

Abstract
Glucocorticoids exert an opposing rapid regulation of glutamate and GABA synaptic inputs to hypothalamic magnocellular neurons via the activation of postsynaptic membrane-associated receptors and the release of retrograde messengers. Glucocorticoids suppress synaptic glutamate release via the retrograde release of endocannabinoids and facilitate synaptic GABA release via an unknown retrograde messenger. Here, we show that the glucocorticoid facilitation of GABA inputs is due to the retrograde release of neuronal nitric oxide and that glucocorticoid-induced endocannabinoid synthesis and nitric oxide synthesis are mediated by divergent G-protein signaling mechanisms. While the glucocorticoid-induced, endocannabinoid-mediated suppression of glutamate release is dependent on activation of the Gαs G-protein subunit and cAMP–cAMP-dependent protein kinase activation, the nitric oxide facilitation of GABA release is mediated by Gβγ signaling that leads to activation of neuronal nitric oxide synthase. Our findings indicate, therefore, that glucocorticoids exert opposing rapid actions on glutamate and GABA release by activating divergent G-protein signaling pathways that trigger the synthesis of, and glutamate and GABA synapse-specific retrograde actions of, endocannabinoids and nitric oxide, respectively. The simultaneous rapid stimulation of nitric oxide and endocannabinoid synthesis by glucocorticoids has important implications for the impact of stress on the brain as well as on neural-immune interactions in the hypothalamus.
Keywords: glucocorticoid, Gβγ, Gα, GABA, nitric oxide, endocannabinoid, magnocellular neuron
Introduction
Glucocorticoids are adrenocorticosteroid hormones that are secreted in response to activation of the hypothalamic–pituitary–adrenal (HPA) axis in a circadian pattern under baseline conditions and at high levels as part of the physiological response to stress. Glucocorticoids exert negative feedback effects on HPA axis activation via both rapid and delayed inhibitory actions at the level of the paraventricular nucleus of the hypothalamus (PVN), the pituitary and the hippocampus (Sapolsky et al., 1990). In addition to these negative feedback effects, glucocorticoids also regulate other neuroendocrine systems, including the hypothalamic-neurohypophysial system responsible for the secretion of the neuropeptides vasopressin and oxytocin from magnocellular neurons of the PVN and hypothalamic supraoptic nucleus (SON) (de Kloet, 2000).
In addition to the classical actions of glucocorticoids responsible for the delayed transcriptional effects that are mediated by the known intracellular corticosteroid receptors and genomic regulation, there is increasing evidence for rapid glucocorticoid actions triggered by the activation of membrane-associated receptors and nongenomic signaling mechanisms (de Kloet, 2000; Haller et al., 2008; Tasker et al., 2006). Our recent studies suggest that glucocorticoids elicit a rapid suppression of glutamatergic excitatory synaptic inputs to neuroendocrine cells of the PVN and SON via the activation of a postsynaptic, membrane-associated receptor and the G-protein-dependent synthesis of retrograde endocannabinoid messengers (Di et al., 2003, 2005b). The glucocorticoid-induced endocannabinoid synthesis is dependent on the Gαs G-protein subunit and the cAMP–cAMP-dependent protein kinase (PKA) signaling pathway (Malcher-Lopes et al., 2006). We also found that glucocorticoids elicit an opposite action on GABA synapses in hypothalamic magnocellular neurons, leading to a facilitation of GABA release via the postsynaptic, G-protein-dependent synthesis of an unknown retrograde messenger (Di et al., 2005b) (for review, see Di and Tasker, 2008).
In the present study, we determined that the glucocorticoid facilitation of GABA release is not dependent on the Gαs-cAMP–PKA pathway, but is caused by the Gβγ-mediated activation of neuronal nitric oxide (NO) synthase and the synthesis of NO. We show, therefore, that glucocorticoids activate divergent G-protein signaling pathways, via the Gαs and Gβγ subunits, respectively, to produce different retrograde messengers, endocannabinoids and NO, which each act in a synapse-specific manner to suppress excitatory synaptic inputs and facilitate inhibitory synaptic inputs, respectively. This glucocorticoid-induced suppression of synaptic excitation and facilitation of synaptic inhibition in the same cells would be expected to cause a rapid functional inhibition of the magnocellular neuroendocrine systems during the stress response.
Materials and Methods
Slice preparation.
Male Sprague Dawley rats (4–6 weeks old, Charles River) were used in these experiments according to a protocol approved by the Tulane University Institutional Animal Care and Use Committee. Hypothalamic slices containing the SON were prepared as described previously (Di et al., 2003). Briefly, rats were decapitated under deep halothane anesthesia and the brain was quickly removed and submerged in a cooled (∼1°C), oxygenated modified artificial CSF (aCSF), in which NaCl was replaced by an equimolar concentration of sucrose, which was used to improve neuronal viability. The standard aCSF contained the following (in mm): 140 NaCl, 3 KCl, 1.3 MgSO4, 1.4 NaH2PO4, 2.4 CaCl2, 11 glucose, and 5 HEPES, bubbled with 100% O2. The hypothalamus was then blocked and, in the same ice-cold, oxygenated, modified aCSF, two coronal hypothalamic slices (300 μm) containing the SON were sectioned and bisected along the midline (i.e., at the third ventricle). The hemi-slices were maintained submerged in a holding chamber in oxygenated standard aCSF at room temperature for ≥1.5 h before being transferred to a submersion recording chamber on a fixed-stage upright microscope (Olympus; BX50WI).
Electrophysiological methods.
Following a 15 min equilibration period in the recording chamber, the slices were visualized using infrared illumination and differential interference contrast (IR-DIC) optics, and whole-cell patch-clamp recordings were performed at 32–34°C using electrodes with a tip resistance of 3–4 MΩ. The Pipette solution used to record EPSCs contained (in mm) 120 K-gluconate, 10 KCl, 1 NaCl, 1 MgCl2, 1 CaCl2, 10 EGTA, 2 Mg-ATP, 0.3 Na-GTP, and 10 HEPES. The internal solution used to record IPSCs contained either (in mm) 110 d-gluconic acid, 110 CsOH, 10 CsCl, 1 MgCl2, 1 CaCl2, 10 EGTA, 2 Mg-ATP, 0.3 Na-GTP, and 10 HEPES; or 120 CsCl, 2 MgCl2, 1 CaCl2, 11 EGTA, 2 Mg-ATP, 0.3 Na-GTP, and 30 HEPES. The latter solution was used for the majority of recordings of IPSCs to reverse the GABAA receptor-mediated currents and increase the Cl− driving force. All recordings were performed in voltage-clamp mode using a Multiclamp 700 amplifier and pCLAMP 9 software (Molecular Devices). Data were low-pass filtered at 2 kHz, digitized at 5–10 kHz and recorded for off-line analysis. To isolate miniature IPSCs (mIPSCs), tetrodotoxin (TTX, 1 μm) was added to the aCSF to block spike-mediated transmitter release, and 6,7-dinitroquinoxaline-2,3-dione (DNQX; 20 μm) and d-(-)-2-amino-5-phosphonopentanoic acid (AP-5; 50 μm) were included to block ionotropic glutamate receptor-mediated synaptic currents. To isolate miniature EPSCs (mEPSCs), TTX (1 μm) and the GABAA receptor antagonist bicuculline methiodide (10 μm) were added to the aCSF. Magnocellular neurons were voltage clamped at either 0 mV or −60 mV (with the high [Cl−]-containing patch solution) to record IPSCs and at −60 mV to record EPSCs. To focus on the fast, nongenomic actions of glucocorticoids, 3-min epochs of mIPSCs and mEPSCs were recorded at 7–10 min of steroid treatment and were compared with 3-min control epochs acquired just before the treatment; mIPSCs and mEPSCs were analyzed for changes in mean frequency, peak amplitude and decay time (defined as the time from peak to 63% decay of the PSCs) using the Minianalysis 6.0 program (Synaptosoft).
Evoked synaptic responses were elicited by stimulation (0.2–0.5 mA, 0.25 ms, 0.05 Hz) with a concentric bipolar electrode placed just dorsal-medial to the SON. Evoked IPSCs (eIPSCs) were recorded in the presence of DNQX (20 μm) and AP-5 (50 μm), and evoked EPSCs (eEPSCs) were recorded in the presence of picrotoxin (50 μm). Mean ePSC amplitudes recorded at 50–70% of maximal amplitude were compared under baseline conditions (3 min before drug treatment) and at 7–10 min of drug application. Evoked IPSCs were also confirmed as GABA-dependent by their blockade with the GABAA receptor antagonist picrotoxin (50 μm).
Magnocellular neurons were identified visually by their relatively large somatic size and their position in the SON under IR-DIC visualization. In a few cells, intracellular labeling with biocytin and post hoc immunohistochemical double labeling with antisera to oxytocin- and vasopressin-associated neurophysins were performed to identify oxytocinergic and vasopressinergic subpopulations of the SON. Since the few identified oxytocin neurons (n = 2) and vasopressin neurons (n = 2) showed similar responses, and the majority of unidentified cells, which were recorded randomly throughout the SON, i.e., with an equal probability of sampling from oxytocinergic and vasopressinergic subpopulations, also responded in a similar manner to dexamethasone (Dex), data from all magnocellular neurons were pooled for analysis.
Nitric oxide measurements.
Magnocellular neurons were loaded individually with an NO-sensitive dye, 4-amino-5-methylamino-2′,7′-difluorofluorescein (DAF-FM; 21 μm; Invitrogen), via the patch pipette. The patch solution was the same as that used to quantify IPSCs, except that the EGTA was replaced by 0.1 mm bis-fura-2. The bath contained DNQX (20 μm) and AP-5 (50 μm) to block glutamate receptors, and dexamethasone (1 μm) and/or l-NAME (50 μm) was applied in the bath during the course of the experiment. Images were recorded at 1 s intervals with a QuantEM:512c camera (Photometrics) in EM gain mode using IP-lab 4.0 software. The magnocellular neuron soma was defined as the region of interest (ROI) with the software and was excited at 488 nm with a DG4 (Sutter) xenon light source for 100 ms of each 1 s cycle, and emitted light was filtered through a 510–550 nm bandpass filter.
Images were analyzed off line with Image J (NIH) using the Time Series Analyzer's recentering plugin to correct for slight translations in cell position during recording. Background fluorescence was subtracted from a similarly sized ROI adjacent to the cell. Mean somatic fluorescence intensity for each time point (F) was divided by the average intensity of a 3-min stable baseline period just before treatment with drugs (F0), and multiplied by 100% to give the percentage change from baseline, ΔF/F0 (%). For presentation, each minute of data was averaged for each cell, then averaged for all cells within a treatment group and plotted (±SEM).
Drug application.
We used the water-soluble form of dexamethasone ((2-hydroxypropyl)-β-cyclodextrin-conjugated dexamethasone) (Sigma-Aldrich) for these experiments. We have characterized the effects of dexamethasone and corticosterone in previous studies and found them to have similar effects (Di et al., 2005b). The EC50 of dexamethasone is ∼500 nm; here we used a saturating dose of dexamethasone, 1 μm. The following drugs were stored as stock solutions at −20°C and were dissolved to their final concentrations in aCSF before their application in the bath perfusion, including TTX (Sigma-Aldrich), the glutamate receptor antagonists DNQX and AP-5 (Tocris), leptin (Sigma), the CB1 receptor antagonists O-2050 (Tocris) and SR141716 (kindly provided by the NIMH Chemical Synthesis and Drug Supply Program), the NO precursor l-arginine and the NO donor SNAP (S-nitroso-N-acetylpenicillamine), the nonselective NO synthase (NOS) inhibitor l-NAME (Nω-Nitro-l-arginine methyl ester hydrochloride) and the selective neuronal NOS (nNOS) inhibitor N-propyl-l-arginine (NPLA) (Tocris), and the N-myristoylated G-protein βγ-binding peptide, mSIRK (myr-SIRKALNILGYPDYD-OH) and its negative control mSIRK(L9A) (myr-SIRKALNIAGYPDYD-OH) (Santa Cruz Biotechnology). The nonhydrolyzable guanylyl nucleotide GDP-β-S (Sigma-Aldrich), the cAMP–PKA antagonist (R)-adenosine, cyclic 3′, 5′-(hydrogenphosphorothioate) triethylammonium (cAMPS-Rp) (Tocris), and the Gαs and Gβ polyclonal antibodies (Santa Cruz Biotechnology) were included in the patch solution for intracellular application.
Data analysis.
All data are expressed as means ± SEs. Statistical comparisons of electrophysiological data were performed using the Student's paired t test for within-group cell comparisons and the Student's unpaired t test for between-group comparisons. The one-way repeated measures ANOVA was used for multiple comparisons and followed by the Student-Newman–Keuls test as needed. Probability values <0.05 were considered significant for all comparisons.
Results
Glucocorticoid facilitation of synaptic inhibition
Bath application of the synthetic glucocorticoid agonist Dex (1 μm) caused an increase in the amplitude of IPSCs evoked by electrical stimulation (eIPSCs) in 7 of 7 SON neurons tested (134.7 ± 37.9% of baseline; p < 0.05; n = 7). In contrast, Dex (1 μm) caused a decrease in the amplitude of eEPSCs in 5 of 6 SON neurons tested (75.8 ± 3.3% of baseline; p < 0.05; n = 5) (Fig. 1A,B). Dexamethasone (1 μm) also caused an increase in the frequency of mIPSCs (127.1 ± 5.9% of baseline; p < 0.01; n = 6) within 3–5 min of introduction into the bath perfusion, without affecting mIPSC amplitude or decay time (Fig. 1C,D). Dex (1 μm) caused a significant decrease in the frequency of mEPSCs (65.6 ± 11.4% of baseline; p < 0.05; n = 7) without affecting mEPSC amplitude or decay time (data not shown). This confirmed our previous findings of glucocorticoid facilitation of presynaptic GABA release and suppression of glutamate release (Di et al., 2005b).
Figure 1.

Opposing rapid actions of glucocorticoids on glutamate and GABA release onto magnocellular neurons. A, Dex (1 μm) caused an increase in the amplitude of evoked IPSCs and a decrease in the amplitude of evoked EPSCs. Traces are averages of 7–9 consecutive evoked PSCs over 3 min before and after 7 min of Dex application. GABA currents are inward due to high intracellular [Cl−] (see Materials and Methods). B, Histogram of mean evoked IPSC and EPSC amplitudes (Amp) before and after 7 min of Dex (1 μm). Numbers in parentheses represent numbers of cells analyzed in this and the following figures. *p < 0.05; **p < 0.01 in this and subsequent figures. C, Recording of mIPSCs before and after 7–10 min of Dex application (1 μm) showing Dex-induced increase in mIPSC frequency. Lower inset traces are time expanded from designated segments of upper traces. GABA currents are outward. D, Normalized running average (1 min bin width) of mIPSC frequency (Freq), amplitude, and decay time (n = 6). Dexamethasone induced a rapid increase in mIPSC frequency, but had no effect on mIPSC amplitude and decay time.
Glucocorticoid facilitation of synaptic inhibition is not mediated by endocannabinoids
We previously reported paradoxical findings that glucocorticoid facilitation of GABA release was blocked by the selective CB1 receptor antagonist AM251 (1 μm), but it was not mimicked by the CB1 receptor agonists WIN 55,212-2, anandamide or arachidonoylglycerol (Di et al., 2005b), indicating an AM251-sensitive, cannabinoid-insensitive mechanism. Here, we tested two other selective CB1 antagonists, SR141716 and O-2050, on the glucocorticoid-induced facilitation of GABA release. Bath application of SR141716 (1 μm) increased the basal frequency of mIPSCs to 115.0 ± 5.6% of baseline (p < 0.05; n = 6), but failed to block the glucocorticoid effect, since subsequent bath application of Dex (1 μm) in the presence of SR141716 further increased the mIPSC frequency to 131.3 ± 6.3% compared with baseline (p < 0.01) and to 114.2 ± 1.2% compared with the frequency in SR141716 (p < 0.01; n = 6) (Fig. 2). Bath application of O-2050 (1 μm) showed a similar effect, increasing basal mIPSC frequency to 114.9 ± 5.0% of baseline (p < 0.05; n = 5) and failing to block the Dex-induced increase in mIPSC frequency (128.0 ± 7.4% vs baseline; p < 0.01; 112.6 ± 3.9% vs O-2050; p < 0.05; n = 5) (Fig. 2). These results indicate that the glucocorticoid-induced facilitation of GABA release is mediated by an endocannabinoid-independent mechanism, and suggest that AM251 has nonspecific actions on glucocorticoid-induced GABA release that do not depend on CB1 activation.
Figure 2.

Glucocorticoid effect on GABA synaptic inputs was not mediated by endocannabinoids. Summary histograms show the mIPSC frequency (Freq) response to Dex in the presence of cannabinoid receptor antagonists. The CB1 antagonists SR141716 (1 μm) and O-2050 (1 μm) failed to inhibit the Dex-induced increase in mIPSC frequency. The CB1 antagonists increased the basal mIPSC frequency, suggesting an endocannabinoid inhibitory tone on GABA release.
For the purpose of comparison, we repeated previous experiments to confirm the sensitivity of the glucocorticoid-induced suppression of synaptic excitation to blockade of CB1 receptors (Di et al., 2005b). Bath application of the CB1 receptor antagonist SR141716 (1 μm) abolished the Dex-induced decrease in mEPSC frequency (data not shown). Our results show, therefore, that unlike the glucocorticoid-induced suppression of glutamate release, the glucocorticoid-induced facilitation of GABA release is not mediated by an endocannabinoid-dependent mechanism.
Glucocorticoid activation of divergent G-protein signaling pathways
We showed previously that both the rapid glucocorticoid-induced facilitation of synaptic inhibition and the rapid glucocorticoid-induced suppression of synaptic excitation in magnocellular neurons are mediated by the activation of one or more membrane-associated glucocorticoid receptors (Di et al., 2005b). Both effects are dependent on G-protein activation, since they are abolished by blocking postsynaptic G-protein activity. We also showed that the glucocorticoid-induced suppression of glutamate release is mediated by activation of a Gs-cAMP–PKA signaling pathway (Malcher-Lopes et al., 2006). Here, we determined whether the opposing action of glucocorticoids on GABA release relies on the same downstream G-protein-dependent signaling pathway. We first tested for the G-protein subunit dependence of the rapid Dex facilitation of GABA release. Intracellular infusion through the patch pipette of an antiserum against the Gαs protein subunit (1:1000, Santa Cruz Biotechnology) failed to block the Dex (1 μm)-induced increase in mIPSC frequency (115.6 ± 10.6% of control; p < 0.05; n = 7) (Fig. 3A,B). This suggested that the Gαs subunit is not necessary for the glucocorticoid regulation of GABA release onto magnocellular neurons. We then tested whether the glucocorticoid facilitation of GABA release is coupled to the G-protein βγsubunit. A Gβ antiserum introduced through the recording pipette (1:1000; Santa Cruz Biotechnology) blocked the Dex-induced increase in mIPSCs (100.6 ± 2.4% of control; p = 0.63; n = 6) (Fig. 3A,B). Additionally, selective activation of the Gβγ subunit by bath application of the G-protein βγ-binding peptide mSIRK (0.5 μm) in the absence of glucocorticoid elicited an increase in mIPSC frequency (117.5 ± 3.7%; p < 0.5; n = 8) similar to the Dex-induced increase in mIPSC frequency (Fig. 3B). It also occluded the Dex-induced facilitation of GABA release, since subsequent Dex application (1 μm) in the continued presence of mSIRK failed to further increase mIPSC frequency (104.8 ± 3.0% of mSIRK value, p = 0.15, n = 8) (Fig. 3B). The specificity of the mSIRK effect was confirmed with application of the inactive isomer, mSIRK(L94), used as a negative control. mSIRK(L94) (0.5 μm) had no effect on the baseline mIPSC frequency by itself (101.6 ± 3.7%; n = 5) and did not block the Dex-induced facilitation of mIPSC frequency (119.1 ± 4.9% in the presence of mSIRK(L94); p < 0.05; n = 4) (Fig. 3B). We further tested for a PKA dependence of the rapid glucocorticoid facilitation of inhibitory synaptic inputs. Intracellular application through the patch pipette of a PKA antagonist, cAMPS-Rp (50 μm), failed to block the Dex (1 μm)-induced increase in mIPSC frequency (113.1 ± 3.2% of control; p < 0.05; n = 11) (Fig. 3B). The Dex effect on mIPSC frequency appeared to be partially attenuated by the PKA inhibitor; however, the difference in the Dex effect with and without the internal PKA blocker was not significant. Therefore, the rapid glucocorticoid regulation of GABA release is not mediated by activation of a Gαs-cAMP–PKA signaling pathway.
Figure 3.

Distinct G-protein subunit dependence of glucocorticoid regulation of GABA synaptic inputs. A, Normalized running average of mIPSC frequency (Freq). The increase in mIPSC frequency elicited by Dex (1 μm) using a control pipette solution (n = 6) was abolished when the intracellular pipette solution contained a Gβ antibody (n = 6), but was unaffected by intracellular application of a Gαs antibody (n = 7). B, Summary histogram of the effect of Dex on mean mIPSC frequency in the presence of G-protein subunit antagonists, agonist and a PKA inhibitor. The Dex-induced increase in mIPSC frequency was not blocked by the Gαs antibody, but was abolished by the Gβ antibody. The selective Gβ-binding protein, mSIRK (0.5 μm), mimicked and occluded the Dex effect on mIPSC frequency, and the inactive mSIRK isomer, mSIRK(L94) (0.5 μm), had no effect on basal mIPSC frequency or on the Dex facilitation of mIPSC frequency. The PKA inhibitor cAMPS-Rp (50 μm) had no effect on the Dex-induced increase in mIPSC frequency. C, Summary histogram of the effect of Dex on mean mEPSC frequency in the presence of G-protein subunit antibodies and a PKA inhibitor. The Dex-induced decrease in mEPSC frequency was not affected by intracellular application of the Gβ antibody, but was abolished by the Gαs antibody and PKA inhibitor cAMPS-Rp.
In contrast, intracellular application of the Gαs antiserum, as well as a PKA antagonist, cAMPS-Rp, blocked the Dex (1 μm)-induced decrease in mEPSC frequency (99.4 ± 6.1% of control, n = 6; and 95.0 ± 4.7% of control; n = 8, respectively) (Fig. 3C), confirming our previous findings (Malcher-Lopes et al., 2006), whereas intracellular application of the Gβ antiserum did not block the Dex effect on mEPSCs (74.1 ± 6.0% of baseline; p < 0.05; n = 5) (Fig. 3C). These results demonstrated, therefore, that the Gαs-cAMP–PKA signaling pathway is necessary for the glucocorticoid regulation of glutamate release on to magnocellular neurons. Together, our findings indicate that the rapid glucocorticoid suppression of glutamate release and facilitation of GABA release are dependent on different G-protein subunits and suggest a divergence in the G-protein signaling pathways responsible for the rapid, synapse-specific glucocorticoid regulation of excitatory and inhibitory synaptic inputs to SON neurons.
The glucocorticoid-induced suppression of glutamate release is blocked by leptin, which is mediated by phosphodiesterase 3B inhibition of cAMP activity (Malcher-Lopes et al., 2006). Here we tested the effects of leptin on the glucocorticoid-induced facilitation of inhibitory synaptic inputs. Bath application of leptin (10 nm) had no effect on basal mIPSCs (102.26 ± 1.5% of baseline, p = 0.19; n = 6) and it failed to block the Dex-induced increase in mIPSC frequency (116.6 ± 2.4% vs leptin, 118.6 ± 5.4% vs baseline; p < 0.05; n = 6) (Fig. 4A,B). In contrast, leptin (10 nm) completely blocked the glucocorticoid-induced decrease in mEPSC frequency (99.6 ± 8.0% of baseline; p = 0.18; n = 5) (Fig. 4A), confirming our previous findings (Malcher-Lopes et al., 2006). Because leptin inhibition of the glucocorticoid-induced suppression of mEPSCs is mediated by inhibition of cAMP production, the lack of inhibitory effect of leptin on the glucocorticoid-induced facilitation of mIPSCs further indicates that the glucocorticoid-induced facilitation of GABA release works independently of the Gs/cAMP/PKA signaling mechanism.
Figure 4.

Distinct sensitivity to leptin of glucocorticoid modulation of glutamate release and GABA release. A, Normalized running average (1 min bin width) of mPSC frequency (Freq). Leptin (10 nm) blocked the Dex (1 μm)-induced decrease in mEPSC frequency (n = 5), but failed to block the Dex-induced increase in mIPSC frequency (n= 6). B, Summary histogram of mean mIPSC frequency response to leptin and Dex. Leptin had no effect on either the basal mIPSC frequency or the Dex-induced increase in mIPSC frequency.
Glucocorticoid-induced facilitation of GABA release is mediated by nitric oxide
Nitric oxide has been shown to act as a retrograde messenger in the regulation of GABA inputs to magnocellular neurons (Bains and Ferguson, 1997; Stern and Ludwig, 2001), and the PVN and SON express high levels of the NO synthetic enzyme, NOS (Nylen et al., 2001; Stern and Zhang, 2005). We tested, therefore, for a role of NO in the rapid glucocorticoid facilitation of GABA release. Bath application of l-NAME (50 μm), a subtype-nonselective NOS inhibitor, was without effect by itself on eIPSCs (102.7 ± 4.3% of baseline, n = 5) but completely blocked the increase in the eIPSC amplitude by Dex (1 μm) (95.4 ± 6.9% of baseline value; 92.9 ± 5.5% of l-NAME value; p = 0.38; n = 5) (Fig. 5A,B). Similarly, l-NAME had no effect on basal mIPSC frequency, but it completely blocked the increase in mIPSC frequency induced by Dex (1 μm) (92.8 ± 3 0.9% of baseline value; 102.1 ± 3.5% of l-NAME value, n = 8) (Fig. 5C,D,E). The NO production in response to Dex appeared to be of neuronal origin because the Dex effect on mIPSCs was also blocked by NPLA (1 μm), a selective neuronal NOS inhibitor (96.2 ± 7.7% of baseline value; 97.6 ± 1.7% of NPLA value, n = 4) (Fig. 5E). The same application of l-NAME (50 μm), however, did not block the Dex-induced decrease in mEPSC frequency (72.9 ± 4.9%; p < 0.5; n = 7) (data not shown) which corroborates our previous findings of the NO independence of the glucocorticoid suppression of glutamate release (Di et al., 2003).
Figure 5.

Glucocorticoid-induced facilitation of GABA release is mediated by nitric oxide. A, The Dex (1 μm)-induced increase in amplitude (Amp) of eIPSCs was blocked by previous bath application of the NOS inhibitor l-NAME (50 μm). Traces are averages of 7–9 consecutive eIPSCs during the 3 min before l-NAME application (Control), during the last 3 min of a 10 min application of l-NAME, and during the last 3 min of a subsequent 10 min application of Dex and l-NAME. l-NAME had no effect by itself, but abolished the Dex-induced increase in eIPSC amplitude. B, Summary histogram of mean evoked IPSC amplitudes in Dex, l-NAME, and Dex + l-NAME (n = 8). C, Recording of mIPSCs 2 min before l-NAME application (Control) and following 10 min in l-NAME and 8 min in Dex (1 μm) + l-NAME. GABA currents are outward. D, Normalized running average of mean mIPSC frequency (Freq) response to Dex and l-NAME. Dexamethasone (1 μm) had no effect on mIPSC frequency in the presence of l-NAME (50 μm). E, Summary histogram of mean mIPSC frequency response to Dex in the presence of NOS antagonists. l-NAME and the nNOS-selective inhibitor NPLA (1 μm) were without effect on basal mIPSC frequency, but abolished the Dex-induced increase in mIPSC frequency. F, Summary histogram of mean mIPSC frequency response to Dex in the absence and presence of NO precursor l-arginine (L-arg, 500 μm). l-Arginine and Dex had similar, nonadditive facilitatory effects on mIPSC frequency. The effects were not influenced by the order of application of the two compounds.
In addition, the NO precursor l-arginine mimicked and occluded the facilitatory effect of Dex on GABA-mediated mIPSCs (Fig. 5F). Thus, bath application of l-arginine (500 μm) caused an increase in mIPSC frequency (128.6 ± 6.5% of baseline, p < 0.05, n = 7) that was similar to that elicited by Dex, without affecting mIPSC amplitude or decay time. Following 10 min of l-arginine application, the addition of Dex (1 μm) to the perfusion bath failed to elicit any additional increase in mIPSC frequency (96.9 ± 2.7% of the l-arginine value, n = 7). Similarly, when the order of application of Dex and l-arginine was reversed, i.e., l-arginine was applied following a 10-min application of Dex, there was not an additive effect of the drugs on mIPSC frequency. These results together suggest that glucocorticoids and NO act on GABA release via a common mechanism.
We showed previously that postsynaptic G-protein inactivation blocked the glucocorticoid-induced facilitation of GABA release (Di et al., 2005b). If NO synthesis is downstream from G-protein activation, stimulating NO production exogenously should mimic the glucocorticoid effect on GABA release despite blockade of G-protein activity. We tested this first by blocking G-protein activity with GDP-β-S (500 μm) applied intracellularly through the patch pipette, followed by bath application of the NO precursor l-arginine to stimulate NO synthesis. As shown above, blocking G-protein activity abolished the Dex-induced increase in mIPSC frequency, but the mIPSC frequency was subsequently increased following the addition of l-arginine (500 μm) to the perfusion bath (115.8 ± 5.5% of the baseline, p < 0.05, n = 6) (Fig. 6). Similarly, intracellular infusion of an antibody to the G-protein β subunit blocked the Dex-induced increase in mIPSC frequency, but subsequent bath application of the NO donor SNAP (100 μm) elicited an increase in mIPSC frequency (124.6 ± 5.1%, p < 0.05, n = 6) (Fig. 6).
Figure 6.

Nitric oxide facilitation of GABA release is downstream of G-protein activation. Intracellular application of a G-protein blocker, GDP-β-S (500 μm), or the Gβ antibody blocked the Dex-induced increase in mIPSC frequency. Subsequent addition of the NO precursor l-arginine (l-arg) (500 μm) or the NO donor SNAP (100 μm) reinstated the facilitation of GABA-mediated mIPSCs.
We also measured the glucocorticoid-induced changes in NO levels in SON magnocellular neurons directly using fluorescence measurements in individual neurons loaded via the patch pipette with the NO indicator DAF-FM (21 μm) (Stern and Zhang, 2005; Sheng and Braun, 2007). Bath application of Dex (1 μm) caused a 12 ± 4% increase in the intensity of DAF fluorescence in SON somata within 5–10 min (ΔF/F0; p < 0.05; n = 8) (Fig. 7), suggesting a glucocorticoid-induced increase in NO production in magnocellular neurons. The NO donor SNAP (100 μm) was applied as a positive control for DAF sensitivity to NO. The Dex-induced increase in DAF fluorescence was abolished by bath application of l-NAME (50 μm) for 10 min before Dex application (ΔF/F0 = 100 ± 3% of baseline; n = 9) (Fig. 7).
Figure 7.
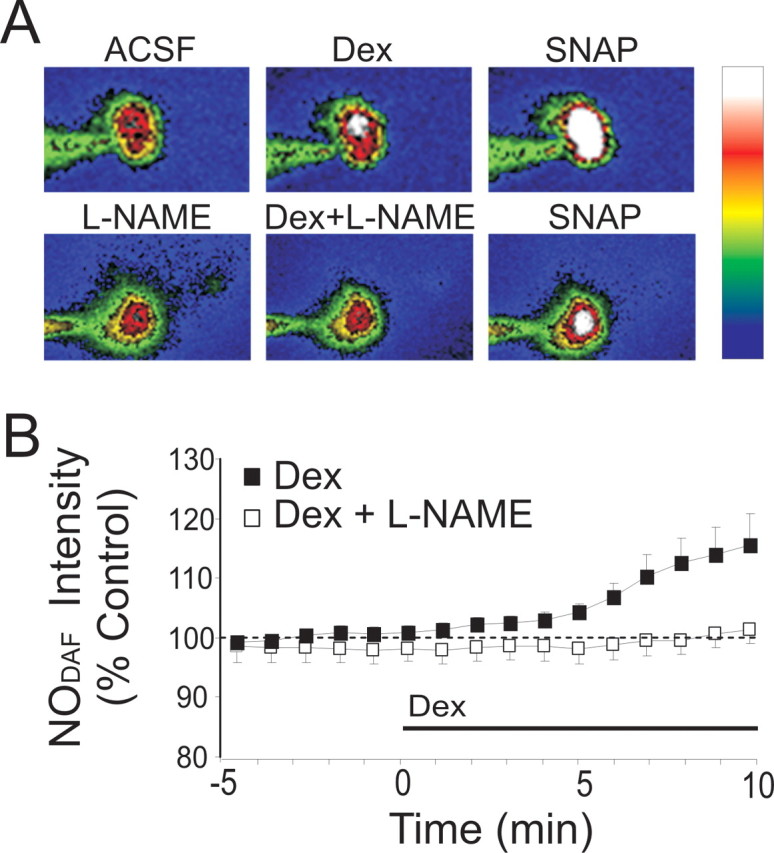
Glucocorticoid-induced increase in NO synthesis. A, Top (left to right), Images of the relative fluorescence of the NO-sensitive dye DAF-FM (NODAF) in a magnocellular neuron recorded 3 min before Dex application (ACSF), 7 min after the onset of Dex application (1 μm), and 7 min following further exposure to an NO donor (SNAP, 100 μm) applied as a positive control. Bottom, Same experiment as in top, except that l-NAME was present throughout the experiment to prevent NO production via NOS. In the pseudocolor image scale (right), white corresponds to the highest fluorescence, blue the lowest. B, Running average of DAF fluorescence normalized to control level measured during 3 min before Dex application. Dexamethasone (1 μm) elicited an increase in intracellular NO intensity within 3–5 min of bath application (n = 8). Ten minute preapplication of l-NAME (50 μm) abolished the Dex-induced increase in NO intensity (n = 9).
Discussion
Our previous findings demonstrated an opposing rapid glucocorticoid regulation of excitatory and inhibitory synaptic inputs to magnocellular neurons of the SON and PVN via the activation of postsynaptic, membrane-associated glucocorticoid receptors and the release of one or more retrograde messengers (Di et al., 2005a,b; Di and Tasker, 2008). The rapid glucocorticoid action at excitatory synapses is dependent on a postsynaptic Gαs-cAMP–PKA signaling pathway (Malcher-Lopes et al., 2006) that leads to the synthesis and retrograde release of endocannabinoids and the presynaptic CB1 receptor-dependent suppression of glutamate release (Di et al., 2005b). Until now, the molecular mechanisms and the retrograde messenger responsible for the rapid glucocorticoid facilitation of GABA release, however, were not known. Here we show that the opposing glucocorticoid modulation of glutamate and GABA release is mediated by distinct G-protein signaling mechanisms, which stimulate the synthesis and release of different retrograde messengers, endocannabinoids and NO, at glutamate and GABA synapses. A model of the divergent signaling downstream from the putative membrane glucocorticoid receptors is presented in Figure 8. In this model, the steroid binds to a postsynaptic membrane G-protein-coupled receptor, or to a receptor associated with a G-protein-coupled receptor (Kelly and Rønnekleiv, 2008; Micevych and Mermelstein, 2008), which stimulates a Gαs/cAMP/PKA-dependent synthesis of endocannabinoids and a Gβγ-dependent activation of nNOS and synthesis of NO. The endocannabinoids act as a retrograde messenger at excitatory synapses to suppress glutamate release and NO acts at inhibitory synapses to facilitate GABA release.
Figure 8.

Model of rapid glucocorticoid signaling in SON magnocellular neurons. According to this model, glucocorticoids (CORT) bind to a membrane-associated, G-protein-coupled receptor (mbGR) and activate divergent intracellular G-protein signaling pathways. Activation of Gαs leads to cAMP/PKA-dependent endocannabinoid (eCB) synthesis; the activated Gβγ dimer leads downstream to nNOS activation and NO synthesis. Endocannabinoids then act in a retrograde manner to suppress glutamate release from excitatory synaptic terminals via CB1 activation, and NO feeds back onto inhibitory synaptic terminals to facilitate GABA release onto SON magnocellular neurons. The combined suppression of synaptic excitation and facilitation of synaptic inhibition by glucocorticoids should cause a reduced excitability of the magnocellular neurons and a decrease in oxytocin (OXY) and vasopressin (AVP) release into the blood.
Glucocorticoids activate divergent signaling pathways
Steroid hormones are capable of eliciting effects in target cells that do not rely on the transcriptional activity of classical intracellular steroid receptors, but rather are mediated by membrane-associated actions and signal transduction mechanisms. Several lines of evidence from our work suggest that the rapid glucocorticoid effects on hypothalamic neuroendocrine cells are mediated by a membrane-associated receptor and G-protein-dependent mechanism(s) (Di et al., 2003, 2005b; Malcher-Lopes et al., 2006). Specific G-protein subunits have been implicated in the nongenomic actions of other steroids. Blocking Gα and Gβγ subunit activity, for example, prevents estrogen-induced nongenomic src/ERK and NO synthase activation in COS-7 and endothelial cells (Kumar et al., 2007), and the rapid action of androgen requires Gβγ activation (Zagar et al., 2004). Here, we report the novel finding that the membrane glucocorticoid receptor activates both G-protein components to stimulate divergent signaling pathways to endocannabinoid and NO production. The Gαs subunit was shown previously to mediate the glucocorticoid inhibition of glutamate release in both magnocellular and parvocellular neuroendocrine cells (Malcher-Lopes et al., 2006). We have confirmed those findings in magnocellular neurons in the current study, and we have provided new evidence showing that the G-protein Gβγ subunit, but not the Gαs subunit, is necessary for the glucocorticoid-induced increase in mIPSC frequency in magnocellular neurons, indicating a Gβγ-dependent mechanism of glucocorticoid-induced facilitation of GABA release.
Leptin inhibits the glucocorticoid-induced endocannabinoid synthesis and endocannabinoid suppression of glutamate release from excitatory synapses by blocking postsynaptic cAMP production (Malcher-Lopes et al., 2006). The failure of leptin to block the glucocorticoid-induced facilitation of GABA release from inhibitory synapses described here suggests that the split in the membrane glucocorticoid receptor signaling pathway that leads to endocannabinoid and NO production occurs upstream from cAMP production. This further supports our thesis that glucocorticoids differentially regulate excitatory and inhibitory synaptic inputs to magnocellular neurons through separate Gαs and Gβγ pathways.
Glucocorticoid facilitation of GABA release is mediated by nitric oxide
We reported previously that the glucocorticoid-induced facilitation of GABA release in magnocellular neurons was blocked by the CB1 antagonist AM251, but was not mimicked by any of the cannabinoid agonists tested, anandamide, 2-AG, and WIN 55,212-2 (Di et al., 2005b; Tasker et al., 2006). In contrast, here we found that the glucocorticoid facilitation of GABA release was not blocked by two other CB1 receptor antagonists, SR141716 (rimonobant) and O-2050. The glucocorticoid-induced suppression of glutamate release, however, is blocked by both AM251 and SR141716 (Di et al., 2005b). These findings together indicate that the rapid glucocorticoid modulation of GABA release is not mediated by a retrograde endocannabinoid messenger, and that AM251 exerts a CB1-independent effect on glucocorticoid-induced GABA release.
Our current findings suggest that NO is the retrograde messenger involved in the rapid glucocorticoid facilitation of GABA release in magnocellular neuroendocrine cells. Nitric oxide is an important activity-dependent modulator of the magnocellular neuroendocrine response to physiological stimuli such as stress, dehydration and lactation (Srisawat et al., 2000; Stern, 2004; Ryu et al., 2008). Neurons of the PVN and SON show robust NOS expression (Stern and Zhang, 2005; Yuan et al., 2006), and the application of NO precursors or NO donors preferentially facilitates synaptic GABA release (Stern and Ludwig, 2001; Li et al., 2002, 2003), consistent with a close functional and anatomical coupling of NO and GABA in the SON (Stern and Zhang, 2005). Although there is recent evidence for NO facilitation of glutamate release in the SON in vivo (Gillard et al., 2007), NO regulation of synaptic activity in the hypothalamus is primarily implicated in the facilitation of GABA release to modulate neuronal excitability (Zhang and Patel, 1998; Krukoff, 1999; Yang et al., 2007). Our observation of a NO dependence selectively of the glucocorticoid modulation of GABA release in the SON is consistent with these findings.
The glucocorticoid-induced facilitation of GABA release was blocked by both the nonspecific NOS inhibitor l-NAME and the nNOS-selective antagonist NPLA, suggesting that NO synthesis is mediated by nNOS, and that the source of the NO, therefore, is neuronal. In our experiments, both NMDA and AMPA glutamate receptors were pharmacologically blocked, indicating that the glucocorticoid-induced NO production was mediated by an NMDA receptor-independent mechanism (Amir, 1994; Bains and Ferguson, 1997; Kiss and Vizi, 2001). Determining the NO signaling pathway downstream from Gβγ requires further study.
Nitric oxide has been implicated in the mechanisms of other rapid, nongenomic steroid actions. Estrogen exerts rapid vasoprotective actions via enhanced NO release that is mediated by activation of both PI3K/Akt and MAPK signaling pathways (Wyckoff et al., 2001; Lu et al., 2004; Mantione, 2008). Aldosterone has also been reported to induce NO-dependent rapid nongenomic effects on the renal vasculature in humans (Chun and Pratt, 2006). Furthermore, glucocorticoids have been reported to activate NOS via a nongenomic PI3K/Akt pathway, leading to NO-dependent vasorelaxation in vivo (Hafezi-Moghadam et al., 2002). Similarly, a nongenomic, NO-dependent neuro-protective effect of glucocorticoids was reported in a cerebral ischemia model (Limbourg et al., 2002).
Glutamate and GABA synapse specificity
An interesting observation stemming from our findings is the strict spatial segregation of the glucocorticoid-induced endocannabinoid and NO actions at glutamate and GABA synapses, respectively. Several mechanisms may contribute to this synapse specificity. First, the presynaptic effector mechanisms responsive to cannabinoid and NO signaling may be selectively expressed at either glutamate or GABA terminals, respectively. Second, the postsynaptic synthesis and release of each of the messengers may be highly localized to either glutamate or GABA synapses. Third, extracellular diffusion barriers (e.g., astrocytes) may limit the actions of the messengers to one synapse type or the other. Although NO modulation of glutamate release has not been described in magnocellular neurons (although see Gillard et al., 2007), functional presynaptic CB1 receptors are expressed at both excitatory and inhibitory synapses in the SON (Di et al., 2005b), such that GABA terminals also possess cannabinoid signaling capability. Whereas it might be expected, and indeed it has been reported (Wilson and Nicoll, 2002), that endocannabinoids have a relatively limited spatial range of diffusion, the spatial restriction of NO diffusion is less likely, since as a gas its spread would be expected to be more promiscuous, reaching volumes with a radius of up to 100 μm (Philippides et al., 2000). It is likely, therefore, that the mechanism responsible for the synapse-specific actions of endocannabinoids and NO involves a combination of localized release and extracellular constraints on diffusion, such as barriers formed by astrocytic coverage of synapses (Piet et al., 2004).
Functional significance
The glucocorticoid regulation of neurohypophysial neurons is characterized primarily by an inhibition of PVN and SON magnocellular neuron activation and of oxytocin and vasopressin secretion. Glucocorticoids rapidly modulate the overall responsiveness of vasopressin and oxytocin neurons, serving potentially to coordinate the activity of the HPA and hypothalamic neurohypophysial systems (Lauand et al., 2007; Ruginsk et al., 2007). Stress and stress levels of glucocorticoids inhibit oxytocin release and milk ejection in lactating animals, delay parturition in late pregnancy (Papanek et al., 1997; Douglas and Russell, 2001; Russell et al., 2001), and inhibit vasopressin release during hemorrhage (Darlington et al., 1989, 1990).
The rapid glucocorticoid regulation of glutamatergic and GABAergic synaptic inputs to magnocellular neurons shown in this study are likely to have robust functional consequences for the release of vasopressin and oxytocin. Decreasing glutamate release and facilitating GABA release would be expected to tip the excitation-inhibition balance significantly toward inhibition in these cells. This overall inhibitory effect of glucocorticoids is likely to underlie the negative feedback effect of stress-induced glucocorticoids on neurohypophysial hormone release. These rapid inhibitory feedback actions of glucocorticoids during the stress response may play an integrative role in the regulation of neuroendocrine function as well as a neuroprotective role in the hypothalamus, since both endocannabinoids and NO are known to have anti-inflammatory actions in the brain (Malcher-Lopes et al., 2008).
Footnotes
This work was supported by National Institutes of Health Grants MH066958 and MH069879. We thank Katalin Halmos for her expert technical assistance.
References
- Amir S. N-Methyl-D-aspartate receptor-mediated signaling in the supraoptic nucleus involves activation of a nitric oxide-dependent pathway. Brain Res. 1994;645:330–334. doi: 10.1016/0006-8993(94)91668-3. [DOI] [PubMed] [Google Scholar]
- Bains JS, Ferguson AV. Nitric oxide regulates NMDA-driven GABAergic inputs to type I neurones of the rat paraventricular nucleus. J Physiol. 1997;499:733–746. doi: 10.1113/jphysiol.1997.sp021965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun TY, Pratt JH. Nongenomic renal effects of aldosterone: dependency on NO and genomic actions. Hypertension. 2006;47:636–637. doi: 10.1161/01.HYP.0000205225.88721.2c. [DOI] [PubMed] [Google Scholar]
- Darlington DN, Keil LC, Dallman MF. Potentiation of hormonal responses to hemorrhage and fasting, but not hypoglycemia in conscious adrenalectomized rats. Endocrinology. 1989;125:1398–1406. doi: 10.1210/endo-125-3-1398. [DOI] [PubMed] [Google Scholar]
- Darlington DN, Chew G, Ha T, Keil LC, Dallman MF. Corticosterone, but not glucose, treatment enables fasted adrenalectomized rats to survive moderate hemorrhage. Endocrinology. 1990;127:766–772. doi: 10.1210/endo-127-2-766. [DOI] [PubMed] [Google Scholar]
- de Kloet ER. Stress in the brain. Eur J Pharmacol. 2000;405:187–198. doi: 10.1016/s0014-2999(00)00552-5. [DOI] [PubMed] [Google Scholar]
- Di S, Tasker JG. Rapid synapse-specific regulation of hypothalamic magnocellular neurons by glucocorticoids. Prog Brain Res. 2008;170:379–388. doi: 10.1016/S0079-6123(08)00431-7. [DOI] [PubMed] [Google Scholar]
- Di S, Malcher-Lopes R, Halmos KC, Tasker JG. Nongenomic glucocorticoid inhibition via endocannabinoid release in the hypothalamus: a fast feedback mechanism. J Neurosci. 2003;23:4850–4857. doi: 10.1523/JNEUROSCI.23-12-04850.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di S, Boudaba C, Popescu IR, Weng FJ, Harris C, Marcheselli VL, Bazan NG, Tasker JG. Activity-dependent release and actions of endocannabinoids in the rat hypothalamic supraoptic nucleus. J Physiol. 2005a;569:751–760. doi: 10.1113/jphysiol.2005.097477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di S, Malcher-Lopes R, Marcheselli VL, Bazan NG, Tasker JG. Rapid glucocorticoid-mediated endocannabinoid release and opposing regulation of glutamate and gamma-aminobutyric acid inputs to hypothalamic magnocellular neurons. Endocrinology. 2005b;146:4292–4301. doi: 10.1210/en.2005-0610. [DOI] [PubMed] [Google Scholar]
- Douglas AJ, Russell JA. Endogenous opioid regulation of oxytocin and ACTH secretion during pregnancy and parturition. Prog Brain Res. 2001;133:67–82. doi: 10.1016/s0079-6123(01)33006-6. [DOI] [PubMed] [Google Scholar]
- Gillard ER, Coburn CG, de Leon A, Snissarenko EP, Bauce LG, Pittman QJ, Hou B, Currás-Collazo MC. Vasopressin autoreceptors and nitric oxide-dependent glutamate release are required for somatodendritic vasopressin release from rat magnocellular neuroendocrine cells responding to osmotic stimuli. Endocrinology. 2007;148:479–489. doi: 10.1210/en.2006-0995. [DOI] [PubMed] [Google Scholar]
- Hafezi-Moghadam A, Simoncini T, Yang Z, Limbourg FP, Plumier JC, Rebsamen MC, Hsieh CM, Chui DS, Thomas KL, Prorock AJ, Laubach VE, Moskowitz MA, French BA, Ley K, Liao JK. Acute cardiovascular protective effects of corticosteroids are mediated by non-transcriptional activation of endothelial nitric oxide synthase. Nat Med. 2002;8:473–479. doi: 10.1038/nm0502-473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haller J, Mikics E, Makara GB. The effects of non-genomic glucocorticoid mechanisms on bodily functions and the central neural system. A critical evaluation of findings. Front Neuroendocrinol. 2008;29:273–291. doi: 10.1016/j.yfrne.2007.10.004. [DOI] [PubMed] [Google Scholar]
- Kelly MJ, Rønnekleiv OK. Membrane-initiated estrogen signaling in hypothalamic neurons. Mol Cell Endocrinol. 2008;290:14–23. doi: 10.1016/j.mce.2008.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss JP, Vizi ES. Nitric oxide: a novel link between synaptic and nonsynaptic transmission. Trends Neurosci. 2001;24:211–215. doi: 10.1016/s0166-2236(00)01745-8. [DOI] [PubMed] [Google Scholar]
- Krukoff TL. Central actions of nitric oxide in regulation of autonomic functions. Brain Res Brain Res Rev. 1999;30:52–65. doi: 10.1016/s0165-0173(99)00010-7. [DOI] [PubMed] [Google Scholar]
- Kumar P, Wu Q, Chambliss KL, Yuhanna IS, Mumby SM, Mineo C, Tall GG, Shaul PW. Direct interactions with G alpha i and G betagamma mediate nongenomic signaling by estrogen receptor alpha. Mol Endocrinol. 2007;21:1370–1380. doi: 10.1210/me.2006-0360. [DOI] [PubMed] [Google Scholar]
- Lauand F, Ruginsk SG, Rodrigues HL, Reis WL, de Castro M, Elias LL, Antunes-Rodrigues J. Glucocorticoid modulation of atrial natriuretic peptide, oxytocin, vasopressin and Fos expression in response to osmotic, angiotensinergic and cholinergic stimulation. Neuroscience. 2007;147:247–257. doi: 10.1016/j.neuroscience.2007.04.021. [DOI] [PubMed] [Google Scholar]
- Li DP, Chen SR, Pan HL. Nitric oxide inhibits spinally projecting paraventricular neurons through potentiation of presynaptic GABA release. J Neurophysiol. 2002;88:2664–2674. doi: 10.1152/jn.00540.2002. [DOI] [PubMed] [Google Scholar]
- Li Y, Zhang W, Stern JE. Nitric oxide inhibits the firing activity of hypothalamic paraventricular neurons that innervate the medulla oblongata: role of GABA. Neuroscience. 2003;118:585–601. doi: 10.1016/s0306-4522(03)00042-3. [DOI] [PubMed] [Google Scholar]
- Limbourg FP, Huang Z, Plumier JC, Simoncini T, Fujioka M, Tuckermann J, Schütz G, Moskowitz MA, Liao JK. Rapid nontranscriptional activation of endothelial nitric oxide synthase mediates increased cerebral blood flow and stroke protection by corticosteroids. J Clin Invest. 2002;110:1729–1738. doi: 10.1172/JCI15481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Q, Pallas DC, Surks HK, Baur WE, Mendelsohn ME, Karas RH. Striatin assembles a membrane signaling complex necessary for rapid, nongenomic activation of endothelial NO synthase by estrogen receptor alpha. Proc Natl Acad Sci U S A. 2004;101:17126–17131. doi: 10.1073/pnas.0407492101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malcher-Lopes R, Di S, Marcheselli VS, Weng FJ, Stuart CT, Bazan NG, Tasker JG. Opposing crosstalk between leptin and glucocorticoids rapidly modulates synaptic excitation via endocannabinoid release. J Neurosci. 2006;26:6643–6650. doi: 10.1523/JNEUROSCI.5126-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malcher-Lopes R, Franco A, Tasker JG. Glucocorticoids shift arachidonic acid metabolism toward endocannabinoid synthesis: a non-genomic anti-inflammatory switch. Eur J Pharmacol. 2008;583:322–339. doi: 10.1016/j.ejphar.2007.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantione KJ. Estrogen's actions transcend a sole reproductory function in cell signaling. Med Sci Monit. 2008;14:SC1–SC3. [PubMed] [Google Scholar]
- Micevych PE, Mermelstein PG. Membrane estrogen receptors acting through metabotropic glutamate receptors: an emerging mechanism of estrogen action in brain. Mol Neurobiol. 2008;38:66–77. doi: 10.1007/s12035-008-8034-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nylen A, Skagerberg G, Alm P, Larsson B, Holmqvist BI, Andersson KE. Detailed organization of nitric oxide synthase, vasopressin and oxytocin immunoreactive cell bodies in the supraoptic nucleus of the female rat. Anat Embryol (Berl) 2001;203:309–321. doi: 10.1007/s004290100164. [DOI] [PubMed] [Google Scholar]
- Papanek PE, Sladek CD, Raff H. Corticosterone inhibition of osmotically stimulated vasopressin from hypothalamic-neurohypophysial explants. Am J Physiol. 1997;272:R158–R162. doi: 10.1152/ajpregu.1997.272.1.R158. [DOI] [PubMed] [Google Scholar]
- Philippides A, Husbands P, O'Shea M. Four-dimensional neuronal signaling by nitric oxide: a computational analysis. J Neurosci. 2000;20:1199–1207. doi: 10.1523/JNEUROSCI.20-03-01199.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piet R, Vargová L, Syková E, Poulain DA, Oliet SH. Physiological contribution of the astrocytic environment of neurons to intersynaptic crosstalk. Proc Natl Acad Sci U S A. 2004;101:2151–2155. doi: 10.1073/pnas.0308408100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruginsk SG, Oliveira FR, Margatho LO, Vivas L, Elias LL, Antunes-Rodrigues J. Glucocorticoid modulation of neuronal activity and hormone secretion induced by blood volume expansion. Exp Neurol. 2007;206:192–200. doi: 10.1016/j.expneurol.2007.04.012. [DOI] [PubMed] [Google Scholar]
- Russell JA, Douglas AJ, Ingram CD. Brain preparations for maternity–adaptive changes in behavioral and neuroendocrine systems during pregnancy and lactation. An overview. Prog Brain Res. 2001;133:1–38. doi: 10.1016/s0079-6123(01)33002-9. [DOI] [PubMed] [Google Scholar]
- Ryu V, Lee JH, Um JW, Yoo SB, Lee J, Chung KC, Jahng JW. Water-deprivation-induced expression of neuronal nitric oxide synthase in the hypothalamic paraventricular nucleus of rat. J Neurosci Res. 2008;86:1371–1379. doi: 10.1002/jnr.21598. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM, Armanini MP, Packan DR, Sutton SW, Plotsky PM. Glucocorticoid feedback inhibition of adrenocorticotropic hormone secretagogue release. Relationship to corticosteroid receptor occupancy in various limbic sites. Neuroendocrinology. 1990;51:328–336. doi: 10.1159/000125357. [DOI] [PubMed] [Google Scholar]
- Sheng JZ, Braun AP. Small- and intermediate-conductance Ca2+-activated K+ channels directly control agonist-evoked nitric oxide synthesis in human vascular endothelial cells. Am J Physiol Cell Physiol. 2007;293:C458–C467. doi: 10.1152/ajpcell.00036.2007. [DOI] [PubMed] [Google Scholar]
- Srisawat R, Ludwig M, Bull PM, Douglas AJ, Russell JA, Leng G. Nitric oxide and the oxytocin system in pregnancy. J Neurosci. 2000;20:6721–6727. doi: 10.1523/JNEUROSCI.20-17-06721.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern JE. Nitric oxide and homeostatic control: an intercellular signaling molecule contributing to autonomic and neuroendocrine integration? Prog Biophys Mol Biol. 2004;84:197–215. doi: 10.1016/j.pbiomolbio.2003.11.015. [DOI] [PubMed] [Google Scholar]
- Stern JE, Ludwig M. NO inhibits supraoptic oxytocin and vasopressin neurons via activation of GABAergic synaptic inputs. Am J Physiol Regul Integr Comp Physiol. 2001;280:R1815–R1822. doi: 10.1152/ajpregu.2001.280.6.R1815. [DOI] [PubMed] [Google Scholar]
- Stern JE, Zhang W. Cellular sources, targets and actions of constitutive nitric oxide in the magnocellular neurosecretory system of the rat. J Physiol. 2005;562:725–744. doi: 10.1113/jphysiol.2004.077735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasker JG, Di S, Malcher-Lopes R. Minireview: rapid glucocorticoid signaling via membrane-associated receptors. Endocrinology. 2006;147:5549–5556. doi: 10.1210/en.2006-0981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA. Endocannabinoid signaling in the brain. Science. 2002;296:678–682. doi: 10.1126/science.1063545. [DOI] [PubMed] [Google Scholar]
- Wyckoff MH, Chambliss KL, Mineo C, Yuhanna IS, Mendelsohn ME, Mumby SM, Shaul PW. Plasma membrane estrogen receptors are coupled to endothelial nitric-oxide synthase through Galpha (i) J Biol Chem. 2001;276:27071–27076. doi: 10.1074/jbc.M100312200. [DOI] [PubMed] [Google Scholar]
- Yang Q, Chen SR, Li DP, Pan HL. Kv1.1/1.2 channels are downstream effectors of nitric oxide on synaptic GABA release to preautonomic neurons in the paraventricular nucleus. Neuroscience. 2007;149:315–327. doi: 10.1016/j.neuroscience.2007.08.007. [DOI] [PubMed] [Google Scholar]
- Yuan Q, Scott DE, So KF, Wu W. Developmental changes of nitric oxide synthase expression in the rat hypothalamoneurohypophyseal system. Anat Rec A Discov Mol Cell Evol Biol. 2006;288:36–45. doi: 10.1002/ar.a.20271. [DOI] [PubMed] [Google Scholar]
- Zagar Y, Chaumaz G, Lieberherr M. Signaling cross-talk from Gbeta4 subunit to Elk-1 in the rapid action of androgens. J Biol Chem. 2004;279:2403–2413. doi: 10.1074/jbc.M309132200. [DOI] [PubMed] [Google Scholar]
- Zhang K, Patel KP. Effect of nitric oxide within the paraventricular nucleus on renal sympathetic nerve discharge: role of GABA. Am J Physiol. 1998;275:R728–R734. doi: 10.1152/ajpregu.1998.275.3.R728. [DOI] [PubMed] [Google Scholar]