

Abstract
Classical protocols for carbonyl allylation, propargylation and vinylation typically rely upon the use of preformed allyl metal, allenyl metal and vinyl metal reagents, respectively, mandating stoichiometric generation of metallic byproducts. Through transfer hydrogenative C-C coupling, carbonyl addition may be achieved from the aldehyde or alcohol oxidation level in the absence of stoichiometric organometallic reagents or metallic reductants. Here, we review transfer hydrogenative methods for carbonyl addition, which encompass the first cataltyic protocols enabling direct C–H functionalization of alcohols.

Keywords: Ruthenium, Iridium, Transfer Hydrogenation, Diene, Enyne, Allylation, Propargylation, Vinylation, Catalytic, Cross Coupling, Reductive Coupling
Introduction to Carbonyl Allylation
Enantioselective carbonyl allylation ranks among the most broadly utilized methods in organic synthesis.[1] In seminal reports, Mikhailov and Bubnov (1964) and Hosomi and Sakurai (1976) described the first carbonyl allylations employing isolable allyl boron reagents and isolable allyl silanes, respectively.[2] Subsequently, Hoffmann (1978) devised the first chirally modified allyl metal reagent, an allylborane derived from camphor.[3a,b] These studies inspired the development of numerous protocols for asymmetric carbonyl allylation based on chirally modified allyl metal reagents, including those developed by Kumada (1982),[3c] Brown (1983),[3d] Roush (1985),[3e] Reetz (1988),[3f] Masamune (1989),[3g] Corey (1989),[3h] Seebach (1987),[3i] Duthaler (1989),[3j] Panek (1991),[3k] Leighton (2002),[3l,m] and Soderquist (2005).[3n] By virtue of these efforts, highly enantioselective carbonyl and imine allylation is now possible for nearly every imaginable substrate class. Nevertheless, the effort required to prepare chirally modified allyl metal reagents poses a significant barrier to their use. Further, stoichiometric quantities of chiral inducing element and stoichiometric byproduct generation detracts from the utility of such reagents (Figure 1).
Figure 1.

Chirally modified allyl metal reagents for use in asymmetric carbonyl allylation.
The aforementioned limitation shave not gone unaddressed. Following groundbreaking work by Yamamoto (1991),[4a] highly enantioselective chiral Lewis acid catalyzed carbonyl allylations were described by Umani-Ronchi and Keck (1993).[4b,c] Elegant studies by Denmark (1994), demonstrate that catalytic quantities of chiral Lewis base also promote enantioselective carbonyl allylation.[4d,e] These methods are highly effective, but do not circumvent the use of preformed allyl metal reagents. For example, allyl stannanes employed in the Umani-Ronchi-Keck allylation mandate stoichiometric generation of tin byproducts. Further, such allyl metal reagents typically are prepared from the organomagnesium or organolithium compounds, which, in turn, are prepared from the allyl halides, meaning that the carbon atom of the allyl donor is activated stoichiometrically three times in advance of C-C coupling. Naturally, such preactivation increases cost and contributes to excessive waste generation (Scheme 1).
Scheme 1.

Chirally modified Lewis acid and Lewis base catalyzed enantioselective carbonyl allylation.
Another major approach to carbonyl allylation involves the reduction of metallo-π-allyls derived from allylic alcohols, allylic carboxylates or allylic halides. Here, stoichiometric quantities of metallic reductants, such as SmI2, SnCl2 and Et2Zn are required for catalytic turnover.[5–8] Finally, byproduct-free carbonyl allylation can be achieved via carbonyl-ene processes, however, enantioselective variants of these processes are limited to highly activated electrophiles.[9,10]
We have found that diverse π-unsaturated reactants engage in reductive C-C coupling under the conditions of catalytic hydrogenation.[11] Specifically, by exploiting unsaturates as latent carbanion equivalents, highly regio- and stereoselective carbonyl and imine vinylation, aldol and Mannich coupling, and acyl substitution are achieved.[12] One can easily envision hydrogenative carbonyl allylations, wherein allenes, dienes and allylic acetates serve as allyl donors. This concept is extended further through C-C bond forming transfer hydrogenation, wherein hydrogen embedded within an alcoholic reactant, typically isopropanol, mediates reductive C-C coupling. Of greater significance, an alcohol may serve dually as hydrogen donor and precursor to the carbonyl electrophile. In this way, carbonyl addition may be achieved directly from the alcohol oxidation level in the absence of preformed organometallic reagents or metallic reductants (Scheme 2).[13,14]
Scheme 2.

Conceptual framework for hydrogenative and transfer hydrogenative carbonyl allylation.
Hydrogenative carbonyl allylation employing allenes as allyl donors
The feasibility of hydrogenative carbonyl allylation was established in studies on the reductive coupling of allenes to aldehydes and activated ketones employing hydrogen as terminal reductant. Specifically, iridium catalyzed hydrogenation of commercially available 1,1-dimethylallene in the presence of carbonyl electrophiles delivers products of reverse prenylation as single regioisomers in good to excellent yield (Table 1).[15] Functional groups often considered “hydrogen-labile,” for example, aryl halides, benzylic ethers and nitroarenes, remain intact under the conditions of hydrogenative coupling. The combination of a cationic iridium complex in conjunction with Li2CO3 as a basic additive prevents over-reduction of the olefinic product. Notably, all atoms of each reactant, including hydrogen, are incorporated into the product, preventing stoichiometric generation of byproducts.
Table 1.
Iridium catalyzed hydrogenative coupling of 1,1-dimethylallene to carbonyl compounds.
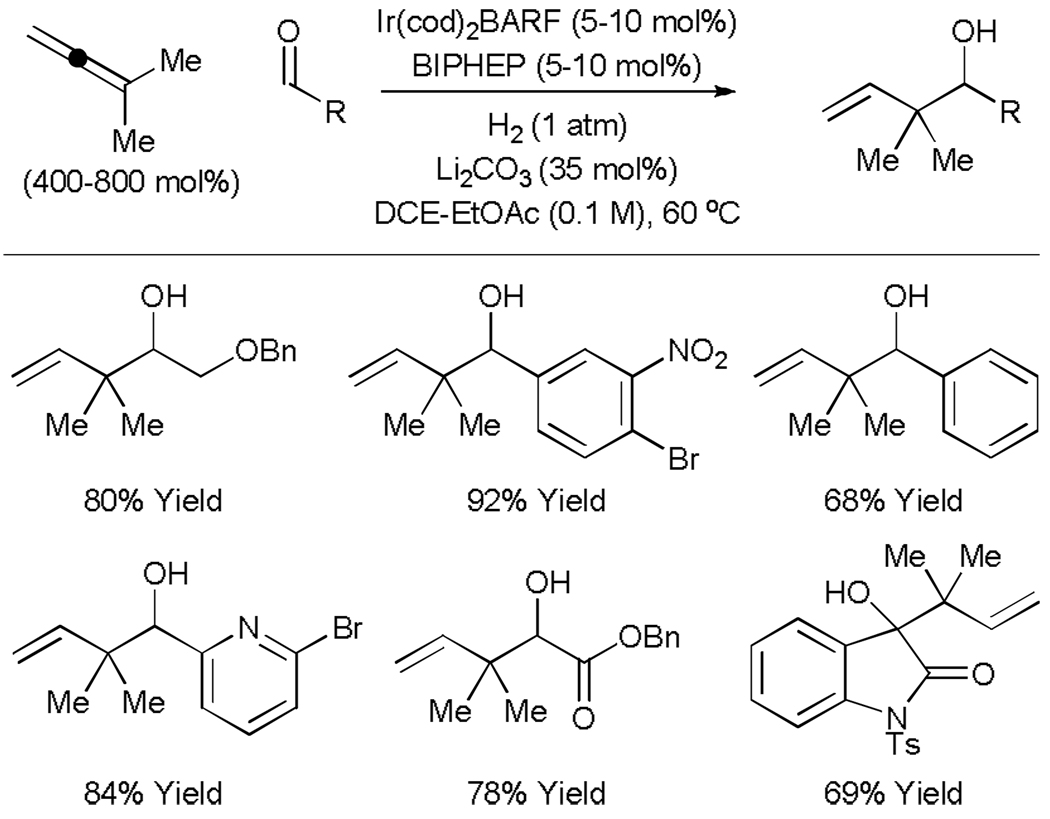 |
Hydrogenative coupling of 1,1-dimethylallene to 5-nitro-2-furancarboxaldehyde under an atmosphere of elemental deuterium provides a product of reverse prenylation incorporating deuterium solely at the interior vinylic position (80% 2H), as revealed by 2H NMR spectroscopic analysis. This result is consistent with a catalytic mechanism involving base-mediated heterolytic hydrogen activation to furnish an iridium monohydride followed by allene hydrometallation to deliver the primary σ-allyl species A. Allyl addition to the carbonyl electrophile through a closed six-centred transition structure B provides iridium alkoxide C, which upon hydrogenolysis releases the alcohol product and an iridium monohydride to close the cycle. Incomplete deuterium incorporation is likely due to β-hydride elimination of the tertiary σ-allyl haptomer of A to form isoprene. The results of isotopic labelling cannot exclude alternative pathways involving allene-aldehyde oxidative coupling (Scheme 3).
Scheme 3.

Iridium catalyzed hydrogenative coupling of 1,1-dimethylallene to an aldehyde under an atmosphere of deuterium.
Unlike reverse prenylation, the parent allylation employing gaseous allene was accompanied by over-reduction of the coupling product. It was postulated that the transfer of hydrogen from an alcoholic reductant rather, as opposed to direct use of elemental hydrogen, would enable more precisely controlled introduction of hydrogen into the catalytic system. Indeed, by simply substituting isopropanol for elemental hydrogen under conditions nearly identical to those previously employed, allene-aldehyde reductive coupling occurs to provide products of reverse prenylation in good to excellent yield, as well as products of allylation and crotylation, which are generated without any detectable over-reduction (Table 2, top).[16]
Table 2.
Iridium catalyzed transfer hydrogenative coupling of allenes to aldehydes and alcohols.
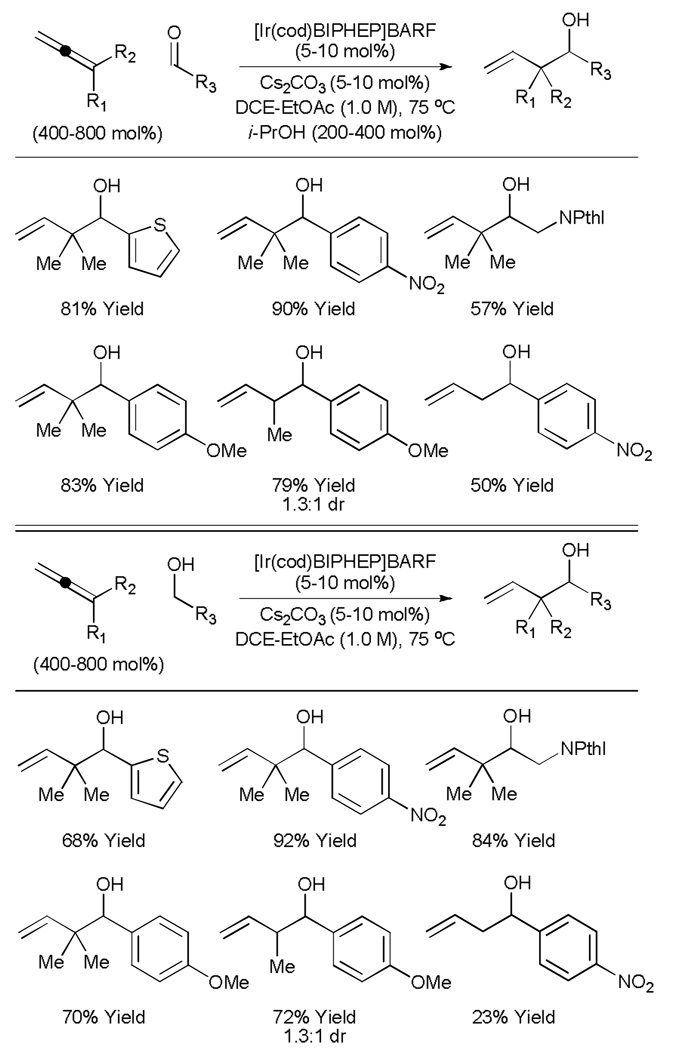 |
Direct C-allylation of alcohols is potentially achieved in transfer hydrogenative C-C couplings in which an alcoholic reactant serves as both hydrogen donor and aldehyde precursor. Indeed, under conditions employing a cationic iridium precatalyst and basic additive, alcohols couple directly to 1,1-dimethylallene to provide an identical set of carbonyl reverse prenylation products in good to excellent yield.[16] The reaction is broadly tolerant of the electronics of the alcohol, allowing direct carbinol C–H functionalization of benzylic and even aliphatic alcohols. Carbonyl allylation and crotylation employing gaseous allene and methyl allene, respectively, occurs without over-reduction of the olefinic product. In the case of allylation, lower efficiency is attributed to contamination of commerical allene gas with propyne (Table 2, bottom).
Exposure of 1,1-dimethylallene to benzaldehyde under standard conditions employing d8-isopropanol as reductant results in deuterium transfer to the vinylic position (85% 2H) of the resulting adduct. Similary, exposure of d2-benzyl alcohol to the reaction conditions results in transfer of the benzylic deuteride to the internal vinylic position of the product (85% 2H). (Scheme 4, top). These data are consistent with a hydrometallative mechanism, but cannot exclude catalytic mechanisms involving allene-aldehyde oxidative coupling. Crossover experiments involving exposure of 1,1-dimethylallene to equimolar quantities of p-nitrobenzyl alcohol and benzaldehyde under standard coupling conditions result in formation the indicated p-nitrophenyl- and phenyl-containing adducts in a 4:1 ratio, respectively. In a related experiment involving exposure of 1,1-dimethylallene to equimolar quantities of p-nitrobenzaldehyde and benzyl alcohol under standard coupling conditions, an identical product distribution is observed. These data establish fast and reversible alcohol dehydrogenation to form non-metal-bound aldehyde in advance of C-C coupling (Scheme 4, bottom). In preliminary studies, a chiral iridium complex modified by (R)-C3-TUNEPHOS was found to promote the coupling of 1,1-dimethylallene to p-nitrobenzyl alcohol in 55% isolated yield and 76% enantiomeric excess.[17] Erosion of enantiomeric excess is not observed under the coupling conditions, indicating that the product of carbonyl allylation, a secondary alcohol, is not subject to redox equilibration.
Scheme 4.

Top: Isotopic labeling experiments in iridium catalyzed transfer hydrogenative couplings of 1,1-dimethylallene. Bottom: Crossover experiments establish rapid redox equilibration in advance of C-C coupling.
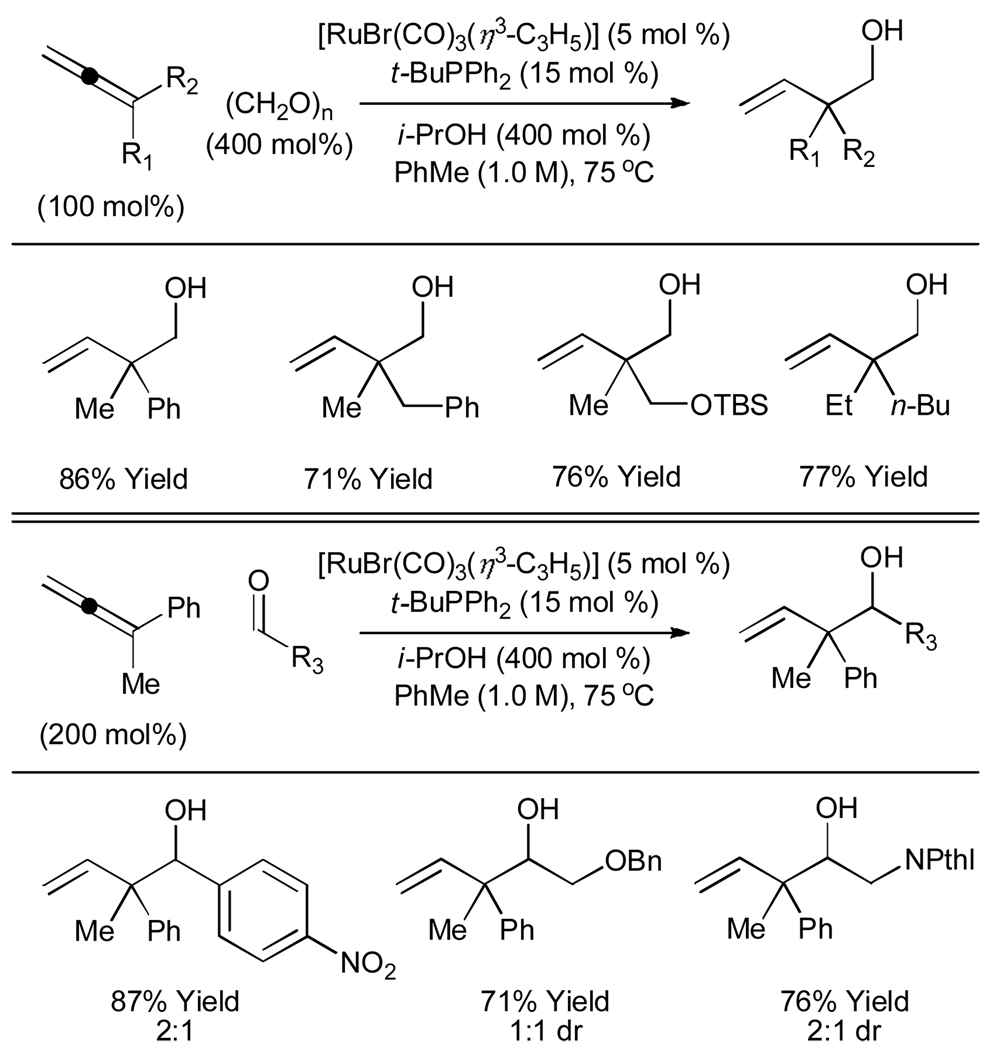
Ruthenium catalyzed transfer hydrogenation is one of the most powerful methods for the reduction of carbonyl compounds.[18] Surprisingly, reductive C-C bond formations catalyzed by ruthenium are highly uncommon.[19–21] Under the conditions of ruthenium catalyzed transfer hydrogenation employing isopropanol as the terminal reductant, 1,1-disubstituted allenes engage in reductive coupling to paraformaldehyde and higher aldehydes.[22] Coupling occurs with branched regioselectivity to deliver homoallylic alcohols bearing all-carbon quaternary centers (Table 3). Ruthenium catalyzed allene-alcohol transfer hydrogenative coupling is currently under investigation.
Table 3.
Ruthenium catalyzed transfer hydrogenative coupling of allenes to paraformaldehyde and higher aldehydes.
 |
Hydrogenative carbonyl allylation employing 1,3-dienes as allyl donors
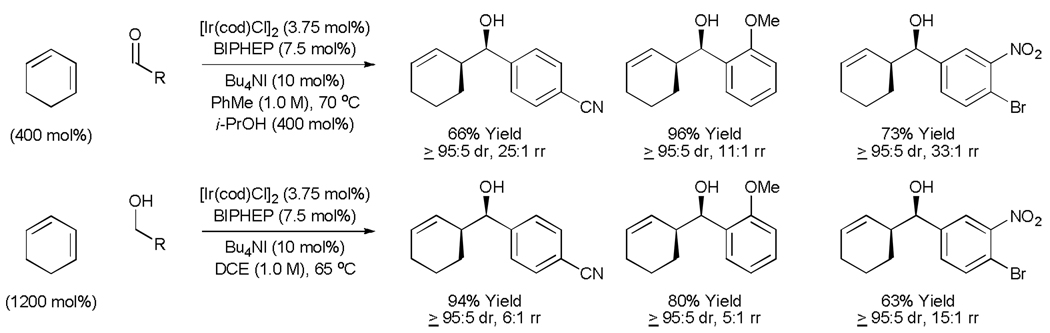
The hydrometallation of conjugated dienes represents an alternate method for the generation of allyl metal species. Under the conditions of iridium catalyzed hydrogenative coupling employing isopropanol as the terminal reductant, 1,3-cyclohexadiene couples to diverse aryl aldehydes to provide products of carbonyl cyclohexenylation in good to excellent yield and with high levels of diastereocontrol.[23] Under nearly identical conditions but in the absence of isopropanol, 1,3-cyclohexadiene couples directly to benzylic alcohols to furnish the very same products of carbonyl cyclohexenylation. Thus, carbonyl addition is achieved with equal facility from the aldehyde or alcohol oxidation level. Regioisomeric 1,5-olefinic adducts are formed as minor byproducts in all cases. Deuterium labelling studies corroborate a catalytic mechanism wherein alcohol dehydrogenation provides an iridium hydride, which upon diene hydrometallation produces an allyl metal nucleophile. These data do not exclude alternate pathways involving diene-aldehyde oxidative coupling (Table 4).
Table 4.
Iridium catalyzed transfer hydrogenative coupling of 1,3-cyclohexadiene to aldehydes and alcohols.
 |
Under the conditions of transfer hydrogenation employing RuHCl(CO)(PPh3)3 as precatalyst, the acylic conjugated dienes butadiene, isoprene and 2,3-dimethylbutadiene couple to benzylic alcohols to furnish products of carbonyl crotylation, carbonyl isoprenylation, and carbonyl reverse 2-methyl-prenylation, respectively.[24a] In these reactions, the presence of an acid cocatalyst (m-NO2BzOH) is essential as only trace quantities of product are observed otherwise. Additionally, exogenous acetone and phosphine ligand are found to have benefical effects upon the efficiency of the reaction. In all cases, efficient coupling is observed using only 250 mol% of the diene (Table 5, Left). This first generation catalytic system also promotes coupling to simple unactivated aliphatic alcohols, as demonstrated by the coupling of isoprene to 1-nonanol in 65% isolated yield (Scheme 5). Related diene-aldehyde couplings proceed efficiently using either isopropanol or formic acid as terminal reductants (Table 5, Left). The branched regioselectivity observed in these processes complements the linear regioselectivity observed in related Ni-catalyzed diene-aldehyde reductive couplings.[26,27] Transfer hydrogenative diene-alcohol or diene-aldehyde couplings employing the more highly coordinatively unsaturated ruthenium catalyst, Ru(TFA)2(CO)(PPh3)2,[28] induce further oxidation of the initially formed homoallylic alcohol to furnish β,γ-unsaturated ketones (Table 5, Right).[24b,25] Thus, all oxidations levels of substrate (alcohol or aldehyde) and product (homoallyl alcohol or β,γ-unsaturated ketone) are accessible (Scheme 5).
Table 5.
Left: Ruthenium catalyzed transfer hydrogenative coupling of acyclic dienes to alcohols and aldehydes to furnish homoallylic alcohols.a Right: Ruthenium catalyzed transfer hydrogenative coupling of acyclic dienes to alcohols and aldehydes to furnish β, γ-unsaturated ketones.b
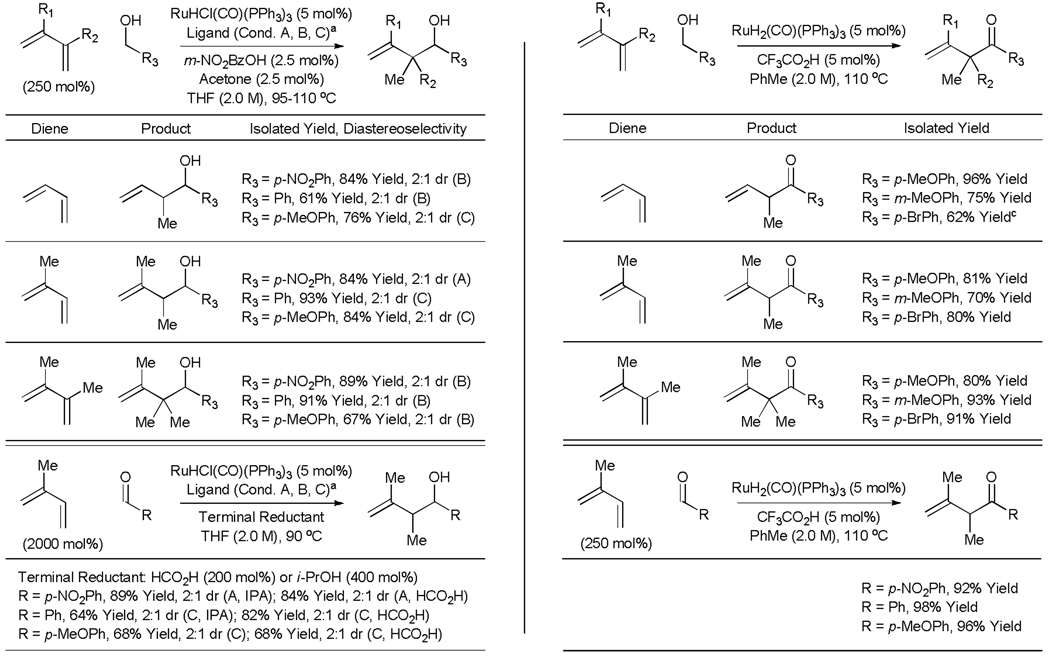 |
Conditions A employ no added ligand: Conditions B employ (p-MeOPh)3P (15 mol%) as ligand; Conditions C employ rac-BINAP (5 mol%) as ligand
Butadiene (800 mol%), isoprene (250 mol%), 2,3-dimethylbutadiene (300 mol%)
The reaction product was contaminated with approximately 10% of the α, β-unsaturated ketone.
Scheme 5.

Ruthenium catalyzed transfer hydrogenative coupling of isoprene to an unactivated aliphatic alcohol.
The coupling of isoprene to d2-benzyl alcohol results in transfer of a benzylic deuteride to the allylic methyl (19% 2H) and allylic methine (32% 2H). These data suggest reversible hydrometallation of the less substituted olefin to form the secondary σ-allyl species. Isomerization to the more stable primary σ-allyl haptomer precedes carbonyl addition, which occurs with allylic inversion through a six-centred transition state to deliver, upon protonolysis, the product of carbonyl allylation. Similarly, in aldehyde couplings employing d8-isopropanol as the terminal reductant, deuterium incorporation is observed at the allylic methyl (19%2 H) and allylic methine (10% 2H) (Scheme 6).
Scheme 6.

Isotopic labeling experiments in ruthenium catalyzed transfer hydrogenative couplings of isoprene.
Hydrogenative carbonyl allylation employing allyl acetate as an allyl donor
Prevailing protocols for carbonyl allylation employing allylic alcohols, allylic carboxylates or allylic halides as allyl donors require stoichiometric quantities of metallic reductants, such as SmI2, SnCl2 and Et2Zn for catalytic turnover.[5–8] The facility of alcohol dehydrogenation under the aforementioned transfer hydrogenative coupling conditions suggests the feasibility of catalytic allyl acetate mediated carbonyl allylations employing sacrificial alcohols as terminal reductant. Again, one may envision related processes in which an alcohol serves both as reductant and aldehyde precursor, thus enabling catalytic carbonyl allylation from the alcohol oxidation level. The outcome of such transformations was rendered uncertain by reports of alcohol-allyl acetate coupling to form enones under the conditions of ruthenium catalysis,[29] and the fact that allyl acetates reacts with alcohols to deliver products of O-allylation upon exposure to iridium catalysts (Scheme 7).[30]
Scheme 7.

Reversal of reactivity in the metal catalyzed coupling of allyl acetates to alcohols.
In the presence of an iridium complex derived from [IrCl(cod)]2 and (-)-TMPTP or (R)-Cl,MeO-BIPHEP, allyl acetate reductively couples to aryl aldehydes, enals and aliphatic aldehydes to furnish products of C-allylation with exceptional levels of asymmetric induction (Table 6, top).[31] In these processes, isopropanol functions as the terminal reductant. At most, only trace quantities (≤ 5%) of O-allylation product are observed. Remarkably, an identical set of carbonyl allylation products are accessible from the corresponding alcohols. High levels of asymmetric induction are achieved using chiral iridium catalysts modifed by (R)-BINAP or (R)-Cl,MeO-BIPHEP (Table 6, bottom). Thus, highly enantioselective catalytic carbonyl allylation is achieved from the aldehyde or alcohol oxidation level in the absence of allyl metal reagents.
Table 6.
Iridium catalyzed carbonyl allylation from the aldehyde or alcohol oxidation level employing allyl acetate.a
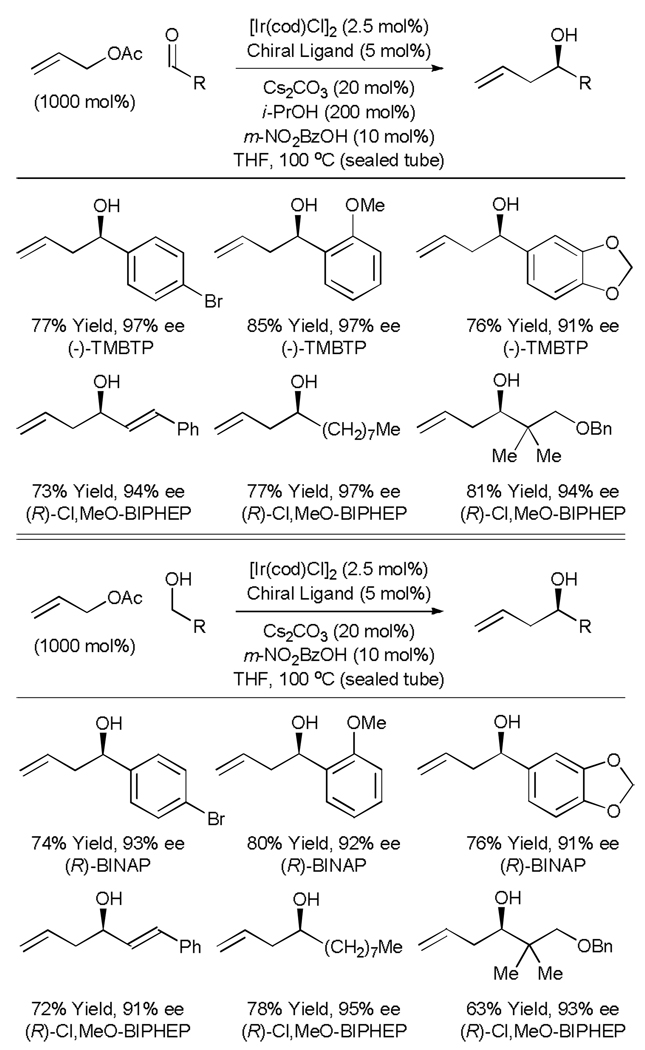 |
(-)-TMPTP = (-)-4,4’-bis(diphenylphosphino)-2,2’,5,5’-tetramethyl-3,3’-bithiophene.
Experiments aimed at illuminating key features of the catalytic mechanism reveal that the active catalyst is cyclometallated complex I, which arises upon ortho-C-H insertion of iridium onto m-nitrobenzoic acid. The BINAP derivative of complex I has been characterized by single crystal X-ray diffraction, and has been established as a catalytically competent species. The results of isotopic labeling are consistent with intervention of symmetric iridium π-allyl intermediates or rapid interconversion of σ-allyl haptomers through the agency of a symmetric π-allyl. Competition experiments demonstrate rapid and reversible hydrogenation-dehydrogenation of the carbonyl partner in advance of C-C coupling. Notably, the coupling products, which are homo-allylic alcohols, experience very little erosion of optical purity by way of redox equilibration under the coupling conditions, yet isopropanol, a secondary alcohol, may serve as terminal reductant (Scheme 8).
Scheme 8.

Left: Iridium catalyzed transfer hydrogenative coupling of allyl acetate to an alcohol employing isotopically labeled allyl acetate. Right: catalytically competent cyclometallated complex I.
Hydrogenative carbonyl propargylation employing 1,3-enynes as propargyl donors
Like carbonyl allylation, much effort has been devoted to the development of efficient methods for diastereo- and enantioselective carbonyl propargylation.[32] As early as 1950, Prévost showed that allenic Grignard reagents participate in carbonyl additions to generate mixtures of β-acetylenic and α-allenic carbinols, which evoked the term “propargylic transposition.”[33a,b] Relative stereocontrol in such additions was later demonstrated by Chodkiewicz (1969).[33c] Lequam and Guillerm (1973)[33d] reported that preformed allenic stannanes enable carbonyl propargylation when exposed to chloral. Subsequently, Mukaiyama (1981) showed that stannanes generated in situ from propargyl iodides and stannous chloride provide mixtures of β-acetylenic and α-allenic carbinols upon reaction with aldehydes.[33e] Related propargylations employing allenylboron reagents were first reported by Favre and Gaudemar (1966).[33f] Propargylations employing allenylsilicon reagents were first reported by Danheiser (1980).[33g]
Initially developed asymmetric propargylation protocols relied upon chirally modified allenyl metal reagents. For example, Yamamoto (1982)[33h] and Corey (1990)[33i] have shown that allenylboron reagents with chiral modifiers at boron engage in asymmetric carbonyl propargylation with useful levels of enantioselectivity. Similary, allenylstannanes chirally modified at the tin center engage in asymmetric carbonyl propargylation, as first reported by Mukaiyama (1987).[33j] Axially chiral allenylstannanes, allenylsilanes and allenylboron reagents propargylate aldehydes enantiospecifically, as first described by Marshall (1991, 2001),[33k,l] and Hayashi (1993),[33m] respectively (Figure 2). More recently, chiral Lewis acid and chiral Lewis base catalyzed asymmetric aldehyde propargylations employing allenylmetal reagents have been reported by Keck (1994)[33n] and Denmark (2001),[33o] respectively.
Figure 2.

Chirally modified allenyl metal reagents for use in asymmetric carbonyl propargylation.
We envision an alternative approach to carbonyl propargylation based on C-C bond forming transfer hydrogenation, wherein conjugated enynes serve as surrogates to preformed allenyl metal reagents. However, the feasibility of such processes was uncertain as related 1,3-enyne-carbonyl reductive couplings catalyzed by rhodium[34] and nickel[35–37] promote C-C coupling at the acetylenic terminus of the enyne. Despite this unfavorable precedent, enyne-alcohol transfer hydrogenative coupling delivers the desired products of carbonyl propargylation as single regioisomers using a catalyst prepared in situ from RuHCl(CO)(PPh3)3 and DPPF.[38] Iridium complexes also catalyze this process, but ruthenium catalysts were found to be superior.
This first generation catalytic system for enyne-mediated propargylation is applicable to benzylic alcohols, allylic alcohols and unactivated aliphatic alcohols. Additionally, 1,3-enynes possessing aryl, heteroaryl, alkyl and heteroalkyl groups at the alkyne terminus are tolerated. In all cases, products are formed in good to excellent isolated yields with complete levels of regioselection (Table 7, top). Under related transfer hydrogenation conditions employing isopropanol as the terminal reductant, carbonyl propargylation may be conducted from the aldehyde oxidation level in good to excellent yield (Table 7, bottom). Thus, carbonyl propargylation is achieved in the absence of preformed allenyl metal reagents from the alcohol or aldehyde oxidation level.
Table 7.
Ruthenium catalyzed transfer hydrogenative coupling of 1,3-enynes to alcohols and aldehydes.
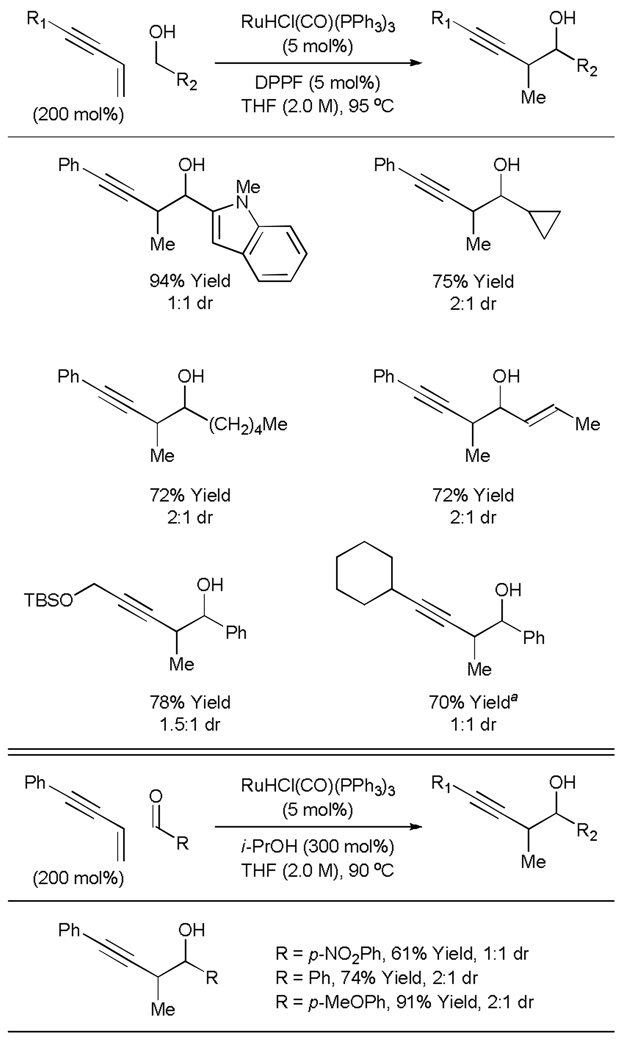 |
m-NO2BzOH (5 mol%) employed as additive.
Ruthenium catalyzed enyne coupling to d2-benzyl alcohol results in transfer of a benzylic deuteride to the allylic methyl (56% 2H) and allylic methine (24% 2H). Deuterium is completely retained at the benzylic methine of the coupling product (Scheme 9). These results are consistent with a mechanism involving alcohol dehydrogenation to generate a ruthenium hydride followed by reversible enyne hydrometallation to furnish an allenylruthenium intermediate. The aldehyde-allenylmetal nucleophile-electrophile pair thus formed engages in carbonyl addition with propargylic transposition to deliver the product of carbonyl propargylation. The ability to bypass barriers imposed by oxidition level, coupled with the accessibility of diverse enynes, makes the development of stereocontrolled enyne-mediated propargylations an important goal of ongoing research.
Scheme 9.

Isotopic labeling in the ruthenium catalyzed transfer hydrogenative coupling of a 1,3-enyne.
Hydrogenative carbonyl vinylation employing 1,3-enynes as propargyl donors
The synthetic utility of allylic alcohols has led to the development of diverse methods for their preparation. Among existing protocols, carbonyl vinylation represents an effective and convergent means of preparing allylic alcohols. Following seminal studies of Oguni (1984) and Noyori (1986),[39] enantioselective catalytic addition of vinylzinc reagents to aldehydes were developed by Oppolzer (1992) and Wipf (1994).[40,41,42] Generation of the vinylzinc reagent relies upon alkyne hydroboration or hydrozirconation with subsequent transmetallation to zinc employing using ZnMe2, meaning successive use of four stoichiometric organometallic reagents is required to pre-activate the alkyne as a vinyl carbanion equivalent. Consequently, molar equivalents of multiple metallic byproducts are generated (Scheme 10).
Scheme 10.

Carbonyl vinylation via stoichiometric alkyne hydrometallation.
Direct alkyne-carbonyl reductive coupling bypasses the use of multiple stoichiometric organometallic reagents. As reported by Ojima (1994), Crowe (1995) and Montgomery (1997), this pattern of reactivity was first observed in the cyclization of acetylenic aldehydes catalyzed by rhodium, titanium and nickel, respectively.[43,44,45] Intermolecular variants of the nickel catalyzed alkyne-carbonyl reductive couplings soon followed.[37,45] However, while reductive couplings of this type signal a departure from stoichiometric organometallic reagents, they exploit terminal reductants such as hydrosilanes, hydrostannanes, organozinc reagents, organoboron reagents or chromium(II) chloride, which generate molar equivalents of chemical byproducts. Under the conditions of rhodium and iridium catalyzed hydrogenation, byproduct-free alkyne-carbonyl and imine-carbonyl reductive coupling may be achieved in a highly regio-and stereoselective fashion (Scheme 11).[11, 12a–c,34]
Scheme 11.

Selected examples of rhodium and iridium catalyzed hydrogenative coupling of alkynes to carbonyl compounds and imines.
By exploiting alcohols as both aldehyde precursors and sources of hydrogen, it should be possible to promote direct byproduct-free carbonyl vinylation in the absence of any stoichiometric reductant. Accordingly, transfer hydrogenative alkyne-alcohol coupling was explored under the conditions of ruthenium catalysis. In an initial set of experiments, it was found that Ru(O2CCF3)2(CO)(PPh3)2 catalyzes alkyne-alcohol coupling to provide the desired allylic alcohols, representing a direct C-H vinylation of the alcohol (Table 8).[47] Thus, simple nonconjugated alkynes are activated as vinyl anion equivalents in carbonyl addition from the alcohol oxidation level under mild conditions. Ruthenium catalyzed alkyne-aldehyde transfer hydrogenative coupling is currently under investigation.
Table 8.
Ruthenium catalyzed transfer hydrogenative coupling of alkynes to alcohols.
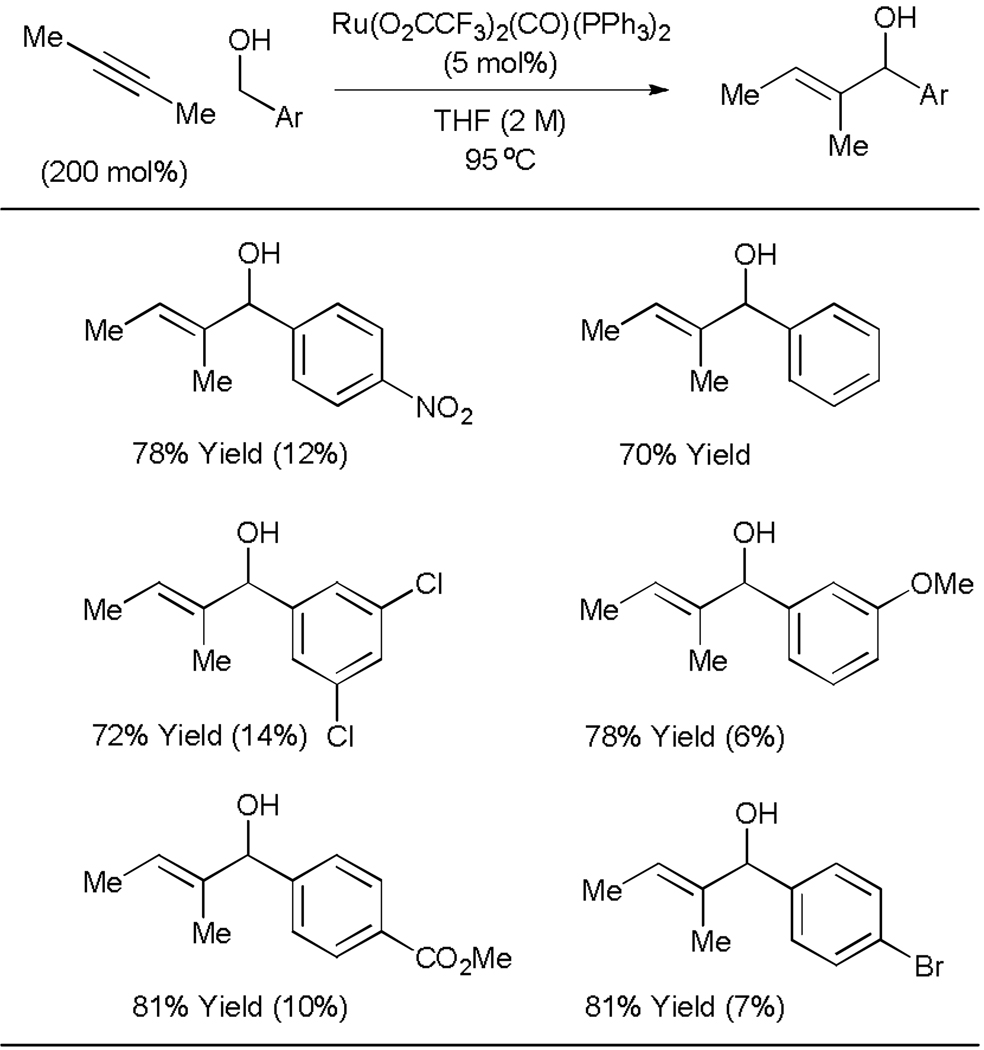 |
Isolated yield of enone side-product indicated in parentheses.
Conclusions and outlook
Through hydrogenative and transfer hydrogenative C-C coupling, nonstabilized carbanion equivalents may be generated from unsaturates, enabling carbonyl allylation, propargylation and vinylation from the aldehyde or alcohol oxidation level in the absence of preformed organometallic reagents or metallic reductants. Further, the alcohol-unsaturate couplings described in this account represent a byproduct-free method for direct C–H functionalization of alcohols, evoking numerous avenues of exploration. For example, ethylene-alcohol transfer hydrogenative coupling would dispense with the requirement of utilizing diethylzinc, a pyrophoric liquid, in carbonyl ethylation. The coupling of ethylene to (bio)ethanol would provide an efficient means of preparing (bio)butanol. Amine-unsaturate coupling would enable imine addition from the amine oxidation level, providing efficient access to pharmaceutical building blocks. These and many other challenges remain - just as classical carbanion chemistry is broad, so is the potential to generate their carbanion equivalents via hydrogenation and transfer hydrogenation.
Footnotes
Acknowledgment is made to Merck, Umicore, the Robert A. Welch Foundation, the ACS-GCI Pharmaceutical Roundtable, the Korea Research Foundation (KRF-2007-356-E00037) and the NIHNIGMS (RO1-GM069445) for partial support of this research for partial support of this research. Dr. Oliver Briel of Umicore is thanked for the generous donation of iridium and ruthenium salts. Professor Xumu Zhang is thanked for the generous donation of (R)-C3-TUNEPHOS. Drs. Ian Davies, Kevin Campos and Scott Shultz of Merck are thanked for the generous donation of (-)-TMBTP.
References
- 1.For reviews on enantioselective carbonyl allylation, see: Ramachandran PV. Aldrichim. Acta. 2002;35:23–35. Kennedy JWJ, Hall DG. Angew. Chem. 2003;115:4880–4887. Angew. Chem. Int. Ed. 2003;42:4732–4739. doi: 10.1002/anie.200301632. Denmark SE, Fu J. Chem. Rev. 2003;103:2763–2793. doi: 10.1021/cr020050h. Yu C-M, Youn J, Jung H-K. Bull. Korean Chem. Soc. 2006;27:463–472. Marek I, Sklute G. Chem. Commun. 2007:1683–1691. doi: 10.1039/b615042j. Hall DG. Synlett. 2007:1644–1655.
- 2.a) Mikhailov BM, Bubnov YN. Izv. Akad. Nauk SSSR, Ser. Khim. 1964:1874–1876. [Google Scholar]; b) Hosomi A, Sakurai H. Tetrahedron Lett. 1976;17:1295–1298. [Google Scholar]; c) Sakurai H. Pure Appl. Chem. 1982;54:1–22. [Google Scholar]
- 3.Chirally modified allyl metal reagents: Herold T, Hoffmann RW. Angew. Chem. 1978;90:822–823. Angew. Chem. Int. Ed. Engl. 1978;17:768–769. Hoffmann RW, Herold T. Chem. Ber. 1981;114:375–383. Hayashi T, Konishi M, Kumada M. J. Am. Chem. Soc. 1982;104:4963–4965. Brown HC, Jadhav PK. J. Am. Chem. Soc. 1983;105:2092–2093. Roush WR, Walts AE, Hoong LK. J. Am. Chem. Soc. 1985;107:8186–8190. Reetz M. Pure Appl. Chem. 1988;60:1607–1614. Short RP, Masamune S. J. Am. Chem. Soc. 1989;111:1892–1894. Corey EJ, Yu C-M, Kim SS. J. Am. Chem. Soc. 1989;111:5495–5496. Seebach D, Beck AK, Imwinkelzied R, Roggo S, Wonnacott A. Helv. Chim. Acta. 1987;70:954–974. Riediker M, Duthaler RO. Angew. Chem. 1989;101:488–490. Angew. Chem., Int. Ed. Engl. 1989;28:494–495. Panek JS, Yang M. J. Am. Chem. Soc. 1991;113:6594–6600. Kinnaird JWA, Ng PY, Kubota K, Wang X, Leighton JL. J. Am. Chem. Soc. 2002;124:7920–7921. doi: 10.1021/ja0264908. Hackman BM, Lombardi PJ, Leighton JL. Org. Lett. 2004;6:4375–4377. doi: 10.1021/ol0480731. Burgos CH, Canales E, Matos K, Soderquist JA. J. Am. Chem. Soc. 2005;127:8044–8049. doi: 10.1021/ja043612i.
- 4.Catalytic asymmetric carbonyl allylation employing allyl metal reagents: Furuta K, Mouri M, Yamamoto H. Synlett. 1991:561–562. Costa AL, Piazza MG, Tagliavini E, Trombini C, Umani-Ronchi A. J. Am. Chem. Soc. 1993;115:7001–7002. Keck GE, Tarbet KH, Geraci LS. J. Am. Chem. Soc. 1993;115:8467–8468. Denmark SE, Coe DM, Pratt NE, Griedel BD. J. Org. Chem. 1994;59:6161–6163. doi: 10.1021/jo052202p. Denmark SE, Fu J. J. Am. Chem. Soc. 2001;123:9488–9489. doi: 10.1021/ja016552e.
- 5.For selected reviews covering carbonyl allylation via umpolung of π-allyls, see: Tamaru Y. In: Handbook of Organopalladium Chemistry for Organic Synthesis. Negishi E-i, de Meijere A., editors. Vol. 2. New York: Wiley; 2002. pp. 1917–1943. Tamaru Y. In: Perspectives in Organopalladium Chemistry for the XXI Century. Tsuji J, editor. Amsterdam: Elsevier; 1999. pp. 215–231. Kondo T, Mitsudo T-a. Curr. Org. Chem. 2002;6:1163–1179.
- 6.For selected examples of reactions involving nucleophilic π-allyls, see: Palladium: Tabuchi T, Inanaga J, Yamaguchi M. Tetrahedron Lett. 1986;27:1195–1196. Takahara JP, Masuyama Y, Kurusu Y. J. Am. Chem. Soc. 1992;114:2577–2586. Kimura M, Ogawa Y, Shimizu M, Sueishi M, Tanaka S, Tamaru Y. Tetrahedron Lett. 1998;39:6903–6906. Kimura M, Shimizu M, Shibata K, Tazoe M, Tamaru Y. Angew. Chem. 2003;115:3514–3517. doi: 10.1002/anie.200351182. Angew. Chem. Int. Ed. 2003;42:3392–3395. doi: 10.1002/anie.200351182. Zanoni G, Gladiali S, Marchetti A, Piccinini P, Tredici I, Vidari G. Angew. Chem. 2004;116:864–867. doi: 10.1002/anie.200352743. Angew. Chem. Int. Ed. 2004;43:846–849. doi: 10.1002/anie.200352743. Rhodium. Masuyama Y, Kaneko Y, Kurusu Y. Tetrahedron Lett. 2004;45:8969–8971. Ruthenium. Tsuji Y, Mukai T, Kondo T, Watanabe Y. J. Organomet. Chem. 1989;369:C51–C53. Kondo T, Ono H, Satake N, Mitsudo T-a, Watanabe Y. Organometallics. 1995;14:1945–1953.
- 7.For selected examples of carbonyl allylation via catalytic Nozaki-Hiyama-Kishi coupling, see: Fürstner A, Shi N. J. Am. Chem. Soc. 1996;118:2533–2534. Bandini M, Cozzi PG, Umani-Ronchi A. Polyhedron. 2000;19:537–539. McManus HA, Cozzi PG, Guiry PJ. Adv. Synth. Catal. 2006;348:551–558. Hargaden GC, Müller-Bunz H, Guiry PJ. Eur. J. Org. Chem. 2007:4235–4243. Hargaden GC, O'Sullivan TP, Guiry PJ. Org. Biomol. Chem. 2008;6:562–566. doi: 10.1039/b715834c.
- 8.For a recent review of catalytic Nozaki-Hiyama-Kishi coupling, see: Hargaden GC, Guiry PJ. Adv. Synth. Catal. 2007;349:2407–2424.
- 9.For reviews on carbonyl-ene reactions, see: Mikami K, Shimizu M. Chem. Rev. 1992;92:1021–1050. Berrisford DJ, Bolm C. Angew. Chem. 1995;107:1862–1864. Angew. Chem. Int. Ed. 1995;34:1717–1719. Johnson JS, Evans DA. Acc. Chem. Res. 2000;33:325–335. doi: 10.1021/ar960062n.
- 10.For nickel catalyzed carbonyl-ene reactions, see: Ho C-Y, Ng S-S, Jamison TF. J. Am. Chem. Soc. 2006;128:5362–5363. doi: 10.1021/ja061471+. Ng S-S, Ho C-Y, Jamison TF. J. Am. Chem. Soc. 2006;128:11513–11528. doi: 10.1021/ja062866w.
- 11.Reviews on hydrogen-mediated C-C coupling: Jang H-Y, Krische MJ. Acc. Chem. Res. 2004;37:653–661. doi: 10.1021/ar020108e. Ngai M-Y, Kong J-R, Krische MJ. J. Org. Chem. 2007;72:1063–1072. doi: 10.1021/jo061895m. Iida H, Krische MJ. Top. Curr. Chem. 2007;279:77–104. Skucas E, Ngai M-Y, Komanduri V, Krische MJ. Acc. Chem. Res. 2007;40:1394–1401. doi: 10.1021/ar7001123.
- 12.For recent examples, see: C=X Vinylation: Skucas E, Kong JR, Krische MJ. J. Am. Chem. Soc. 2007;129:7242–7243. doi: 10.1021/ja0715896. Barchuk A, Ngai M-Y, Krische MJ. J. Am. Chem. Soc. 2007;129:8432–8433. doi: 10.1021/ja073018j. Ngai M-Y, Barchuk A, Krische MJ. J. Am. Chem. Soc. 2007;129:12644–12645. doi: 10.1021/ja075438e. Aldol and Mannich Addition. Jung C-K, Garner SA, Krische MJ. Org. Lett. 2006;8:519–522. doi: 10.1021/ol052859x. Jung C-K, Krische MJ. J. Am. Chem. Soc. 2006;128:17051–17056. doi: 10.1021/ja066198q. Garner SA, Krische MJ. J. Org. Chem. 2007;72:5843–5846. doi: 10.1021/jo070779w. Bee C, Iida H, Han SB, Hassan A, Krische MJ. J. Am. Chem. Soc. 2008;130:2746–2747. doi: 10.1021/ja710862u. Acyl Substitution. Hong Y-T, Barchuk A, Krische MJ. Angew. Chem. 2006;118:7039–7042. Angew. Chem. Int. Ed. 2006;128:6885–6888.
- 13.For reviews of related hydrogen auto-transfer processes, which result in formal substitution of the hydroxyl moiety rather than carbonyl addition, see: Guillena G, Ramón DJ, Yus M. Angew. Chem. 2007;119:2410–2416. doi: 10.1002/anie.200603794. Angew. Chem. Int. Ed. 2007;46:2358–2364. doi: 10.1002/anie.200603794. Hamid MHSA, Slatford PA, Williams JMJ. Adv. Synth. Catal. 2007;349:1555–1575.
- 14.Processes that enable the direct catalytic functionalization of carbinol C-H bonds are highly uncommon. For an isolated report, see: Shi L, Tu Y-Q, Wang M, Zhang F-M, Fan C-A, Zhao Y-M, Xia WJ. J. Am. Chem. Soc. 2005;127:10836–10837. doi: 10.1021/ja0528331.
- 15.Skucas E, Bower JF, Krische MJ. J. Am. Chem. Soc. 2007;129:12678–12679. doi: 10.1021/ja075971u. [DOI] [PubMed] [Google Scholar]
- 16.Bower JF, Skucas E, Patman RL, Krische MJ. J. Am. Chem. Soc. 2007;129:15134–15135. doi: 10.1021/ja077389b. [DOI] [PubMed] [Google Scholar]
- 17.Bower JF, Shibahara F, Krische MJ. Unpublished Results. [Google Scholar]
- 18.For selected reviews of ruthenium catalyzed transfer hydrogenation, see: Zassinovich G, Mestroni G, Gladiali S. Chem. Rev. 1992;92:1051–1069. Noyori R, Hashiguchi S. Acc. Chem. Res. 1997;30:97–102. Noyori R, Yamakawa M, Hashiguchi S. J. Org. Chem. 2001;66:7931–7944. doi: 10.1021/jo010721w. Noyori R, Ohkuma T. Angew. Chem. 2001;113:40–75. Angew. Chem. Int. Ed. 2001;40:40–73. Noyori R. Angew. Chem. 2002;114:2108–2123. Angew. Chem. Int. Ed. 2002;41:2008–2022. Noyori R. Adv. Synth. Catal. 2003;345:15–32. Muñiz K. Angew. Chem. 2005;117:6780–6785. Angew. Chem., Int. Ed. 2005;44:6622–6627. doi: 10.1002/anie.200501787. Noyori R. Chem. Commun. 2005:1807–1811. doi: 10.1039/b502713f. Gladiali S, Alberico E. Chem. Soc. Rev. 2006;35:226–236. doi: 10.1039/b513396c. Ikariya T, Blacker AJ. Acc. Chem. Res. 2007;40:1300–1308. doi: 10.1021/ar700134q.
- 19.For a review of ruthenium catalyzed alkene hydroformylation, see: Kalck P, Peres Y, Jenck J. Adv. Organomet. Chem. 1991;32:121–146.
- 20.For ruthenium catalyzed reductive C-C bond formations beyond alkene hydroformylation, see: Yu C-M, Lee S, Hong Y-T, Yoon S-K. Tetrahedron Lett. 2004;45:6557–6561. Also see references 6g an 6h.
- 21.For selected reviews of ruthenium catalyzed C-C coupling, see: Trost BM, Toste FD, Pinkerton AB. Chem. Rev. 2001;101:2067–2096. doi: 10.1021/cr000666b. Kondo T, Mitsudo T-a. Curr. Org. Chem. 2002;6:1163–1179. Derien S, Monnier F, Dixneuf PH. Top. Organomet. Chem. 2004;11:1–44.
- 22.Ngai M-Y, Skucas E, Krische MJ. Org. Lett. 2008;10:2705–2708. doi: 10.1021/ol800836v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bower JF, Patman RL, Krische MJ. Org. Lett. 2008;10:1033–1035. doi: 10.1021/ol800159w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.a) Shibahara F, Bower JF, Krische MJ. J. Am. Chem. Soc. 2008;130:6338–6339. doi: 10.1021/ja801213x. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Shibahara F, Bower JF, Krische MJ. Submitted for Publication. [Google Scholar]
- 25.For a related ruthenium catalyzed hydroacylation of 1,3-dienes employing aldehydes as acyl donors, see: Kondo T, Hiraishi N, Morisaki Y, Wada K, Watanabe Y, Mitsudo T-a. Organometallics. 1998;17:2131–2134.
- 26.For intermolecular nickel-catalyzed diene-aldehyde reductive coupling, see: Kimura M, Ezoe A, Shibata K, Tamaru Y. J. Am. Chem. Soc. 1998;120:4033–4034. Takimoto M, Hiraga Y, Sato Y, Mori M. Tetrahedron Lett. 1998;39:4543–4546. Kimura M, Fujimatsu H, Ezoe A, Shibata K, Shimizu M, Matsumoto S, Tamaru Y. Angew. Chem. 1999;111:410–413. doi: 10.1002/(SICI)1521-3773(19990201)38:3<397::AID-ANIE397>3.0.CO;2-Y. Angew. Chem. Int. Ed. 1999;38:397–400. doi: 10.1002/(SICI)1521-3773(19990201)38:3<397::AID-ANIE397>3.0.CO;2-Y. Kimura M, Shibata K, Koudahashi Y, Tamaru Y. Tetrahedron Lett. 2000;41:6789–6793. Kimura M, Ezoe A, Tanaka S, Tamaru Y. Angew. Chem. 2001;113:3712–3714. doi: 10.1002/1521-3773(20011001)40:19<3600::aid-anie3600>3.0.co;2-n. Angew. Chem. Int. Ed. 2001;40:3600–3602. doi: 10.1002/1521-3773(20011001)40:19<3600::aid-anie3600>3.0.co;2-n. Loh T-P, Song H-Y, Zhou Y. Org. Lett. 2002;4:2715–2717. doi: 10.1021/ol026216i. Sato Y, Sawaki R, Saito N, Mori M. J. Org. Chem. 2002;67:656–662. doi: 10.1021/jo0106086. Bareille L, Le Gendre P, Moïse C. Chem. Commun. 2005:775–777. doi: 10.1039/b414322a. Kimura M, Ezoe A, Mori M, Iwata K, Tamaru Y. J. Am. Chem. Soc. 2006;128:8559–8568. doi: 10.1021/ja0608904. Yang Y, Zhu S-F, Duan H-F, Zhou C-Y, Wang L-X, Zhou Q-L. J. Am. Chem. Soc. 2007;129:2248–2249. doi: 10.1021/ja0693183. Sato Y, Hinata Y, Seki R, Oonishi Y, Saito N. Org. Lett. 2007;9:5597–5599. doi: 10.1021/ol702543m.
- 27.For reviews encompassing nickel-catalyzed diene-aldehyde reductive coupling, see: Tamaru Y. J. Organomet. Chem. 1999;576:215–231. Ikeda S-i. Angew. Chem. 2003;115:5276–5278. Angew. Chem. Int. Ed. 2003;42:5120–5122. doi: 10.1002/anie.200301673. Montgomery J. Angew. Chem. 2004;116:3980–3988. Angew. Chem. Int. Ed. 2004;43:3890–3908. doi: 10.1002/anie.200300634. Tamaru Y, editor. Modern Organo Nickel Chemistry. Weinheim, Germany: Wiley-VCH; 2005. Kimura M, Tamaru Y. Top. Curr. Chem. 2007;279:173–207.
- 28.For rhodium-catalyzed reductive diene-aldehyde reductive coupling employing elemental hydrogen as the terminal reductant, see: Jang H-Y, Huddleston RR, Krische MJ. Angew. Chem. 2003;115:4208–4211. doi: 10.1002/anie.200351986. Angew. Chem. Int. Ed. 2003;42:4074–4077. doi: 10.1002/anie.200351986. Dobson A, Robinson SD, Uttley MF. J. Chem. Soc. Dalton Trans. 1975:370–377.
- 29.Kondo T, Mukai T, Watanabe Y. J. Org. Chem. 1991;56:487–489. [Google Scholar]
- 30.a) Lopez F, Ohmura T, Hartwig JF. J. Am. Chem. Soc. 2003;125:3426–3427. doi: 10.1021/ja029790y. [DOI] [PubMed] [Google Scholar]; b) Nakagawa H, Hirabayashi T, Sakaguchi S, Ishii Y. J. Org. Chem. 2004;69:3474–3477. doi: 10.1021/jo049828k. [DOI] [PubMed] [Google Scholar]; c) Shu C, Hartwig JF. Angew. Chem. 2004;116:4898–4901. [Google Scholar]; Angew. Chem. Int. Ed. 2004;43:4794–4797. doi: 10.1002/anie.200460214. [DOI] [PubMed] [Google Scholar]; d) Roberts JP, Lee C. Org. Lett. 2005;7:2679–2682. doi: 10.1021/ol0508328. [DOI] [PubMed] [Google Scholar]; e) Ueno S, Hartwig JF. Angew. Chem. 2008;120:1954–1957. [Google Scholar]; Angew. Chem. Int. Ed. 2008;47:1928–1931. doi: 10.1002/anie.200705267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim IS, Ngai M-Y, Krische MJ. J. Am. Chem. Soc. 2008;130:6340–6341. doi: 10.1021/ja802001b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.For reviews encompassing carbonyl propargylation employing allenyl metal reagents, see: Moreau J-L. In: The Chemistry of Ketenes, Allenes and Related Compounds. Patai S, editor. New York: Wiley; 1980. pp. 363–413. Marshall JA. Chem. Rev. 1996;96:31–48. doi: 10.1021/cr950037f. Gung BW. Org. React. 2004;64:1–113. Marshall JA, Gung BW, Grachan ML. In: Modern Allene Chemistry. Krause N, Hashmi ASK, editors. Weinheim, Germany: Wiley-VCH; 2004. pp. 493–592. Marshall JA. J. Org. Chem. 2007;72:8153–8166. doi: 10.1021/jo070787c.
- 33.For selected milestones in carbonyl propargylation, see: Prévost C, Gaudemar M, Honigberg J. Compt. Rend. 1950;230:1186–1188. Wotiz JH. J. Am. Chem. Soc. 1950;72:1639–1642. Karila M, Capmau ML, Chodkiewicz W. Compt. Rend. 1969;269:342–345. Lequan M, Guillerm G. J. Organomet. Chem. 1973;54:153–164. Mukaiyama T, Harada T. Chem. Lett. 1981:621–624. Favre E, Gaudemar M. Compt. Rend. 1966;263:1543–1545. Danheiser RL, Carini DJ. J. Org. Chem. 1980;45:3925–3927. Haruta R, Ishiguro M, Ikeda N, Yamamoto H. J. Am. Chem. Soc. 1982;104:7667–7669. Corey EJ, Yu C-M, Lee D-H. J. Am. Chem. Soc. 1990;112:878–879. Minowa N, Mukaiyama T. Bull. Chem. Soc. Jpn. 1987;60:3697–3704. Marshall JA, Wang X-J. J. Org. Chem. 1991;56:3211–3213. Marshall JA, Maxson K. J. Org. Chem. 2000;65:630–633. doi: 10.1021/jo991543y. Matsumoto Y, Naito M, Uozumi Y, Hayashi T. J. Chem. Soc., Chem. Commun. 1993:1468–1469. Keck GE, Krishnamurthy D, Chen X. Tetrahedron Lett. 1994;35:8323–8324. Denmark SE, Wynn T. J. Am. Chem. Soc. 2001;123:6199–6200. doi: 10.1021/ja016017e.
- 34.For rhodium catalyzed reductive coupling of 1,3-enynes to carbonyl compounds and imines, see: Jang H-Y, Huddleston RR, Krische MJ. J. Am. Chem. Soc. 2004;126:4664–4668. doi: 10.1021/ja0316566. Kong J-R, Cho C-W, Krische MJ. J. Am. Chem. Soc. 2005;127:11269–11276. doi: 10.1021/ja051104i. Kong J-R, Ngai M-Y, Krische MJ. J. Am. Chem. Soc. 2006;128:718–719. doi: 10.1021/ja056474l. Komanduri V, Krische MJ. J. Am. Chem. Soc. 2006;128:16448–16449. doi: 10.1021/ja0673027. Hong Y-T, Cho C-W, Skucas E, Krische MJ. Org. Lett. 2007;9:3745–3748. doi: 10.1021/ol7015548.
- 35.For nickel catalyzed reductive coupling of 1,3-enynes to carbonyl compounds, see: Miller KM, Luanphaisarnnont T, Molinaro C, Jamison TF. J. Am. Chem. Soc. 2004;126:4130–4131. doi: 10.1021/ja0491735. Miller KM, Jamison TF. Org. Lett. 2005;7:3077–3080. doi: 10.1021/ol051075g. Miller KM, Colby EA, Woodin KS, Jamison TF. Adv. Synth. Catal. 2005;347:1533–1536.
- 36.For seminal contributions to nickel catalyzed alkyne-carbonyl reductive coupling, see reference 45a
- 37.For reviews encompassing nickel catalyzed alkyne-carbonyl reductive coupling, see: Montgomery J. Angew. Chem. 2004;116:3980–3998. Angew. Chem. Int. Ed. 2004;43:3890–3908. doi: 10.1002/anie.200300634. Montgomery J, Sormunen GJ. Top. Curr. Chem. 2007;279:1–23. Moslin RM, Miller-Moslin K, Jamison TF. Chem. Commun. 2007:4441–4449. doi: 10.1039/b707737h.
- 38.Patman RL, Williams VM, Bower JF, Krische MJ. Angew. Chem. 2008;120:5298–5301. doi: 10.1002/anie.200801359. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2008;47:5220–5223. doi: 10.1002/anie.200801359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.a) Oguni N, Omi T. Tetrahedron Lett. 1984;25:2823–2824. [Google Scholar]; b) Kitamura M, Suga S, Kawai K, Noyori R. J. Am. Chem. Soc. 1986;108:6071–6072. doi: 10.1021/ja00279a083. [DOI] [PubMed] [Google Scholar]
- 40.For enantioselective catalytic addition of vinylzinc reagents to aldehydes, see: Oppolzer W, Radinov R. Helv. Chim. Acta. 1992;75:170–173. Oppolzer W, Radinov R. J. Am. Chem. Soc. 1993;115:1593–1594. Soai K, Takahashi KJ. Chem. Soc., Perkin Trans.1. 1994:1257–1258. Wipf P, Xu W. Tetrahedron Lett. 1994;35:5197–5200. Oppolzer W, Radinov RN, De Brabander J. Tetrahedron Lett. 1995;36:2607–2610. Wipf P, Ribe S. J. Org. Chem. 1998;63:6454–6455. Oppolzer W, Radinov RN, El-Sayed E. J. Org. Chem. 2001;66:4766–4770. doi: 10.1021/jo000463n. Dahmen S, Bräse S. Org. Lett. 2001;3:4119–4122. doi: 10.1021/ol016954r. Chen YK, Lurain AE, Walsh PJ. J. Am. Chem. Soc. 2002;124:12225–12231. doi: 10.1021/ja027271p. Ji J-X, Qiu L-Q, Yip CW, Chan ASC. J. Org. Chem. 2003;68:1589–1590. doi: 10.1021/jo026551k. Lurain AE, Walsh PJ. J. Am. Chem. Soc. 2003;125:10677–10683. doi: 10.1021/ja035213d. Ko D-H, Kang S-W, Kim KH, Chung Y, Ha D-C. Bull. Korean. Chem. Soc. 2004;25:35–36. Sprout CM, Richmond ML, Seto CT. J. Org. Chem. 2004;69:6666–6673. doi: 10.1021/jo049160+. Jeon S-J, Chen YK, Walsh PJ. Org. Lett. 2005;7:1729–1732. doi: 10.1021/ol050255n. Lauterwasser F, Gall J, Hoefener S, Bräse S. Adv. Synth. Catal. 2006;348:2068–2074. Jeon S-J, Fisher EL, Carroll PJ, Walsh PJ. Am. Chem. Soc. 2006;128:9618–9619. doi: 10.1021/ja061973n. Salvi L, Jeon S-J, Fisher EL, Carroll PJ, Walsh PJ. J. Am. Chem. Soc. 2007;129:16119–16125. doi: 10.1021/ja0762285.
- 41.For reviews encompassing catalytic enantioselective aldehyde vinylation using organozinc reagents, see: Wipf P, Kendall C. Chem. Eur. J. 2002;8:1778–1784. doi: 10.1002/1521-3765(20020415)8:8<1778::aid-chem1778>3.0.co;2-h. Wipf P, Nunes RL. Tetrahedron. 2004;60:1269–1279.
- 42.For catalytic enantioselective ketone vinylation using organozinc reagents, see: Li H, Walsh PJ. J. Am. Chem. Soc. 2004;126:6538–6539. doi: 10.1021/ja049206g. Li H, Walsh PJ. J. Am. Chem. Soc. 2005;127:8355–8361. doi: 10.1021/ja0425740. Jeon S-J, Li H, Garcia C, La Rochelle LK, Walsh PJ. J. Org. Chem. 2005;70:448–455. doi: 10.1021/jo048683e.
- 43.Ojima I, Tzamarioudaki M, Tsai C-Y. J. Am. Chem. Soc. 1994;116:3643–3644. [Google Scholar]
- 44. Crowe WE, Rachita MJ. J. Am. Chem. Soc. 1995;117:6787–6788. b) For an aligned study, see: Kablaoui NM, Buchwald SL. J. Am. Chem. Soc. 1995;117:6785–6786.
- 45.For intramolecular nickel catalyzed alkyne-carbonyl reductive coupling, see: Oblinger E, Montgomery J. J. Am. Chem. Soc. 1997;119:9065–9066. Tang X-Q, Montgomery J. J. Am. Chem. Soc. 1999;121:6098–6099. Tang X-Q, Montgomery J. J. Am. Chem. Soc. 2000;122:6950–6954. Knapp-Reed B, Mahandru GM, Montgomery J. J. Am. Chem. Soc. 2005;127:13156–13157. doi: 10.1021/ja054590i.
- 46.For intermolecular nickel catalyzed alkyne-carbonyl reductive coupling, see: Huang W-S, Chan J, Jamison TF. Org. Lett. 2000;2:4221–4223. doi: 10.1021/ol006781q. Miller KM, Huang W-S, Jamison TF. J. Am. Chem. Soc. 2003;125:3442–3443. doi: 10.1021/ja034366y. Takai K, Sakamoto S, Isshiki T. Org. Lett. 2003;5:653–655. doi: 10.1021/ol0272996. Mahandru GM, Liu G, Montgomery J. J. Am. Chem. Soc. 2004;126:3698–3699. doi: 10.1021/ja049644n.
- 47.Patman RL, Chaulagain MR, Williams VM, Krische MJ. Manuscript in Preparation. [Google Scholar]