aspects of the pathogenesis of Parkinson’s disease in animals and exacerbate ROS production by isolated brain mitochondria oxidizing complex I substrates [6,38,39]. The addition of a rotenone concentration that results in ~50% inhibition of oxygen consumption in wild type (4 nM) and Hq (3 nM) brain mitochondria with the complex I-linked substrates malate and glutamate led to a ~5 fold stimulation in net ROS production that was similar between the two mitochondrial populations (Fig. 4A). The subsequent addition of 20 nM rotenone, a concentration sufficient to inhibit respiration by >90% in both types of mitochondria (Fig. 1D), led to a robust (>10 fold) enhancement in ROS production to rates that did not differ significantly between WT and Hq mitochondria (Fig 4A, p=0.13, n=4). Similar results were observed with maximal complex I inhibition using the parkinsonian toxin MPP+ (Fig. 4B). The deficit in complex I was confirmed by immunoblot in the same samples analyzed for ROS production (Fig. 2A–C and Fig 4C). Because MPP+ and rotenone are known to promote calpain activation and AIF redistribution in cells, assays were conducted in the presence of 10 μM CaCl2 to allow detection of any influence of calpain-cleaved AIF on mitochondrial ROS release. The calcium uniport inhibitor ruthenium red was included to prevent mitochondrial damage due to calcium uptake. Complex I inhibition alone did not lead to AIF cleavage in isolated brain mitochondria (Fig. 4C), and rotenone-stimulated ROS generation was similar in both types of mitochondria in the absence of calcium (data not shown).
Fig. 4.
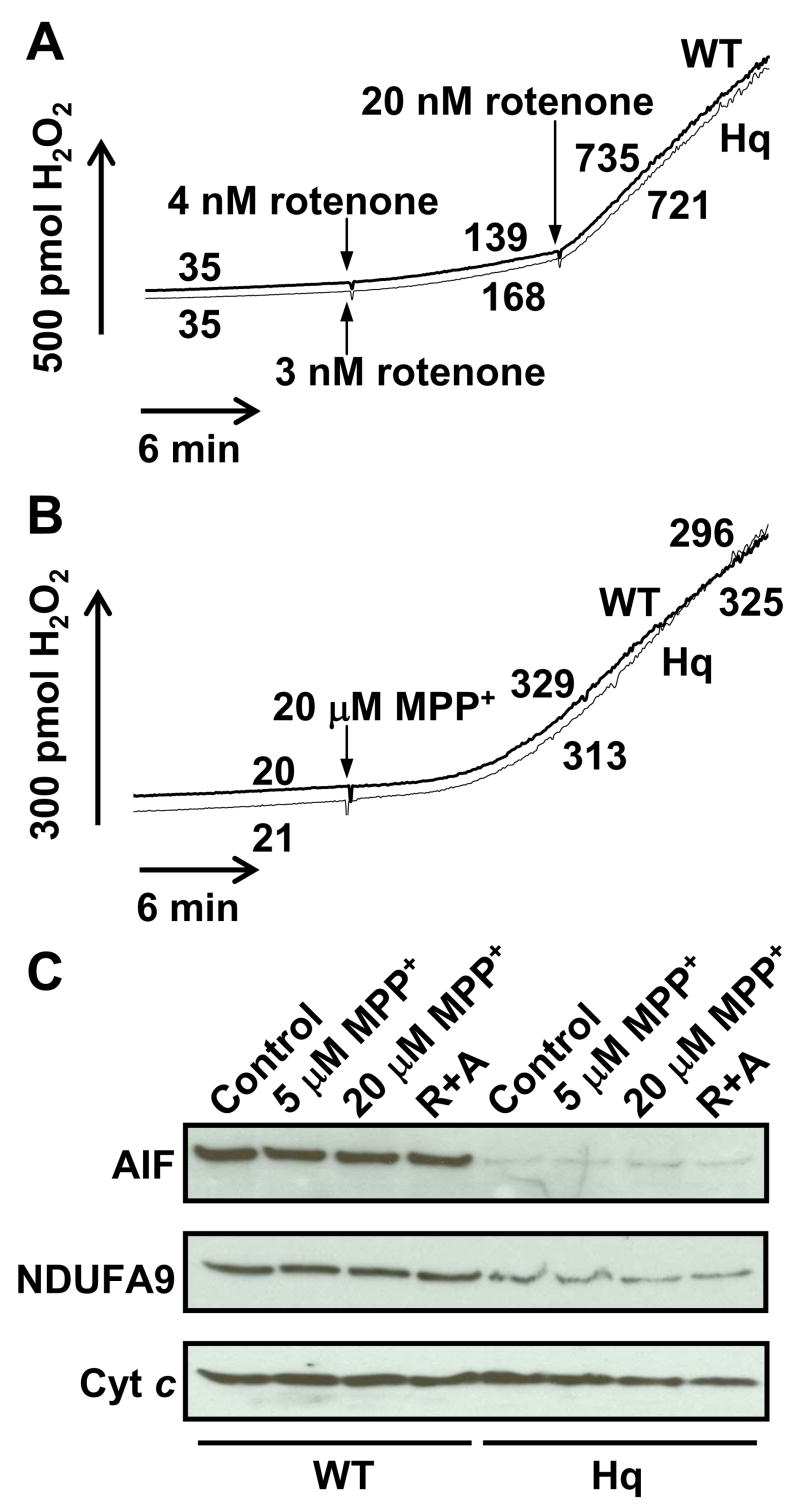
Rotenone or MPP+ stimulates ROS production to the same extent in WT and Hq brain mitochondria. (A,B) Representative traces of H2O2 release by WT and Hq brain mitochondria (0.1 mg/ml) incubated in parallel under the malate/glutamate conditions described in Experimental Procedures. In (A), 4 nM rotenone was added to WT mitochondria and 3 nM rotenone was added to Hq mitochondria after 10 min (arrows). An arrow denotes the addition of 20 nM rotenone to both types of mitochondria after an additional 10 min. In (B), 20 μM MPP+ was added to both WT and Hq brain mitochondria after 10 min (arrow). Numbers indicate rates in pmol H2O2/min/mg mitochondrial protein. Because mitochondrial MPP+ uptake and complex I inhibition are slow compared to rotenone, traces are non-linear. Therefore MPP+ rates were calculated at both 10 and 18 min. after MPP+ addition. (C) Levels of AIF (~62 kD), NDUFA9 (~39 kD), and cytochrome c (cyt c, ~13 kD) retained in mitochondria following a 20 min treatment with MPP+ (5 or 20 μM, Fig. 4B), vehicle (Fig. 3A), or 2 μM rotenone and 2 μM antimycin A in the presence of succinate (R+A, Fig. 5B). Cytochrome c, AIF, and NDUFA9 were not detected in the supernatant, indicating that none of these proteins were released from mitochondria under the various treatment conditions.
In contrast to the low basal rate of ROS production when electron transfer is initiated through complex I (Fig. 3A), mitochondria produce relatively high amounts of superoxide when electrons are donated to complex II via succinate oxidation (Fig. 5A). This ROS was hypothesized to originate from complex I following reverse electron transfer from coenzyme Q [13,20,27,29,54,59] and was inhibited by rotenone in both types of mitochondria (Fig. 5A). ROS production by Hq mitochondria in the presence of succinate was approximately half (51 ± 4.5%) of the WT rate (Fig. 5A, p<0.05, n=4), consistent with a complex I origin for superoxide under these experimental conditions. The rates of H2O2 release when electrons were donated by succinate in the presence of rotenone and when complex III was inhibited by antimycin A did not differ between WT and Hq brain mitochondria (Fig. 5B). These results suggest that AIF deficiency does not alter ROS production by the electron transport chain at sites that lie downstream of complex I.
Fig. 5.

Succinate-linked ROS production is impaired in Hq brain mitochondria in the absence but not in the presence of rotenone. (A) Representative traces of H2O2 release by WT and Hq brain mitochondria (0.1 mg/ml) incubated in parallel under the succinate conditions described in Experimental Procedures. Rotenone (2 μM) was added to both WT and Hq mitochondria after 10 min to inhibit reverse electron transfer to complex I. (B) Representative traces of H2O2 release as in A, but in the added presence of rotenone (2 μM). Antimycin A (2 μM) was added after 10 min to stimulate ROS production from complex III. Results are representative of 4 independent experiments.
To examine how our findings obtained with isolated brain mitochondria oxidizing defined substrates translate to in situ mitochondria, H2O2 release was compared in intact nerve terminals (synaptosomes) isolated from pre-symptomatic WT and Hq mice. In situ mitochondria within synaptosomes reside in an intracellular milieu similar to the intact cell. Consequently, their function can be evaluated in the presence of endogenous substrates downstream of externally supplied glucose [37,47,53]. We found no difference in the basal H2O2 release rates between WT and Hq synaptosomes (Fig. 6A, p=0.26, n=4), consistent with our findings using isolated brain mitochondria oxidizing complex I substrates (Fig. 3A) and in contrast to results obtained with brain mitochondria oxidizing succinate (Fig. 5A). Similar to the situation with isolated brain mitochondria in the presence of complex I substrates (Fig. 4A), complete inhibition of complex I oxidation by rotenone significantly increased the rate of H2O2 release by synaptosomes (Fig. 6A). The rate of H2O2 release by WT synaptosomes in the presence of 1 μM rotenone was 218 ± 3% of the basal rate, consistent with previous reports [47,53]. H2O2 release by Hq synaptosomes was stimulated to 220 ± 21% of the basal rate by rotenone, an amount not significantly different from WT (p=0.93, n=4). The complex I deficit in Hq synaptosomes was confirmed by immunoblot for NDUFA9 (Fig. 6A inset, 6B and C). The amount of MnSOD or cytochrome c per mg synaptosomal protein was not significantly different between Hq and WT synaptosomes. This finding suggests that Hq and WT synaptosomes contain the same number of mitochondria (Fig. 6B and C, n=3).
Fig. 6.
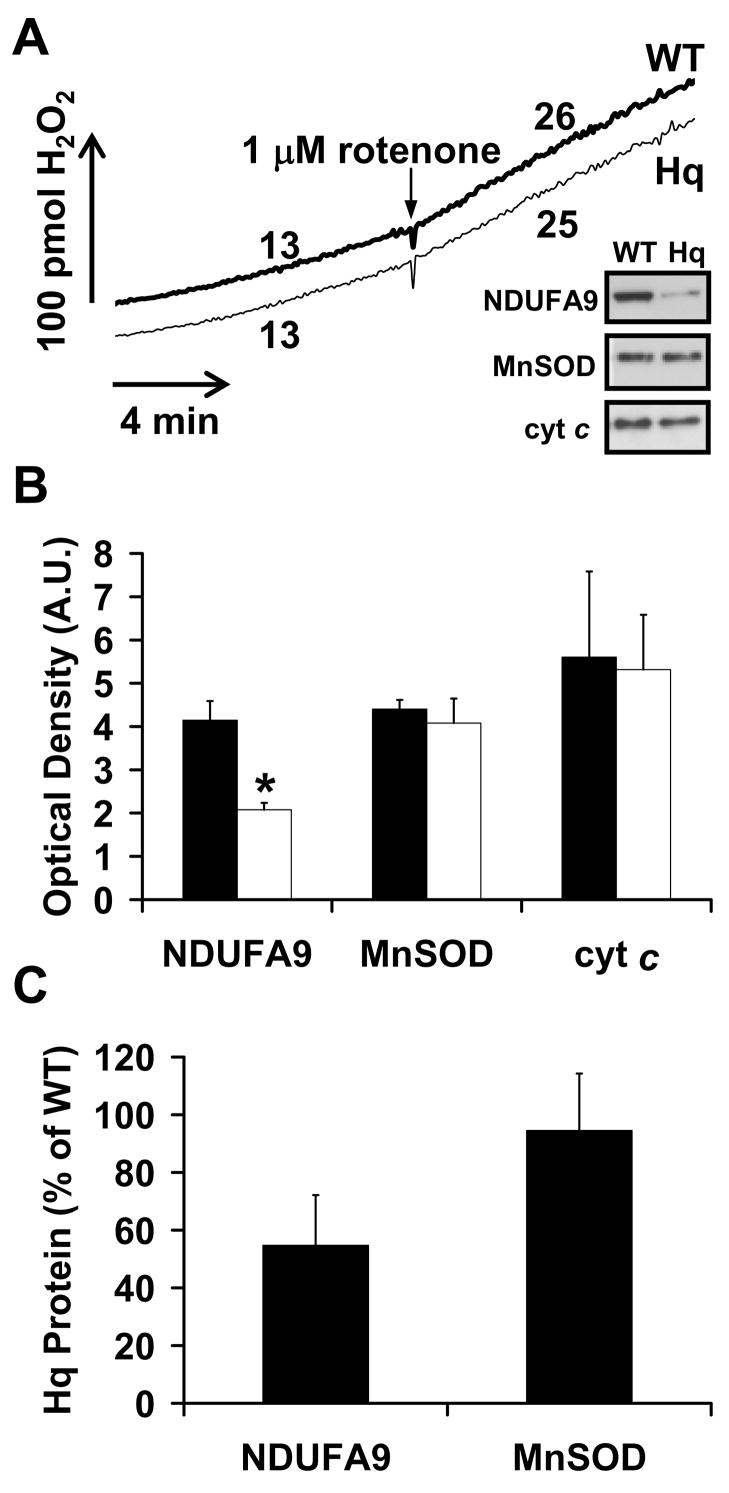
Basal and rotenone-stimulated rates of ROS release do not differ between WT and Hq synaptosomes that contain the same number of mitochondria. (A) WT and Hq synaptosomes (0.5 mg/ml) were incubated in parallel as described in Experimental Procedures and H2O2 release was measured using Amplex® UltraRed. Rotenone (1 μM) was added to both WT and Hq synaptosomes when indicated. Numbers represent H2O2 release rates in pmol H2O2/min/mg synaptosomal protein. Results are representative of 4 independent experiments. The inset is an immunoblot for NDUFA9 from the same experiment depicted, with MnSOD and cytochrome c (cyt c) loading controls. (B) NDUFA9, MnSOD, and cytochrome c were detected by immunoblot in WT (closed bars) vs. Hq mitochondria (open bars), then quantified by densitometry (n=3). The asterisk indicates a significant difference from WT (p<0.05). (C) Proteins in Hq mitochondria expressed as a percentage of WT levels after values were normalized to cytochrome c as a loading control.
To further address the possibility that the AIF or complex I deficiency contributes to mitochondrial oxidative stress in vivo in pre-symptomatic Hq mice, oxidative protein carbonyl modifications in freshly isolated brain mitochondria were examined. We found that the pattern and intensity of protein carbonyl staining in mitochondria from Hq animals was similar to WT (Fig. 7A, Hq=103.0 ± 10.9% of WT, p=0.95, n=3). The deficit in AIF and complex I in these samples was confirmed by immunoblot with cytochrome c as a loading control (Fig 7A). No immunostaining was detected when mitochondria were incubated with a control derivatization solution rather than 2,4 dinitrophenylhydrazine (DNPH), demonstrating the specificity of the 2,4 dinitrophenylhydrazone (DNP) immunodetection for derivatized protein carbonyl modifications.
Fig. 7.

Oxidative protein carbonyl modifications are similar in WT and Hq brain mitochondria (A), synaptosomes (B), and brain homogenates (C). Using the Oxyblot Kit (Chemicon), protein carbonyl groups were derivatized with DNPH (20 μg protein) and detected using an antibody to DNP. Membranes in (A) were reprobed for AIF and NDUFA9 to confirm the AIF and complex I deficit in Hq mitochondria. Immunodetection for cytochrome c (cyt c) controlled for protein loading. Membranes in (B) were reprobed for MnSOD or cytochrome c (see Fig. 6) to confirm that WT and Hq synaptosomes contained equal numbers of mitochondria. Membranes in (C) were reprobed for MnSOD and β-actin to control for loading. Bands present in the lane marked by the asterisk in (C) are non-specific since this sample was incubated with the control derivatization solution in place of DNPH. All results are representative of 3 independent experiments.
It is possible that we failed to detect a difference in protein carbonyl staining between Hq and WT mitochondria because a subset of oxidatively damaged mitochondria was excluded during isolation or because oxidative damage was restricted to unrepresented synaptosomal mitochondria. However, as with isolated non-synaptosomal mitochondria, the pattern and intensity of protein carbonyl staining was similar in synaptosomes isolated from WT or Hq forebrain (Fig. 7B, Hq=88.9 ± 13.0% of WT, p=0.52, n=3).
To control for the possibility that oxidative protein modifications during the isolation of mitochondria or synaptosomes masked any endogenous differences in WT vs. Hq mouse forebrain, we also assessed the level of oxidative damage to proteins in acutely prepared homogenates. Similar to the results with isolated mitochondria or synaptosomes, there was no difference in the pattern or intensity of protein carbonyl staining in homogenates of the cerebellum (Hq=99.9 ± 3.4% of WT, p=0.95, n=3), cortex (Hq=105.2 ± 3.8% of WT, p=0.29, n=3), or striatum (Hq=99.9 ± 3.5% of WT, p=0.94, n=3) derived from pre-symptomatic Hq mice compared to WT controls (Fig. 7C). A triplet of bands migrating between 10–15 kD that was present in the non-derivatized control sample (Fig. 7C, asterisk) was not specific for protein carbonyls and was excluded from the densiometric analysis. Immunodetection for MnSOD and β-actin confirmed equal loading (Fig. 7C). Taken together, our measurements of protein carbonyl staining indicate that there is no global difference in oxidative damage to proteins between WT and AIF- and complex I-depleted Hq mouse brains prior to the onset of neurodegeneration.
Discussion
AIF plays a well-established role in mediating apoptotic nuclear changes once it is released from mitochondria [24,26]. However its essential mitochondrial function(s) remain enigmatic. Because AIF is a flavoprotein that shares significant homology with prokaryotic oxido-reductases [34,62], many of the recent efforts to uncover the mitochondrial function of AIF have focused on the putative ability of AIF to regulate ROS. Nevertheless, despite several investigations in this arena, to date the role of AIF in mitochondrial ROS regulation has been ambiguous, with indications for stimulation [55] and inhibition [3,23,57,58] in various studies.
The accompanying down-regulation in complex I as well as non-mitochondrial mechanisms of ROS regulation have contributed to the difficulty in defining the putative modulation of mitochondrial ROS by AIF in cellular models of AIF deficiency [3,5,8,41,55,56]. Here we exploited isolated brain mitochondria from harlequin mice as a model system for assessing the effects of both AIF and complex I on ROS production in the presence of defined mitochondrial substrates and inhibitors. Conclusions from studies with isolated mitochondria were supported by measurements of ROS release from synaptosomes containing in situ mitochondria and by analysis of in vivo protein oxidation in brain mitochondria, synaptosomes, and homogenates.
Because coupling influences mitochondrial ROS production [7], we initially compared the respiratory characteristics of WT and Hq mitochondria. AIF-deficient non-synaptosomal Hq brain mitochondria were well-coupled and respiratory control ratios and purity did not differ significantly from WT (Fig. 1, Table 1). A threshold of 72% complex I inhibition before respiratory impairment becomes detectable in non-synaptosomal brain mitochondria was previously reported [14], possibly accounting for our failure to detect a difference in complex I-linked state 3 respiration. However, we were able to distinguish WT and Hq brain mitochondria polarographically by measuring their sensitivity to respiratory inhibition by the complex I inhibitor rotenone (Fig. 1D).
Although WT and Hq brain mitochondria were coupled to the same extent, H2O2 release in the presence of the complex II-linked substrate succinate was significantly attenuated in Hq brain mitochondria (Fig. 5A). The amount of H2O2 production was proportional to the level of the complex I subunit NDUFA9 rather than AIF and was blocked in both WT and Hq mitochondria by the complex I inhibitor rotenone (Fig. 5). Thus we were able to demonstrate that the complex I deficit in Hq brain mitochondria was sufficient to significantly impair ROS production under conditions where complex I is established to be the major source of superoxide [13,20,27,29,54,59].
In contrast to mitochondria oxidizing succinate, surprisingly the basal rate of ROS release by mitochondria oxidizing complex I substrates was not proportional to the level of complex I (Fig. 3). Respiration by synaptosomal mitochondria is reportedly more sensitive to complex I inhibition compared to non-synaptosomal mitochondria that are derived from the cell bodies of both neurons and glia [14,15]. However, we also found no difference in basal ROS release rates from WT or Hq synaptosomes metabolizing glucose (Fig. 6). Thus, both non-synaptosomal Hq brain mitochondria oxidizing complex I-linked substrates and in situ synaptosomal Hq mitochondria oxidizing endogenous substrates exhibited WT levels of ROS production despite a ~90% reduction in AIF and a level of complex I deficiency that inhibited succinate-linked reverse electron transfer-mediated ROS production by ~50%.
To further evaluate the role of AIF and complex I in mitochondrial ROS production, WT and Hq mitochondria were treated with rotenone or MPP+ in the presence of complex I-linked substrates. Net ROS generation was stimulated >10 fold by complete complex I inhibition in both WT and Hq brain mitochondria (Fig. 4A and B). No AIF release was observed under these conditions. Rates of rotenone-stimulated ROS production were not significantly different even though Hq mitochondria contained approximately half the WT amount of complex I and presumably half the number of sites within complex I available for ROS production. The finding that rotenone-stimulated ROS production rates are the same between WT and Hq brain mitochondria oxidizing complex I substrates was substantiated by measuring H2O2 release from intact synaptosomes containing in situ mitochondria (Fig. 6). One interpretation of these results is that complex I is normally present in excess and mitochondrial ROS production rates are limited by the entry of electrons from substrates into the complex. An alternative explanation is that the major site of basal and rotenone-stimulated superoxide production by brain mitochondria lies outside complex I.
It is increasingly recognized that there are mitochondrial matrix enzymes influenced by the NAD(P)H/NAD(P)+ ratio that are capable of producing ROS. A prime source is the common E3 component of the α-ketoglutarate and pyruvate dehydrogenase complexes, dihydrolipoamide dehydrogenase (Dld) [2,18,50,52]. This enzyme generates the most ROS in the presence of a high NADH/NAD+ ratio [18,52], a condition met by complex I inhibition. Using Dld-depleted non-synaptosomal brain mitochondria isolated from mice heterozygous for a Dld null mutation (Dld+/−), Starkov and colleagues demonstrated a reduction in both basal State 4 ROS production and rotenone-stimulated ROS production with α-ketoglutarate as substrate [50]. Since the partial complex I deficiency of Hq brain mitochondria did not have a significant influence on rotenone or MPP+-stimulated ROS generation, we hypothesize that the Dld component of the α-ketoglutarate dehydrogenase complex was a major source of the ROS measured in our experiments. The role of complex I in physiological and pathological ROS generation should become clearer as additional genetic models of complex I deficiency become available. Basal mitochondrial ROS levels were also unaffected in Ndufs4-deficient neurons lacking complex I activity [10]. This finding, while requiring additional characterization, provides additional support for the hypothesis that complex I is not the principle source of physiological ROS produced by brain mitochondria.
It has been debated whether succinate oxidation can contribute to mitochondrial ROS production from complex I in vivo. A recent article reported that succinate promotes H2O2 release from brain mitochondria even in the presence of complex I substrates [65]. However, ROS production mediated by reverse electron transfer is inhibited by physiological concentrations of ADP or endogenous fatty acids that produce a depolarization on the order of 10 mV in isolated mitochondria [54,59]. The finding that ROS production by in situ mitochondria within synaptosomes [53] or cerebellar granule neurons [21] is not sensitive to “mild uncoupling” suggests that mitochondria within neurons do not have a sufficient protonmotive force to favor the thermodynamics of reverse electron transfer. We now add genetic evidence to support this conclusion. The finding that reverse-electron transfer mediated ROS production by complex I-depleted isolated mitochondria was inhibited by ~50% (Fig. 5A) while ROS release from synaptosomes was unaffected (Fig. 6A) suggests that reverse electron transfer does not contribute to physiological ROS production by brain mitochondria under normal (glucose replete) conditions.
The observation that AIF-deficient Hq cerebellar granule neurons are more sensitive to H2O2-induced cell death formed part of the basis for the original hypothesis that AIF serves an anti-oxidant role [23]. However, WT and Hq brain mitochondria scavenged exogenous H2O2 with a similar efficiency (Fig. 3B) and we did not detect a difference in oxidative protein carbonyl modifications at a stage prior to the onset of neurodegeneration in several brain regions including the cerebellum (Fig. 7).
Nevertheless, while our data are consistent with the hypothesis that complex I and AIF have a limited direct role in regulating ROS production by brain mitochondria, it is possible that these factors modulate ROS in vivo under conditions that remain to be tested. For example, decreased complex I activity is expected to result in increased NADH that can then be converted into NADPH. Thus it is possible that the inability to detect signs of elevated ROS in the AIF-deficient Hq brain can be explained by more efficient H2O2 detoxification by the NADPH-linked glutathione peroxidase/reductase system. Because elevated lipid hydroperoxides were detected in Hq brains [23] and AIF was implicated in 12/15-lipoxygenase-dependent cell death [43], we also do not exclude the important possibility that AIF specifically regulates lipid oxidation or the removal of cells with damaged lipids.
Collectively, our results favor the hypothesis that oxidative stress is a consequence rather than a cause of in vivo mitochondrial dysfunction in the AIF-depleted Hq brain. However, further investigation is clearly needed to understand the precise biochemical functions of AIF and identify the proximal causes of neurodegeneration in the harlequin mouse. It will also be important to examine the extent to which our observations made with brain mitochondria can be extrapolated to the mitochondria of other tissues, as well as to determine whether calpain processing alters the biochemical function of AIF.
In summary, we reached three important conclusions from our study of AIF- and complex I-depleted Hq brain mitochondria. First, AIF itself does not appear to directly regulate ROS production by brain mitochondria. Second, reverse electron transfer-mediated ROS production is unlikely to contribute to physiological ROS production by brain mitochondria in situ. Third, basal and rotenone or MPP+-stimulated rates of ROS production by mitochondria oxidizing complex I substrates are not proportional to the amount of complex I. This final conclusion raises the important possibility that some or all of these ROS do not originate from complex I. The development of additional genetic models with specific electron transport chain deficiencies should yield further insight into both the origin and role of ROS in neurodegenerative pathology.
Acknowledgments
The authors would like to thank Drs. Martin D. Brand and Gary Fiskum for critically reading the manuscript and helpful discussions. This work was supported by an American Parkinson Disease research grant to S.J.C., NIH grant AG025901 to J.K.A. and D.G.N, and NIH grants NS054764 and ES012077 Pilot to B.M.P.
Abbreviations
- AIF
apoptosis-inducing factor
- Dld
dihydrolipoamide dehydrogenase
- DNP
2,4 dinitrophenylhydrazone
- DNPH
2,4 dinitrophenylhydrazine
- DTNB
5,5′-dithiobis-(2-nitrobenzoic acid)
- Hq
harlequin
- MPP+
1-methyl-4-phenylpyridinium
- SOD
superoxide dismutase
- WT
wild type
- X/XO
xanthine/xanthine oxidase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Andersen JK. Oxidative stress in neurodegeneration: cause or consequence? Nat Med. 2004;10(Suppl):S18–S25. doi: 10.1038/nrn1434. [DOI] [PubMed] [Google Scholar]
- 2.Andreyev AY, Kushnareva YE, Starkov AA. Mitochondrial metabolism of reactive oxygen species. Biochemistry (Mosc) 2005;70:200–214. doi: 10.1007/s10541-005-0102-7. [DOI] [PubMed] [Google Scholar]
- 3.Apostolova N, Cervera AM, Victor VM, Cadenas S, Sanjuan-Pla A, varez-Barrientos A, Esplugues JV, McCreath KJ. Loss of apoptosis-inducing factor leads to an increase in reactive oxygen species, and an impairment of respiration that can be reversed by antioxidants. Cell Death Differ. 2006;13:354–357. doi: 10.1038/sj.cdd.4401776. [DOI] [PubMed] [Google Scholar]
- 4.Barja G. Mitochondrial oxygen radical generation and leak: sites of production in states 4 and 3, organ specificity, and relation to aging and longevity. J Bioenerg Biomembr. 1999;31:347–366. doi: 10.1023/a:1005427919188. [DOI] [PubMed] [Google Scholar]
- 5.Benit P, Goncalves S, Dassa EP, Briere JJ, Rustin P. The Variability of the Harlequin Mouse Phenotype Resembles that of Human Mitochondrial-Complex I-Deficiency Syndromes. PLoS ONE. 2008;3:e3208. doi: 10.1371/journal.pone.0003208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- 7.Brookes PS. Mitochondrial H(+) leak and ROS generation: an odd couple. Free Radic Biol Med. 2005;38:12–23. doi: 10.1016/j.freeradbiomed.2004.10.016. [DOI] [PubMed] [Google Scholar]
- 8.Brown D, Yu BD, Joza N, Benit P, Meneses J, Firpo M, Rustin P, Penninger JM, Martin GR. Loss of Aif function causes cell death in the mouse embryo, but the temporal progression of patterning is normal. Proc Natl Acad Sci U S A. 2006;103:9918–9923. doi: 10.1073/pnas.0603950103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burns RS, Chiueh CC, Markey SP, Ebert MH, Jacobowitz DM, Kopin IJ. A primate model of parkinsonism: selective destruction of dopaminergic neurons in the pars compacta of the substantia nigra by N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Proc Natl Acad Sci U S A. 1983;80:4546–4550. doi: 10.1073/pnas.80.14.4546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choi WS, Kruse SE, Palmiter RD, Xia Z. Mitochondrial complex I inhibition is not required for dopaminergic neuron death induced by rotenone, MPP+, or paraquat. Proc Natl Acad Sci U S A. 2008;105:15136–15141. doi: 10.1073/pnas.0807581105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chu CT, Zhu JH, Cao G, Signore A, Wang S, Chen J. Apoptosis inducing factor mediates caspase-independent 1-methyl-4-phenylpyridinium toxicity in dopaminergic cells. J Neurochem. 2005;94:1685–1695. doi: 10.1111/j.1471-4159.2005.03329.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Churbanova IY, Sevrioukova IF. Redox-dependent changes in molecular properties of mitochondrial apoptosis inducing factor. J Biol Chem. 2007 doi: 10.1074/jbc.M709147200. [DOI] [PubMed] [Google Scholar]
- 13.Cino M, Del Maestro RF. Generation of hydrogen peroxide by brain mitochondria: the effect of reoxygenation following postdecapitative ischemia. Arch Biochem Biophys. 1989;269:623–638. doi: 10.1016/0003-9861(89)90148-3. [DOI] [PubMed] [Google Scholar]
- 14.Davey GP, Clark JB. Threshold effects and control of oxidative phosphorylation in nonsynaptic rat brain mitochondria. J Neurochem. 1996;66:1617–1624. doi: 10.1046/j.1471-4159.1996.66041617.x. [DOI] [PubMed] [Google Scholar]
- 15.Davey GP, Peuchen S, Clark JB. Energy thresholds in brain mitochondria. Potential involvement in neurodegeneration. J Biol Chem. 1998;273:12753–12757. doi: 10.1074/jbc.273.21.12753. [DOI] [PubMed] [Google Scholar]
- 16.El GV, Csaba Z, Olivier P, Lelouvier B, Schwendimann L, Dournaud P, Verney C, Rustin P, Gressens P. Apoptosis-Inducing Factor Deficiency Induces Early Mitochondrial Degeneration in Brain Followed by Progressive Multifocal Neuropathology. J Neuropathol Exp Neurol. 2007;66:838–847. doi: 10.1097/NEN.0b013e318148b822. [DOI] [PubMed] [Google Scholar]
- 17.Fiskum G, Starkov A, Polster BM, Chinopoulos C. Mitochondrial mechanisms of neural cell death and neuroprotective interventions in Parkinson’s disease. Ann N Y Acad Sci. 2003;991:111–119. doi: 10.1111/j.1749-6632.2003.tb07469.x. [DOI] [PubMed] [Google Scholar]
- 18.Gazaryan IG, Krasnikov BF, Ashby GA, Thorneley RN, Kristal BS, Brown AM. Zinc is a potent inhibitor of thiol oxidoreductase activity and stimulates reactive oxygen species production by lipoamide dehydrogenase. J Biol Chem. 2002;277:10064–10072. doi: 10.1074/jbc.M108264200. [DOI] [PubMed] [Google Scholar]
- 19.Herrero A, Barja G. Localization of the Site of Oxygen Radical Generation inside the Complex I of Heart and Nonsynaptic Brain Mammalian Mitochondria. J Bioenerg Biomembr. 2000;32:609–615. doi: 10.1023/a:1005626712319. [DOI] [PubMed] [Google Scholar]
- 20.Hinkle PC, Butow RA, Racker E, Chance B. Partial resolution of the enzymes catalyzing oxidative phosphorylation. XV. Reverse electron transfer in the flavin-cytochrome beta region of the respiratory chain of beef heart submitochondrial particles. J Biol Chem. 1967;242:5169–5173. [PubMed] [Google Scholar]
- 21.Johnson-Cadwell LI, Jekabsons MB, Wang A, Polster BM, Nicholls DG. ‘Mild Uncoupling’ does not decrease mitochondrial superoxide levels in cultured cerebellar granule neurons but decreases spare respiratory capacity and increases toxicity to glutamate and oxidative stress. J Neurochem. 2007;101:1619–1631. doi: 10.1111/j.1471-4159.2007.04516.x. [DOI] [PubMed] [Google Scholar]
- 22.Joza N, Oudit GY, Brown D, Benit P, Kassiri Z, Vahsen N, Benoit L, Patel MM, Nowikovsky K, Vassault A, Backx PH, Wada T, Kroemer G, Rustin P, Penninger JM. Muscle-specific loss of apoptosis-inducing factor leads to mitochondrial dysfunction, skeletal muscle atrophy, and dilated cardiomyopathy. Mol Cell Biol. 2005;25:10261–10272. doi: 10.1128/MCB.25.23.10261-10272.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klein JA, Longo-Guess CM, Rossmann MP, Seburn KL, Hurd RE, Frankel WN, Bronson RT, Ackerman SL. The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature. 2002;419:367–374. doi: 10.1038/nature01034. [DOI] [PubMed] [Google Scholar]
- 24.Krantic S, Mechawar N, Reix S, Quirion R. Apoptosis-inducing factor: a matter of neuron life and death. Prog Neurobiol. 2007;81:179–196. doi: 10.1016/j.pneurobio.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 25.Kristian T, Fiskum G. A fluorescence-based technique for screening compounds that protect against damage to brain mitochondria. Brain Res Brain Res Protoc. 2004;13:176182. doi: 10.1016/j.brainresprot.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 26.Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- 27.Kushnareva Y, Murphy AN, Andreyev A. Complex I-mediated reactive oxygen species generation: modulation by cytochrome c and NAD(P)+ oxidation-reduction state. Biochem J. 2002;368:545–553. doi: 10.1042/BJ20021121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kussmaul L, Hirst J. The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc Natl Acad Sci U S A. 2006;103:7607–7612. doi: 10.1073/pnas.0510977103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lambert AJ, Brand MD. Inhibitors of the quinone-binding site allow rapid superoxide production from mitochondrial NADH:ubiquinone oxidoreductase (complex I) J Biol Chem. 2004;279:39414–39420. doi: 10.1074/jbc.M406576200. [DOI] [PubMed] [Google Scholar]
- 30.Lapidus RG, Sokolove PM. Spermine inhibition of the permeability transition of isolated rat liver mitochondria: an investigation of mechanism. Arch Biochem Biophys. 1993;306:246–253. doi: 10.1006/abbi.1993.1507. [DOI] [PubMed] [Google Scholar]
- 31.Lim ML, Mercer LD, Nagley P, Beart PM. Rotenone and MPP+ preferentially redistribute apoptosis-inducing factor in apoptotic dopamine neurons. Neuroreport. 2007;18:307–312. doi: 10.1097/WNR.0b013e32801b3ca6. [DOI] [PubMed] [Google Scholar]
- 32.Liou AK, Zhou Z, Pei W, Lim TM, Yin XM, Chen J. BimEL up-regulation potentiates AIF translocation and cell death in response to MPTP. FASEB J. 2005;19:1350–1352. doi: 10.1096/fj.04-3258fje. [DOI] [PubMed] [Google Scholar]
- 33.Lipton SA, Bossy-Wetzel E. Dueling activities of AIF in cell death versus survival: DNA binding and redox activity. Cell. 2002;111:147–150. doi: 10.1016/s0092-8674(02)01046-2. [DOI] [PubMed] [Google Scholar]
- 34.Mate MJ, Ortiz-Lombardia M, Boitel B, Haouz A, Tello D, Susin SA, Penninger J, Kroemer G, Alzari PM. The crystal structure of the mouse apoptosis-inducing factor AIF. Nat Struct Biol. 2002;9:442–446. doi: 10.1038/nsb793. [DOI] [PubMed] [Google Scholar]
- 35.Miramar MD, Costantini P, Ravagnan L, Saraiva LM, Haouzi D, Brothers G, Penninger JM, Peleato ML, Kroemer G, Susin SA. NADH oxidase activity of mitochondrial apoptosis-inducing factor. J Biol Chem. 2001;276:16391–16398. doi: 10.1074/jbc.M010498200. [DOI] [PubMed] [Google Scholar]
- 36.Murphy MP, Krueger MJ, Sablin SO, Ramsay RR, Singer TP. Inhibition of complex I by hydrophobic analogues of N-methyl-4-phenylpyridinium (MPP+) and the use of an ion-selective electrode to measure their accumulation by mitochondria and electron-transport particles. Biochem J. 1995;306(Pt 2):359–365. doi: 10.1042/bj3060359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nicholls DG. Bioenergetics and transmitter release in the isolated nerve terminal. Neurochem Res. 2003;28:1433–1441. doi: 10.1023/a:1025653805029. [DOI] [PubMed] [Google Scholar]
- 38.Panov A, Dikalov S, Shalbuyeva N, Taylor G, Sherer T, Greenamyre JT. Rotenone model of Parkinson disease: multiple brain mitochondria dysfunctions after short term systemic rotenone intoxication. J Biol Chem. 2005;280:42026–42035. doi: 10.1074/jbc.M508628200. [DOI] [PubMed] [Google Scholar]
- 39.Perier C, Tieu K, Guegan C, Caspersen C, Jackson-Lewis V, Carelli V, Martinuzzi A, Hirano M, Przedborski S, Vila M. Complex I deficiency primes Bax-dependent neuronal apoptosis through mitochondrial oxidative damage. Proc Natl Acad Sci U S A. 2005;102:19126–19131. doi: 10.1073/pnas.0508215102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Polster BM, Basanez G, Etxebarria A, Hardwick JM, Nicholls DG. Calpain I induces cleavage and release of apoptosis-inducing factor from isolated mitochondria. J Biol Chem. 2005;280:6447–6454. doi: 10.1074/jbc.M413269200. [DOI] [PubMed] [Google Scholar]
- 41.Pospisilik JA, Knauf C, Joza N, Benit P, Orthofer M, Cani PD, Ebersberger I, Nakashima T, Sarao R, Neely G, Esterbauer H, Kozlov A, Kahn CR, Kroemer G, Rustin P, Burcelin R, Penninger JM. Targeted Deletion of AIF Decreases Mitochondrial Oxidative Phosphorylation and Protects from Obesity and Diabetes. Cell. 2007;131:476–491. doi: 10.1016/j.cell.2007.08.047. [DOI] [PubMed] [Google Scholar]
- 42.Schilling B, Bharath MMS, Row RH, Murray J, Cusack MP, Capaldi RA, Freed CR, Prasad KN, Andersen JK, Gibson BW. Rapid purification and mass spectrometric characterization of mitochondrial NADH dehydrogenase (Complex I) from rodent brain and a dopaminergic neuronal cell line. Mol Cell Proteomics. 2005;4:84–96. doi: 10.1074/mcp.M400143-MCP200. [DOI] [PubMed] [Google Scholar]
- 43.Seiler A, Schneider M, Forster H, Roth S, Wirth EK, Culmsee C, Plesnila N, Kremmer E, Radmark O, Wurst W, Bornkamm GW, Schweizer U, Conrad M. Glutathione Peroxidase 4 Senses and Translates Oxidative Stress into 12/15-Lipoxygenase Dependent- and AIF-Mediated Cell Death. Cell Metab. 2008;8:237–248. doi: 10.1016/j.cmet.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 44.Shepherd D, Garland PB. Citrate synthase from rat liver. Methods Enzymol. 1969;13:11–16. doi: 10.1042/bj1140597. Ref Type: Generic. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sherer TB, Betarbet R, Testa CM, Seo BB, Richardson JR, Kim JH, Miller GW, Yagi T, Matsuno-Yagi A, Greenamyre JT. Mechanism of toxicity in rotenone models of Parkinson’s disease. J Neurosci. 2003;23:10756–10764. doi: 10.1523/JNEUROSCI.23-34-10756.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sims NR. Rapid isolation of metabolically active mitochondria from rat brain and subregions using Percoll density gradient centrifugation. J Neurochem. 1990;55:698–707. doi: 10.1111/j.1471-4159.1990.tb04189.x. [DOI] [PubMed] [Google Scholar]
- 47.Sipos I, Tretter L, dam-Vizi V. Quantitative relationship between inhibition of respiratory complexes and formation of reactive oxygen species in isolated nerve terminals. J Neurochem. 2003;84:112–118. doi: 10.1046/j.1471-4159.2003.01513.x. [DOI] [PubMed] [Google Scholar]
- 48.Srere PA. Citrate synthase. Methods Enzymol. 2008;13:3–11. Ref Type: Generic. [Google Scholar]
- 49.Starkov AA, Fiskum G. Regulation of brain mitochondrial H2O2 production by membrane potential and NAD(P)H redox state. J Neurochem. 2003;86:1101–1107. doi: 10.1046/j.1471-4159.2003.01908.x. [DOI] [PubMed] [Google Scholar]
- 50.Starkov AA, Fiskum G, Chinopoulos C, Lorenzo BJ, Browne SE, Patel MS, Beal MF. Mitochondrial alpha-ketoglutarate dehydrogenase complex generates reactive oxygen species. J Neurosci. 2004;24:7779–7788. doi: 10.1523/JNEUROSCI.1899-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Starkov AA, Polster BM, Fiskum G. Regulation of hydrogen peroxide production by brain mitochondria by calcium and Bax. J Neurochem. 2002;83:220–228. doi: 10.1046/j.1471-4159.2002.01153.x. [DOI] [PubMed] [Google Scholar]
- 52.Tretter L, dam-Vizi V. Generation of reactive oxygen species in the reaction catalyzed by alpha-ketoglutarate dehydrogenase. J Neurosci. 2004;24:7771–7778. doi: 10.1523/JNEUROSCI.1842-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tretter L, dam-Vizi V. Uncoupling is without an effect on the production of reactive oxygen species by in situ synaptic mitochondria. J Neurochem. 2007;103:1864–1871. doi: 10.1111/j.1471-4159.2007.04891.x. [DOI] [PubMed] [Google Scholar]
- 54.Tretter L, Mayer-Takacs D, dam-Vizi V. The effect of bovine serum albumin on the membrane potential and reactive oxygen species generation in succinate-supported isolated brain mitochondria. Neurochem Int. 2007;50:139–147. doi: 10.1016/j.neuint.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 55.Urbano A, Lakshmanan U, Choo PH, Kwan JC, Ng PY, Guo K, Dhakshinamoorthy S, Porter A. AIF suppresses chemical stress-induced apoptosis and maintains the transformed state of tumor cells. EMBO J. 2005;24:2815–2826. doi: 10.1038/sj.emboj.7600746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vahsen N, Cande C, Briere JJ, Benit P, Joza N, Larochette N, Mastroberardino PG, Pequignot MO, Casares N, Lazar V, Feraud O, Debili N, Wissing S, Engelhardt S, Madeo F, Piacentini M, Penninger JM, Schagger H, Rustin P, Kroemer G. AIF deficiency compromises oxidative phosphorylation. EMBO J. 2004;23:4679–4689. doi: 10.1038/sj.emboj.7600461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van EV, Bertrand AT, van der NR, Kostin S, Doevendans PA, Crijns HJ, de WE, Sluiter W, Ackerman SL, De Windt LJ. Downregulation of apoptosis-inducing factor in harlequin mutant mice sensitizes the myocardium to oxidative stress-related cell death and pressure overload-induced decompensation. Circ Res. 2005;96:e92–e101. doi: 10.1161/01.RES.0000172081.30327.28. [DOI] [PubMed] [Google Scholar]
- 58.van EV, Bertrand AT, van Oort RJ, van der NR, Engelen M, van Rijen HV, Doevendans PA, Crijns HJ, Ackerman SL, Sluiter W, De Windt LJ. EUK-8, a superoxide dismutase and catalase mimetic, reduces cardiac oxidative stress and ameliorates pressure overload-induced heart failure in the harlequin mouse mutant. J Am Coll Cardiol. 2006;48:824–832. doi: 10.1016/j.jacc.2006.02.075. [DOI] [PubMed] [Google Scholar]
- 59.Votyakova TV, Reynolds IJ. DeltaPsi(m)-Dependent and -independent production of reactive oxygen species by rat brain mitochondria. J Neurochem. 2001;79:266–277. doi: 10.1046/j.1471-4159.2001.00548.x. [DOI] [PubMed] [Google Scholar]
- 60.Yadava N, Nicholls DG. Spare respiratory capacity rather than oxidative stress regulates glutamate excitotoxicity after partial respiratory inhibition of mitochondrial complex I with rotenone. J Neurosci. 2007;27:7310–7317. doi: 10.1523/JNEUROSCI.0212-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yadava N, Potluri P, Smith EN, Bisevac A, Scheffler IE. Species-specific and mutant MWFE proteins. Their effect on the assembly of a functional mammalian mitochondrial complex I. J Biol Chem. 2002;277:21221–21230. doi: 10.1074/jbc.M202016200. [DOI] [PubMed] [Google Scholar]
- 62.Ye H, Cande C, Stephanou NC, Jiang S, Gurbuxani S, Larochette N, Daugas E, Garrido C, Kroemer G, Wu H. DNA binding is required for the apoptogenic action of apoptosis inducing factor. Nat Struct Biol. 2002;9:680–684. doi: 10.1038/nsb836. [DOI] [PubMed] [Google Scholar]
- 63.Zhu C, Wang X, Huang Z, Qiu L, Xu F, Vahsen N, Nilsson M, Eriksson PS, Hagberg H, Culmsee C, Plesnila N, Kroemer G, Blomgren K. Apoptosis-inducing factor is a major contributor to neuronal loss induced by neonatal cerebral hypoxia-ischemia. Cell Death Differ. 2007;14:775–784. doi: 10.1038/sj.cdd.4402053. [DOI] [PubMed] [Google Scholar]
- 64.Zoccarato F, Cavallini L, Alexandre A. Respiration-dependent removal of exogenous H2O2 in brain mitochondria: inhibition by Ca2+ J Biol Chem. 2004;279:4166–4174. doi: 10.1074/jbc.M308143200. [DOI] [PubMed] [Google Scholar]
- 65.Zoccarato F, Cavallini L, Bortolami S, Alexandre A. Succinate modulation of H2O2 release at NADH:ubiquinone oxidoreductase (Complex I) in brain mitochondria. Biochem J. 2007;406:125–129. doi: 10.1042/BJ20070215. [DOI] [PMC free article] [PubMed] [Google Scholar]