

Abstract
Purpose
Clinical observations have suggested that shedding of the MHC class I chain-related molecule (MIC) may be one of the mechanisms by which tumors evade host immune surveillance and progress. However, this hypothesis has never been proven. In this study, we tested this hypothesis using a prostate tumor model and investigated the impact of shedding of MIC on tumor development.
Experimental Design
We generated a shedding-resistant non-cleavable form of MICB (MICB.A2). We overexpressed MICB.A2, the wild-type MICB, and the recombinant soluble MICB (rsMICB) in mouse prostate tumor TRAMP-C2 (TC2) cells and implanted these cells into severe combined immunodeficiency (SCID) mice.
Results
No tumors were developed in animals that were implanted with TC2-MICB.A2 cells; whereas all the animals that were implanted with TC2, TC2-MICB, or TC2-rsMICB cells developed tumors. When a NKG2D-specific antibody CX5 or purified rsMICB was administrated to animals prior to tumor implantation, all animals that were implanted with TC2-MICB.A2 cells developed tumors. In vitro cytotoxicity assay revealed the loss of NKG2D-mediated NK cell function in these pre-challenged animals suggesting that persistent levels of soluble MICB in the serum can impair NK cell function and thus allow tumor growth.
Conclusions
These data suggest that MIC shedding may contribute significantly to tumor formation by transformed cells and that inhibition of MIC shedding to sustain the NKG2D receptor-MIC ligand recognition may have potential clinical implication in targeted cancer treatment.
Introduction
Expression of murine NKG2D ligands on tumor cells has been shown to be effective in activating NK-mediated tumor elimination experimentally (1-4). In murine systems, identified NKG2D ligands include the retinoic acid early inducible family of proteins RAE-1 (1, 2), the minor histocompatibility antigen H60 (1, 2), and the murine ULBP-like transcript 1 (MULT1; 4, 5). Cells expressing these molecules are sensitive to the cytotoxicity of mouse NK cells. Ectopic expression of RAE-1 and H-60 results in rejection of tumor cell lines expressing normal levels of MHC I molecule (2, 3, 4). Immune depletion and other experiments showed that the tumor rejection is due to NK cells and CD8 T cells (2, 3). NKG2D neutralization in vivo enhances host sensitivity to carcinogen-induced spontaneous tumor initiation (6). These studies have proven the principle function of the NKG2D ligand-receptor mediated NK cell immunity in tumor rejection.
In humans, the MHC class I chain-related molecule A (MICA) and MICB (generally termed as MIC) are the most investigated NKG2D ligands which were proposed to play roles in tumor rejection (7-9). MIC is rarely expressed by normal human tissues but induced in most human epithelial tumors (10-13). Expression of MIC on the tumor cell surface can markedly enhance the sensitivity of tumor cells to NK cells in vitro and has been shown to inhibit the growth of human gliomas or small lung carcinomas in experimental models (14, 15). These studies suggested that NK cell can potentially eliminate MIC-positive tumor cells in cancer patients. However, as clinically observed, most of the human epithelial tumors are found to be MIC+ rather than MIC- (10-13), which suggests the functional compromise of the MIC ligand -NKG2D receptor system in cancer patients to permit the growth of MIC+ tumor cells. We and others have shown that tumor-derived soluble MIC (sMIC) as a result of tumor shedding is one of the factors causing the ineffectiveness of NKG2D–mediated immunity in cancer patients (13, 16-21). In vitro studies have shown that engagement of soluble MICA to NKG2D results in marked reduction in surface NKG2D expression on NK cells and T cells (13, 16, 21). Thus, sMIC is believed to induce down-modulation of NKG2D expression on systemic and tumor infiltrated NK and T cells and thus result in functional impairment of NK and T cells in MIC+ cancer patients (13, 16, 17). Reduction in the density of MIC expressed on the tumor cell surface due to MIC shedding from tumors is also proposed to be one of the mechanisms for tumor evasion (21).
These compelling clinical data suggest that MIC shedding from tumor cells is likely associated with tumor progression, which has prompted the hypothesis that tumor shedding of MIC is the mechanism by which MIC-positive tumors evade NK cell immune surveillance and progress in cancer patients. However, it is impossible to test this hypothesis clinically. Taking the advantage that human MICB can be recognized by mouse NKG2D (22, 23) and that only the extracellular α1α2 domain of MIC interacts with NKG2D (24-26), here we test the hypothesis experimentally that shedding of MIC permits tumor growth and that sustained interaction between NKG2D and membrane-integrated form of MIC can cause tumor rejection. Using a well-characterized prostate tumor model TRAMP-C2 (27), we demonstrate for the first time that expression of the shedding-resistant but not the natural form of MICB prevents tumor formation by transformed cells.
Material and Methods
Cell
TRAMP-C2 (TC2) cell line (gift of Dr. NM. Greenberg, Fred Hutchinson Cancer Research Center, WA) was maintained in DMEM medium as described (27). RMA-Rae-1β cells (gift of Dr. D. Raulet, Berkeley) was maintained in RPMI1640 supplemented with 10% FCS. Eco-phoenix cells (Orbigen, San Diego, CA) were maintained in DMEM supplemented with 10% FBS.
DNA construction, transfection, and transduction
cDNA encoding full-length of human MICB (allele 0101, 28) was kindly provided by Dr. A. Steinle (University of Tubingen, Tubingen, Germany) and subcloned into the retroviral vector pBMNZ-IRES-GFP (Orbigen, San Diego, CA). To generate recombinant soluble MICB (rsMICB), cDNA encoding the extracellular domain of MICB was amplified by PCR. To generate a putative shedding-resistant form of MICB (designated as MICB.A2), aa 215-274 of MICB was replaced with the comparable sequence of the α3 domain of HLA-A2 using recombinant PCR of the cDNA sequences (29). rsMICB-FLAG fusion peptide was generated by tagging the cDNA sequence of FLAG (DKYDDDK) to the 3-end of the rsMICB cDNA using PCR. Error-free amplified cDNAs were identified by sequencing and subcloned into the retroviral vector pBMNZ-IRES-GFP (Orbigen). Plasmids were transfected into Eco-pheonix packaging cells to generate retrovirus. TC2 cells were transduced with respective retrovirus. Stable GFP-positive cell population was isolated by drug selection and sorted by flow cytometry.
Affinity purification of rsMICB and rsMICB-FLAG peptides
The HiTrip NHS-activated column (GE Healthcare) was conjugated with the mAb 6D4.6 (Santa Cruz Biotechnology, Santa Cruz, CA) before loaded with conditioned media from TC2-rsMICB or TC2-rsMICB.FLAG cells. After washing, rsMICB or rsMICB.FLAG was eluted with 100 mM sodium citrate, pH2.5 and neutralized immediately with 1.5M Tris buffer (pH 8.8).
Immunoprecipitation and Western blotting
Supernatant was collected from TC2-MICB cell culture and passed through a 0.45μm filter to remove cell debris. Cells were washed and lyzed with lysis buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 1% Triton X-100). Clear supernatant and lysates were incubated with the mAb 6D4.6. Immunocomplexes were collected using protein A/G-agarose (Pierce, Rockford, IL). PNGase F (New England Biolabs, Beverly, MA) treatment was carried overnight at 37°C. Immunocomplexes were separated on a 4-15% SDS-PAGE, blotted onto a nitrocellulose membrane, and probed with goat anti-MICB antibody AF1599 (R&D systems, Minneapolis, MN). Immunoreactive proteins were detected by incubating the blot with a horseradish peroxidase-conjugated secondary antibody (Pharmacia, Piscataway, NJ) and ECL reagents (Pharmacia).
MICB shedding assay and (s)MICB ELISA
Cells were seeded at the density of 4X105 cells/well in a 6-well plate in complete media overnight and replaced with 1 ml/well serum-free media for 6 hrs. Supernatant was collected and filtered through 0.45μ filter. Cells were lyzed with 1ml lysis buffer. Amount of soluble MICB in the supernant and MICB in the cell lysates was measured using human MICB DuoSet sandwich ELISA kit (R&D Systems). For measuring mouse serum levels of soluble MICB, serum was diluted 1:2 with PBS for ELISA assay.
In vivo study
Animal studies were approved by Institutional Animal Care and Use Committee. Six to 10 of 6-week-old SCID male mice (Harlan Sprague Dawley, IN) were used in each group. 1x106/mouse of the following cells were s.c. injected into respective group of animals: TC2, TC2-MICB, TC2-MICB.A2, and TC2-rsMICB. All animals were monitored for tumor growth for up to 12 weeks. Tumor volume was estimated using the formula: volume = L X W2/2. Animals were euthanized when tumor volumes reached 1000 mm3. Tumors, spleens, and peripheral blood were terminally collected. Serum was separated by centrifugation and used for rsMICB ELISA.
In vivo NKG2D blocking or neutralization
To block NKG2D receptor, 100 μg of the functional grade of anti-NKG2D blocking antibody CX5 (ebiosciences) was injected i.p. on the day before and the day after tumor implantation and thereafter every three days. Blocking was confirmed by flow cytometry of peripheral lymphocytes collected from orbital sinus bleeding with PE-conjugated CX5 (ebiosciences). To modify NKG2D function, animals were injected i.p. with 50ng of purified rsMICB prior to implantation of TC2-MICB.A2 cells and thereafter twice per week for four weeks. Blood was collected once a week from sinus orbital bleeding and serum levels of rsMICB was measured by ELISA.
Flow cytometry
For detection of cell surface expression of NKG2D ligands, TC2 and its derivative cells were trypsinized, blocked with anti-mouse CD16/32 (eBiosciences, San Diego, CA), and incubated with anti-MICA/B mAb 6D4.6 or anti-MICB MAB1599 (R&D systems) or anti-pan-RAE-1 mAb17582 (R&D systems) followed by a PE-conjugated secondary reagent. For detection of rsMICB expression, the BD Cytofix/Cytoperm kit (BD Sciences) was used. Briefly, TC2-rsMICB cells were cultured in the presence of BD GolgiPlug for 3 hr to prevent the secretion of rsMICB before harvesting. Cells were resuspended in BD Fixation/permeablization solution for 20 min at 4°C and incubated with 6D4.6 followed with PE-conjugated secondary reagents. For mouse NKG2D binding assay, cells were incubated with 10 μg/ml of the fusion protein of recombinant soluble mouse NKG2D and human Fc (smNKG2D-Fc, R&D Systems) followed by PE-conjugated F(ab)2 goat-anti-human IgG. For H-2Kb expression, cells were incubated with Alex647-conjugated anti-H-2Kb/Db mAb (Biolegend, San Diego, CA).
Single cell suspensions of splenocytes were prepared as described (30). Cells were stained with FITC-conjugated mAb DX5 (ebiosciences) and PE-conjugated anti-mouse NKG2D mAb CX5 (ebioscience) or A10 (ebiosciences) and analyzed using a BD FACscan or LSRII. For ex vivo rsMICB competitive binding assay, freshly isolated splenocytes were incubated with 10 ng/μl of rsMICB-FLAG followed with FITC-conjugated mAb M2 (Sigma-Aldrich) and PE-conjugated mAb DX5 (ebioscience). Data were analyzed using the BD CellQuestPro (BD Biosciences) or FlowJo software (Tree Star Inc, Asland, OR).
Cytotocixity assay
Fresh NK cells were prepared using Spinsep murine NK enrichment cocktail (Stem Cell Technology, Vancouver, BC) and were > 90% DX5+. LAK cells were prepared by culturing NK cells for 4-7 days in 1000 U/ml of rhIL-2. Cytotoxicity was performed in triplicates using the standard 4h 51Cr release assay (13). Antibody blocking were done by pre-incubating effector cells with 30 μg/ml NKG2D blocking mAb CX5 (ebiosciences) or pre-incubating target cells with 100 μg/ml of anti-pan RAE-1 pAb at 37°C for 1hr (31).
Statistical analysis
Data were analyzes using JMP software. Significance between two animal groups was determined by student's t-test. P < 0.05 was considered significant.
Results
Putative cleavage region of MIC(B) in TRAMP-C2 tumor cells
TC2 is a mouse prostate tumor cell line generated from the TRAMP (TRansgenic Adenocarcinoma Mouse Prostate) mouse (27), which does not express any homologous molecules to human MIC (28). TC2 cells were transduced with retroviruses that carry cDNAs of human MICB and GFP. Transduced cells stably expressing high levels of MICB (designated as TC2-MICB cells) were generated by puromycin selection and multiple-rounds of flow cytometry cell sorting for GFP-positive cells.
In order to generate a shedding-resistant form of MICB, we first performed experiments to predict putative cleavage region of MICB by tumor cells. Soluble MICB resulted from TC2 shedding (designated as ssMICB) was immunoprecipitated from supernatant of TC2-MICB cells with a mouse monoclonal antibody (mAb) 6D4.6 specific to the α1α2 ectodomain of MICA/B (10). The full length MICB was immunoprecipitated from cell lysates with the same antibody. Immunecomplexes were separated and immunoblotted with a goat polyclonal antibody AF1599 specific to the ectodomain of MICB. After N-glycosidase (PNGaseF) treatment, ssMICB yield two bands of molecular mass approximately 31-33 kDa (Fig. 1a). The molecular mass is consistent with other studies showing soluble MICB and soluble MICA released by human tumor cells (32, 33). When samples were treated with dinitrothiocyanobenzene (DNTB), a disulfide isomerase inhibitor, only a single band of soluble MICB was revealed (data not shown), suggesting that the two bands of soluble MICB released by TC2 cells are the reduced and non-reduced forms of ssMICB. Similar observation of soluble MICA shed by human tumor cell lines was shown by Kaiser et al (33). The deglycosylated full-length MICB is shown to be approximately 41 kDa in the cell lysates (Fig. 1a), consistent with other studies (34). Although the precise cleavage site can not be determined, these data suggest that MICB was cleaved at the alpha-3 domain proximal to the transmembrane region to generate ssMICB (Fig. 1b). Similar cleavage region is also predicted for human tumor cell lines to generate soluble MICA (33).
Figure 1.
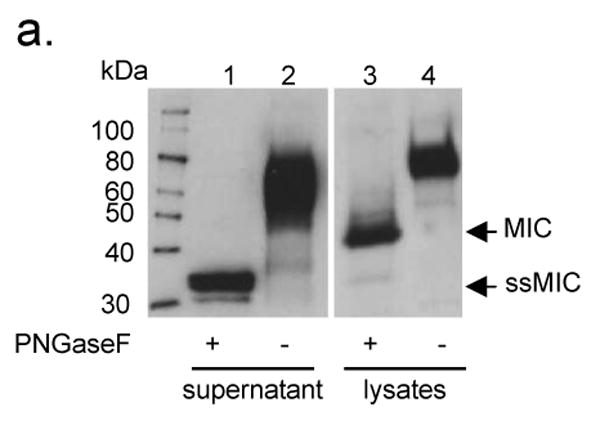

Putative MICB cleaved site(s) in TC2 cells. a. Western-blot showing the predicted size of cleaved soluble MICB in TC2 cells. Supernatant and lysates of TC2-MICB cells were immunoprecipitated with anti-MIC mAb 6D4.6. The immune complexes were treated with PNGase F and resolved on SDS-PAGE. Proteins were transferred to nitrocellulose membrane and blotted with goat anti-MICB polyclonal Ab. Lane 1 and 2, detection of sMICB from TC2-MICB supernatant. The molecular mass of the deglycosylated cleaved sMICB is estimated to be 31-33 kDa. Lane 3 and 4, detection of full-length MICB from TC2-MICB cell lysates. The full-length deglycosylated MICB is estimated to be 41 kDa on 4-15% SDS-PAGE. b. A diagram depicted the putative MICB cleavage site(s).
Generation of tumor cell lines expressing the putative shedding-resistant MICB.A2 and recombinant soluble MICB
To study the impact of MIC shedding on tumor formation and growth in vivo, we generated two forms of MICB, the recombinant secretable form of MICB (rsMICB) and a putative shedding-resistant form of MICB (MICB.A2). rsMICB was generated by deletion of the transmembrane and cytoplasmic domain. MICB.A2 was generated by replacing part of the α3 domain of MICB (aa 215-274) with the corresponding residues from HLA-A2 (Fig. 2a). Since NKG2D only interacts with the α1α2 domain of MIC (24), MICB.A2 would presumably continue to recognize NKG2D. rsMICB and MICB.A2 were overexpressed in TC2 cells using the GFP retroviral system described above. Positive-expressing clones were selected by puromycin and repeated sorting by flow cytometry for GFP-positive cells. The expression level of cellular rsMICB and surface MICB.A2 in TC2 cells were confirmed by flow cytometry with the anti-MIC mAb 6D4.6 (Fig. 2b).
Figure 2.

Construction and expression of the shedding-resistant non-cleavable and soluble recombinant forms of MICB (rsMICB) in TC2 cell lines. a. Diagram showing generation of the non-cleavable form MICB.A2 by replacing aa 215-274 of the MICB α3 domain with the corresponding sequence of HLA-A2. rsMICB was generated by deletion of the entire transmembrane and cytoplasmic region of MICB. b. Flow cytometry showing expression levels of MICB, MICB.A2, and rsMICB in TC2 cell lines. cDNAs of MICB, MICB.A2, or rsMICB were inserted into a IRES-GFP retroviral vector pBMNZ. TC2 cells were transduced with respective retrovirus. GFP-positive cells were sorted by flow cytometry. For detection of MICB and MICB.A2 expression, cells were directly incubated with anti-MIC 6D4.6 antibody followed a PE-conjugated secondary reagent. For detection of the secretable rsMICB expression, TC2-rsMICB cells were cultured in the presence of BD GolgiPlug for 3 hr to prevent the secretion of rsMICB before harvesting. Cells were resuspended in BD Fixation/permeablization solution for 20 min at 4°C and incubated with 6D4.6 followed with a PE-conjugated secondary reagent. c. MICB.A2 is shedding-resistant. Top graph showing amount of shed soluble MICB in the culture supernatant and MICB in the cell lysates. 4x105 cells/well were plated on a 6-well plate o/n. Media were removed and replaced with 1ml serum-free media. 6hrs later, Media were collected and filtered. Cells were lysed with 1 ml lysis buffer. 50ul of culture spernatant and cell lysates were used for (s) MICB ELISA assay. Bar, SE. Bottom graph represents the degree of shedding as calculated by molars of soluble MICB in the supernatant vs. total molars of soluble MICB and MICB (a sum of supernatant and cell lysates). The final results were normalized by cell numbers at the time of the assay. Results represent three independent experiments. *, P < 0.001.
Partial replacing the a3 domain of MICB protects from tumor cell shedding
An ELISA assay was used to assess the degree of shedding of MICB and MICB.A2 in TC2 cell lines. Both the capture and the detection antibodies are specific to the extracellular domain of MICB and can also detect MICB.A2 by western blotting (data not shown). With a given number of cells, the amount of cleaved soluble MIC in the culture supernatant and the amount of MIC in the lysates were measured by a sandwich ELISA assay (Fig. 2c). The degree of MIC shedding was estimated by the molar percentage of soluble MICB released into the supernatant. Approximately 30% of MICB was cleaved into the media in TC2 cells, whereas no cleaved form of MICB.A2 was detectable in the culture supernatant (Fig. 2c). This indicates that MICB.A2 can not be cleaved into soluble forms by tumor cells and is shedding-resistant.
Expression of MICB.A2 in TC2 cells stimulates mouse NK cell cytolytic activity
Human MICB can be recognized by mouse NKG2D and activate mouse NK cells (22, 23). To test whether overexpressing MICB.A2 can also activate mouse NK cells, we first addressed the physical interaction of MICB.A2 with soluble mouse NKG2D-Fc (smNKG2D-Fc) fusion protein by flow cytometry analyses. As measured by mean fluorescence intensity (Fig. 3a), smNKG2D-Fc was more prominently bound by TC2-MICB and TC2-MICB.A2 cells and only weakly bound by TC2 and TC2-rsMICB cells. Accordingly, in vitro cytotocixity assay revealed marked increase in sensitivity of TC2 cells to IL-2 activated mouse NK (LAK) cells when MICB or MICB.A2 was overexpressed (Fig. 3b, p < 0.01). The increased susceptibility of TC2-MICB and TC2-MICB.A2 cells to LAK cells can be inhibited by pre-incubation of LAK cells with the NKG2D-specific blocking antibody CX5 (35; Fig 3b), suggesting a NKG2D-dependent LAK cell killing effect. Thus, the shedding-resistant MICB.A2 maintained the functional property of MICB to be recognized by mouse NKG2D.
Figure 3.
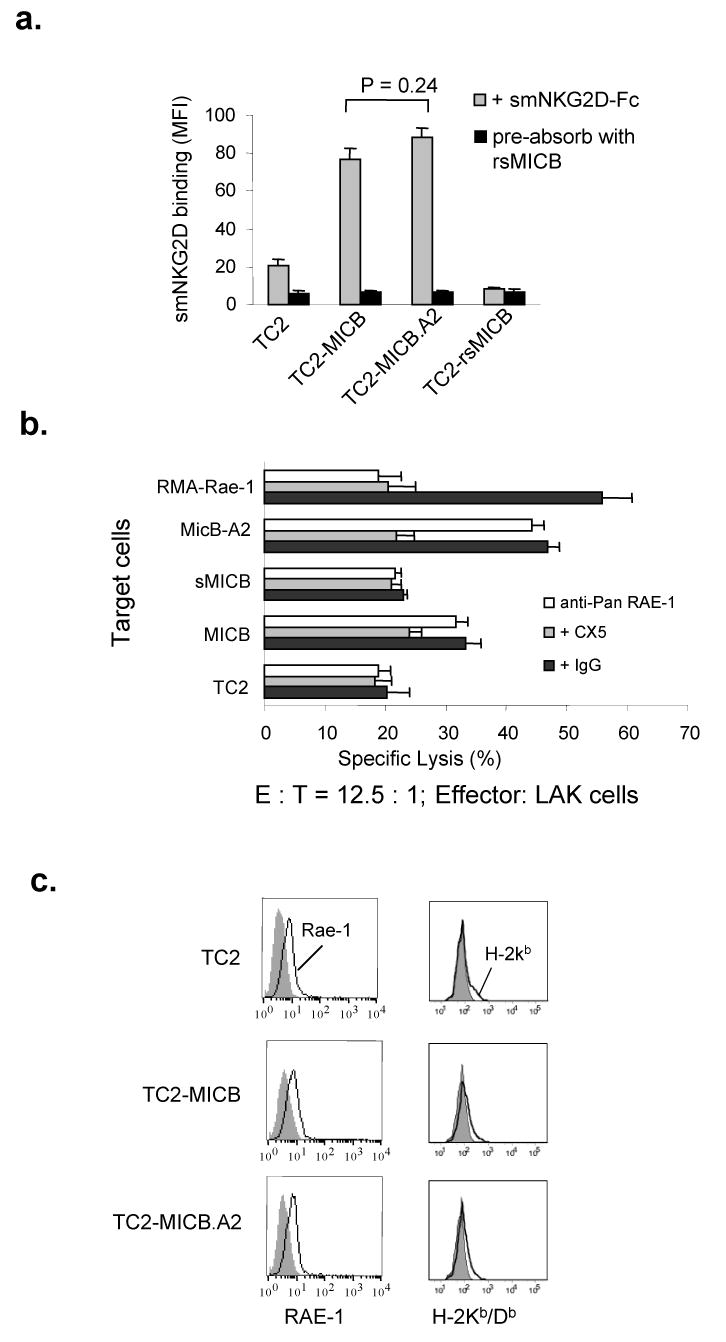
Overexpression of MICB and MICB.A2 increases the sensitivity of TC2 cells to NK cell killing. a. Binding of MICB and MICB.A2 by mouse NKG2D. Cells were incubated with the chimeric soluble mouse NKG2D-humanFc (smNKG2D-Fc) followed by PE-conjugated anti-human IgG. Cells were analyzed by flow cytometry. Data shown are mean fluorescence intensity (MFI). When smNKG2D-Fc was pre-absorbed with rsMICB, no binding was seen in any of the cell lines. No significant difference was shown in the binding ability between MICB and MICB.A2 (p = 0.24). b. Sensitivity of various MICB-expressing TC2 cells to mouse NK cells. NK cells were isolated from SCID mice and cultured in complete media with 1000 U/ml of IL-2 for 4 days before used as effectors in standard 4-h 51Cr release assay. For blocking NKG2D receptor, effector cells were pre-incubated with 30 μg/ml of CX5 antibody. For blocking RAE-1, target cells were pre-incubated with 100 ug/ml of anti-pan RAE-1 pAb for 1hr prior to the assay. RMA-Rae-1 cells were used positive controls for blocking antibodies. E:T, effector:target ratio. Bar, SE. *, P < 0.01 compared to TC2 cells as targets. c. Flow cytometry histograms showing surface expression of RAE-1 (left panel) and H-2Kb/Db (right panel) in TC2, TC2-MICB, and TC2-MICB.A2 cells. Filled histograms, cells were stained with control isotype antibodies. Open histograms, cells were stained with specific antibodies. Results represent three independent experiments.
Although not expressing any MIC homolog, TC2 cells express some levels of endogenous NKG2D ligand RAE-1 variants, but not H60 (22, 36). However, the level of endogenous NKG2D ligands is not sufficient to stimulate LAK cell in vitro cytotoxicity (Fig. 3b). To address whether the increased sensitivity of TC2-MICB and TC2-MICB.A2 cells to LAK cell killing is possibly due to increased expression of RAE-1, we analyzed endogenous RAE-1 expression on these cell lines by flow cytometry with a rat anti-pan RAE-1 monoclonal antibody. A consistency of RAE-1 expression among TC2, TC2-MICB, and TC2-MICB.A2 cell lines is shown in Fig 3c. Furthermore, pre-incubation of target cells with an anti-pan RAE-1 blocking antibody (30) did not significantly reduce the susceptibility of TC2-MICB or TC2-MICB.A2 cells to LAK cells, whereas the sensitivity of the control RMA-Rae-1β cells to LAK cells was significantly reduced (Fig. 3b). These suggest that the increased killing of TC2-MICB or TC2-MICB.A2 cells by LAK cells is not due to increased RAE-1 expression.
TC2 cells express a very low level of H-2Kb/Db (37), which is a potential ligand for inhibitory Ly49 receptor families. We analyzed H-2Kb/Db expression on these cell lines by flow cytometry. Consistent levels of H-2Kb/Db expression were found in TC2 and cell lines expressing MICB or MICB.A2 (Fig. 3c), suggesting that the increased sensitivity of TC2-MICB and TC2-MICB.A2 cells to LAK cells was not attributed to a reduced level of H-2Kb/Db expression.
The shedding-resistant MICB.A2 but not the natural MICB prevents TC2 tumor formation in vivo
In three independent experiments, when SCID animals were implanted with TC2-rsMICB, TC2-MICB, or TC2-MICB.A2 cells, none of animals that were implanted with the TC2-MICB.A2 cells developed tumors with a 12-week follow-up observation period, whereas all the animals that were implanted with TC2-rsMICB or TC2-MICB cells developed tumors within three weeks (Fig. 4a and 4b). In addition, no significant difference in tumor growth was observed among TC2, TC2-rsMICB, and TC2-MICB-originated tumors (Fig. 4a). To address whether the failure to reject TC2-MICB tumors is due to the large dose (1x 106) of tumor cells injected, we repeated the experiment with TC2-MICB and TC2-MICB.A2 cells using smaller numbers of inoculated cells. A tenfold (1x105) and a 100-fold (1x104) decrease in the number of inoculated tumor cells did not change the outcome (Fig. 4c). We also examined MICB expression in the TC2-MICB-originated tumor cells extracted from SCID animals by flow cytometry. All the extracted tumor cells expressed the similar levels of MICB as prior to implantation (Fig. 4d). This suggests that tumor growth in animals that were implanted with TC2-MICB cells is not due to NK cells selectively eliminating MICB-positive cells.
Figure 4.

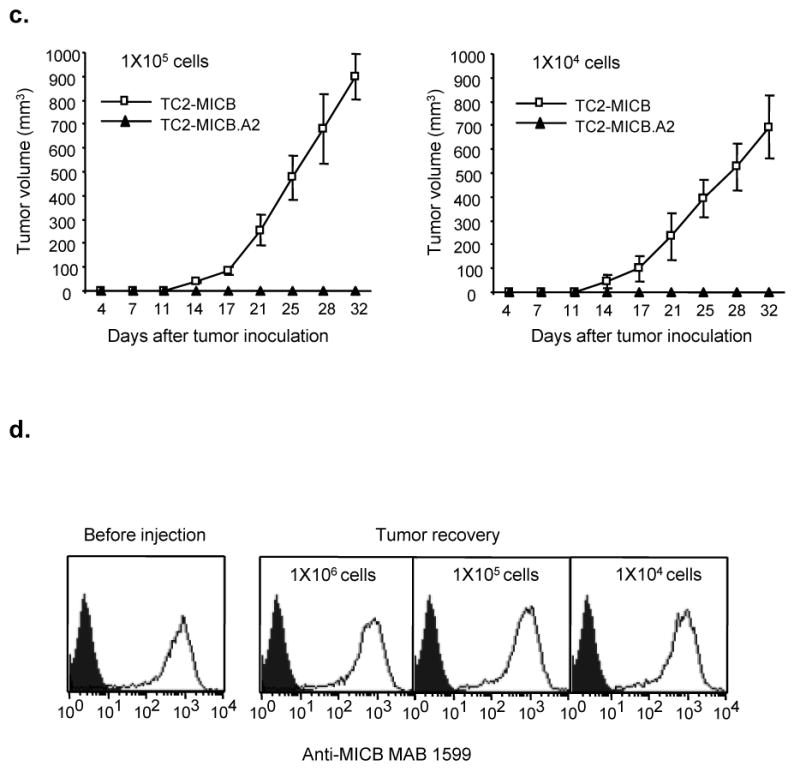
Expression of MICB.A2 but not the cleavable MICB prevents tumor formation in vivo. Six animals were used in each group. Tumor growth was monitored twice weekly. Tumor volume was estimated by the formula Volume = L2xW/2. a. Tumor growth of various MICB expressing TC2 cells in SCID mice. b. Rate of tumor formation of various MICB expressing TC2 cells in SCID mice. 1x106 cells were injected s.c. into each animal in (a) and (b). c. Tumor growth of TC2-MICB cells when injected at lower doses (1x105 and 1x104 cells/animal). d. Representative flow cytometry histoplots showing MICB expression in tumor cells extracted from animals inoculated with TC2-MICB cells compared to prior inoculation. The MICB-specific antibody MAB1599 (R & D systems) was used as primary antibody. Filled histogram, MICB expression in TC2 cells. Open histogram, MICB expression in TC2-MICB cells or tumor cells. Results represent three independent experiments.
Shedding of MICB by TC2 cells allows TC2-MICB tumor growth in mice
In 4-h in vitro cytotocixity assays, both TC2-MICB and TC2-MICB.A2 cells were sensitive to LAK cells (Fig. 3b). However, NK tumor immunity was effective only in animals when the non-cleavable MICB.A2 was expressed on tumor cells. We propose that the discrepancy of in vivo and in vitro observation is attributed to tumor cell shedding of MICB in vivo which accumulatively compromises NK cell function in animals implanted with MICB-expressing tumor cells. To test this hypothesis, we measured serum levels of soluble MICB in all the animals four weeks after tumor implantation using a sandwich ELISA assay. A significant level of soluble MICB was detected in the sera of animals that were implanted with tumor cells expressing rsMICB and MICB; whereas no soluble MICB was detectable in animals implanted with tumor cells expressing MICB.A2 (Fig. 5a). To address why TC2-MICB cells were sensitive to LAK cell in vitro, LAK cells were incubated with the supernatant of TC2-MICB cells for various time periods and used as effector cells to kill target TC2-MICB.A2 cells. Only after 8-h incubation, LAK cell killing ability was significantly affected. Therefore, in the 4-h in vitro cytotoxicity assay, the killing ability of LAK cells was not significantly affected by soluble MICB resulted from target TC2-MICB cells (data not shown). We further examined NK cell tumor killing ability from these animals. For this purpose, freshly isolated splenic NK cells were used as effector cells for in vitro cytotoxicity assay. NK cells from mice bearing MICB and rsMICB-expressing tumors had a significant reduction in cytotoxicity against TC2-MICB.A2 target cells in comparison to those from TC2 tumor-bearing or tumor-free animals (P<0.01, Fig. 5b). The cytotoxicity of these NK cells was inhibited by pre-incubating with a NKG2D-specific inhibitory mAb CX5 (Fig. 5b), suggesting a NKG2D-dependent effect. Together, these results suggest that persistent presence of soluble MICB in vivo due to tumor cell shedding of MICB compromised NKG2D-mediated NK cell lytic activity and thus permitted the growth of MICB-expressing tumor cells.
Figure 5.

Shedding of MICB by TC2-MICB cells compromises NK cell activity in vivo. a. Serum levels of sMICB in all the tumor-bearing animals. b. Reduced NKG2D-dpenedent NK cells cytotoxicity of splenic NK cells from animals bearing TC2-sMICB and TC2-MICB tumors. Freshly isolated NK cells were used as effectors; TC2-MICB.A2 cells were used as target cells. **, P < 0.01 compared to which of TC2 or TC2-MICB.A2. Results represent three independent experiments.
Persistent presence of soluble MICB blocks the NKG2D-mediated NK cell recognition of target cells
When animals were treated with the CX5 antibody to block NKG2D receptor, injection of TC2-MICB.A2 cells gave rise to tumor formation in all the SCID animals (Fig. 6a). This suggests that the inhibition of TC2-MICB.A2 tumor formation in SCID animals is NKG2D-dependent. To test the effect of presence of soluble MICB on tumor formation of MICB.A2-expressing cells, we injected animals with purified rsMICB (50 ng) before and after implanting TC2-MICB.A2 cells. Under this experimental condition, implantation of TC2-MICB.A2 cells gave 100% tumor formation (Fig. 6a). Tumor cells extracted from these animals were shown to be GFP-positive and express MICB.A2 by flow cytometry analyses (data not shown). NK cells isolated from these animals showed very little cytolytic activity against TC2-MICB.A2 target cells (data not shown). These data suggest that persistent presence of soluble MICB compromises the cytotoxicity of NK cells against TC2-MICB.A2 cells.
Figure 6.

Soluble MICB induced NK cell dysfunction. a. In vivo blocking NKG2D with CX5 antibody or neutralization the function of NKG2D with rsMICB enables TC2-MICB.A2 cells to form tumors. To block NKG2D receptor in vivo, 100 μg of NKG2D-specific antibody CX5 was injected i.p. on the day before and the day after tumor implantation and thereafter every three days. To modify NKG2D function, animals were i.p. injected with 50 ng of purified rsMICB prior to implantation of TC2-MICB.A2 cells and there after twice a week for four weeks. b. Measurements of NKG2D expression shown as mean fluorescence intensity (MFI) on splenic NK cells freshly isolated from SCID animals (n=6) injected with various tumor cells. bar, mean value of MFI. c, Compatitive binding assay indicating saturation of NKG2D receptor by sMICB in animals bearing TC2-MICB and TC2-rsMICB tumors. Freshly isolated splenocytes were incubated with 10 ng/μl of rsMICB-FLAG followed by FITC-conjugated anti-FLAG mAb M2 and PE-conjugated mAb DX5. Data shown are measurements of mean fluorescence intensity (MFI) of M2 staining from six animals of each experimental group. Bar, standard error.*, p < 0.01 when compared to animals injected with TC2 or TC2-MICB.A2 tumor cells.
We sought the mechanisms by which tumor shedding-derived soluble MICB would diminish NK cell activity. Soluble MICB may down-modulate surface NKG2D expression on NK cells (16) or block the recognition of NK cells to target cells by physical occupancy of the NKG2D receptor. To distinguish these two mechanisms, we first analyzed NKG2D expression on splenic NK cells freshly isolated from animals injected with various TC2 tumor cells using flow cytometry analyses with a non-blocking NKG2D antibody A10 (29). There was no significant difference in surface NKG2D expression on NK cells from mice bearing TC2-MICB and TC2-sMICB tumors compared to those from animals bearing TC2 tumors or tumor-free animals (Fig. 6b), suggesting that the suppressive effect of soluble MICB on NK cell activity was not through down-modulation of surface NKG2D receptor. We further examined the occupancy of NKG2D receptor on NK cells by tumor-derived sMICB using competitive binding assay. Freshly isolated splenocytes were incubated with purified rsMICB-Flag and NK cell binding ability to rsMICB-Flag was measured by flow cytometry using the anti-Flag mAb M2. NK cells from animals inoculated with TC2-MICB or TC2-rsMICB cells had significantly reduced binding to rsMICB-Flag compared to those from animals inoculated with TC2 or TC2-MICB.A2 cells (p < 0.01, Fig. 6c). Together, these data suggest that soluble MICB dampens NKG2D-dependent NK cell activity mainly by masking the NKG2D receptor and thus blocking the interaction of NKG2D with target molecules.
Discussion
This study has provided conclusive evidence supporting the hypothesis that shedding of MIC by transformed cells can promote tumor growth. In this study, we generated a shedding-resistant NKG2D ligand MICB.A2 by partially modifying the α3 domain of MICB and demonstrated that overexpressing MICB.A2 prevented tumor formation by the mouse prostate tumor cell line TC2. We also demonstrated that, when soluble MICB was persistently present, expression of the shedding-resistant MICB.A2 on the tumor cell surface did not prevent or delay tumor formation in vivo. Our study signifies the impact of MIC shedding on tumor formation and the magnitude of sustained MIC-induced NKG2D immunity in preventing early tumor development.
Although the mechanisms of MIC shedding is still under investigation (19, 33, 38), clinical evidence has demonstrated that shedding of MIC is common in MIC-positive cancers, such as prostate, colon, breast adenocarcinomas, and melanomas (10-13). In these patients, the function of NK and/or CD8 T cells was compromised due to soluble MIC-induced internalization of the NKG2D receptor (13, 16-18). Thus, it was hypothesized that MIC shedding in tumors can promote tumor immune evasion and progress to advanced disease. Recent studies have demonstrated that MIC expression is not restricted in tumor cells and that MIC can be induced in cells in response to DNA damage (39), a prior event to transformation. Therefore, the current study indicates that, in the event of malignant transformation, inhibiting shedding of MIC from MIC-positive transformed cells can prevent the initiation of tumor formation.
We chose to overexpress human MICB rather than mouse NKG2D ligands in this study for the following rationales. First, MIC has been shown to be shed by tumor cells in cancer patients, thus the study is clinically relevant. Secondly, MICB has been shown to interact with mouse NKG2D and MICB-positive cells are sensitive to mouse NK cells (22, 23). We also have shown that MICB was shed by the mouse prostate cell line TC2 in the same pattern as MIC shedding in prostate cancer patients (Wu, unpublished). Thirdly, although mouse NKG2D ligands are functionally similar to human MIC in NK cell activation, these molecules are structurally different and may have different physiological roles. Mouse NKG2D ligands lack the α3 domain and are mostly GPI-linked proteins (9); in addition, little is know about what controls the expression of mouse NKG2D ligands in vivo and whether they would shed in a similar fashion to MIC in human tumor cells. Lastly, different from human NKG2D ligands, studies have shown that naturally expressed mouse NKG2D ligands on tumor cells may not cause tumor rejection, largely due to insufficient levels of the ligand expression (2) or low affinity of binding to NKG2D (40). In this study, although TC2 cells express some levels of mouse NKG2D ligand RAE-1 variants, TC2 tumors were palpable in SCID mice within a week after implantation and grew aggressively (Fig. 4), suggesting that the levels of activating RAE-1 variants expressed by TC2 cells are too low to induce anti-tumor immunity. This was also supported by the low binding ability of soluble mouse NKG2D to TC2 cells (Fig. 3a). Therefore, it could be challenging to define an optimal level of mouse NKG2D ligand expression for tumor rejection. Together, the choice of MICB makes the in vivo study described here more clinically relevant to human cancers.
In activated mouse NK cells, due to alternative DNA splicing, two isoforms of NKG2D couple with two intracellular adaptors, DAP10 and DAP12, which trigger phosphatidyl inositols kinase (PI3K) and Syk family protein tyrosine kinase, respectively (40, 41). In human, NKG2D only associates with DAP10. However, in mouse NK cells lacking DAP12 or Syk family kinases, DAP10-PI3K pathway alone is sufficient to initiate ligand-induced NKG2D-mediated killing of target cells (41). Thus, regardless that signaling via mouse NKG2D is more complex than human NKG2D, impact of NKG2D ligand shedding on tumor formation as found in the current study would be significant in both species.
Most of the in vitro evidence suggests that engagement of tumor cell surface MIC to NKG2D can activate NK cell immunity against tumor cells. Thus, expression of MIC on tumor cells is proposed to activate host protective anti-tumor immune response. However, most of the epithelial originated human cancer cells were found to have MIC expressed on the surface, suggesting the ineptness of MIC-induced NK cell immunity. Consistent with clinical observations, we also show that overexpressing the natural cleavable form of MICB in TC2 cells has no significant affect on tumor growth in vivo. Although overexpressing the non-cleavable shedding-resistant MICB.A2 can cause TC2 tumor rejection, this effect can be inhibited by the persistent presence of soluble MICB (Fig. 6a). Together, our data suggest that the role of MIC in host tumor immune surveillance is determined by whether MIC is all or partially membrane-bound. If all the MIC molecules sustain to be membrane-bound and non-cleavable, expression of MIC activates NK cell-mediated host immunity. In contrast, if a portion of the MIC molecules is cleaved and becomes soluble, tumor cells can not be targeted by NK cells due to soluble MIC-mediated masking and possible down-regulation of the receptor NKG2D, regardless of abundant MIC remaining on the tumor cell surface as observed in many cancer patients (10-13).
In summary, our data provide the first in vivo conclusive evidence of the impact of MIC shedding on tumor growth and of the importance of sustained MIC ligand -NKG2D receptor interaction in control of tumor growth. In addition, our results show no significant difference in tumor growth among animals whether the natural form of MICB or soluble recombinant MICB was expressed. This observation implies that wild-type MIC expression in established tumors may have very little effect on inducing host NK cell activation, due to shedding of MIC by tumor cells and the consequent dampening of host immunity. Together, our results suggest that strategies to sustain the recognition of NKG2D receptor and tumor MIC ligand may have potential anti-cancer therapeutic implications.
Acknowledgments
Supported by DOD-USMRC new Investigators grant W81XWH-04-1-0577, DOD-USMRC IDEA Development Award W81XWH-06-1-0014, NW Prostate SPORE Program, and NIH Temin Award 1K01CA116002 (J. Wu). We thank Michael Tao for assistant with animal tissue collections and Dr. Norman M. Greenberg for helpful discussions. The authors have no conflicting financial interests.
Non-standard abbreviation
- MIC
MHC class I chain-related molecule
- TC2
TRAMP-C2
- RAE
retinoic acid early inducible family of protein
- LAK
IL-2 activated NK
Footnotes
Statement of Clinical Relevance In humans, the MHC class I chain-related molecules MICA and MICB (generally termed MIC) are frequently found expressed on epithelial-originated tumor cells. MIC is a ligand for the activating natural killer (NK) cell receptor NKG2D. Engagement of tumor-expressed MIC to NKG2D can activate NK cell tumor-lytic activity in vitro. Thus, expression of MIC on tumor cells is proposed to play a significant role in tumor immune surveillance. However, as observed in many cancer patients, majority of the tumors remain MIC-positive, suggesting the ineptness of the NKG2D-mediated NK cell function. We and others have demonstrated that shedding of MIC by tumor cells can impair NK cell function in cancer patients. These clinical studies prompted the hypothesis that tumor shedding of MIC may be one of the mechanisms by which MIC-positive tumors evade NK cell immune surveillance and progress. This study tested this hypothesis in vivo using a prostate tumor model and for the first time demonstrated that interfering with shedding of MIC could prevent tumor formation. Our study suggests that shedding of MIC may contribute significantly to tumor formation by transformed cells and thus inhibiting tumor shedding of MIC may have potential therapeutic implication in targeted cancer therapy.
References
- 1.Cerwenka A, Bakker AB, McClanahan T, et al. Retinoic acid early inducible genes define a ligand family for the activating NKG2D receptor in mice. Immunity. 2000;12:721–7. doi: 10.1016/s1074-7613(00)80222-8. [DOI] [PubMed] [Google Scholar]
- 2.Diefenbach A, Jensen ER, Jamieson AM, Raulet DH. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature. 2001;413:165–71. doi: 10.1038/35093109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cerwenka A, Baron JL, Lanier LL. Ectopic expression of retinoic acid early inducible-1 gene (RAE-1) permits natural killer cell-mediated rejection of a MHC class I-bearing tumor in vivo. Proc Natl Acad Sci U S A. 2001;98:11521–6. doi: 10.1073/pnas.201238598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Diefenbach A, Hsia JK, Hsiung MY, Raulet DH. A novel ligand for the NKG2D receptor activates NK cells and macrophages and induces tumor immunity. Eur J Immunol. 2003;33:381–91. doi: 10.1002/immu.200310012. [DOI] [PubMed] [Google Scholar]
- 5.Carayannopoulos LN, Naidenko OV, Fremont DH, Yokoyama WM. Cutting edge: murine UL16-binding protein-like transcript 1: a newly described transcript encoding a high-affinity ligand for murine NKG2D. J Immunol. 2002;169:4079–83. doi: 10.4049/jimmunol.169.8.4079. [DOI] [PubMed] [Google Scholar]
- 6.Smyth MJ, Swann J, Cretney E, Zerafa N, Yokoyama WM, Hayakawa Y. NKG2D function protects the host from tumor initiation. J Exp Med. 2005;202:583–8. doi: 10.1084/jem.20050994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Long EO. Tumor cell recognition by natural killer cells. Semin Cancer Biol. 2002;12:57–61. doi: 10.1006/scbi.2001.0398. [DOI] [PubMed] [Google Scholar]
- 8.Raulet DH. Roles of the NKG2D immunoreceptor and its ligands. Nat Rev Immunol. 2003;3:781–90. doi: 10.1038/nri1199. [DOI] [PubMed] [Google Scholar]
- 9.Cerwenka A, Lanier LL. NKG2D ligands: unconventional MHC class I-like -molecules exploited by viruses and cancer. Tissue Antigens. 2003;61:335–43. doi: 10.1034/j.1399-0039.2003.00070.x. [DOI] [PubMed] [Google Scholar]
- 10.Groh V, Rhinehart R, Secrist H, Bauer S, Grabstein KH, Spies T. Broad tumor-associated expression and recognition by tumor-derived gamma delta T cells of MICA and MICB. Proc Natl Acad Sci U S A. 1999;96:6879–84. doi: 10.1073/pnas.96.12.6879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vetter CS, Groh V, thor Straten P, Spies T, Brocker EB, Becker JC. Expression of stress-induced MHC class I related chain molecules on human melanoma. J Invest Dermatol. 2002;118:600–5. doi: 10.1046/j.1523-1747.2002.01700.x. [DOI] [PubMed] [Google Scholar]
- 12.Jinushi M, Takehara T, Tatsumi T, et al. Expression and role of MICA and MICB in human hepatocellular carcinomas and their regulation by retinoic acid. Int J Cancer. 2003;104:354–61. doi: 10.1002/ijc.10966. [DOI] [PubMed] [Google Scholar]
- 13.Wu JD, Higgins LM, Steinle A, Cosman D, Haugk K, Plymate SR. Prevalent expression of the immunostimulatory MHC class I chain-related molecule is counteracted by shedding in prostate cancer. J Clin Invest. 2004;114:560–8. doi: 10.1172/JCI22206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Friese MA, Platten M, Lutz SZ, et al. MICA/NKG2D-mediated immunogene therapy of experimental gliomas. Cancer Res. 2003;63:8996–9006. [PubMed] [Google Scholar]
- 15.Busche A, Goldmann T, Naumann U, Steinle A, Brandau S. Natural killer cell-mediated rejection of experimental human lung cancer by genetic overexpression of major histocompatibility complex class I chain-related gene A. Hum Gene Ther. 2006;17:135–46. doi: 10.1089/hum.2006.17.135. [DOI] [PubMed] [Google Scholar]
- 16.Groh V, Wu J, Yee C, Spies T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature. 2002;419:734–8. doi: 10.1038/nature01112. [DOI] [PubMed] [Google Scholar]
- 17.Doubrovina ES, Doubrovin MM, Vider E, et al. Evasion from NK cell immunity by MHC class I chain-related molecules expressing colon adenocarcinoma. J Immunol. 2003;171:6891–9. doi: 10.4049/jimmunol.171.12.6891. [DOI] [PubMed] [Google Scholar]
- 18.Raffaghello L, Prigione I, Airoldi I, et al. Downregulation and/or release of NKG2D ligands as immune evasion strategy of human neuroblastoma. Neoplasia. 2004;6:558–68. doi: 10.1593/neo.04316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salih HR, Rammensee HG, Steinle A. Cutting edge: down-regulation of MICA on human tumors by proteolytic shedding. J Immunol. 2002;169:4098–102. doi: 10.4049/jimmunol.169.8.4098. [DOI] [PubMed] [Google Scholar]
- 20.Holdenrieder S, Stieber P, Peterfi A, Nagel D, Steinle A, Salih HR. Soluble MICA in malignant diseases. Int J Cancer. 2006;118:684–7. doi: 10.1002/ijc.21382. [DOI] [PubMed] [Google Scholar]
- 21.Marten A, von Lilienfeld-Toal M, Buchler MW, Schmidt J. Soluble MIC is elevated in the serum of patients with pancreatic carcinoma diminishing gammadelta T cell cytotoxicity. Int J Cancer. 2006;119:2359–65. doi: 10.1002/ijc.22186. [DOI] [PubMed] [Google Scholar]
- 22.Diefenbach A, Jamieson AM, Liu SD, Shastri N, Raulet DH. Ligands for the murine NKG2D receptor: expression by tumor cells and activation of NK cells and macrophages. Nat Immunol. 2000;1:119–26. doi: 10.1038/77793. [DOI] [PubMed] [Google Scholar]
- 23.Dunn C, Chalupny NJ, Sutherland CL, et al. Human cytomegalovirus glycoprotein UL16 causes intracellular sequestration of NKG2D ligands, protecting against natural killer cell cytotoxicity. J Exp Med. 2003;197:1427–39. doi: 10.1084/jem.20022059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li P, Morris DL, Willcox BE, Steinle A, Spies T, Strong RK. Complex structure of the activating immunoreceptor NKG2D and its MHC class I-like ligand MICA. Nat Immunol. 2001;2:443–51. doi: 10.1038/87757. [DOI] [PubMed] [Google Scholar]
- 25.Holmes MA, Li P, Petersdorf EW, Strong RK. Structural studies of allelic diversity of the MHC class I homolog MIC-B, a stress-inducible ligand for the activating immunoreceptor NKG2D. J Immunol. 2002;169:1395–400. doi: 10.4049/jimmunol.169.3.1395. [DOI] [PubMed] [Google Scholar]
- 26.Strong RK. Asymmetric ligand recognition by the activating natural killer cell receptor NKG2D, a symmetric homodimer. Mol Immunol. 2002;38:1029–37. doi: 10.1016/s0161-5890(02)00032-9. [DOI] [PubMed] [Google Scholar]
- 27.Foster BA, Gingrich JR, Kwon ED, Madias C, Greenberg NM. Characterization of prostatic epithelial cell lines derived from transgenic adenocarcinoma of the mouse prostate (TRAMP) model. Cancer Res. 1997;57:3325–30. [PubMed] [Google Scholar]
- 28.Bahram S, Spies T. Nucleotide sequence of a human MHC class I MICB cDNA. Immunogenetics. 1996;43:230–3. doi: 10.1007/BF00587305. [DOI] [PubMed] [Google Scholar]
- 29.Horton RM, Cai ZL, Ho SN, Pease LR. Gene splicing by overlap extension: tailor-made genes using the polymerase chain reaction. Biotechniques. 1990;8:528–35. [PubMed] [Google Scholar]
- 30.Ho EL, Carayannopoulos LN, Poursine-Laurent J, et al. Costimulation of multiple NK cell activation receptors by NKG2D. J Immunol. 2002;169:3667–75. doi: 10.4049/jimmunol.169.7.3667. [DOI] [PubMed] [Google Scholar]
- 31.Masuda H, Saeki Y, Nomura M, et al. High levels of RAE-1 isoforms on mouse tumor cell lines assessed by anti-“pan” RAE-1 antibody confer tumor susceptibility to NK cells. Biochem Biophys Res Commun. 2002;290:140–5. doi: 10.1006/bbrc.2001.6165. [DOI] [PubMed] [Google Scholar]
- 32.Salih HR, Goehlsdorf D, Steinle A. Release of MICB molecules by tumor cells: mechanism and soluble MICB in sera of cancer patients. Hum Immunol. 2006;67:188–95. doi: 10.1016/j.humimm.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 33.Kaiser BK, Yim D, Chow IT, et al. Disulphide-isomerase-enabled shedding of tumour-associated NKG2D ligands. Nature. 2007;447:482–6. doi: 10.1038/nature05768. [DOI] [PubMed] [Google Scholar]
- 34.Wu J, Chalupny NJ, Manley TJ, Riddell SR, Cosman D, Spies T. Intracellular retention of the MHC class I-related chain B ligand of NKG2D by the human cytomegalovirus UL16 glycoprotein. J Immunol. 2003;170:4196–200. doi: 10.4049/jimmunol.170.8.4196. [DOI] [PubMed] [Google Scholar]
- 35.Ogasawara K, Hamerman JA, Hsin H, et al. Impairment of NK cell function by NKG2D modulation in NOD mice. Immunity. 2003;18:41–51. doi: 10.1016/s1074-7613(02)00505-8. [DOI] [PubMed] [Google Scholar]
- 36.Diefenbach A, Raulet DH. The innate immune response to tumors and its role in the induction of T-cell immunity. Immunol Rev. 2002;188:9–21. doi: 10.1034/j.1600-065x.2002.18802.x. [DOI] [PubMed] [Google Scholar]
- 37.Grossmann ME, Wood M, Celis E. Expression, specificity and immunotherapy potential of prostate-associated genes in murine cell lines. World J Urol. 2001;19:365–70. doi: 10.1007/pl00007104. [DOI] [PubMed] [Google Scholar]
- 38.Le Maux Chansac B, Misse D, Richon C, et al. Potentiation of NK cell-mediated cytotoxicity in human lung adenocarcinoma: role of NKG2D-dependent pathway. Int Immunol. 2008;20:801–10. doi: 10.1093/intimm/dxn038. [DOI] [PubMed] [Google Scholar]
- 39.Gasser S, Orsulic S, Brown EJ, Raulet DH. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature. 2005;436:1186–90. doi: 10.1038/nature03884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Diefenbach A, Tomasello E, Lucas M, et al. Selective associations with signaling proteins determine stimulatory versus costimulatory activity of NKG2D. Nat Immunol. 2002;3:1142–9. doi: 10.1038/ni858. [DOI] [PubMed] [Google Scholar]
- 41.Zompi S, Hamerman JA, Ogasawara K, et al. NKG2D triggers cytotoxicity in mouse NK cells lacking DAP12 or Syk family kinases. Nat Immunol. 2003;4:565–72. doi: 10.1038/ni930. [DOI] [PubMed] [Google Scholar]