

Abstract
Spinocerebellar ataxia type 31 (SCA31) is an adult-onset autosomal-dominant neurodegenerative disorder showing progressive cerebellar ataxia mainly affecting Purkinje cells. The SCA31 critical region was tracked down to a 900 kb interval in chromosome 16q22.1, where the disease shows a strong founder effect. By performing comprehensive Southern blot analysis and BAC- and fosmid-based sequencing, we isolated two genetic changes segregating with SCA31. One was a single-nucleotide change in an intron of the thymidine kinase 2 gene (TK2). However, this did not appear to affect splicing or expression patterns. The other was an insertion, from 2.5–3.8 kb long, consisting of complex penta-nucleotide repeats including a long (TGGAA)n stretch. In controls, shorter (1.5–2.0 kb) insertions lacking (TGGAA)n were found only rarely. The SCA31 repeat insertion's length inversely correlated with patient age of onset, and an expansion was documented in a single family showing anticipation. The repeat insertion was located in introns of TK2 and BEAN (brain expressed, associated with Nedd4) expressed in the brain and formed RNA foci in the nuclei of patients' Purkinje cells. An electrophoretic mobility-shift assay showed that essential splicing factors, serine/arginine-rich splicing factors SFRS1 and SFRS9, bind to (UGGAA)n in vitro. Because (TGGAA)n is a characteristic sequence of paracentromeric heterochromatin, we speculate that the insertion might have originated from heterochromatin. SCA31 is important because it exemplifies human diseases associated with “inserted” microsatellite repeats that can expand through transmission. Our finding suggests that the ectopic microsatellite repeat, when transcribed, might cause a disease involving the essential splicing factors.
Introduction
Autosomal-dominant cerebellar degenerative disorders are generally referred to as spinocerebellar ataxia (SCA).1 Clinically, progressive cerebellar ataxia is the cardinal neurological symptom, and it is often accompanied by variable extracerebellar neurological features, such as pyramidal tract signs, extrapyramidal signs, ophthalmoparesis, and sensory disturbances. Neuropathologically, the cerebellum and its related systems, such as the brainstem, spinal cord, and basal ganglia, can be involved to various degrees.
Nearly 30 genetic loci have been identified. Of these, expansions of tri-nucleotide (CAG) repeats are the causes of SCA1 (MIM #164400); SCA2 (MIM #183090); SCA3, or Machado-Joseph disease (MJD) (MIM #109150); SCA6 (MIM #183086), SCA7 (MIM #164500); SCA17 (MIM #607136); and dentatorubral-pallidoluysian atrophy (DRPLA) (MIM #125370). These disorders, together with Huntington disease (HD) (MIM +143100) and spinal and bulbar muscular atrophy (MIM #313200), are called polyglutamine diseases2 because the CAG repeats, which are expanded in patients, reside in the coding regions and are translated into polyglutamine tracts. SCA8 (MIM #608768), SCA10 (MIM #603516), and SCA12 (MIM #604326) are caused by expansions of bidirectionally transcribed CTG and CAG; ATTCT; and CAG repeats, respectively, in the non-coding regions of the responsible genes. These disorders, together with myotonic dystrophy type 1 (DM1) (MIM #160900), DM2 (MIM #602668), HD-like disease type 2 (HDL2) (MIM #606438), and Fragile X tremor/ataxia syndrome (FXTAS) (MIM #300623), caused by RNA-mediated gain-of-function mechanisms, are called noncoding repeat expansion disorders3. These are dynamic repeat-expansion disorders, but some forms of SCA are caused by static mutations (e.g., missense, frameshift, or deletion) in functionally important genes,4 such as β-III spectrin (SPTBN2) (MIM #604985) in SCA5 (MIM #600224), tau tubulin kinase 2 (TTBK2) (MIM #611695) in SCA11 (MIM #604432), voltage-gated potassium channel (KCNC3) (MIM #176264) in SCA13 (MIM #605259), protein kinase C γ (PRKCG) (MIM #176980) for SCA14 (MIM #605361), inositol 1,4,5-triphosphate receptor type 1 (ITPR1) (MIM #147265) in SCA15 (MIM #606658), and fibroblast growth factor 14 (FGF14) (MIM #601515) in SCA27 (MIM #609307). The fact that so many mutations result in the SCA phenotype in humans suggests that the cerebellum and its related nervous systems have particularly sophisticated and vulnerable neurobiological characteristics.
We previously mapped a form of SCA (previously called “chromosome 16q22.1-linked autosomal dominant cerebellar ataxia [ADCA]”; MIM #117210 and newly termed as “SCA31” in this report) to human chromosome 16q22.1.5,6 This locus was already known for a clinically distinct ataxia, SCA47 (MIM %600223). SCA31 presents as pure cerebellar ataxia with an average age of onset of 61.2 years and thus is the latest to appear of all SCAs.6,8 The Purkinje cell, the only projecting neuron in the cerebellar cortex, is predominantly affected.8 SCA31 is estimated to rank as the third most frequent SCA in Japan, after SCA3 (MJD) and SCA6. In SCA4, in contrast, cerebellar ataxia is always accompanied by both sensory axonal neuropathy and reduced or absent tendon reflexes, and Babinski's signs may be also seen.7,9 The age of onset is usually in the third or fourth decade, and neuropathologically, the degeneration of multiple systems is observed.10 Only two families have been genetically confirmed as having SCA4: one is the original SCA4 family that is of Scandinavian origin but resides in the United States,7 and the other is a German family.9 Discovery of the mutations is needed to settle the question as to whether SCA4 and SCA31 are allelic diseases.11
We previously reported that a single-nucleotide change (−16C > T) in the PLEKHG4 (puratrophin-1) gene (MIM ∗609526) is tightly associated with SCA31.6 However, two patients who did not have this change were subsequently found,12,13 indicating that the −16C > T change in PLEKHG4 is a marker in a strong linkage disequilibrium with SCA31 but is not the cause of this disease. Conducting fine SNP typing allowed the SCA31 critical region to be tracked to a 900 kb “founder” chromosome lying between rs11640843 (SNP0413) and −16C > T in PLEKHG4.6 Whereas all SCA31 patients shared a single haplotype for this critical region, none of the controls (800 Japanese and 60 white American control chromosomes) had it, indicating that SCA31 is caused by a single founder mutation.13 Previous attempts to find this mutation by PCR-based sequencing in all annotated coding exons and expressed sequence tags (ESTs) failed. The strong founder effect in SCA31 made a mutation search complicated because segregating genetic changes are not always causative. Therefore, we needed to identify all the genetic changes and then investigate which one was the causative mutation.
In this study, we carried out a comprehensive mutation search, including Southern blot analysis to detect any chromosomal rearrangements and BAC- and fosmid-based complete genome sequencing to identify all the genomic changes in the 900 kb critical region. Here we show that SCA31 is associated with an inserted sequence that consists of complex penta-nucleotide repeats containing (TGGAA)n.
Material and Methods
Human Samples
Study Subjects
We collected blood samples after obtaining informed consent from all involved SCA31 families, a small nuclear family of American SCA4 kindred7, and controls. The study conformed to the tenets of the Declaration of Helsinki, and the ethics were approved by the institutional review board of Tokyo Medical and Dental University, Tokyo, Japan.
Genomic DNA was extracted on the basis of a standard protocol.6 The SCA31 subjects studied were 160 affected individuals from 98 SCA31 families, consisting of the previously described 125 affected individuals from 64 families13, an individual from the “U09” family without the −16C > T PLEKHG4 change13, and 34 newly recruited individuals from 33 families. Normal controls consisted of 400 Japanese and 30 white American individuals, in whom no personal or family histories of ataxia or any inherited disorders had been documented. Five individuals from the original SCA4 kindred (kindred 18757), including three with typical SCA4 symptoms and SCA4 disease-haplotypes, were also studied. In addition, the previously described 21 individuals13 who had a similar clinical phenotype but did not carry the SCA31 founder haplotype were also included as disease controls for mutation analysis.
Among the SCA31 individuals, one homozygous patient in family P214 who harbored two identical SCA31 haplotypes between D16S3094 and D16S3095, covering the SCA31 critical interval, was chosen for a complete BAC- and fosmid-based genomic sequencing of the SCA31 critical region. The same homozygous patient, a heterozygous SCA31 patient in family P145, and a normal control (control 1) were chosen for investigation by Southern blotting, quantitative genomic PCR, and array-based comparative genomic hybridization (aCGH) analyses. Mutation candidates found through these analyses were then screened in the remaining SCA31 and control individuals.
The penta-nucleotide repeat insertion (see Results) was tested either by Southern blotting, PCR, or both in all SCA31 individuals, five individuals from an SCA4 family, and all controls (430 normal controls and 21 disease controls). Thirty-nine SCA31 heterozygous patients, from whom we could obtain detailed clinical information and ages of onset, were analyzed for the correlation between insert length and age of onset. One affected SCA4 individual and ten disease controls were screened for mutations in the critical genes, BEAN (brain expressed, associated with Nedd4) (MIM ∗612051) and TK2 (thymidine kinase 2) (MIM ∗188250), and in EST FLJ27243 (see Results) by PCR and direct sequencing.
Brain Tissue Samples
Frozen brain tissues of the cerebellar cortex were used for gene expression analyses (i.e., RT-PCR, TaqMan quantitative RT-PCR analyses, and fluorescence in situ hybridization [FISH]). In addition to the cerebellar cortex, the cerebral white matter (frontal lobe), the frontal cortex, hippocampus, thalamus, and the midbrain from a control individual were studied for RT-PCR analysis. Both control and SCA31 brains were obtained during an autopsy performed under their families' written consent and approved by each institutional ethics committee. These brains were immediately frozen and stored at −80°C until use. Four control frozen brains were studied: two were from patients with sporadic amyotrophic lateral sclerosis (ages at death: 70 and 78 years), one was from a patient with SCA3/MJD (age at death: 65 years), and one was from a patient who had autosomal-dominant progressive external ophthalmoplegia (adPEO) (MIM #157640) and a heterozygous missense mutation in the nuclear-encoded DNA polymerase-γ gene (age at death: 72 years). Neuropathological examinations of these patients did not show obvious neuronal losses in the cerebella. For SCA31, two patients were studied (ages at death: 74 and 78 years).
Mutation screening
Southern Blotting
We screened for genomic rearrangement by performing Southern blot analysis and using cosmid clones for probe synthesis. The method has been previously described.6,15 In brief, we generated cosmid clones, tandemly covering the SCA31 critical region, by subcloning from the BAC contig constructed on the basis of a control human genome.16 Probes radiolabeled with 32P were then generated from each cosmid clone. Genomic DNA extracted from lymphoblastoid cell lines of three individuals (one control individual, an SCA31 homozygote, and a heterozygote) was digested with a restriction enzyme and subjected to Southern blot analysis. The analysis was undertaken with five different restriction enzymes (BamHI, EcoRI, EcoRV, HindIII and XbaI). When needed to confirm results, another five SCA31 subjects and ten normal controls were similarly investigated.
Altered restriction enzyme fragment patterns were observed with the cosmid probe detecting the genomic region between 65,083,571 and 65,124,051 on NCBI Build 36.3. We confirmed the results by employing PCR products as a probe that had been obtained by amplification of a 3009 bp segment (from 65,079,127–65,082,135) within this genomic region and radiolabeled with 32P. This PCR reaction was carried out with primers Ins-long3.0k-F (5′-GCTTCTCTGCTTCTGTCATCAGCTCAC-3′) and Ins-long3.0k-R (5′-ATCTTTCCACACTACCATCCCATCCAG-3′); control genomic DNA was used as a template.
Sequencing of the 900 kb SCA31 Critical Region
This was performed by a modified version of the methods used in Chromosome 21 genome sequencing.17,18
Construction of a BAC Library of the Genomic DNA from the Lymphoblastoid Cell Line Derived from a Homozygous SCA31 Patient. The lymphoblastoid cells derived from the homozygous SCA31 patient14 were embedded in agarose gel plugs, and high-molecular-weight genomic DNA was extracted. The DNA was partially digested with HindIII, and fragments from 100–150 kb were selected by pulse field gel electrophoresis (PFGE). The collected DNA fragments were ligated with pBAC-lac18 and subsequently used for transformation of DH10B cells by electroporation.
Transformed E. coli were spread on lysogeny broth (LB) plates with 12.5 × 10−3 g/liter of chloramphenicol, X-gal (5-bromo-4-chloro-3-indolyl-b-D-galactopyranoside), and IPTG (isopropyl β-D-1-thiogalactopyranoside). Positive colonies were selected by color, picked up, and separately stored in LB liquid medium with 7.5% (v/v) glycerol and 12.5 × 10−3 g/liter chloramphenicol (LB-glycerol-Cm) prepared in 384-well plates. A total of approximately 115,000 clones were obtained. In order to evaluate the quality of the BAC library, we randomly selected 200 clones, isolated their BAC DNA by the alkaline-SDS (sodium dodecyl sulfate) method, and digested it with NotI. Their insert sizes were measured by PFGE. Approximately 90% of BAC clones had DNA inserts of 80–140 kb in length, and the mean size was approximately 110 kb.
Construction of a Fosmid Library of the Genomic DNA of the Same Homozygous Patient. Genomic DNA extracted from the lymphoblastoid cell line from the same homozygous patient14 was sheared, and fragments ranging in length from 35–50 kb were collected. The DNA fragments were cloned into pCC1FOS vector (Epicenter) with the CopyControl Fosmid Library Production Kit (Epicenter) according to the manufacturer's instructions. Approximately 250,000 clones were collected in LB-glycerol-Cm pools, each of which contained 2,000–10,000 clones. Examination of 96 randomly selected clones confirmed that each clone harbored a DNA insertion of approximately 35–45 kb.
Identification of BAC and Fosmid Clones Covering the SCA31 Critical Region. We spotted approximately 115,000 BAC clones in a grid pattern on GeneScreen Plus membranes (PerkinElmer) with a 4 × 4 double offset pattern by using a BioGrid robot (BioRobotics) to make high-density replica (HDR) filters. We created probes by amplifying the genomic DNA of the same patient by PCR to detect every 50 kb segment in the critical region. After labeling the probes with 32P, we hybridized the HDR filters with them. Fifty-two BAC clones were identified as positive clones. The insert ends of these 52 BAC clones were sequenced, and 48 clones were mapped to the 900 kb critical region, whereas the remaining fell into different chromosomal regions.
We constructed a fosmid library to clone the genomic region (approximately 42.3 kb in length) that was missed in the BAC library. Four sites in the region were chosen as PCR targets, and primers that would allow their specific amplification were used. By serial PCR, the pools identified as harboring the desired clones were divided into smaller pools, and subsequently three fosmid clones covering the 42.3 kb region were isolated.
Complete Sequencing of the BAC and Fosmid Clones Covering the 900 kb Critical Region. Twelve BAC and three fosmid clones formed a contig that covered the entire 900 kb critical region and thus were rendered for sequencing (Table 1). The complete base sequences of all but one BAC clone (Ca0215J24) and one fosmid clone (CaFos003) were determined by shotgun sequencing as previously described.17 In brief, every BAC and fosmid clone was sheared into short fragments of approximately 4 kb, treated with shrimp alkaline phosphatase, and then subcloned into pCR-blunt II vector (Invitrogen). The plasmid was used for transforming DH10B by electroporation. Seven hundred and sixty-eight subclones from each BAC clone and 384 subclones from each fosmid clone were picked up. Each was inoculated into 160 μl of LB with glycerol and kanamycin, grown overnight, and then rendered for rolling-circle amplification (RCA) with the Templiphi DNA Amplification Kit (GE Healthcare Bioscience). Amplified subclone plasmids were sequenced with M13 primers and the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems) in ABI PRISM 3730 and 3100 (Applied Biosystems). The raw sequence data were analyzed and assembled with phred, phrap, and consed. The remaining two clones, Ca0215J24 and CaFos003, were sequenced so that the gaps between Ca0217F15 and Ca0262A14 and between CaFos001 and CaFos002, respectively, could be filled.
Table 1.
Genomic Regions Cloned by 12 BACs and Three Fosmids that Were Rendered for Sequencing
| BAC or Fosmid Clone | Cloned region on NCBI Build 36.3 |
|
|---|---|---|
| from | to | |
| Ca0109N14 | 64,935,119 | 65,053,675 |
| Ca0159N04 | 65,052,547 | 65,166,347 |
| Ca0041G15 | 65,163,955 | 65,252,631 |
| Ca0080M24 | 65,215,426 | 65,328,653 |
| Ca0303C23 | 65,265,426 | 65,358,989 |
| CaFos001 | 65,345,373 | 65,382,103 |
| CaFos003 | 65,369,225 | 65,410,300 |
| CaFos002 | 65,385,244 | 65,430,086 |
| Ca0151F23 | 65,401,149 | 65,500,053 |
| Ca0312C05 | 65,485,609 | 65,608,342 |
| Ca0238A16 | 65,530,876 | 65,635,257 |
| Ca0217F15 | 65,634,014 | 65,724,706 |
| Ca0215J24 | 65,692,848 | 65,788,160 |
| Ca0262A14 | 65,726,364 | 65,829,738 |
| Ca0154D03 | 65,815,462 | 65,917,148 |
Together, the 12 BACs and three fosmids formed a contig covering the entire 900 kb critical region.
Finally, the obtained nucleotide sequence information in the SCA31 critical interval of the homozygous patient was compared with a public database (NCBI, build 36.3). For each discordant nucleotide sequence, 20 pilot control individuals were examined so that it could be determined whether such discordance was simply due to polymorphism. If the discordance was not proven to be polymorphism, a larger number of controls (n = 430) and SCA31 patients were analyzed. With this approach, nucleotide sequences that segregate with SCA31 in the entire 900 kb critical region were searched.
Quantitative Genomic PCR and aCGH
Quantitative genomic PCR was performed with TaqMan probes (Applied Biosystems) designed to measure two exons of each gene within the SCA31 critical region as previously described.19 Gene dosage was also analyzed by custom-designed, high-definition array-based comparative genomic hybridization (aCGH) microarrays (Agilent Technologies). Oligonucleotide probes were designed by two of us (Y.T. and S.T.) according to the published method.20,21 A 1.88 Mb genomic region between D16S3031 and D16S3107 (NCBI, build 36.3), completely covering the 900 kb SCA31 critical region, was included in the microarray, and probes were designed with an average interval of 100 bp. Genomic DNA samples of three individuals (one homozygous patient,14 one heterozygous patient, and one control subject) were tested and compared with each other.
Characterizing the Penta-Nucleotide Repeat Insertion
Analysis of the Penta-Nucleotide Repeat Length by PCR and Agarose-Gel Electrophoresis
The genomic DNA of the patients was amplified by PCR with primers 1.5k-ins-F (5′- ACTCCAACTGGGATGCAGTTTCTCAAT-3′) and 1.5k-ins-R (5′- TGGAGGAAGGAAATCAGGTCCCTAAAG-3′). Each PCR reaction was performed in a final volume of 10 μl, containing 0.25 μM of each primer, 400 μM (each) of dNTP, 1.5 mM of MgCl2, 0.25 U of LA Taq HS polymerase (Takara Bio), and 50–100 ng of genomic DNA. Thermal cycles were as follows: initial denaturing at 95°C for 5 min followed by 30 cycles of denaturing at 95°C for 20 s and annealing and extension at 68°C for 8 min. Five microliters (5 μl) of each PCR product was digested with HaeIII and then run through an 0.8% agarose gel at 30 V (V) for 15 hr with 1 kb DNA Ladder (Takara Bio). When multiple members were collected, differences in the length of insertion were analyzed in the same gel.
The lengths (L = length) of HaeIII-digested PCR products (L/kb) were calculated as below. The electrophoretic migration distance (d; in millimeters) of each sample was measured and then introduced into the formula: d = alnL + b, in which “a” and “b” were determined by the standard line obtained from the data of the size markers. As the HaeIII-digested PCR product has 193-bp sequences flanking the SCA31 penta-nucleotide repeat insertion, the length of the insertion was calculated as L – 0.19/kb. The correlation between the length of the repeat insertion and the age of onset was analyzed via calculation of Pearson's product-moment correlation coefficient (r).
Sequencing of the Penta-Nucleotide Repeat in SCA31 Patients and Two Controls
PCR products including the penta-nucleotide repeat were obtained as described in the previous section. We first analyzed the repeat sequences in five SCA31 patients by direct sequencing of the gel-extracted, purified PCR product with primers 010-1F (5′-CATAGTGGCACATGCATGTAGTC-3′) and 10R (5′-CCCAGGCTGGAGTGCAGTGAC-3′) to see the configurations of the insertion sequences. Using the same method, we investigated nucleotide sequences at the insertion site in five normal controls so that we could see the (TAAAA)n repeat numbers (see Results). Because we found two exceptional controls (controls 1 and 2) harboring smaller insertions, we also sequenced these insertions by the same method.
Because the simple repeat sequences in the insertion extended too long to be read through, shotgun sequencing was also performed in the homozygous patient and on the allele with an insertion from control 1. For this shotgun sequencing, purified PCR products were directly sheared by HydroShear (Genomic Solutions). Ninty-six colonies were picked up for each PCR product and sequenced as done in BAC and fosmid shotgun sequencing. The obtained sequence data were assembled and analyzed with phred, phrap, and consed.
Analysis of Gene Expression
Poly-A+ RNA obtained from frozen human cerebellar tissues was reverse-transcribed with SuperScript III (Invitrogen). Because the insertion site was not annotated in the public database as being associated with any genes, we independently examined whether the repeat insertion could be transcribed. We designed primers around the repeat insertion site with 300–1000 bp intervals and attempted to amplify the site by PCR with various pairings of primers. If amplification was successful, we cloned the PCR products into pCR2.1-TOPO (Invitrogen) and cycle-sequenced them to find previously unidentified transcripts.
Gene expression in various human tissues was examined by PCR with the primers listed in Table S1. Human Multiple Tissue Panels I and II (Clontech, BD Bioscience) as well as various brain regions obtained from control brains at autopsy were used. For the identification of full-length transcripts, 5′- and 3′-rapid cloning of the cDNA ends (RACE) and RT-PCR analysis of control human cerebellar cDNA were performed. Expressions of newly identified transcripts were also confirmed by PCR screening of a human cerebellar cDNA library (Takara Bio, Inc.). When necessary, strand-specific RT-PCR was performed according to the method previously described.22
Quantitative RT-PCR
Quantitative PCR of cDNA was carried out by the TaqMan expression chemistry protocol with an ABI Prism 7700 Sequence Detection System (Applied Biosystems). Primers and probes used in this study were designed by the manufacturer (Table S2). Quantities measured by analysis were adjusted for glyceraldehyde 3-phosphate dehydrogenase (GAPDH) with TaqMan GAPDH Control Reagents (Applied Biosystems). Analyses were repeated three times for each sample, and results were compared between the patient (n = 2) and control (n = 4) groups.
Fluorescence In Situ Hybridization for Detecting RNA Foci
The detection of the repeat transcripts in Purkinje cells by FISH was carried out as previously described23 with digoxigenin (DIG)-labeled, locked nucleic acid (LNA) probes (Exiqon) (Table S3). The brain samples of the two SCA31 patients and four control subjects were analyzed in this part of the study. Frozen cerebellar cortex samples obtained at autopsy were stored at −80°C and sectioned at 10 μm thickness in a cryostat. Sections were fixed for 30 min at room temperature in 4% paraformaldehyde in PBS. After fixation, sections were washed and treated with 50% formamide, 2× SSC (300 mM NaCl and 30 mM sodium citrate [pH 7.0]) for 10 min at room temperature. Sections were then hybridized overnight at 37°C with a solution containing probe (1 ng/μl), 40% formamide, 2× SSC, 0.2% bovine serum albumin (BSA), 10% dextran sulfate, 2 mM vanadyl adenosine complex, and 1 mg/ml each of yeast transfer RNA and salmon sperm DNA. After being washed three times for 30 min at 45°C with 50% formamide and 2× SSC, the probe was detected with anti-DIG antibody Fab conjugated with alkaline phosphatase (Roche) and visualized with HNPP/FastRed (Roche) according to the manufacture's protocol. After nuclei were stained with 4′, 6-diamidino-2-phenylindole (DAPI), sections were mounted with an aqueous mounting kit. The specificity of the RNA foci was confirmed both by detection of foci with different stringencies in experiments and by verification that they disappeared with RNase A treatment, but not with DNase. At least ten Purkinje cells were observed in each section.
Recombinant Glutathione-S-Transferase-Fused Splicing Factor Protein Synthesis and the Electrophoretic Mobility-Shift Assay
To further clarify the pathogenic implication of transcribed repeat insertion, we looked for proteins that could potentially bind to the transcribed repeats. Based on the fact that the SCA31 repeat sequence was related to satellite sequence in heterochromatin (see Results), and also based on consultation with ESEfinder 3.0, we found two serine/arginine-rich splicing factor proteins (SR proteins), SFRS1 and SFRS9, that were good candidates for binding to transcribed SCA31 insertions.
Recombinant GST-fused SR proteins were synthesized as follows. The cDNA of the full-length coding regions of SR proteins (SFRS1-1: NM_006924.4; SFRS1-2: NM_001078166.1; and SFRS9: NM_003769.2; all in NCBI) were amplified by PCR from a cDNA library of human cerebellum (Takara Bio) with custom primers (Table S4). SFRS1-1-R corresponds to SFRS1 isoform 1 (SFRS1-1) and SFRS1-2-R to SFRS isoform 2 (SFRS1-2). The PCR products were double digested with EcoRI and SalI and cloned in frame into the EcoRI-SalI site of the pGEX-6P1 vector (GE Healthcare Bioscience). The SR-protein constructs were first introduced into JM109, amplified, and then used for transforming the BL21 strain of E. coli.
The GST-fused SR proteins were harvested by a modified version of the previously described method.24 In brief, 500 ml of transformed BL21 cell cultures was induced with 0.5–1.0 mM of IPTG for 3 hr, and the collected cells were suspended in 50 mM Tris-HCl (pH 8.0) and 150 mM NaCl. The E. coli were sonicated and treated with 0.02% DNase, 1 mg/ml lysozyme, and 1 mM PMSF. GST-fused proteins were purified from the soluble fraction with Glutathione Sepharose 4 Fast Flow (GE Healthcare Bioscience) and eluted with 50 mM Tris-HCl (pH 8.0) and 20 mM glutathione. After dialysis, the concentrations of the proteins were measured by BCA assay. GST-SFRS1-1 and GST-SFRS9 were successfully synthesized, whereas GST-SFRS1-2 was poorly collected in the soluble fraction. GST-SFRS1-1 was used as the representative of GST-SFRS1 in the subsequent study.
For electrophoretic mobility-shift assays (EMSAs), the synthetic RNA oligonucleotides, (UGGAA)8, (UAGAA)8, and (UAGAAUAAAA)4, were labeled with digoxigenin (DIG). The RNA probes and the specific competitors, unlabeled (UGGAA)8, (UAGAA)8, and (UAGAAUAAAA)4, were denatured at 94°C and immediately used for the following procedures. RNA probes (1.5 pmoles) were mixed with 2.4 × 10−7 g of either synthetic GST-fused protein or GST alone, different concentrations (0- to 100-fold the concentration of the probe) of one of the specific competitors, and 4 nM of poly[d(A-T)] in a 20 μl solution of 20 mM HEPES (pH 7.6), 1 mM EDTA, 10 mM (NH4)2SO4, 1 mM DTT, 0.2% (w/v) Tween 20, and 30 mM KCl. The mixtures were incubated for 25 min at room temperature and then placed on ice. After dilution of the mixtures with 0.5× TBE buffer to give one-fifth of the original concentrations, the protein-bound probes were separated from the free forms by being run through 6% native polyacrylamide gels at 30 V in 0.5× TBE buffer. The separated RNA probes were transferred to positively charged nylon membranes by a semi-dry method and then detected with the DIG Luminescent Detection Kit (Roche) according to the manufacturer's instructions.
In Silico Location Search of Penta-Nucleotide Repeats
To reveal the origin of complex penta-nucleotide insertion, we searched locations in the human genome where repeat sequences (TGGAA)n, (TAGAA)n, and (TAAAATAGAA)n were abundantly present. For this purpose, repeat sequences were set unmasked, and then locations of (TGGAA)40, (TAGAA)40, and (TAAAATAGAA)3 were searched for in the “reference only” human genome by BLAST. The best-matched 500 locations with E value < 0.0001 were selected and were shown in “genome view” according to the degree to which they matched. We then checked the matched sequences manually to confirm that pure stretches had been correctly selected. The resultant locations were shown in chromosome figures.
Mutation Screening in the SCA4 Family and 21 Disease Controls
To investigate the genetic relationship between SCA31 and SCA4, we screened five DNA samples from the American SCA4 family7 by performing PCR-based direct sequencing of all coding exons and exon-intron boundaries of BEAN, TK2, and FLJ27243 by using 40 primer pairs (shown upon request). Southern blot analysis in the genomic region encompassing BEAN and TK2 (positions from 65,160,201–65,932,756 on chromosome 16, NCBI build 36.3) was also performed similarly to the SCA31 mutation search.
The 21 disease controls13 who did not have the SCA31 founder haplotypes were also tested for the insertion. Ten out of these 21 individuals were additionally screened for any mutations in BEAN, TK2, or in FLJ27243 with the same 40 primer pairs as those used for SCA4.
Results
Southern Blot Analysis Revealed a 2.5–3.8 Insertion Consisting of Complex Pentanucleotide Repeats Containing (TGGAA)n in SCA31 Patients
Figure 1 shows a comprehensive physical map of the SCA31 critical region. Screening of this 900 kb critical region by Southern blotting allowed us to identify an insertion at nucleotide number 65,081,803 on human chromosome 16 on NCBI build 36.3 (Figures 2A and 2B). There were no other gene rearrangements in the entire critical region. The insertion was found in all 160 affected individuals from 98 SCA31 families, including the individual in the U9 family previously described,13 and ranged in length from 2.5–3.8 kb. PCR amplification followed by direct sequencing or shotgun sequencing of the insert disclosed that the insertion consisted of a preceding four nucleotides, “TCAC,” and the three penta-nucleotide repeat components (TGGAA)n, (TAGAA)n, and (TAAAA)n in all SCA31 patients tested (Figure 2C). In the homozygous patient's repeat, which was sequenced by the shotgun method, a pure (TGGAA)n stretch extended for at least 110 repeats, presumably for more than 1 kb in light of the size of the sheared DNA fragment. Pure (TAAAATAGAA)n also stretched for more than 112 repeats. These pure repeat sequences were separated by a bridging sequence and (TAGAA)46.
Figure 1.
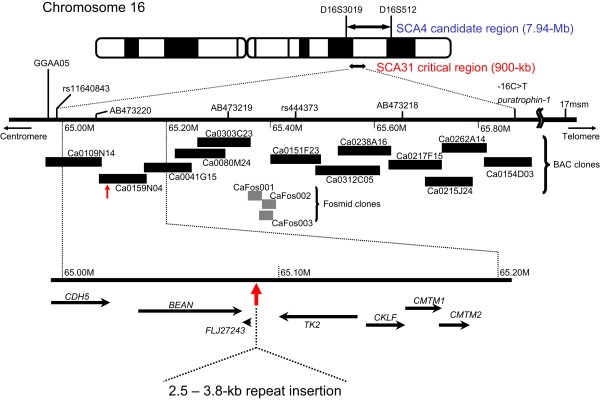
A Comprehensive Physical Map of the 900 kb SCA31 Critical Interval between rs11640843 and −16C > T in the PLEKHG4 Gene
This region was entirely covered without gaps by 12 BAC and three fosmid clones derived from a SCA31 homozygous patient. These clones were sequenced. An insertion ranging in length from 2.5–3.8 kb was found at nucleotide number 65,081,803 on human chromosome 16 on NCBI build 36.3 between BEAN and TK2.
Figure 2.

Identification of Complex Pentanucleotide Repeat Insertions in SCA31 Patients
(A) Southern blot analysis showing the SCA31 insertion. The left-hand panel shows EcoRI-digested genomic fragments detected with a cosmid probe for the region between nucleotides 65,083,571 and 65,124,051. A rare 1.5 kb insertion and an unusual 0.7 kb expanded (TAAAA)n (both shown with solid black arrows) were observed in one control (control 1). SCA31 insertions in two patients are shown with a red arrow. “Hom.” and “Het.” designate the homozygous patient and heterozygous patient, respectively. The dotted arrow indicates normal chromosomes without insertions. The thick 5.8 kb bands common in the three subjects show fragments outside the insertion site. The right-hand panel shows aberrant EcoRI-digested 9–10 kb genomic fragments (a red arrow) that completely segregated with SCA31 patients (P). All heterozygous patients (P) and controls (C) have “normal” 6 kb fragments (dotted arrows). Radiolabeled PCR products obtained by amplifying the 3009 bp genomic segment between nucleotides 65,079,127 and 65,082,135 on NCBI build 36.3 were used as probes.
(B) Sequences around the SCA31 insertion (chromosome16: nucleotides 65,081,260–65,082,786 on NCBI build 36.3). Flanking primers for PCR amplification (underlined in red) of insertion and flanking HaeIII recognition sites (in shaded boxes) are shown. The AluSx sequence30 is shown with a green underline. Without an insertion, PCR amplification with flanking primers and a subsequent HaeIII digestion will produce a DNA fragment 193 bp in length.
(C) The components of the SCA31 insertion in the homozygous patient. The SCA31 insertion consists of a preceding 4 bp TCAC and three different penta-nucleotides, (TGGAA)n, (TAGAA)n, and (TAAAA)n. (TGGAA)n is the patient-specific repeat (shown in red), and both (TGGAA)n and (TAAAATAGAA)n are pure stretches too long to be read through. The bridging sequence between (TGGAA)n and (TAGAA)46 is underlined in red.
(D) The sequence of the insertion in control 1. Rare insertions were observed in controls at the same position as the SCA31 insertion, but with shorter length and different components. The insertion in control 1 consisted of a preceding 4 bp TCAC and two pentanucleotide components, (TAGAA)n and (TAAAA)n. The (TGGAA)n was not detected.
In contrast to SCA31 chromosomes, the vast majority of controls (99.77% among 800 Japanese and 60 white American chromosomes) did not have any insertions. The 21 disease controls and SCA4 subjects also did not have this insertion. Very rarely, however, insertions were observed in two individuals (2/860 chromosomes: 0.23%) (controls 1 and 2) (Figure 2A, left-hand panel). PCR amplification and sequencing showed that the inserts were in the same position as in the SCA31 patients, and their lengths were 1.5 kb in control 1 and 2.0 kb in control 2. Sequencing analysis of controls 1 and 2 disclosed that they both had inserts with a preceding 4 base “TCAC,” (TAAAA)n, (TAGAA)n, and (TAAAATAGAA)n stretch (Figure 2D). However, no (TGGAA) sequences were observed. Because these individuals did not manifest any cerebellar signs or have any documented histories of inherited diseases in their families, we considered that these inserts with complex penta-nucleotide repeats were not pathogenic, or at least did not have enough toxicity to develop a disease during the human lifespan. Therefore, we concluded that the insertions in SCA31 patients exerted their toxicity either because of their lengths (> = 2.5-kb) or because of their (TGGAA)n component, which made a clear distinction between SCA31 and the rare control insertions.
The insertion site was identical for all insertions, and the junction point of the insertion was located at the 3′-tail of an AluSx25 (Figure 2B). Alu variants have previously been linked to certain disease loci, such as DM1, HD, Friedreich ataxia (FRDA) (MIM #229300), and fragile X syndrome (FRAXA) (MIM #300624).26 There is a related short penta-nucleotide repeat of (TAAAA)n immediately downstream of the insertion site (Figure 2B). This (TAAAA)n was polymorphic (“n” usually ranged from 8–21), and a very rare expansion up to approximately 140 repeats was observed in one out of 860 control chromosomes (frequency: 0.12%) (control 1, shorter allele; see Figure 2A, left-hand panel). Both (TAAAA)n and the SCA31 founder insertion are polypurine tracts interrupted with thymidines. SCA31 is similar to Friedreich's ataxia (FRDA) in that they both contain “GAA.”
The Length of the SCA31 Insertion Is Inversely Correlated with the Age of Onset
The SCA31 penta-nucleotide repeat insertion ranged from 2.5–3.8 kb in length (Figure 3A). Although SCA31 is a disease with a strong founder effect, the fact that the length of the insertion varied by ∼1.3 kb among families suggests that the insertion was not completely stable during multiple transmissions from one or a few principal ancestors. Importantly, a significant correlation was observed in that patients with longer repeats show earlier disease onset (r = −0.41, p = 0.010, n = 39) (Figure 3B). Very mild anticipation (younger age of onset in future generations) is sometimes observed in SCA318, which suggests that the insertion might have a propensity for expansion. Indeed, we detected a subtle expansion of the inserted repeat within one SCA31 family (Figure 3C).
Figure 3.
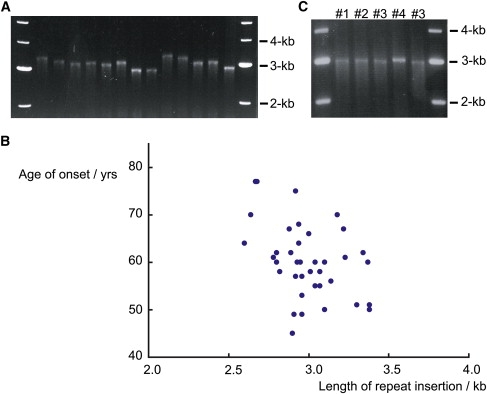
The Length of Insertion Inversely Correlates with Age of Onset in SCA31
(A) PCR amplification and agarose gel electrophoresis showing that the length of the insertion differs among SCA31 families.
(B) A scatter plot showing an inverse correlation between the length of the SCA31 insertion and the age of onset. The length of the repeat insertion was inversely correlated with age of onset (Pearson's product-moment correlation coefficient r = −0.41, p = 0.010, n = 39.).
(C) A slight expansion of the SCA31 insertion observed in one SCA31 family. Individual #4 has a slightly longer insertion than the others (#1–#3) in the same SCA31 family. This individual #4 is in the youngest generation among them.
Complete Genomic Sequencing of the 900 kb Critical Region Unveiled Only Two Mutation Candidates
Because of the strong founder effect in SCA31, we needed to detect all genetic changes in the critical region. For this reason, we also performed BAC- and fosmid-based shotgun sequencing over the entire 900 kb SCA31 critical region. Upon completing entire sequencing in the homozygous patient, we initially found 336 sites annotated differently from the reference sequence (NCBI build 36.3). However, most of these 336 changes were also found in the controls, allowing us to exclude them as mutation candidates. In the course of this effort, we also investigated 34 new SCA31 patients and found that two independent SCA31 patients shared the disease-specific haplotype only between AB473214 and AB473219 (Table 2). As a result, we finally found that the penta-nucleotide repeat insertion and a single-nucleotide change (AB473217) at 65,114,245 are the only genetic changes segregating with the disease. This single-nucleotide change (AB473217) is in an intron of the TK2 (thymidine kinase 2) gene, 4,964 nucleotides distant from the nearest splice junction. RT-PCR analysis did not indicate the presence of aberrant transcripts in SCA31 patients (data shown upon request). Quantitative genomic PCR and aCGH did not show copy-number variations in the SCA31 critical region (data shown upon request). Taking all these data together, we considered that the complex penta-nucleotide repeat insertion containing (TGGAA)n was the only mutation that could plausibly cause SCA31. Further efforts were focused on this repeat insertion.
Table 2.
The Haplotypes of Representative SCA31 Patients and Control Subjects
The SCA31 Repeat Insertion Is Located in Newly Indentified Introns of BEAN and TK2
According to the NCBI database (build 36.3), the insertion was located between two genes, BEAN and TK2, and also upstream of an EST, FLJ27243 (Figure 1). However, we found previously unidentified downstream exons for BEAN and TK2, demonstrating that the insertion is in introns of these two genes transcribed in opposite directions (Figure 4). We confirmed by RT-PCR that the extended versions of BEAN and TK2, which we named BEAN-EXT and TK2-EXT, respectively, and FLJ27243 are all expressed at low levels in the brain (Figures 5A–5C). Notably, BEAN-EXT, as well as BEAN, is expressed exclusively in the brain. However, neither ordinary nor quantitative RT-PCR proved that the repeat insertion caused splicing abnormalities or alterations in the expression levels of BEAN, TK2, or other nearby genes (Figure 5D).
Figure 4.
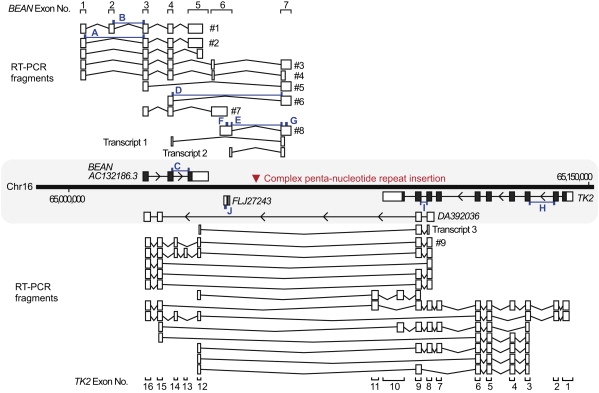
Various Transcripts Spanning the SCA31 Repeat Insertion Site
The locations of BEAN, TK2, FLJ27243, and the SCA31 insertion (red arrowhead) are shown on the physical map of the chromosomal region between nucleotides 65,000,000 and 65,150,000 on NCBI build 36.3. Exons registered in the NCBI database (shown in a shaded area) are shown with black boxes, and 5′- and 3′-UTRs are shown with white boxes attached to them. Although the SCA31 insertion is located in the intergene region between BEAN and TK2 on the NCBI database, various newly identified transcripts of these genes (shown with white boxes with their exon numbers) were detected by RT-PCR, and some of them encompassed the SCA31 insertion. The insertion appeared to be located in introns of BEAN and TK2, two genes transcribed in opposite directions. DA392036 annotated in the NCBI database seemed to be a part of TK2-EXT. Transcripts 1–3 correspond to the transcripts detected by RT-PCR in Figures 5A and 5B (Table S1). The primer pairs for RT-PCR are shown with small blue boxes (A–J; Figure 5D and Tables S1 and S2).
Figure 5.
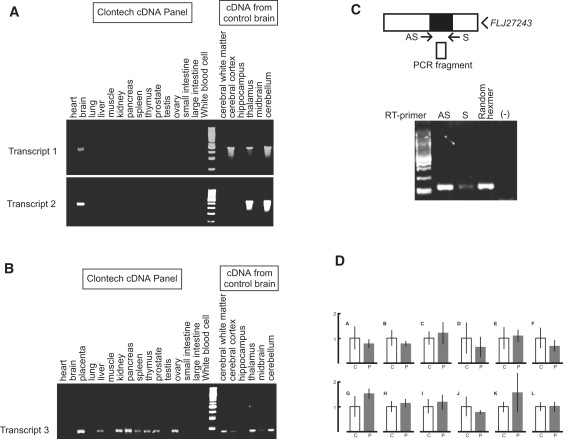
Gene Expression of BEAN-EXT, TK2-EXT, and FLJ27243 in Humans
(A) RT-PCR analysis for BEAN-EXT mRNA (transcripts 1 and 2 in Figure 5) showing its brain-specific expression.
(B) RT-PCR analysis for TK2-EXT mRNA (transcript 3 in Figure 5) showing higher expression in various systemic organs than in the brain.
(C) RT-PCR of FLJ27243 mRNA in the human cerebellum. Strand-specific RT-PCR shows expression of FLJ27243, represented by “RT with AS primer,” in the brain. The “S primer” represents transcription in the orientation of BEAN, and “Random Hexamer” indicates transcripts in both directions. The specificity of this strand-specific RT-PCR is confirmed by negative amplification when reverse transcriptase is omitted [(-)].
(D) Quantitative RT-PCR on BEAN, TK2, FLJ27243, and CKLF mRNAs in controls' (n = 4) and patients' (n = 2) cerebella. The locations of RT-PCR primer and probe sets (A–J) are indicated in Figure 5 (C: controls; P: patients; the scale bar represents 1 SD) (see Table S2 for probe sequences). No consistent difference was found in the expression levels of BEAN (including BEAN-EXT; probe sets: A–G), TK2 (probe sets H and I), FLJ27243 (probe set J), or CKLF (probe sets K–L) mRNAs compared in the control versus SCA31 patient groups.
The Transcribed SCA31 Insertion Forms Nuclear Foci in Purkinje Cells
We next performed in situ hybridization to see whether transcribed repeat sequences form aggregates (“RNA foci”) in nuclei, as in RNA-mediated noncoding repeat-expansion disorders,3 such as DM123 and DM2.27 Using an LNA probe targeting (UAAAAUAGAA)n, we detected RNA foci in approximately 30%–50% of the nuclei of SCA31 patients' Purkinje cells (Figure 6). Such RNA foci were never observed in controls, allowing a clear distinction. This might indicate not only that the insertion is transcribed as BEAN-EXT transcript in SCA31 brains but also that the insertion transcribed in the direction of BEAN-EXT forms abnormal RNA aggregates in Purkinje cells, the primary target of SCA31. Foci were never observed with a probe for anti-sense (UUCUAUUUUA)n repeats corresponding to the TK2-EXT transcripts in either SCA31 or control brains.
Figure 6.
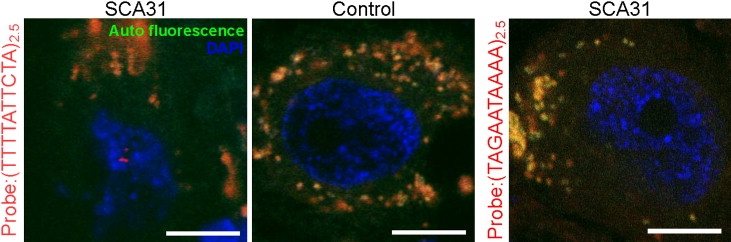
Presence of RNA Foci in SCA31 Purkinje Cells
RNA foci (red dots) seen in a nucleus (stained with DAPI; blue) of an SCA31 Purkinje cell with an LNA-(TTTTATTCTA)2.5 probe targeting the transcripts containing the (UAAAAUAGAA)n repeat (autofluorescence; orange). In controls, foci were completely negative. Anti-sense transcripts, searched with an LNA-(TAGAATAAAA)2.5 probe, did not appear as RNA foci. Scale bars represent 10 μm.
(UGGAA) n, the Disease-Specific Transcribed Component of the SCA31 Insertion, binds to SFRS1 and SFRS9 In Vitro
We next searched for proteins that could potentially bind to the transcribed SCA31 insertion, particularly to the (UGGAA)n sequence. Accumulating evidence increasingly suggests that satellite III (SatIII), the paracentromeric repetitive sequences rich in (TGGAA)n, are transcribed under stress28–30 to form nuclear stress bodies (nSBs) and play an important role in regulating the splicing machinery by recruiting certain splicing factors,31 such as serine/arginine-rich splicing factor (SFRS) 1 (also known as ASF/SF2 or SRp30a) and SFRS9 (SRp30c), to nSBs. On the basis of this fact, we performed EMSAs and found that SFRS1 and SFRS9 directly bind to (UGGAA)n, the transcribed sequence of (TGGAA)n, in vitro (Figure 7).
Figure 7.
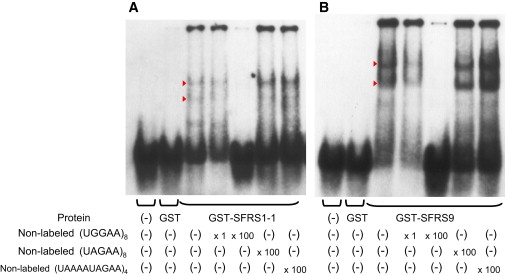
The Pentanucleotide (TGGAA)n Binds to Splicing Factors SFRS1 and SFRS9 In Vitro
EMSA showing specific binding of SFRS1 isoform 1 (SFRS1-1) (A) and SFRS9 (B) to RNA oligonucleotide (UGGAA)8. Shifted bands (arrowheads) were observed in mixtures of digoxigenin(DIG)-(UGGAA)8 and either GST-SFRS1-1 or GST-SFRS-9. The shifted bands disappeared with the addition of nonlabeled (UGGAA)8, whereas the addition of excess amounts of nonlabeled (UAGAA)8 or (UAAAAUAGAA)4 did not interfere with the band shift. No shift was seen when DIG-(UGGAA)8 was mixed with GST alone.
(TGGAA)n Is Abundant in Heterochromatin, Particularly in Centromeric Regions
In order to see the distribution of (TGGAA)n in the human genome, we also performed an in silico search for (TGGAA)n and found that these penta-nucleotide repeats are indeed abundant in the centromeres of chromosomes 2, 4, 7, 10, 16, 17, 20, and Y (Figure 8A). On the other hand, (TAGAA)n is observed in euchromatins and telomeres (Figure 8B). (TAAAATAGAA)n was not detected in the human genome.
Figure 8.
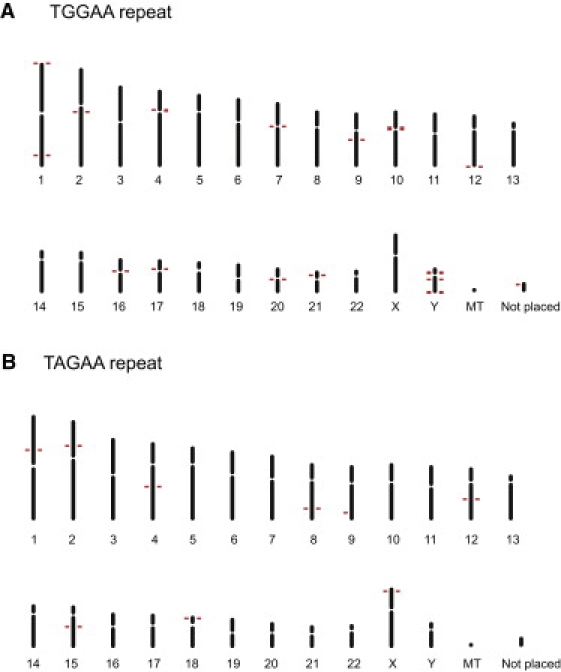
(TGGAA)n Is Particularly Abundant in Centromeric Regions
(A) (TGGAA)40 sequences are abundant in the centromeres of chromosomes 2, 4, 7, 10, 16, 17, 20 and Y, whereas they are sparse in normal euchromosomes.
(B) (TAGAA)40 sequences are widely observed in euchromatin as well as in telomeres.
SCA4 and SCA31 Are Not Likely to Be Allelic
Neither the SCA31 insertion nor the single-nucleotide change in TK2 (AB473217) was detected in SCA4 individuals. PCR-based direct sequencing of all coding exons and exon-intron boundaries of BEAN, TK2, and FLJ27243 with 40 primer pairs and Southern blot analysis in the genomic region encompassing BEAN and TK2 were also negative. These results did not support the hypothesis that SCA4 and SCA31 were allelic diseases. Similarly, the 21 disease controls13 also did not harbor the insertion. Mutations in BEAN, TK2, and FLJ27243 were not found in the ten disease controls tested.
Discussion
In summary with regard to the results of our mutation search, only two genetic changes were found to completely segregate with the disease: the complex penta-nucleotide repeat insertion at nucleotide 65,081,803 and a single-nucleotide change (AB473217) at nucleotide 65,114,245 in human chromosome 16 (NCBI build 36.3). The single-nucleotide change, located in an intron, did not seem to have any obvious effects on splicing or expression patterns, providing no evidence that this is the causative mutation. On the other hand, the length of the insertion with penta-nucleotide repeats was inversely correlated with age of onset, in agreement with the general rules of repeat-expansion disorders.1–3 We therefore concluded that the penta-nucleotide repeat insertion containing (TGGAA)n is the only likely candidate for the SCA31 mutation. The fact that we did not find any allelic mutations in BEAN, TK2, or FLJ27243 in our cohort of patients (5 SCA4 and 21 disease-control individuals) does not deny this conclusion because none of the repeat-expansion diseases, except SCA6, have allelic mutations.1–3
All the previously known repeat-expansion disorders are caused by expansions of microsatellites present as polymorphic DNA repeats in humans: normal individuals have smaller numbers of repeats, whereas patients harbor longer repeats.26 To our knowledge, SCA31 is the first human disease discovered to be associated with a microsatellite “insertion.” The observation in rare controls of shorter inserted repeats lacking (TGGAA)n suggests that the inserted microsatellite has to have sufficient length (≥ 2.5 kb), the (TGGAA)n stretch, or both to cause the disease. Notably, haplotypes based on flanking markers were not similar in the controls with the rare inserts and the SCA31 patients. Although this might indicate that the insertions in controls and SCA31 patients arose from different insertion events, concluding so is still premature. Both the control and SCA31 inserts had a preceding 4 bp TCAC and also possessed (TAGAA)n, (TAAAA)n, and (TAAAATAGAA)n. In fact, controls with rare premutation alleles in the DM2 repeat were found to have a haplotype similar to that of DM2 patients.32 Thus, further analysis will need to address whether SCA31 and control insertions have different ancestries.
The presence of (TGGAA)n, the characteristic sequence of satellites II and III33, suggests that the SCA31 insertion might be related to heterochromatin; this idea is supported by our in silico search for (TGGAA)n. SCA31 could be the second disease associated with heterochromatin insertion after autosomal-recessive congenital deafness (DFNB10) (MIM #605316),34 caused by a β-satellite sequence insertion into a coding exon. Although the β-satellite sequence insertion causes DFNB10 via a loss-of-function mechanism, SCA31 appears to be associated with intronic repeat insertions that are transcribed to form RNA foci.
How does this insertion cause the disease? Haplo-insufficiency or dominant-negative mechanisms do not appear likely because both BEAN (including BEAN-EXT) and TK2 (including TK2-EXT) were expressed at the same level in SCA31 patients' brains as they were in control brains, at least at mRNA levels. Paracentromeric satellite sequences rich in penta-nucleotide repeats (TGGAA)n are thought to have various essential roles, such as the maintenance of chromatin conformation.33 The expanded repeat sequence of (TGGAA)n, which tends to take non-B DNA structures,33 might induce local chromosomal structural changes that could alter the expressions of other genes, as proposed in FRDA caused by (GAA)n expansion (“sticky DNA”).1
Alternatively, there is a possibility that the transcripts of the repeat insertion convey the pathogenesis (i.e., “RNA-mediated gain-of-function mechanism”).3 Earlier onset in homozygotes than in heterozygotes, as described in a previous study14, appear to support the gain-of-function mechanism. In noncoding repeat expansion disorders, such as DM123, DM227, FXTAS35, HDL236, and SCA822, transcribed repeats form aggregates (“RNA foci”) in the nuclei of affected cells. The sequestration of proteins that bind to these foci, such as muscleblind-like protein 1 and CUG-binding protein (CUG-BP)13 in DM1 and DM2, as well as CUG-BP1 and heterogeneous nuclear ribonucleoprotein (hnRNP)A2 in FXTAS37, are believed to cause dysregulation of alternative splicing. In light of these facts, the presence of nuclear RNA foci in Purkinje cells and in vitro binding of essential splicing factors SFRS1 and SFRS9 to (UGGAA)n imply that SCA31 might also be associated with RNA-mediated gain-of-function mechanisms.
SR proteins, such as SFRS1, play important roles in constitutive splicing, alternative splicing regulation in which they antagonize hnRNPs31,38, and stabilizing mRNAs.38 Suppression of SFRS1 expression results in embryonic lethality in C. elegans39 and death in particular subsets of neurons.40 If the transcripts of the SCA31 insertion should indeed sequester SFRS1 and SFRS9 by forming RNA foci, it might disturb the pre-mRNA processing patterns of various genes41 and ultimately lead to neuronal death. Interestingly, overexpression of hsrω, the Drosophila noncoding RNA gene similar to SatIII, is shown to exacerbate neurodegeneration in a fly model of polyglutamine disease, in which sequestration of transcription factors such as CREB binding protein (CBP) are considered important for pathogenesis.42 Further analysis is clearly needed, not only to dissect how this newly indentified insertion mutation causes a human disease, but also to disclose the roles of highly repeated sequences in heterochromatin.
Supplemental Data
Supplemental Data include four tables and can be found with this article online at http://www.cell.com/AJHG.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
ESEfinder 3.0, http://rulai.cshl.edu/cgi-bin/tools/ESE3/esefinder.cgi?process=home
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/sites/entrez?db=OMIM
Accession Numbers
The GenBank accession numbers for Homo sapiens BEAN-EXT transcripts are AB472390 (#1 in Figure 5), AB472391 (#2), AB472392 (#3), AB47233 (#6), AB472395 (#7), AB472396 (#4), AB472398 (#8), and AB472399 (#5). The GenBank accession number for Homo sapiens TK2-EXT transcript is AB472397 (#9 in Figure 5). The GenBank accession number for human genome single nucleotide polymorphisms (SNPs) are AB473214; 65,024,796 G/A: AB473217; 65,114,245 G/C: AB473218; 65,658,263 T/C: AB473219; 65,337,827 A/G: AB473220; and 65,049,292 G/A. For the insertion site at 65,081,803, the accession number is AB473216; for the control insertion, the accession number is AB473734; and for the insertion in the SCA31 patient, the accession number is AB473733.
Acknowledgments
We would like to thank the families who participated in this study, as well as Iku Sudo, Minori Kono, and Hideko Uno for their technical assistance. This study was funded by numerous grants, particularly from the Health and Labour Sciences Research Grants on Human Genome (KI) and Ataxic Diseases (HM), Ministry of Health, Labour and Welfare, Japan, and the 21st Century COE Program, Brain Integration and its Disorders, the Ministry of Education, Science and Culture, Japan (HM). We also thank Laura P.W. Ranum (University of Minnesota) and Christopher E. Pearson (Hospital for Sick Children, Toronto) for their advice. We thank Eric Sheldon (Scientific Language Editing Team, Tsukuba, Japan) for proofreading our manuscript.
References
- 1.Brice A., Pulst S.-M. Butterworth Heinemann, Elsevier, Inc.; Philadelphia: 2007. Spinocerebellar degenerations: The ataxias and spastic paraplegias. [Google Scholar]
- 2.Zoghbi H.Y., Orr H.T. Glutamine repeats and neurodegeneration. Annu. Rev. Neurosci. 2000;23:217–247. doi: 10.1146/annurev.neuro.23.1.217. [DOI] [PubMed] [Google Scholar]
- 3.Ranum L.P.W., Cooper T.A. RNA-mediated neuromuscular disorders. Annu. Rev. Neurosci. 2006;29:259–277. doi: 10.1146/annurev.neuro.29.051605.113014. [DOI] [PubMed] [Google Scholar]
- 4.Bandmann O., Singleton A.B. Yet another spinocerebellar ataxia: The saga continues. Neurology. 2008;71:542–543. doi: 10.1212/01.wnl.0000323932.90720.55. [DOI] [PubMed] [Google Scholar]
- 5.Nagaoka U., Takashima M., Ishikawa K., Yoshizawa K., Yoshizawa T., Ishikawa M., Yamawaki T., Shoji S., Mizusawa H. A gene on SCA4 locus causes dominantly inherited pure cerebellar ataxia. Neurology. 2000;54:1971–1975. doi: 10.1212/wnl.54.10.1971. [DOI] [PubMed] [Google Scholar]
- 6.Ishikawa K., Toru S., Tsunemi T., Li M., Kobayashi K., Yokota T., Amino T., Owada K., Fujigasaki H., Sakamoto M. An autosomal dominant cerebellar ataxia linked to chromosome 16q22.1 is associated with a single-nucleotide substitution in the 5′ untranslated region of the gene encoding a protein with spectrin repeat and Rho guanine-nucleotide exchange-factor domains. Am. J. Hum. Genet. 2005;77:280–296. doi: 10.1086/432518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Flanigan K., Gardner K., Alderson K., Galster B., Otterud B., Leppert M.F., Kaplan C., Ptácek L.J. Autosomal dominant spinocerebellar ataxia with sensory axonal neuropathy (SCA4): Clinical description and genetic localization to chromosome 16q22.1. Am. J. Hum. Genet. 1996;59:392–399. [PMC free article] [PubMed] [Google Scholar]
- 8.Owada K., Ishikawa K., Toru S., Ishida G., Gomyoda M., Tao O., Noguchi Y., Kitamura K., Kondo I., Noguchi E. A clinical, genetic, and neuropathologic study in a family with 16q-linked ADCA type III. Neurology. 2005;65:629–632. doi: 10.1212/01.wnl.0000173065.75680.e2. [DOI] [PubMed] [Google Scholar]
- 9.Hellenbroich Y., Bubel S., Pawlack H., Opitz S., Vieregge P., Schwinger E., Zühlke C. Refinement of the spinocerebellar ataxia type 4 locus in a large German family and exclusion of CAG repeat expansions in this region. J. Neurol. 2003;250:668–671. doi: 10.1007/s00415-003-1052-x. [DOI] [PubMed] [Google Scholar]
- 10.Hellenbroich Y., Gierga K., Reusche E., Schwinger E., Deller T., de Vos R.A., Zühlke C., Rüb U. Spinocerebellar ataxia type 4 (SCA4): Initial pathoanatomical study reveals widespread cerebellar and brainstem degeneration. J. Neural Transm. 2006;113:829–843. doi: 10.1007/s00702-005-0362-9. [DOI] [PubMed] [Google Scholar]
- 11.Hellenbroich Y., Bernard V., Zühlke C. Spinocerebellar ataxia type 4 and 16q22.1-linked Japanese ataxia are not allelic. J. Neurol. 2008;255:612–613. doi: 10.1007/s00415-008-0771-4. [DOI] [PubMed] [Google Scholar]
- 12.Ohata T., Yoshida K., Sakai H., Hamanoue H., Mizuguchi T., Shimizu Y., Okano T., Takada F., Ishikawa K., Mizusawa H. A −16C>T substitution in the 5′ UTR of the puratrophin-1 gene is prevalent in autosomal dominant cerebellar ataxia in Nagano. J. Hum. Genet. 2006;51:461–466. doi: 10.1007/s10038-006-0385-6. [DOI] [PubMed] [Google Scholar]
- 13.Amino T., Ishikawa K., Toru S., Ishiguro T., Sato N., Tsunemi T., Murata M., Kobayashi K., Inazawa J., Toda T. Redefining the disease locus of 16q22.1-linked autosomal dominant cerebellar ataxia. J. Hum. Genet. 2007;52:643–649. doi: 10.1007/s10038-007-0154-1. [DOI] [PubMed] [Google Scholar]
- 14.Ouyang Y., Sakoe K., Shimazaki H., Namekawa M., Ogawa T., Ando Y., Kawakami T., Kaneko J., Hasegawa Y., Yoshizawa K. 16q-linked autosomal dominant cerebellar ataxia: A clinical and genetic study. J. Neurol. Sci. 2006;247:180–186. doi: 10.1016/j.jns.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 15.Kobayashi K., Nakahori Y., Miyake M., Matsumura K., Kondo-Iida E., Nomura Y., Segawa M., Yoshioka M., Saito K., Osawa M. An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature. 1998;394:388–392. doi: 10.1038/28653. [DOI] [PubMed] [Google Scholar]
- 16.Li M., Ishikawa K., Toru S., Tomimitsu H., Takashima M., Goto J., Takiyama Y., Sasaki H., Imoto I., Inazawa J. Physical map and haplotype analysis of 16q-linked autosomal dominant cerebellar ataxia (ADCA) type III in Japan. J. Hum. Genet. 2003;48:111–118. doi: 10.1007/s100380300017. [DOI] [PubMed] [Google Scholar]
- 17.Hattori M., Fujiyama A., Taylor T.D., Watanabe H., Yada T., Park H.S., Toyoda A., Ishii K., Totoki Y., Choi D.K. The DNA sequence of human chromosome 21. Nature. 2000;405:311–319. doi: 10.1038/35012518. [DOI] [PubMed] [Google Scholar]
- 18.Asakawa S., Abe I., Kudoh Y., Kishi N., Wang Y., Kubota R., Kudoh J., Kawasaki K., Minoshima S., Shimizu N. Human BAC library: Construction and rapid screening. Gene. 1997;191:69–79. doi: 10.1016/s0378-1119(97)00044-9. [DOI] [PubMed] [Google Scholar]
- 19.Jin H., Ishikawa K., Tsunemi T., Ishiguro T., Amino T., Mizusawa H. Analyses of copy number and mRNA expression level of the α-synuclein gene in multiple system atrophy. J. Med. Dent. Sci. 2008;55:145–153. [PubMed] [Google Scholar]
- 20.Barrett M.T., Scheffer A., Ben-Dor A., Sampas N., Lipson D., Kincaid R., Tsang P., Curry B., Baird K., Meltzer P.S. Comparative genomic hybridization using oligonucleotide microarrays and total genomic DNA. Proc. Natl. Acad. Sci. USA. 2004;101:17765–17770. doi: 10.1073/pnas.0407979101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hara K., Shiga A., Nozaki H., Mitsui J., Takahashi Y., Ishiguro H., Yomono H., Kurisaki H., Goto J., Ikeuchi T. Total deletion and a missense mutation of ITPR1 in Japanese SCA15 families. Neurology. 2008;71:547–551. doi: 10.1212/01.wnl.0000311277.71046.a0. [DOI] [PubMed] [Google Scholar]
- 22.Moseley M.L., Zu T., Ikeda Y., Gao W., Mosemiller A.K., Daughters R.S., Chen G., Weatherspoon M.R., Clark H.B., Ebner T.J. Bidirectional expression of CUG and CAG expansion transcripts and intranuclear polyglutamine inclusions in spinocerebellar ataxia type 8. Nat. Genet. 2006;38:758–769. doi: 10.1038/ng1827. [DOI] [PubMed] [Google Scholar]
- 23.Taneja K.L., McCurrach M., Scalling M., Housman D., Singer R.H. Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J. Cell Biol. 1995;128:995–1002. doi: 10.1083/jcb.128.6.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chiodi I., Corioni M., Giordano M., Valgardsdottir R., Ghigna C., Cobianchi F., Xu R.M., Riva S., Biamonti G. RNA recognition motif 2 directs the recruitment of SF2/ASF to nuclear stress bodies. Nucleic Acids Res. 2004;32:4127–4136. doi: 10.1093/nar/gkh759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Batzer M.A., Deininger P.L., Hellmann-Blumberg U., Jurka J., Labuda D., Rubin C.M., Schmid C.W., Zietkiewicz E., Zuckerkandl E. Standardized nomenclature for Alu repeats. J. Mol. Evol. 1996;42:3–6. doi: 10.1007/BF00163204. [DOI] [PubMed] [Google Scholar]
- 26.Cleary J.D., Pearson C.E. The contribution of cis-elements to disease-associated repeat instability: Clinical and experimental evidence. Cytogenet. Genome Res. 2003;100:25–55. doi: 10.1159/000072837. [DOI] [PubMed] [Google Scholar]
- 27.Liquori C.L., Ricker K., Moseley M.L., Jacobsen J.F., Kress W., Naylor S.L., Day J.W., Ranum L.P. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science. 2001;293:864–867. doi: 10.1126/science.1062125. [DOI] [PubMed] [Google Scholar]
- 28.Rizzi N., Denegri M., Chiodi I., Corioni M., Valgardsdottir R., Cobianchi F., Riva S., Biamonti G. Transcriptional activation of a constitutive heterochromatic domain of the human genome in response to heat shock. Mol. Biol. Cell. 2004;15:543–551. doi: 10.1091/mbc.E03-07-0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Valgardsdottir R., Chiodi I., Giordano M., Rossi A., Bazzini S., Ghigna C., Riva S., Biamonti G. Transcription of Satellite III non-coding RNAs is a general stress response in human cells. Nucleic Acids Res. 2008;36:423–434. doi: 10.1093/nar/gkm1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Valgardsdottir R., Chiodi I., Giordano M., Cobianchi F., Riva S., Biamonti G. Structural and functional characterization of noncoding repetitive RNAs transcribed in stressed human cells. Mol. Biol. Cell. 2005;16:2597–2604. doi: 10.1091/mbc.E04-12-1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jolly C., Lakhotia S.C. Human sat III and Drosophila hsr omega transcripts: a common paradigm for regulation of nuclear RNA processing in stressed cells. Nucleic Acids Res. 2006;34:5508–5514. doi: 10.1093/nar/gkl711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liquori C.L., Ikeda Y., Weatherspoon M., Ricker K., Schoser B.G., Dalton J.C., Day J.W., Ranum L.P. Myotonic dystrophy type 2: human founder haplotype and evolutionary conservation of the repeat tract. Am. J. Hum. Genet. 2003;73:849–862. doi: 10.1086/378720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grady D.L., Ratliff R.L., Robinson D.L., McCanlies E.C., Meyne J., Moyzis R.K. Highly conserved repetitive DNA sequences are present at human centromeres. Proc. Natl. Acad. Sci. USA. 1992;89:1695–1699. doi: 10.1073/pnas.89.5.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scott H.S., Kudoh J., Wattenhofer M., Shibuya K., Berry A., Chrast R., Guipponi M., Wang J., Kawasaki K., Asakawa S. Insertion of beta-satellite repeats identifies a transmembrane protease causing both congenital and childhood onset autosomal recessive deafness. Nat. Genet. 2001;27:59–63. doi: 10.1038/83768. [DOI] [PubMed] [Google Scholar]
- 35.Tassone F., Iwahashi C., Hagerman P.J. FMR1 RNA within the intranuclear inclusions of fragile X-associated tremor/ataxia syndrome (FXTAS) RNA Biol. 2004;1:103–105. doi: 10.4161/rna.1.2.1035. [DOI] [PubMed] [Google Scholar]
- 36.Rudnicki D.D., Holmes S.E., Lin M.W., Thornton C.A., Ross C.A., Margolis R.L. Huntington's disease-like 2 is associated with CUG repeat-containing RNA foci. Ann. Neurol. 2007;61:272–282. doi: 10.1002/ana.21081. [DOI] [PubMed] [Google Scholar]
- 37.Iwahashi C.K., Yasui D.H., An H.J., Greco C.M., Tassone F., Nannen K., Babineau B., Lebrilla C.B., Hagerman R.J., Hagerman P.J. Protein composition of the intranuclear inclusions of FXTAS. Brain. 2006;129:256–271. doi: 10.1093/brain/awh650. [DOI] [PubMed] [Google Scholar]
- 38.Cooper T.A., Wan L., Dreyfuss G. RNA and disease. Cell. 2009;136:777–793. doi: 10.1016/j.cell.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Longman D., Johnstone I.L., Caceres J.F. Functional characterization of SR and SR-related gene in Caenorhabditis elegans. EMBO J. 2000;19:1625–1637. doi: 10.1093/emboj/19.7.1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kanadia R.N., Clark V.E., Punzo C., Trimarchi J.M., Cepko C.L. Temporal requirement of the alternative-splicing factor Sfrs1 for the survival of retinal neurons. Development. 2008;135:3923–3933. doi: 10.1242/dev.024620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sanford J.R., Wang X., Mort M., Vanduyn N., Cooper D.M., Mooney S.D., Edenberg H.J., Liu Y. Splicing factor SFRS1 recognizes a functionally diverse landscape of RNA transcripts. Genome Res. 2009;19:381–394. doi: 10.1101/gr.082503.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sengupta S., Lakhotia S.C. Altered expressions of the noncoding hsr-ω gene enhances poly-Q-induced neurotoxicity in Drosophila. RNA Biol. 2006;3:28–35. doi: 10.4161/rna.3.1.2559. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.