

Abstract
The extracellular fibrinogen-binding protein (Efb), an immunosuppressive and anti-inflammatory protein secreted by Staphylococcus aureus, has been identified as a potent inhibitor of complement-mediated innate immunity. Efb functions by binding to and disrupting the function of complement component 3 (C3). In a recent study, we presented a high-resolution co-crystal structure of the complement inhibitory domain of Efb (Efb-C) bound to its cognate domain (C3d) from human C3 and employed a series of structure/function analyses that provided evidence for an entirely new, conformational change-based mechanism of complement inhibition. To better understand the Efb/C3 complex and its downstream effects on C3 inhibition, we investigated the solvent-accessibility and protein interface of Efb(-C)/C3d using a method of lysine acetylation, proteolytic digestion and mass spectrometric analysis. Lysine modification in Efb was monitored by the mass increment of lysine-containing fragments. Besides confirming the binding sites observed in co-crystal structure study, the in-solution data presented here suggest additional contacting point(s) between the proteins that were not revealed by crystallography. The results of this study demonstrate that solution-based analysis of protein-protein interactions can provide important complementary information on the nature of protein-protein interactions.
Keywords: Mass Spectrometry, Complement, Bacterial, Protein-protein interaction
Introduction
In the last two decades, chemical labeling coupled with mass spectrometry has emerged as a powerful approach in structural proteomics research [1]. The availability of robust chemical modification strategies, in combination with sensitive and accurate mass spectrometric techniques have allowed the exploration of protein structure, protein conformational dynamics and the determination of protein-protein interactions in solution. The general principle behind these strategies is that solvent-exposed residues undergo chemical modification more quickly than non-exposed residues. When a target protein is labeled in the presence and absence of a ligand, mass differences found by MS analysis will help determine the residues involved in the contacting sites. Typically, spatial resolution is achieved by proteolytically digesting the protein of interest after chemical modification and analyzing the resulting fragments by MS. By comparing the mass of the resultant proteolytic peptides between unmodified and modified proteins, the location and magnitude of label incorporation can be identified, revealing areas in the protein of increased and decreased solvent exposure. Implicit in these data are details regarding protein-protein interfaces and also regions which undergo conformational change in response to ligand.
One of the most utilized chemical labeling methods is lysine residue modification [2-6]. Lysine residue labeling offers many advantages for studying protein solvent accessibility, one of which is the unique reactivity of the lysine side-chain primary amine. Furthermore, proteins modified at their lysine residues do not rapidly lose incorporated label and do not require special handling. By virtue of these features, the analytical method used to determine the site of modification is not time-sensitive. In the particular case of the extracellular fibrinogen-binding protein (Efb1) and the C-terminal domain of extracellular fibrinogen-binding protein (Efb-C), the relatively large number of lysine residues (20 of 137 residues for Efb; 13 of 75 residues for Efb-C) provides the opportunity for superior sequence coverage via the lysine-labeling approach. In addition, several of the Efb residues which comprise complex interface are either lysine themselves or are in the near vicinity of lysine side-chains.
Staphylococcus aureus is the etiologic agent for a remarkable variety of skin, soft tissue, respiratory, bone, joint, and cardiovascular disorders [7-11]. While many factors are thought to contribute to the ability of S. aureus to cause a range of clinical disease, recent work has indicated that the bacterium can bypass a competent immune response with some degree of efficiency. To this end, S. aureus expresses multiple immunomodulatory proteins that target both innate and adaptive immune responses. By facilitating the successful escape of the microbe from the various elimination mechanisms of immunity [12], the pathogen can eventually colonize the discrete microenvironments within the human host [12-14]. The ultimate manifestation of this process is disseminated bacterial infection and disease.
The complement system is a central component of innate immunity and plays a critical role in both eliminating invading pathogens and activating downstream immune defense mechanisms. Numerous pathogens have evolved sophisticated means to neutralize the complement system, since their survival in host tissues is predicated upon their ability to avoid complement attack [12]. Among the complement proteins, complement component 3 (C3) is central as it can propagate the classical, alternative and mannose-binding lectin (MBL) pathways of complement activation [15, 16]. Several studies have demonstrated that S. aureus infection is associated with the activation of complement through all three pathways [17-20], and the ensuing deposition of C3b on the bacterial surface was found to enhance opsonophagocytic clearance of the pathogen [21]. Sakiniene et al. showed that complement depletion aggravated S. aureus-induced arthritis and septicemia in a murine model, implying a critical role of complement system for protection against S. aureus infections [22]. Subsequently, Lee and colleagues identified a 19-kD Efb secreted by S. aureus that can inhibit the activation of complement and thereby suppress C3b deposition onto sensitized surfaces [23]. In support of this, Liang and coworkers observed that Efb contributed to bacterial adhesion and invasion within the host [24]. Furthermore, S. aureus strains deleted for Efb were found less virulent in a mouse model of wound healing [25]. Together, these results have strongly suggested that Efb-mediated inhibition of complement activation is an important determinant in S. aureus pathogenesis.
To gain insight into the mechanism of Efb function, we recently determined the crystal structure of its C3-inhibitory domain (denoted Efb-C) both free and bound to its cognate C3d subdomain from human C3 [26]. These structures revealed that Efb-C adopts a novel three helix bundle motif involved in complement regulation and provided insight into the protein-protein interaction network that targets Efb to the surface of C3. Subsequent functional studies demonstrated that Efb binding to C3 is required for its complement inhibitory activity, and that Efb blocks generation of C3b by altering the solution conformation of its native C3 precursor.
In that previous work, nearly 10% of the Efb-C polypeptide is disordered in the crystal lattice. These residues are therefore invisible in the refined structure of the Efb-C/C3d complex. As a consequence, the potential contributions of these N-terminal regions to complement inhibition remain largely unexplored. In this particular case, even transiently stable interactions between Efb and C3 may be biologically relevant since binding of Efb-C is known to effect to the solution conformation of C3 [26]. Furthermore, proteins may adopt conformations in solution that differ in small but important aspects from those observed in accurately refined crystal structures. Consequently, it can be very informative to expand upon solid-state structural data through the use of solution approaches.
In the current report, we describe the use of lysine acetylation technology to study the changes in surface accessibility that occur during Efb(-C)/C3d binding. Moreover, we conducted a protein charge state distribution study using ESI MS to investigate whether lysine acetylation induces structural changes on target proteins. In addition to providing a solution perspective to formation of this immune evasion complex, our results have identified additional points of contact between Efb(-C)/C3d that were not visible in the structure of the complex. This work sets the stage for future studies into the contribution of these interactions to complement inhibition by Efb(-C). In addition, they lay a foundation for the use of selective lysine labeling approaches to monitor ligand-induced conformational changes in C3.
Experimental
Materials
Recombinant Efb, Efb-C and C3d were prepared in a manner identical to that used for crystallization and structure/function studies [26]. Chymotrypsin and glutamic-C were purchased from Princeton Separations, Inc. (Adelphia, NJ). Sinapinic acid (SA) and α-cyano-4-hydroxycinnamic acid (CHCA) were purchased from Waters (Milford, MA). Trifluoroacetic acid (TFA) and mass spectrometry grade acetonitrile (AcN) were purchased from Riedel-de Haën (Seelze, Germany). All the water used in the experiments was obtained from a Milli-Q System (Millipore, Bedford, MA). ZipTip® C18 tips were purchased from Millipore (Bedford, MA). MS calibration standards for both the reflectron (poly(ethylene glycol), PEG 700-4000) and linear (Insulin, Cytochrome-C, Haemoglobin, Myoglobin and Trypsinogen) modes were purchased from Sigma-Aldrich (St. Louis, MO). ProteoMass™ P14R MALDI-MS Standard for LockMass (Micromass, UK) was purchased from Sigma-Aldrich.
N-Acetyl-hydroxysuccinimide (NHS-Ac) was synthesized according to established methods [27] using commercially available materials without any further processing. N,N′-Dicyclohexylcarbodiimide (7.64 g, 37.0 mmol) (Sigmal-Aldrich, St. Louis, MO) was slowly added to a stirred room temperature solution containing glacial acetic acid (1.9 mL, 33.2 mmol) (Fisher, Fairlawn, NJ) and N-hydroxysuccinimide (3.80 g, 33.0 mmol) (Sigmal-Aldrich, St. Louis, MO) dissolved in dichloromethane (33 mL) (Fisher, Fairlawn, NJ). A precipitate formed and the reaction was exothermic; an additional 20 mL dichloromethane was added. The reaction was allowed to stir for 12 hours. The mixture was filtered through a medium porosity glass fritted filter funnel, the solids rinsed with excess dicholormethane, and the combined organic filtrates were concentrated by rotary evaporation. The crude product was recrystallized from ethanol. 3.1304 g of a white crystalline solid product was obtained (60% yield). NHS-Ac was analyzed by 1H NMR (400 MHz, (Bruker, Rheinstetten, Germany) in CDCl3: δ 2.84 (s, 4H), δ 2.35 (s, 3H).
MALDI-TOF
All MALDI experiments were carried out using a Waters Micromass MALDI micro MX mass spectrometer with time-of-flight (TOF) analyzer (Waters Micromass, Milford, MA). The instrument is equipped with a N2 UV laser emitting at 337 nm, a pulsed ion extraction source, an electrostatic reflectron of 2.3 m effective path length, a 2 GHz 8-bit transient analog to digital converter with real time peak display and fast dual micro-channel plate detectors. All sample solutions were mixed in a 1:1 volume ratio with a matrix solution. For reflectron mode, the matrix solution consisted of 10mg/mL α-cyano-4-hydroxycinnamic acid in 1:1 AcN:0.1% TFA, while 10mg/mL sinapinic acid in 4:6 AcN:0.1% TFA was used as the matrix solution for linear mode. 0.8 μL of each sample-matrix mixture was spotted on the target plate and allowed to dry under moderate vacuum for approximately 1 minute. The acceleration voltage was set to 20 kV for all experiments, and typically 50 single-shot mass spectra were summed to give a composite spectrum. All data were reprocessed using Waters MassLynx software. The mass scale was calibrated externally using poly(ethylene glycol) (PEG 700-4000) for reflectron mode and a defined peptide mixture (Insulin, Cytochrome-C, Haemoglobin, Myoglobin and Trypsinogen) for linear mode.
LC-MS/MS
For all LC-MS/MS studies, the LC system consisted of an Ultimate nano HPLC system (LC Packings, Amsterdam, The Netherlands), an ABI syringe delivery pump (Applied Biosystems, Foster City, CA), a Rheodyne injection valve (Model 7125) and 3 Rheodyne switch valves (Model 7000) (Rohnert Park, CA), a C8 peptide microtrap (Michrom, Auburn, CA) and a 50mm Vydac 18 column (I.D. 1.0mm, 300Å, 5μm) from Alltech (Deerfield, IL). The Ultimate nano HPLC system was controlled by Ultichrom software (Dionex, Bavel, The Netherlands). After a 30-minute equilibration at 50μL/min flow rate of the LC system with buffer A (99.95% water and 0.05% TFA), 0.3 nmol pre-digested protein was injected into the Rheodyne injection valve and loaded onto the peptide trap at 50μL/min flow rate (delivered by the ABI pump) for three minutes to accomplish sample desalting. Afterwards, manipulation of the switch valve allowed a gradient delivered by the LC Packings system to elute the peptides from the peptide trap onto the C18 analytical column. During the first 30 min buffer B (80% AcN, 20% water, and 0.01% TFA) was increased from 0 to 60%, at which time buffer B was increased to 95% in 1 min and maintained at that percentage for another 5 min. Finally, the system was reconditioned by decreasing buffer B to 0% in 1 min and maintaining this condition for 30 min.
The LC system was interfaced with an ESI-MS by PEEK tubings (I.D. = 0.005 inches). All ESI-MS/MS spectra were acquired with helium as the collision gas on a Finnigan LCQ DUO ion trap mass spectrometer (Finnigan Co., San Jose, CA, USA) equipped with a fritless electrospray interface under the control of the Finnigan Xcalibur program. The LCQ instrument was run under the following conditions: spray needle voltage 4.5 kV, heated capillary temperature 200°C, mass range 200-2000 Da in each full-MS or MS/MS scan. A three-stage tandem MS method was used to obtain MS/MS data: full-MS scan, zoom scan and MS/MS scan. The resulting tandem mass spectra were used for peptide sequence interpretation and database searching using the SEQUEST program. The fragment ion mass accuracy was specified as ±0.5 Da. To identify a peptide, the following criteria were used: (1) cross-correlation score ≥ 1 for single-charged fragments; (2) cross-correlation score ≥ 2 for multi-charged fragments.
ESI-MS
In the charge state distribution study, gas phase protein ions were produced in the positive mode by pneumatically assisted ESI at a flow rate of 4μL/min. The concentration of all protein samples was 5μM. All other instrumental conditions were identical to those described in the MS/MS study.
Sample Preparation
The main procedure of sample preparation is shown in Fig. 1. In the free protein experiments, Efb(-C) or C3d was diluted in reacting buffer (100 mM Na2CO3, pH 8) to 7.3 μM. 90 μL of protein solution was mixed with 90 μL of reacting buffer to make a 180 μL mixture. The mixture was divided into two 90-μL aliquots: aliquot A and aliquot B. 10 μL water was added to aliquot A while 10 μL NHS-Ac solution (2 M NHS-Ac in AcN diluted 10-fold in reaction buffer) was added to aliquot B. Acetylation was allowed to proceed for 20 min at room temperature. At this time, 300 μL of quenching buffer (200 mM NH4HCO3, pH 8) was added, and each sample was incubated for 20 min further at room temperature. 10 μL of each sample was removed and acidified to pH 2. Before subjecting the protein to linear mode MALDI-TOF MS, the protein was desalted and concentrated by a ZipTip® according to manufacturer's suggestions. To the other 390 μL sample, appropriate proteases (20 ng/μL) were added to achieve a protein/protease mass ratio of 50:1. Overnight digestions were performed at 37 °C for chymotrypsin and 28 °C for V8. Following digestion, 10 μL of peptide mixture was desalted by ZipTip® and subjected to reflectron mode MALDI-TOF MS. The remaining peptide mixture was injected into the LC system for the ESI-MS/MS analysis to create the peptide pool.
Fig. 1.
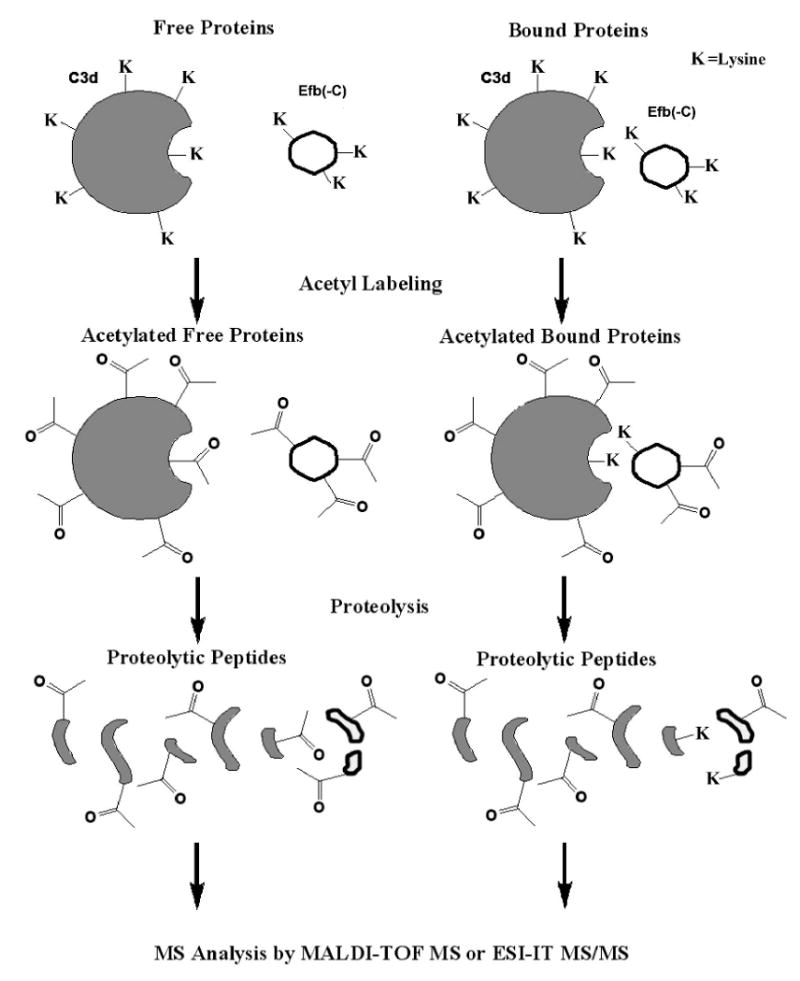
Brief scheme of experimental labeling procedure. C3d (in dark color), Efb(-C) (in light color) or their complex are labeled using the lysine acetylation reagent. After 20 minutes at room temperature, the acetylation reaction is quenched by addition of NH4HCO3 and followed by proteolytic digestion. Both chymotrpsin and glutamic-C V8 were used to achieve the most complete peptide coverage maps.
In experiments with the Efb(-C)/C3d protein complexes, the same protocol as above was applied, except that ESI-MS/MS analysis was omitted as the peptide pool had already been created. All experiments were repeated three times to ensure reproducibility and reliability.
In the charge state study, all samples were prepared in 200 mM NH4HCO3, to ensure a chemical environment identical to the quenching solution following lysine acetylation (see above). The pH of each sample was measured by an accurate pH meter (Denver Instrument, model 220, Göttingen, Germany).and adjusted to the desired value using either hydrochloric acid or ammonium hydroxide.
Results and Discussion
N-acetyl-succinimide is an efficient acetylating reagent and our first goal was to address if it would be a useful tool for analysis of the Efb(-C)/C3d interaction. Optimization of the reaction conditions using Efb-C and C3d suggested that the acetylation studies were best performed with a minimum 200:1 molar ratio of acetylating reagent:lysine for 20 minutes at room temperature.
At a sequence level, Efb and Efb-C are comprised of greater than 14 and 17% lysine residues each. Consequently, such extensive acetylation of reactive lysines might result in alteration or destabilization of the protein structure or changes in overall protein solubility. Analysis of a protein's charge state distribution by ESI is a commonly used method to investigate global structural transitions of proteins in solution [28]. Protein ions generated in the gas phase by electrospray ionization are able to carry multiple charges due to the presence of multiple ionizable groups. Structural changes in proteins lead to changes in both the steric accessibility as well as in the pKa of these protic sites [29]. Since the accessible surface area is expanded in unfolded proteins [30], a well-folded native protein is typically found distributed throughout lower charge states when compared to its unfolded counterpart by ESI. Optical spectroscopy methods have confirmed these results from ESI, and established that this charge state distribution is highly sensitive to tertiary structural changes in the polypeptide chain [31].
To address questions of acetylation-induced structural changes, we monitored the charge state distribution of both free and acetylated Efb and Efb-C at multiple pH values from 8 to 3 under native conditions by ESI mass spectrometry. Examination of the resulting spectra for free (Fig. 2(A, B)) and acetylated Efb (Fig. 2(C, D)) revealed a similar distribution change of charge states between both samples at different pH. As shown in Fig. 2(A, C), the dominant center ion peak of 9+ indicated that both free and acetylated Efb were well-folded at pH 8 and that the two samples were in conformational states indistinguishable through this technique. Furthermore, the fact that additional charge states became populated for both samples as the pH grew more acidic demonstrated that this ESI method was capable of detecting such changes when they occurred. Similar results were obtained with the charge state of Efb-C (see supplemental figure 1). The much smaller size of Efb-C compared with full-length Efb resulted in a complete charge-state pattern. The results show that the acetylated-Efb-C has the same pattern as Efb-C and that it is similar to Efb (Fig. 2). The 6+ is the highest peak for Efb-C followed the much lower 7+ peak. In figure 2, the intensity of the 9+ peak is much higher than 10+, which is again similar to Efb-C's pattern. Thus, the ESI data revealed that lysine acetylation did not perturb the global structural integrity of either Efb or Efb-C.
Fig. 2.
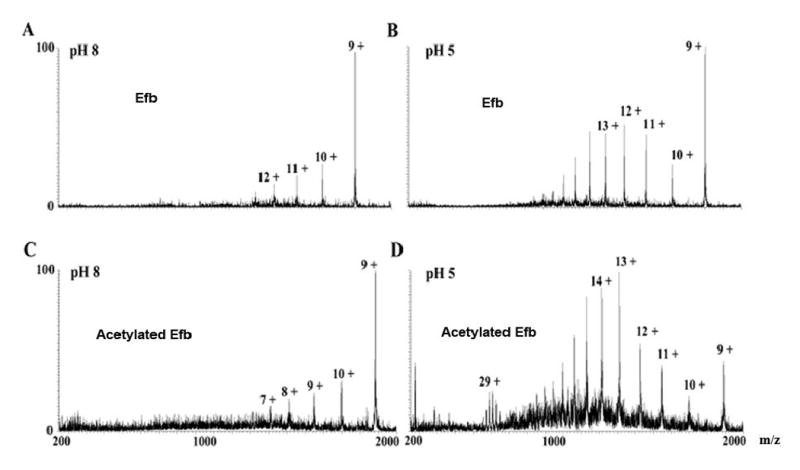
ESI mass spectra of Efb and acetylated Efb recorded at different pH values. A) Efb at pH 8; B) Efb at pH 5; C) acetylated Efb at pH 8, D) acetylated Efb at pH 5. Peaks are labeled according to the corresponding charge states. Note that acetylated Efb displayed the same charge state distribution as native protein at multiple pH values. Similar results were obtained in an ESI charge state distribution analysis of Efb-C (see supplemental figure 1). This suggested that the acetylation reaction itself did not perturb the folding state of Efb(-C).
To gauge the efficiency of the acetylation reaction, free Efb, Efb-C and C3d were acetylated as described above and analyzed by MALDI-TOF MS. The proteins were initially monitored in linear mode, since this allowed for measurement of the entire protein mass. Accordingly, it was possible to determine the number of lysine residues modified by inspection of the observed mass difference between the unlabeled and labeled protein samples. The linear-mode MALDI spectra of unlabeled and labeled Efb-C and Efb are shown in Fig. 3(A), (E) and Fig. 3(B), (F), respectively. For example, the mass differential between Fig. 3(A) and Fig. 3(B) is 546 Da, indicating that there were (546 Da/42 Da/modification, where 42 Da is the molecular weight of acetyl group) 13 modifications in the free Efb-C sample. A similar result was seen in the case of Efb, where 19 lysines were modified (see Fig. 3(E) and (F)). The mass shift between unlabeled and labeled C3d was also determined (data not shown). However, unlike Efb(-C), C3d was not fully acetylated as some lysine residues are buried in the native structure [32]. In addition, we also observed a mixture of C3d molecules that corresponded to variability in the extent of acetyl modifications. Under linear mode, MALDI-TOF MS was not able to resolve these populations adequately and yielded a broad peak that precluded a precise determination of label density. Nevertheless, the centroid value of the C3d peaks suggested that the number of lysines modified was about 14. In the end, Efb and Efb-C were chosen as the major target for the protein-protein interaction study because of the superior resolution afforded in this approach.
Fig. 3.
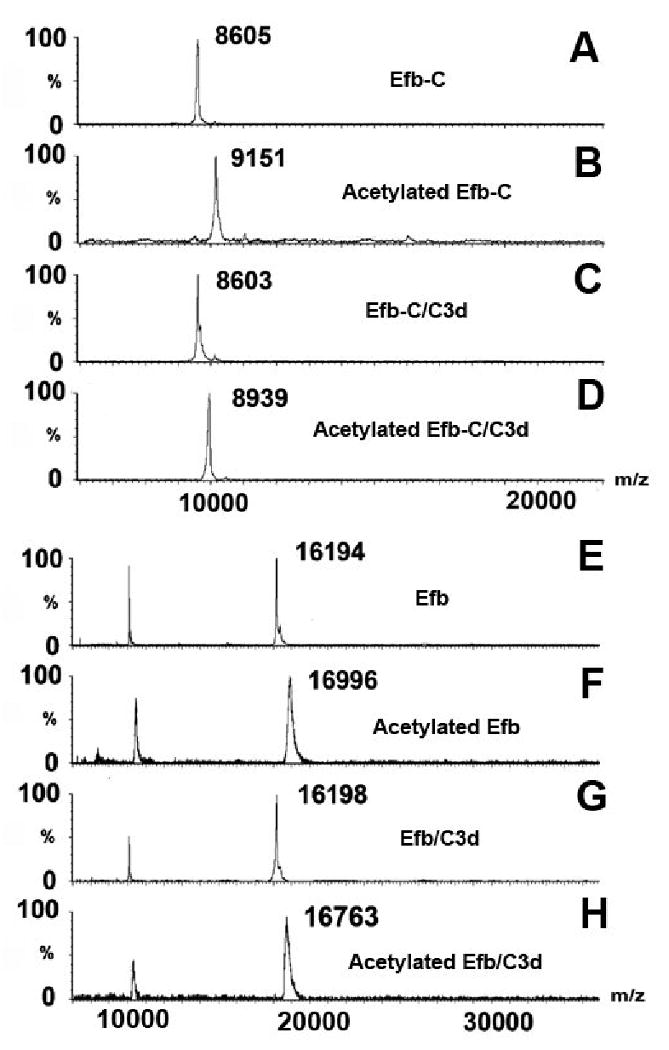
MALDI spectra of Efb-C (A-D) and Efb (E-H) under linear mode. Sample identities are A) free Efb-C without lysine acetylation; B) free Efb-C with lysine acetylation; C) Efb-C/C3d without lysine acetylation; and D) Efb-C/C3d with lysine acetylation; E) free Efb without lysine acetylation; F) free Efb with lysine acetylation; G) Efb /C3d without lysine acetylation; and H) Efb /C3d with lysine acetylation. The mass differential between sample A and B was 546 Da = 9151-8605, indicating that 13 lysine residues were acetylated in the free protein sample. In contrast, the 336 Da (= 8939-8603) difference between complex samples C and D demonstrated that only 8 lysine residues were labeled. The differential labeling between free and bound proteins is caused by the protection of five Efb-C lysine residues in the protein complex. The same number can be obtained from the Efb spectra E-H.
We then examined whether formation of the Efb(-C)/C3d complex might influence the acetylation profile of Efb(-C). To address this question, both Efb and Efb-C were incubated separately in the presence of equimolar concentrations of C3d and subjected to acetylation under identical experimental conditions as those described above. Analysis of bound Efb-C revealed a mass differential after modification of 336 Da, which corresponded to labeling of 8 lysine residues (see Fig. 3(C) and Fig. 3(D)). These results were consistent with protection of 5 lysine residues in Efb-C from acetylation when bound to C3d. A similar mass differential was observed in the case of Efb (see Fig. 3 (G) and (H)).
To investigate the identities of these five lysine residues in detail, spatial resolution was achieved by digestion of the labeled samples with either chymotrypsin or glutamic-C V8. Both of these enzymes have different sequence selectivity, and neither requires a substrate lysine residue for activity. Although the products of chymotrypsin or glutamic-C V8-catalyzed proteolysis can be predicted with some certainty, we nevertheless obtained empirical identification of the resulting proteolytic peptides. To facilitate this, we utilized MALDI-based peptide mass fingerprinting and LC/ESI tandem MS (Table 1).
Table 1. Experimental lysine-containing fragments for Efb and Efb-C.
| Chymotrypsin | ||||
|---|---|---|---|---|
| Sequencea | m/z | Unboundb | Boundc | Obsd |
| 160KQGLVK165 | 672 | N/A | N/A | Efb-C |
| 58QSRPKF63 | 762 | 1 | 1 | Efb |
| 73KHDYNIL79 | 902 | 1 | 1 | Efb |
| 64NSTPKYIKF72 | 1098 | 2 | 2 | Efb |
| 73KHDYNILEF81 | 1179 | 1 | 1 | Efb |
| 58QSRPKFNSTPKY69 | 1453 | 2 | 2 | Efb |
| 152QERIDNVLKQGLVK165 | 1640 | 1 | 1 | Efb |
| 145KVKKMVLQERIDNVL159 | 1813 | 3 | 3 | Efb-C |
| 58QSRPKFNSTPKYIKF72 | 1841 | 3 | 3 | Efb |
| 95NKPAAKTDATIKKEQKL111 | 1884 | 5 | 1 | Efb-C |
| 34GPREKKPVSINHNIVEY50 | 1980 | 1,2 | 1,2 | Efb |
| 34GPREKKPVSINHNIVEYNDGTF55 | 2514 | 1,2 | 1,2 | Efb |
| 89GARPQFNKPAAKTDATIKKEQKL111 | 2540 | 5 | 1 | Efb |
| 95NKPAAKTDATIKKEQKLIQAQNL117 | 2551 | 5 | 1 | Efb-C |
| 87EYGARPQFNKPAAKTDATIKKEQKL111 | 2833 | 5 | 1 | Efb |
| Glutamic-C | ||||
| Sequence | m/z | Unbound | Bound | Obs |
| 144YKVKKMVLQE153 | 1266 | 3 | 2 | Efb |
| 109QKLIQAQNLVRE120 | 1440/1439 | 1 | 1 | Both |
| 88YGARPQFNKPAAKTD102 | 1664 | 2 | 0 | Efb |
| 109QKLIQAQNLVREFE122 | 1716/1715 | 1 | 1 | Both |
| 32GYGPREKKPVSINHNIVE49 | 2037 | 1,2 | 1,2 | Efb |
| 88YGARPQFNKPAAKTDATIKKE108 | 2334 | 4 | 0 | Efb |
| 123KTHTVSAHRKAQKAVNLVSFE143 | 2351 | 3 | 2 | Both |
| 121FEKTHTVSAHRKAQKAVNLVSFE143 | 2627 | 3 | 2 | Both |
| 53GTFKYQSRPKFNSTPKYIKFKHDYNILEFND83 | 3826 | 4 | 4 | Efb |
The peptide sequence identified in peptide pool;
the number of acetylated lysine residues observed in unbound protein by MALDI-TOF MS;
the number of acetylated lysine residues observed in bound protein by MALDI-TOF MS;
Samples wherein the specific peptides were observed.
The accurate mass data provided by MALDI-TOF analysis (within 50 ppm by the external calibration) allowed identification of multiple chymotryptic Efb peptides through the peptide mass fingerprinting (PMF) method, 15 of which contained lysines (see Table 1). To further characterize these fragments, LC/ESI tandem MS was employed. Under the helium collision-induced dissociation (CID) mode, abundant b series and y series ions were produced and these resulted in an independent identification all 15 fragments observed in the MALDI-TOF spectrum. For example, fragment NKPAAKTDATIKKEQKL (95-111, m/z 1884) of Efb(-C) was selected for MS/MS analysis by the ESI. Eight out of 16 possible b series and 10 out of 16 possible y series ions were observed and provided unambiguous identification of the sequence. In this fashion, a 15-member pool of chymotryptic peptides that covered 69% of the Efb sequence was identified. To expand the peptide coverage map of Efb, we carried out an analogous set of experiments using V8 protease. Here, 9 lysine-containing fragments were identified by PMF (see Table 1) and these provided independent sequence coverage of 83%. Fortuitously, this 83% of the sequence contained much of the Efb sequence not obtained in the chymotrypsin analysis; only residues 29-31 and 84-87 were not found in any of the experimental peptides shown in Table 1. Together, we obtained 95% sequence coverage of Efb by integrating both proteolytic peptide maps.
With the ability to identify the proteolytic peptides of Efb(-C) produced after chymotrypsin or V8 digestion, we next analyzed the effects of C3d binding on Efb(-C) acetylation at the peptide level (Table 1). Four lysine-containing peptides were found to have different acetylation levels when comparing their mass spectra following chymotrypsin digestion of Efb(-C) labeled in either the presence or absence of C3d,. The MALDI spectrum of one of these fragments is shown in Fig. 4 (A-D). Fragment NKPAAKTDATIKKEQKLIQ AQNL (95-117, m/z 2551) of Efb-C contains five lysine residues. In the acetylation experiments of free Efb-C, we observed a 210 Da mass increase, from m/z 2551 to m/z 2761; this indicated that all five lysine residues were labeled by acetyl groups. However, when C3d was present we observed a fragment m/z 2593, which represented a monoacetylated adduct. That is to say, only one lysine residue was labeled and the other four were protected by binding to C3d. A similar change was detected for peptide NKPAAKTDATIKKEQKL (95-111), which is a part of the aforementioned NKPAAKTDATIKKEQKLIQAQNL peptide. In both cases, equivalent results were attained for two unique Efb peptides that also covered residues 87-111. Differential lysine protection was not observed for any of the remaining Efb(-C) peptides, nor was any protection observed in peptides comprised of residues 31-93 that are unique to Efb.
Fig. 4.
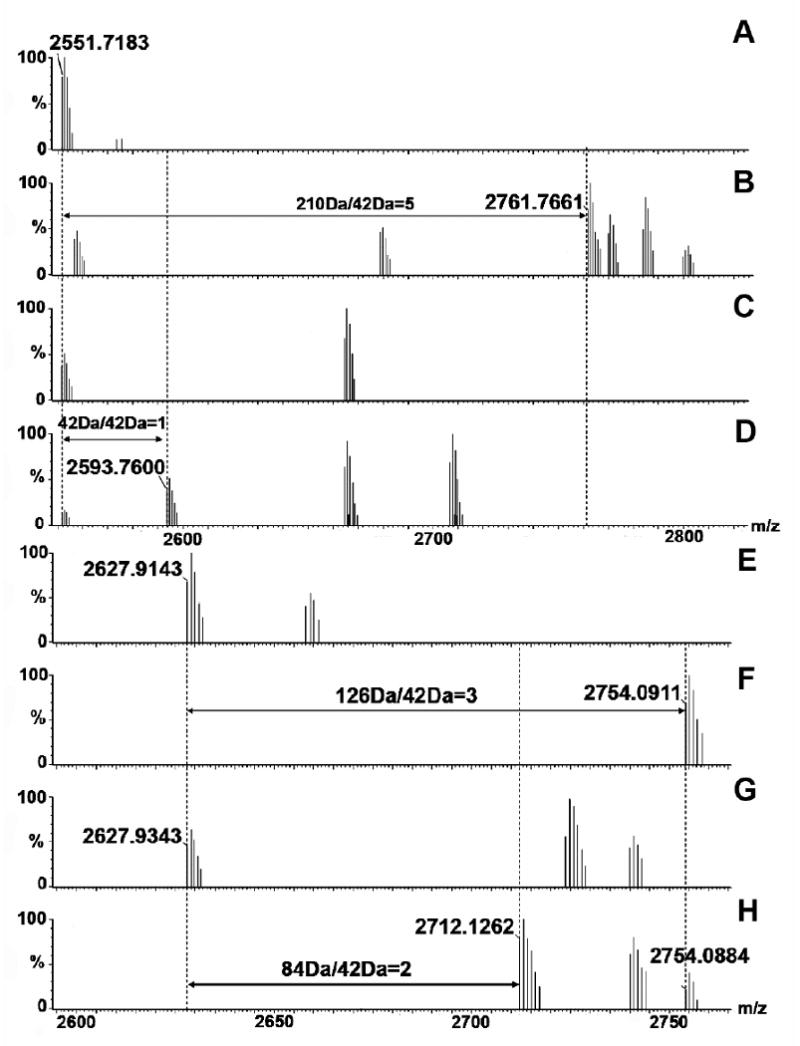
MALDI spectra of peptide fragment NKPAAKTDATIKKEQKLIQAQNL (95-117, m/z 2551, A-D) and fragment FEKTHTVSAHRKAQKAVNLVSFE (120-143, m/z 2627, E-H) of Efb(-C) derived from samples A) free Efb-C without lysine acetylation; B) free Efb-C with lysine acetylation; C) Efb-C/C3d without lysine acetylation; and D) Efb-C/C3d with lysine acetylation. The mass differential of 168 Da (210 Da - 42 Da) in the free protein versus the protein complex indicated that 4 lysine residues in Efb-C were protected upon Efb-C/C3d binding; E) free Efb-C without lysine acetylation; F) free Efb-C with lysine acetylation; G) Efb-C /C3d without lysine acetylation; and H) Efb-C/C3d with lysine acetylation. The mass differential of 42 Da (136 Da - 84 Da) in the free protein versus the protein complex indicated that a single lysine residue in Efb-C was protected upon Efb-C/C3d binding.
In the V8-digestion experiment, we found that five out of 9 lysine-containing fragments from Efb(-C) experienced a reduction in acetylation in the presence of C3d (Table 1). The MALDI spectra of fragment FEKTHTVSAHRKAQKAVNLVSFE (121-143, m/z 2627) are shown in Fig. 4 (E-H). We found that this particular peptide from Efb(-C) was fully acetylated in the absence of C3d, and formed the [M + 3×Acetyl] adduct with a corresponding mass increase to m/z 2754 from m/z 2627. After binding to C3d, we observed a dominant [M + 2×Acetyl] adduct (m/z 2712) in the spectrum, even though a weak [M + 3×Acetyl] peak was still visible. This change demonstrated that one of the three lysine residues in this fragment from Efb(-C) was protected in the presence of C3d. Again, no protection was observed in peptides GYGPREKKPVSINHNIVE (32-49, m/z 2037) or GTFKYQSRPKFNSTPKYIKFKHDYNIL EFND (53-83, m/z 3826) that are comprised of residues unique to Efb.
We used techniques analogous to those described above to perform a reciprocal analysis that monitored the effects of Efb(-C) binding on the acetylation of C3d lysine residues. We observed 18 lysine-containing chymotryptic peptide fragments of C3d by MALDI-TOF. These fragments were characterized by LC-ESI MS/MS sequencing and are shown in Table 2. Two out of 18 fragments experienced a change in their acetylation profile in response to Efb(-C) binding. We found that C3d fragment CGAVKW (1101-1106, m/z 663) was completely protected in the presence of Efb(-C); this same peptide displayed an m/z of 705 Da, which corresponded to the monoacetyl adduct of K1105 in the absence of Efb(-C). For the C3d fragment ISLQEAKDICEEQVNSLPGSITKAGDFL (1149-1176, m/z 3005), we observed an adduct [M + 2×Acetyl] (m/z 3089) that indicated acetylation of both lysine residues when C3d was labeled in the absence of Efb(-C). However, in the presence of Efb(-C), only the monoacetyl adduct [M + 1×Acetyl] (m/z 3047) was detected. In contrast to both of these, a third fragment from C3d, VVKVF (1082-1086, m/z 591), was not acetylated in the either the absence or presence of Efb(-C); this suggested that the lysine residue within this fragment is buried within C3d. Indeed, examination of the C3d crystal structure reveals that this lysine residue is hidden within an insulated pocket [32].
Table 2. Experimental lysine-containing chymotrypsin fragments of C3d.
| Sequencea | m/z | Unboundb | Boundc |
|---|---|---|---|
| 1082VVKVF1086 | 591 | 0 | 0 |
| 1101CGAVKW1106 | 663 | 1 | 0 |
| 1070VKRAPSTW1077 | 944 | 1 | 1 |
| 1038GLEKRQGAL1046 | 971 | 1 | 1 |
| 1070VKRAPSTWL1078 | 1057 | 1 | 1 |
| 1283QKDAPDHQEL1292 | 1180 | 1 | 1 |
| 1038GLEKRQGALEL1048 | 1213 | 1 | 1 |
| 1211LTTAKDKNRW1220 | 1232 | 2 | 2 |
| 1028LDETEQWEKF1037 | 1324 | 1 | 1 |
| 1067AAFVKRAPSTWL1078 | 1346 | 1 | 1 |
| 1107LILEKQKPDGVF1118 | 1386 | 1 | 1 |
| 1070VKRAPSTWLTAY1081 | 1392 | 1 | 1 |
| 1134RNNNEKDMALTAF1146 | 1523 | 1 | 1 |
| 1212TTAKDKNRWEDPGKQL1227 | 1886 | 3 | 3 |
| 1211LTTAKDKNRWEDPGKQL1227 | 2000 | 3 | 3 |
| 1212TTAKDKNRWEDPGKQLY1228 | 2050 | 3 | 3 |
| 1110EKQKPDGVFQEDAPVIHQEMIGGL1133 | 2662 | 2 | 2 |
| 1149ISLQEAKKDICEEQVNSLPGSITKAGDFL1176 | 3005 | 2 | 1 |
The peptide sequence identified in peptide pool;
the number of acetylated lysine residues observed in unbound protein by MALDI-TOF MS;
the number of acetylated lysine residues observed in bound protein by MALDI-TOF MS.
Efb forms a nanomolar-affinity complex with native C3, and in doing so induces a conformational change that blocks formation and deposition of C3b on activated surfaces. According to our previous studies, the C3-inhibitory domain of Efb, or Efb-C, is comprised of three canonical α helices (N-terminal α1 helix from K106 to H125, short loop α2 helix from V127 to L139 and C-terminal α3 helix from K145 to Q161) and a randomly coiled terminus from G162 to R165 [26]. As depicted in Fig. 5, each of the Efb-C helices donates one or more residues that contact C3d. However, there was no interpretable electron density for residues F94 to K100 of Efb-C in the Efb-C/C3d cocrystal, and so this sequence was not present in the refined model. This raised questions as to the structure of the N terminally-directed regions of the Efb molecule, and whether residues in this domain made any contributions to C3d binding. In the present work, we have used a lysine acetylation and mass spectrometry method to characterize the solvent-exposure profile of the Efb(-C)/C3d complex. This study not only identified additional sites of protein-protein interaction involving two lysine residues invisible in the Efb-C/C3d crystal structure, but also provided an independent assessment of the Efb-C/C3d structure in two physical states. Our analysis highlights the complementary nature of liquid- and solid-phase techniques in studying protein structure and interactions.
Fig. 5.
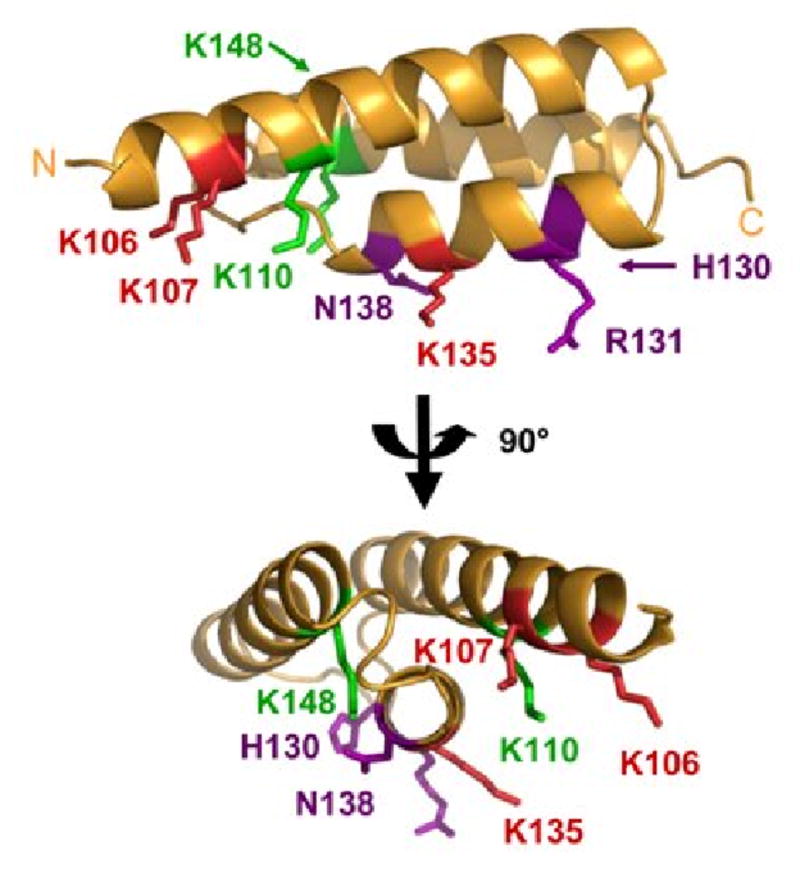
Solution contacts of Efb(-C) and C3d mapped onto the Efb-C crystal structure. The results of differential lysine acetylation studies for free and bound Efb(-C) are superimposed on the Efb-C crystal structure (orange ribbon); two orthogonal views are shown and were generated by rotating in the plane of the page. Residues K106, K110, H130, R131, K135, N138 and K148 of Efb-C were previously identified by x-ray crystallography as contributing to formation of Efb-C/C3d. K110 was solvent accessible in fragment QKLIQAQNLVRE (109-119), and K148 was fully acetylated in the fragment KVKKMVLQERIDNVL (145-159), both of which were derived from the Efb-C/C3d complex. K110 and K148 are therefore shown in green since they do not appear to interact strongly with C3d. Oppositely, K106 was protected in fragment NKPAAKTDATIKKEQKLIQAQNL (95-117) and K135 was protected in the fragment FEKTHTVSAHRKAQKAVNLVSFE (121-143) obtained from the complex. Thus, the five lysine residues that are protected in the protein complex are (shown in red): K96, K100, K106, K107 and K135. Since K96 and K100 lie within a disordered region of the Efb-C structure, these residues cannot be displayed. H130, R131, and N138 are shown in purple since these sidechains do not react with NHS-Ac.
According to the linear MALDI data five Efb(-C) lysine residues were protected upon binding to C3d. The locations of these five lysines were revealed by subsequent proteomic studies. In chymotryptic fragments NKPAAKTDATIKKEQKL (95-111) and NKPAAKTDATIKKEQKLIQAQNL (95-117), 4 out of 5 lysine residues were protected from acetylation in the presence of C3d. Our finding that the V8-derived fragment QKLIQAQNLVRE (109-120) was fully acetylated independently of C3d demonstrated that K110 was the unmodified lysine in these chymotryptic peptides. The remaining protected lysine residue was found in fragments KTHTVSAHRKAQKAVNLVSFE (123-143) and FEKTHTVSAHRKAQKAVNL VSFE (121-143). Of these, residue K135 is the most plausible candidate since its sidechain amine appears to participate in a salt bridge with the D1156 carboxylate of C3d in the crystal structure [26].
Our present work has identified the five Efb(-C) lysine residues protected by C3d binding as K96, K100, K106, K107 and K135. Two of these lysines, K96 and K100, were invisible in the Efb-C/C3d structure. Separately, the we also identified a lysine residue within C3d, namely K1105 of fragment CGAVKW (1101-1106), that was protected from acetylation in the presence of Efb(-C). Together, these results support the hypothesis that additional contacts may form between Efb and C3d. Correlation of the solution mapping studies presented here with the available cocrystal structure suggests that Efb residues 84-101 are in contact with C3d near the vicinity of residue K1105 (see Fig. 6). Given the geometry of the Efb-C/C3d complex, we feel that the most likely explanation for this is that the N-terminally directed region of Efb extends downward to allow binding of Efb residues 84-101 with the residue(s) around K1105, or even with K1105 directly. Such a model appears to explain not only the strong protection from acetylation of Efb(-C) fragment NKPAAKTDATIKKEQKL (95-111), but also the additional protected lysine in C3d following complex formation. While the precise role of these additional contributions remains to be established, it is interesting to note that binding of Efb to C3d has been shown to release additional enthalpy when compared to Efb-C (-9.0 vs. -8.5 kcal/mol; [26]). Thus, even though a portion of the additional residues that bind C3d are found in Efb-C, the dynamics of the truncated protein may be altered in such a way that precludes their forming a stable interface that contributes significantly to binding energy (as mentioned above) [26].
Fig. 6.
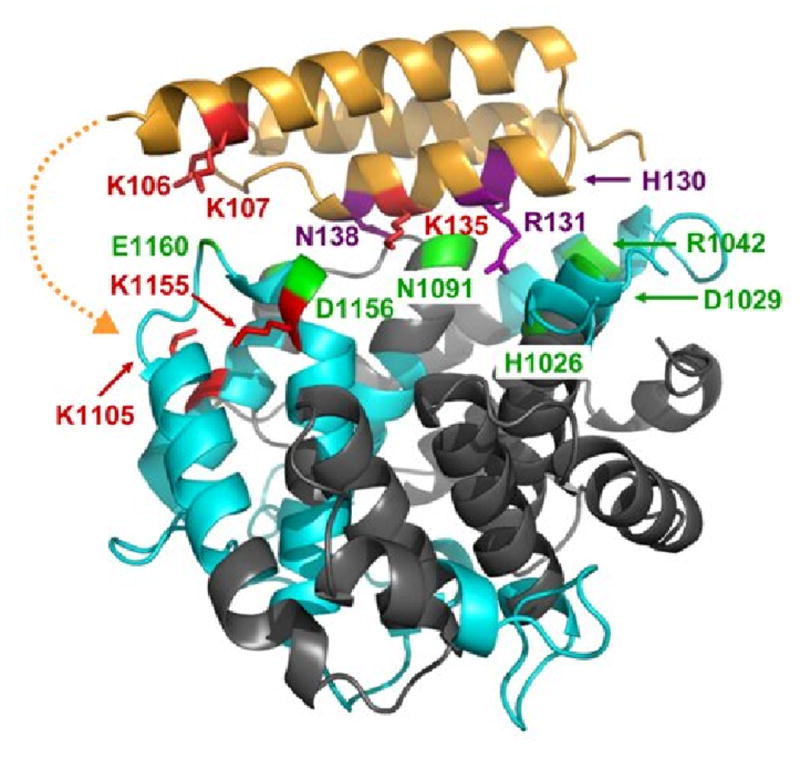
Interaction sites of C3d binding to Efb(-C) as determined by differential lysine acetylation. Efb-C is drawn as in Fig. 6, while C3d is shown in ribbons colored according to regions of peptide coverage as described in Table 2 (blue ribbon) and uncovered regions (slate). Selected contacting residues of C3d which bind to Efb-C according to x-ray crystallography are shown in green. These are H1026, D1029, R1042, N1091, S1097, D1156 and E1160. K1155 in fragment ISLQEAKDICEEQ VNSLPGSITKAGDFL (1149-1176) was protected in Efb-C/C3d, implying that D1156 may contact Efb-C. In addition, K1105 in fragment CGAVKW (1101-1106) was protected from acetylation in the complex. This suggested that portions of Efb(-C) not present in the crystal structure model may also be contacting C3d near residue K1105. The potential location for additional contacts invisible in the Efb-C/C3d crystal structure is depicted by a dashed orange line. No evidence was obtained regarding the role of R1042 in the Efb-C/C3d complex, since K1041 in fragment GLEKRQGAL (1038-1046) was solvent accessible in the complex. Similarly, no further information was available for H1026, D1029 and N1091, as no identified peptides covered this sequence, nor were there any lysine residues in these regions.
Conclusions
By combining the data from the lysine acetylation studies of Efb(-C), C3d and their binary complexes, we have provided new insight on the solvent-accessibility of the full-length Efb protein and how this changes upon formation of a highly specific protein-protein interaction with C3d. The results of current analysis have revealed the presence of additional interactions that further our molecular level understanding of the Efb/C3 complex beyond what was provided by X-ray crystallography. Although the contribution of these additional interactions is a topic of ongoing investigation, it is critical that all such sites of interaction between these proteins are identified if we are to completely understand the structure/function relationships for this class of bacterial immune evasive molecules. Since the mechanism of Efb-mediated inactivation of C3 involves the induction of conformational change, it is entirely possible that apparently subtle and/or transient interactions could have dramatic consequences on the molecular dynamics that are known to contribute greatly to the many complex functions of C3. In this regard, it is worth noting that we have described the potential for additional interactions between the α3 helix of Efb(-C) and the β-chain of native C3 [26], and suggested that these may be important to the conformational effects of Efb(-C) binding on C3 structure. The results we present here have established the method of the lysine acetylation combined with proteolytic digestion and mass spectrometric analysis as a valuable tool for studying the Efb(-C)/C3 interactions in solution. Clearly, analogous studies on the C3 complex will be more involved than those reported here for C3d, since native C3 contains greater than six-times the residues of C3d. However, incorporation of the higher resolution LC-ESI-MS/MS analytical platform is likely to make these differential-labeling approaches more practicable.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health Grant AI-30040 and the School of Biological Sciences of the University of Missouri-Kansas City. We also thank Dr. Elisabetta Fasella who provided 1H NMR analysis of NHS-Ac and Dr. Daniel Riclin for his comments. Representations of protein structures were drawn using PyMol (http://pymol.source forge.net/)
Footnotes
Efb, Extracellular Fibrinogen-binding Protein from S. aureus; Efb-C, the complement inhibitory domain from S. aureus Efb; Efb(-C), Efb and Efb-C collectively; C3, complement component 3; C3d; the thioester-containing domain from C3. MS, mass spectrometry. ESI, electrospray ionization mass spectrometry.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Borch J, Jørgensen TJD, Roepstorff P. Mass spectrometric analysis of protein interactions. Curr Opin Chem Biol. 2005;9:509–516. doi: 10.1016/j.cbpa.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 2.Suckau D, Mak M, Przybylski M. Protein surface topology-probing by selective chemical modification and mass spectrometric peptide mapping. Proc Natl Acad Sci USA. 1992;89:5630–5634. doi: 10.1073/pnas.89.12.5630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kvaratskhelia M, Miller JT, Budihas SR, Pannell LK, Le Grice SF. Identification of specific HIV-1 reverse transcriptase contacts to the viral RNA:tRNA complex by mass spectrometry and a primary amine selective reagent. Proc Natl Acad Sci USA. 2002;99:15988–15993. doi: 10.1073/pnas.252550199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Janecki DJ, Beardsley RL, Reilly JP. Probing Protein Tertiary Structure with Amidination. Anal Chem. 2005;77:7274–7281. doi: 10.1021/ac050891z. [DOI] [PubMed] [Google Scholar]
- 5.Scholten A, Visser NF, van den Heuvel RH, Heck AJ. Analysis of Protein-Protein Interaction Surfaces Using a Combination of Efficient Lysine Acetylation and nanoLC-MALDI-MS/MS Applied to the E9:Im9 Bacteriotoxin—Immunity Protein Complex. J Am Soc Mass Spectrom. 2006;17:983–994. doi: 10.1016/j.jasms.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 6.Edavettal SC, Carrick K, Shah RR, Pedersen LC, Tropsha A, Pope RM, Liu J. A Conformational Change in Heparan Sulfate 3-O-Sulfotransferase-1 Is Induced by Binding to Heparan Sulfate. Biochemistry. 2004;43:4680–4688. doi: 10.1021/bi0499112. [DOI] [PubMed] [Google Scholar]
- 7.Lowy FD. Staphylococcus aureus infections. N Engl J Med. 1998;339:520–532. doi: 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- 8.Sandre RM, Shafran SD. Infective endocarditis: Review of 135 cases over 9 years. Clin Infect Dis. 1996;22:276–286. doi: 10.1093/clinids/22.2.276. [DOI] [PubMed] [Google Scholar]
- 9.Hudson MC, Ramp WK, Frankenburg KP. Staphylococcus aureus adhesion to bone matrix and bone-associated biomaterials. FEMS Microbiol Lett. 1999;173:279–284. doi: 10.1111/j.1574-6968.1999.tb13514.x. [DOI] [PubMed] [Google Scholar]
- 10.Bone RC. Gram-positive organisms and sepsis. Arch Intern Med. 1994;154:26–34. [PubMed] [Google Scholar]
- 11.Romero-Vivas J, Rubio M, Fernandez C, Picazo JJ. Mortality associated with nosocomial bacteremia due to methicillin-resistant Staphylococcus aureus. Clin Infect Dis. 1995;21:1417–1423. doi: 10.1093/clinids/21.6.1417. [DOI] [PubMed] [Google Scholar]
- 12.Lee LY, Miyamoto YJ, McIntyre BW, Hook M, McCrea KW, McDevitt D, Brown EL. The Staphylococcus aureus Map protein is an immunomodulator that interferes with T cell-mediated responses. J Clin Invest. 2002;110:1461–1471. doi: 10.1172/JCI16318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chavakis T, Hussain M, Kanse SM, Peters G, Bretzel RG, Flock JI, Herrmann M, Preissner KT. Staphylococcus aureus extracellular adherence protein (Eap) serves as anti-inflammatory factor by inhibiting the recruitment of host leukocytes. Nat Med. 2002;8:687–693. doi: 10.1038/nm728. [DOI] [PubMed] [Google Scholar]
- 14.Rooijakkers SH, Ruyken M, Roos A, Daha MR, Presanis JS, Sim RB, van Wamel WJ, van Kessel KP, van Strijp JA. Immune evasion by a staphylococcal complement inhibitor that acts on C3 convertases. Nat Immunol. 2005;6:920–927. doi: 10.1038/ni1235. [DOI] [PubMed] [Google Scholar]
- 15.Song W, Sarrias MR, Lambris JD. Complement and innate immunity. Immunopharmacology. 2000;49:187–198. doi: 10.1016/s0162-3109(00)80303-3. [DOI] [PubMed] [Google Scholar]
- 16.Sahu A, Lambris JD. Structure and biology of complement protein C3, a connecting link between innate and acquired immunity. Immunol Rev. 2001;180:35–48. doi: 10.1034/j.1600-065x.2001.1800103.x. [DOI] [PubMed] [Google Scholar]
- 17.Wilkinson BJ, Kim Y, Peterson PK, Quie PG, Michael AF. The key role of peptidoglycan in the opsonisation of Staphylococcus aureus. Infect Immun. 1978;20:388–392. doi: 10.1128/iai.20.2.388-392.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Verbrugh HA, Van Dijk WC, Peters R, Van Der Tol ME, Verhoef J. The role of Staphylococcus aureus cell-wall peptidoglycan, teichoic acid and protein A in the processes of complement activation and opsonization. Immunology. 1979;37:615–621. [PMC free article] [PubMed] [Google Scholar]
- 19.Kawasaki A, Takada H, Kotani S, Inai S, Nagaki K, Matsumoto M, Yokogawa K, Kawata S, Kusumoto S, Shiba T. Activation of the human complement cascade by bacterial cell walls, peptidoglycans, water-soluble peptidoglycan components, and synthetic muramylpeptides--studies on active components and structural requirements. Microbiol Immunol. 1987;31:551–569. doi: 10.1111/j.1348-0421.1987.tb03117.x. [DOI] [PubMed] [Google Scholar]
- 20.Bredius RG, Driedijk PC, Schouten MF, Weening RS, Out TA. Complement activation by polyclonal immunoglobulin G1 and G2 antibodies against Staphylococcus aureus, Haemophilus influenzae type b, and tetanus toxoid. Infect Immun. Infect Immun. 1992;60:4838–4847. doi: 10.1128/iai.60.11.4838-4847.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neth O, Jack DL, Johnson M, Klein NJ, Turner MW. Enhancement of complement activation and opsonophagocytosis by complexes of mannose-binding lectin with mannose-binding lectin-associated serine protease after binding to Staphylococcus aureus. J Immunol. 2002;169:4430–4436. doi: 10.4049/jimmunol.169.8.4430. [DOI] [PubMed] [Google Scholar]
- 22.Sakiniene E, Bremell T, Tarkowski A. Complement depletion aggravates Staphylococcus aureus septicaemia and septic arthritis. Clin Exp Immunol. 1999;115:95–102. doi: 10.1046/j.1365-2249.1999.00771.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee LYL, Liang X, Hook M, Brown EL. Identification and Characterization of the C3 Binding Domain of the Staphylococcus aureus Extracellular Fibrinogen-binding Protein. J Biol Chem. 2004;279:50710–50716. doi: 10.1074/jbc.M408570200. [DOI] [PubMed] [Google Scholar]
- 24.Liang X, Yu C, Sun J, Liu H, Landwehr C, Holmes D, Ji Y. Inactivation of a Two-Component Signal Transduction System, SaeRS, Eliminates Adherence and Attenuates Virulence of Staphylococcus aureus. Infect Immun. 2006;74:4655–4665. doi: 10.1128/IAI.00322-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Palma M, Nozohoor S, Schennings T, Heimdahl A, Flock JI. Lack of the extracellular 19-kilodalton fibrinogen-binding protein from Staphylococcus aureus decreases virulence in experimental wound infection. Infect Immun. 1996;64:5284–5289. doi: 10.1128/iai.64.12.5284-5289.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hammel M, Sfyroera G, Magotti P, Lambris JD, Geisbrecht BV. Structural Basis for a Novel Mechanism of Complement Inhibition by Staphylococcus aureus. Nat Immunol. 2007;8:430–437. doi: 10.1038/ni1450. [DOI] [PubMed] [Google Scholar]
- 27.Seo J, Martasek P, Roman LJ, Silverman RB. Selective l-nitroargininylaminopyrrolidine and l-nitroargininylaminopiperidine neuronal nitric oxide synthase inhibitors. Bioorg Med Chem. 2007;15:1928–1938. doi: 10.1016/j.bmc.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Konermann L, Simmons DA. Protein-folding kinetics and mechanisms studied by pulse-labeling and mass spectrometry. Mass Spectrometry Reviews. 2003;22:1–26. doi: 10.1002/mas.10044. [DOI] [PubMed] [Google Scholar]
- 29.Katta V, Chait BT. Observation of the heme-globin complex in native myoglobin by electrospray-ionization mass spectrometry. J Am Chem Soc. 1991;113:8534–8535. [Google Scholar]
- 30.Fenn JB. Ion formation from charged droplets: Roles of geometry, energy, and time. J Am Soc Mass Spectrom. 1993;4:524. doi: 10.1016/1044-0305(93)85014-O. [DOI] [PubMed] [Google Scholar]
- 31.Konermann L, Douglas DJ. Acid-Induced Unfolding of Cytochrome c at Different Methanol Concentrations: Electrospray Ionization Mass Spectrometry Specifically Monitors Changes in the Tertiary Structure. Biochemistry. 1997;36:12296–12302. doi: 10.1021/bi971266u. [DOI] [PubMed] [Google Scholar]
- 32.Nagar B, Jones RG, Diefenbach RJ, Isenman DE, Rini JM. X-ray crystal structure of C3d: a C3 fragment and ligand for complement receptor 2. Science. 1998;280:1277–1281. doi: 10.1126/science.280.5367.1277. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.