

Abstract
We have discovered a distinct DNA-methylation boundary at a site between 650 and 800 nucleotides upstream of the CGG repeat in the first exon of the human FMR1 gene. This boundary, identified by bisulfite sequencing, is present in all human cell lines and cell types, irrespective of age, gender, and developmental stage. The same boundary is found also in different mouse tissues, although sequence homology between human and mouse in this region is only 46.7%. This boundary sequence, in both the unmethylated and the CpG-methylated modes, binds specifically to nuclear proteins from human cells. We interpret this boundary as carrying a specific chromatin structure that delineates a hypermethylated area in the genome from the unmethylated FMR1 promoter and protecting it from the spreading of DNA methylation. In individuals with the fragile X syndrome (FRAXA), the methylation boundary is lost; methylation has penetrated into the FMR1 promoter and inactivated the FMR1 gene. In one FRAXA genome, the upstream terminus of the methylation boundary region exhibits decreased methylation as compared to that of healthy individuals. This finding suggests changes in nucleotide sequence and chromatin structure in the boundary region of this FRAXA individual. In the completely de novo methylated FMR1 promoter, there are isolated unmethylated CpG dinucleotides that are, however, not found when the FMR1 promoter and upstream sequences are methylated in vitro with the bacterial M-SssI DNA methyltransferase. They may arise during de novo methylation only in DNA that is organized in chromatin and be due to the binding of specific proteins.
Introduction
Epigenetic mechanisms have assumed an important role in molecular biology and medicine.1,2 Much of the work in epigenetics is based on evidence linking sequence-specific modification of DNA by 5-methyldeoxy-cytidine (5-mC)3–6 and of H3 and H4 histones by acetylation and methylation7–9 to long-term gene silencing. Mammalian genomes carry unique imprints of patterns of DNA and histone modifications that can be cell-type specific and, in some instances, interindividually conserved.10,11
The fragile X syndrome (FRAXA, [MIM 300624]) is characterized by a fragile chromosomal site at Xq27.3, mental retardation, attention deficit/hyperactivity disorder, macroorchidism after puberty, plus facial and skeletal dysmorphisms. At the molecular level, the expansion of a CGG repeat located in the 5′-untranslated region (UTR) of the first exon of the FMR1 (fragile X mental retardation) gene [MIM 309550] and the hypermethylation of its promoter region inactivate the FMR1 gene early in human development.12–19 Inactivation or mutations of the FMR1 gene and lack of its gene product during development lead to this syndrome.12–19 However, in rare cases, full amplification of the CGG repeat in the absence of FMR1 promoter methylation does not result in the FRAXA syndrome.20,21
The cause for the instability of the CGG repeats with expansions to < 200 repeats (premutations) or > 200 repeats (full mutations) is unknown. The presence of stable secondary DNA structures as hairpins, triplex, and quadruplex DNA in the repeat have been discussed as possible problems in the normal replication, repair, or recombination reactions of this DNA segment.22 Moreover, properties of the replication fork, repeat length, and CpG methylation are thought to affect repeat stability.23 Furthermore, the presence of an origin of DNA replication in the vicinity of the repeat might contribute to its instability.24–26
Previous analyses of DNA methylation in the FMR1 promoter were limited to a few methylation-sensitive restriction sites18,19 or to bisulfite sequencing in a small segment of the promoter.27,28 The present study provides detailed analyses, previously not available, of the methylation profile in a 5500 base pair (bp) segment of the FMR1 promoter and 5′- upstream sequences in DNA from numerous human cell lines, from normal primary human cells from different tissues, and from FRAXA individuals. The finding of a distinct zone of transition between methylated and unmethylated sequences in the 5′-upstream region of the FMR1 gene has not, to our knowledge, been previously reported and has significance to explain how the CGG repeat can expand in close vicinity to an origin of DNA replication. The methylation boundary is obliterated and altered in DNA from FRAXA individuals. The border sequences, both methylated and unmethylated, are capable of binding specifically to nuclear proteins. A distinct boundary is also observed in the equivalent DNA sequence in several mouse tissues, despite the fact that the nucleotide sequences of human and mouse are only 46.7% identical in the 5′-upstream segment of the FMR1 gene.
Material and Methods
Source of DNA Samples
Because patterns of DNA methylation can differ from cell type to cell type, we analyzed DNA samples from different primary human adult and fetal cell types, from male and female individuals, and from several human cell lines (Table S1, available online). Mouse DNA from different tissues was also studied for documentation of the conservation of this DNA-methylation boundary at this site across mammalian species (Table S1). Cell lines were propagated in culture by standard procedures with the use of Dulbecco's modified Eagle's medium, 10% fetal bovine serum, and a 5% CO2, 95% air atmosphere at 37°C. Human peripheral blood mononuclear cells (PBMCs) were purified by Ficoll (Biochrome) gradient centrifugation.
Extraction of DNA
DNA from cultured cells or from PBMCs was isolated by the Na-perchlorate-chloroform method.29 Several DNA samples were obtained from commercial suppliers (Table S1). The quantity and purity of all DNA preparations was ascertained by absorbance measurements at 260 and 280 nm. Only DNA samples with a 260/280 absorbance ratio of > 1.8 were used for further analyses.
Bisulfite Genomic Sequencing
For bisulfite genomic sequencing,30,31 500 ng of DNA was converted in a total volume of 20 μl of RNase-free H2O with the EpiTect Bisulfite Kit (QIAGEN, Hilden, Germany), with the use of several alternating denaturation and bisulfite conversion steps. The bisulfite conversion of the DNA was performed in a thermal cycler under the following conditions: 99°C for 5 min, 60°C for 25 min, 99°C for 5 min, 60°C for 85 min, 99°C for 5 min, 60°C for 175 min. By repeated 99°C cycling, the EpiTect protocol secures the maintenance of DNA denaturation during bisulfite treatment, which is essential for complete C-to-T conversion. The converted single-stranded DNA was bound to the membrane of an EpiTect spin column without the use of carrier RNA. The DNA was eluted in 40 μl elution buffer and stored at −20°C. In early experiments in this study, the bisulfite-sequencing method was performed as described elsewhere.32 Nucleotide sequences were routinely determined in the service facility of the Institute for Human Genetics, Erlangen University Medical School.
Primers Used in Bisulfite-Sequencing Experiments
Table S2 lists primer sequences and their positions in the 5′-upstream and promoter regions of the FMR1 gene, in both the human and mouse genomes.
Validation of the FMR1 Bisulfite-Sequencing Method
The reliability of the EpiTect protocol was assessed by applying it to the analysis of commercially obtained human DNA (Figures S1D and S1E) or of the plasmid-cloned25 FMR1 promoter sequence (Figures S1F–S1H). Unmethylated DNA (Figures S1D and S1F) and M.Sss I-premethylated DNA with all CpG dinucleotides methylated (Figures S1E and S1G) were found as anticipated. In a 1:1 mixture (Figure S1H), unmethylated and M.Sss I-premethylated DNA molecules were represented in the expected ratio (Figure S1H). An additional precaution was applied, in that sequences in clones with incomplete C-to-U (T) conversion products were not included in the analyses.
Electrophoretic Mobility Shift Assays
For this series of experiments, protocols and reagents from Roche (Mannheim, Germany) were employed.
Preparation of Nuclear Extracts from Human HCT116 Cells
HCT116 cells were grown to confluence in 75 cm2 plastic flasks. Nuclear extracts were prepared by resuspending the extensively washed cells in hypotonic buffer (10 mM 4-(2-hydroxyethyl)-1-piperazine-ethanesulfonic acid [HEPES], pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.2 mM phenylmethylsulfonyl fluoride [PMSF], 0.5 mM dithiothreitol [DTT]) and by allowing the cells to swell on ice for 10 min. Cell membranes were broken in a Dounce homogenizer by ten strokes with a type B pestle. Nuclei were collected by centrifugation and incubated for 1 hr at 4°C in the same buffer adjusted to 0.26 M KCl. The nuclear extract was subsequently dialyzed at 4°C against 20 mM HEPES, pH 7.9, 10% glycerol, 0.1 M KCl, 0.2 mM ethylenediamine tetraacetate (EDTA), 0.2 mM PMSF, 0.5 mM DTT (binding buffer).
Labeling of DNA Probes
PCR was used for generating the 630 bp sequence spanning the methylation-boundary fragment (red bar in Figure 1C) that was digoxygenin (DIG)-labeled or was left unlabeled in competition experiments as described. In some experiments, all CpG dinucleotides in a 630 bp sequence from the same region were in vitro premethylated in a 5 hr reaction with the M.Sss I DNA methyltransferase (New England Biolabs). Complete methylation was confirmed by failure of methylation-sensitive restriction endonucleases to cleave the premethylated DNA fragment. For protein-binding reactions, DNA fragments were digoxygenin labeled as recommended by the manufacturer (Roche, Mannheim, Germany). In brief, 3.85 pmol of the DNA fragment was incubated at 37°C for 15 min in labeling buffer containing 5 mM CoCl2, 0.05 mM digoxygenin-ddUTP, and 400 units of DNA terminal transferase. The reaction was stopped in 0.2 M EDTA and diluted in H2O to 0.155 pmol DNA.
Figure 1.

Methylation Profiles in the FMR1 Promoter and 5′-Upstream Segment in the Human Cell Line HCT116 and in Primary Human Fibroblasts
(A) Ideogram of the human X chromosome.
(B) Partial map shows the first ten exons (vertical bars) and introns of the FMR1 gene plus the upstream genome segment including the CGG repeat.
(C) Map of the 5′-upstream region of the FMR1 gene. The graph presents all CpG dinucleotides (1 to 104) in the region: The arrow indicates the start site of transcription. The promoter and an origin of DNA replication are also indicated. Nucleotide numbering in this and the following graphs was adapted from the NCBI nucleotide nomenclature: NC_000023:146,786,201–146,840,303 H. sapiens FMR1 gene region (nucleotide numbers on the human X = 23rd chromosome). The boundary between unmethylated and methylated CpG dinucleotides is marked by the symbols ○ and ●, respectively, and is designated by a red bar.
(D) In HCT116 DNA, both strands (upper and lower panels) were sequenced with bisulfite between CpG pairs 20 and 83. Bisulfite sequencing in one strand was extended to CpG pair 104 (lower panel). □, unmethylated CpGs; ■, methylated CpGs.
(E) In primary human foreskin fibroblasts, DNA was sequenced with bisulfite between CpG pairs 20 and 104.
DNA-Protein Binding Reaction and Analyses of Complexes by Electrophoretic Mobility Shift Assays
In a total volume of 20 μl binding buffer, 1 μg of poly [d(I-C)], 0.1 μg poly L-lysine, 0.2 ng DIG-labeled DNA fragment, and 2.64 μg protein from nuclear extracts were incubated for 15 min at 37°C. In competition reactions, a > 2000-fold excess of unlabeled DNA fragments from different sources was added. Reaction products were analyzed by electrophoresis in a precast 6% polyacrylamide retardation gel (NOVEX) in 0.5× TBE buffer for 3 hr at 80 V, 10 mA. TBE buffer is 89 mM each of Tris and boric acid, 2 mM EDTA, pH 8.0. After electrophoresis, the reaction products were transferred by electroblotting to a positively charged nylon membrane with 0.5× TBE for 60 min at 30 V, 150 mA. The DNA fragments were cross-linked to the membrane by baking for 30 min at 120°C. Subsequently, the membranes were washed, incubated for 30 min each in blocking buffer, and diluted in anti-digoxygenin-AP conjugate, followed by a 5 min incubation in detection buffer. Finally, 0.1 mg CSPD per ml working solution (Roche) was added and incubation was continued for 5 min at 25°C. The damp membrane was then sealed and incubated for 10 min at 37°C for enhancement of the the CSPD chemiluminescent reaction. The membrane was subsequently exposed to an imaging device (charge-coupled device [CCD] camera, Fuji-LAS-1000).
Results
Sequence Characteristics and Multiple Genetic Signals in the FMR1 Promoter and Its 5′- Upstream Region
The 5′-upstream region of the FMR1 gene (Xq27.3) is close to the functionally relevant CGG repeat and is characterized by a number of important genetic signals (Figure S1C, Figure 1C):
-
(1)
The CGG repeat lies in the 5′-UTR of the FMR1 gene's first exon. This repeat is expanded in FRAXA individuals and is then devoid of the stabilizing interspersed AGG trinucleotides.33
-
(2)
The FMR1 promoter and the initiation site for RNA transcription are located immediately upstream of the CGG repeat. The promoter, aside from a noncanonical TTACA signal,34 lacks a typical TATAA box but contains, in a region of about 1 kb upstream from the site of transcriptional initiation, multiple SpI sites (MIM 189906) and binding motifs for AP2 (MIM 107580), α-PAL/NRF1 (MIM 600879), and myc35 (MIM 190080).
-
(3)
An origin of DNA replication lies about 300 to 450 nucleotides upstream of the transcriptional initiation site.24–26
-
(4)
Between map position 15,060 and a site about 5500 nucleotides 5′- upstream of the CGG repeat, there are 104 CpG dinucleotides (Figure 1C), among them several very closely spaced methylation-sensitive restriction sites.
-
(5)
A boundary between completely methylated and essentially unmethylated CpG dinucleotides in normal individuals has been identified in this study. This methylation boundary is lost and might be altered in FRAXA patients.
Methylation Profile of the Region
In the maps in most figures, CpG dinucleotides in the genome segment investigated are numbered from position 1, closest to (but not within) the CGG repeat, to position 104, most distant from it. From CpG position 104 upstream, there are very few CpG dinucleotides. Because methylation patterns can differ from one human cell type or line to another,10 we analyzed DNA samples from a number of different cell lines and a variety of human cells and tissues, from fetal, adult, male, and female sources (Table S1).
In DNA from the human colorectal carcinoma cell line HCT116 from a male individual without repeat expansion (n = 30),25 one DNA strand was sequenced between CpG dinucleotides 20 and 83, and the opposite strand was sequenced between CpGs 20 and 104 in individual clones after bisulfite treatment (upper and lower panels in Figure 1D, respectively). The DNA-methylation boundary was found at CpG pairs 65 and 66 (upper and lower panels in Figure 1D) about 650 nucleotides upstream of the CGG repeat. The results were essentially identical, regardless of whether the EpiTect method or a protocol used in earlier studies27,32 was applied (data not shown). In DNA from primary foreskin fibroblasts, only one DNA strand was sequenced between CpG dinucleotide positions 20 to 104, and the DNA-methylation border was found in the region between CpG pairs 66 and 70 (Figure 1E). In the region located further upstream, between CpG dinucleotides 70 and 104, most CpGs were methylated (Figure 1E).
DNA samples from PBMCs of two male (Figures 2A and 2B) and two female (Figures 2C and 2D) donors were analyzed in only one strand between CpG dinucleotide positions 20 and 83. In two male DNA samples, the distinct border between the methylated and unmethylated regions was located at CpG pair 70, and CpG pairs between positions 66 and 70 were methylated in only some of the chromosomes analyzed. In the DNA from the female donors, the methylation boundary was apparent at CpG pair 69 or 70 in about half of the DNA molecules. In the region between CpG pairs 20 and 68, some of the chromosomes were methylated; others remained unmethylated, with the ratio of methylated to unmethylated molecules approaching 1. This mosaic pattern probably reflected the disparity of methylation levels on the two female X chromosomes, one being highly methylated and the other being unmethylated or hypomethylated (Figures 2C and 2D). The methylation boundary and a mosaic pattern downstream were also observed in HeLa DNA (data not shown).
Figure 2.

Methylation Boundaries in the FMR1 Upstream Region from Male and Female PBMCs
(A and B) The two male individuals were 28 (A) and 73 (B) years of age.
(C and D) DNA samples from both female individuals revealed the methylation boundary in those molecules that were unmethylated downstream, plus a methylation mosaic in the downstream region between CpG dinucleotides 20 and 70.
(E) FMR1 map as in Figure 1C.
In DNA from brain (Figure S2A) and liver (Figure S2B) of male fetuses or from adult male brain, liver, and muscle (Figures S2C–S2E, respectively), the DNA-methylation divide was again found at the same or a similar location as in the previously described DNA samples.
Mouse DNA
We also interrogated the DNA sequence in the equivalent genome segment of the mouse genome (Figure 3A). The methylation boundary was, as observed in human DNA, conserved at CpG dinucleotide 57 in the mouse genome in DNA from brain (Figure 3B) and liver (Figure 3C). Maintenance of this methylation boundary in the genome of two different mammalian species strengthens the notion that this site serves an important function. The concordance of data from both species is remarkable also in that sequence identity between the two species in the Fmr1 upstream region is only 46.7%.
Figure 3.
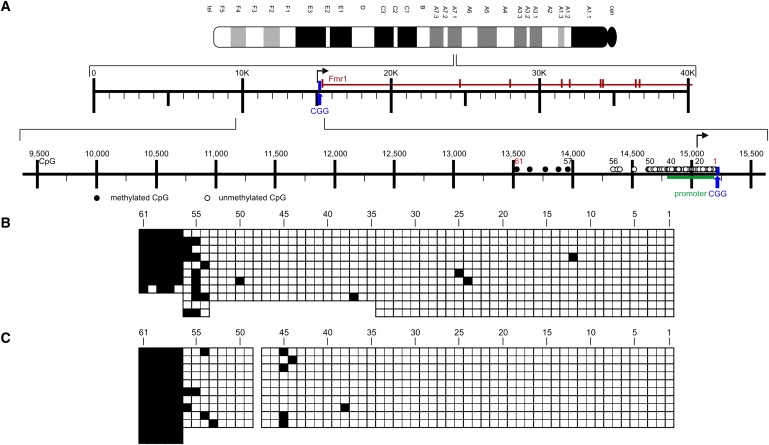
Methylation Boundaries in the Fmr1 Upstream Region in DNA from Adult Mouse Brain and Liver
(A) Ideogram and partial Fmr1 gene map of the mouse. Sequence comparisons between the human and mouse genomes in the Fmr1 upstream region were based on the published mouse genome sequence from the NCBI nucleotide depository for mouse chromosome XA7.1: NC_000086:65,916,730-65,971,138 M. musculus Fmr1 gene region. The mouse Fmr1 map has been arranged similarly to the map in Figure 1C.
(B and C) The methylation boundary in the mouse Fmr1 5′-upstream region maps at CpG dinucleotide 57 in adult mouse brain (B) and liver (C).
In FRAXA Individuals, the Methylation Boundary Is Lost and the FMR1 Promoter Is Heavily Methylated
Primary FRAXA lung fibroblasts from a 22-week-old male fetus (GM07072) and primary FRAXA male fibroblasts (GM05848) were commercially obtained (Table S1), and their DNA was analyzed for its methylation status in the 5′-upstream region of the FMR1 promoter. In both samples, the DNA was completely methylated in almost all CpG dinucleotides investigated between CpG positions 20 and 83 (GM07072) (Figure 4A) or 20 and 88 (GM05848) (Figure 4B), with the exception of a few isle-like CpG pairs that had remained unmethylated in a background of completely methylated DNA (Figures 4A and 4B). Also, in 14,451 DNA from PBMCs of a FRAXA patient, CpG pairs 20 to 83 were all methylated, except for some chromosomes around CpG position 30. The finding of a highly methylated 5′-upstream region in FRAXA individuals corroborates published data from our and other laboratories.27,28 In earlier studies,27 the methylation pattern in the FMR1 5′-upstream region of some FRAXA individuals exhibited mosaic patterns.
Figure 4.
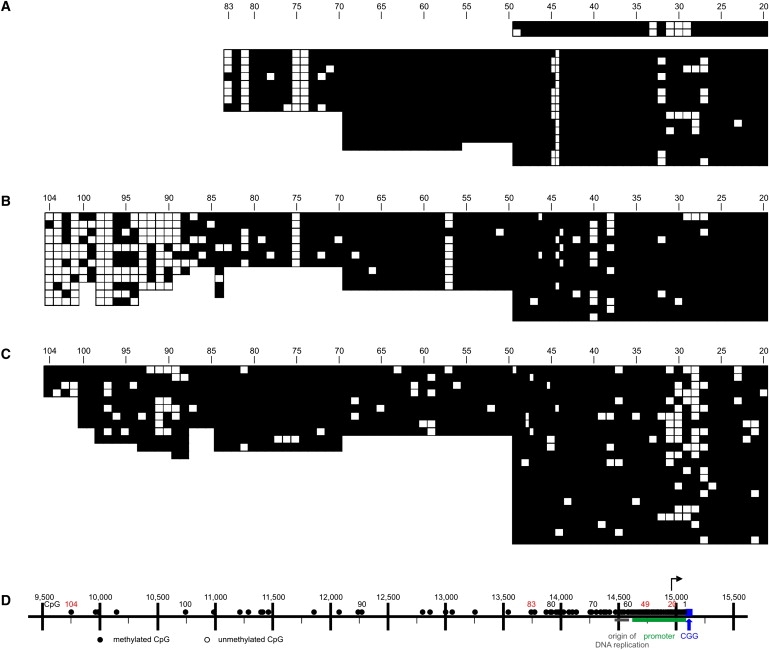
Loss of the Methylation Boundary in Fibroblasts and PBMCs from Male FRAXA Individuals
(A–C) In DNA from FRAXA individuals from commercially obtained fibroblasts GM07072 (A) and GM05848 (B) or from PBMC sample 14,451 (C), the methylation border was absent and methylation was observed throughout the promoter region downstream to the CGG repeat, except for a few unmethylated CpG dinucleotides. In (A), a part of the opposite strand was also sequenced. In (B) and (C), bisulfite sequencing was extended upward to CpG dinucleotide 104.
(D) FMR1 map as in Figure 1C, except that all CpGs are indicated as methylated: ●.
In the DNA from the GM05848 fibroblasts and the 14,451 PBMCs, the bisulfite analysis was extended upward to include CpG dinucleotides 84 to 104. DNA methylation in this far-upstream segment of the boundary was decreased in the fibroblast sample GM05848 (Figure 4B) in comparison to that in non-FRAXA individuals (Figures 1D and 1E). This decrease in methylation density between CpG dinucleotides 89 to 104 in FRAXA DNA sample GM05848 might indicate alterations in the FRAXA boundary region. A comparably marked decrease in DNA methylation in CpG pairs 84 to 104 was not observed in DNA from the 14,451 PBMCs (Figure 4C).
The methylation pattern in the DNA from premutation females (data not shown) was very similar to that of normal female probands (Figures 2C and 2D). On at least one chromosome of premutation carriers, the methylation boundary in the 5′-upstream region of the FMR1 gene was preserved as in male non-FRAXA chromosomes.
Nuclear Proteins Bind Specifically to the Sequence around the Methylation Boundary in Both the Unmetylated and the Methylated States
The nucleotide sequence around the boundary between the fully methylated upstream and the unmethylated downstream DNA segment in the FMR1 upstream region contains CpG dinucleotides 57 to 75 (Figure 5A). Among these 19 CpG dinucleotides, seven are part of a 5′-CGG-3′ triplet. Moreover, the following motifs are present in this DNA segment: three 5′-CCCTC-3′ (GAGGG) motifs, ten 5′-CCTC-3′ (GAGG) sequences, a 5′-(CCAAA)6-3′ repeat, and one AluI site (5′-AGCT-3′) (Figure 5A). The methylation boundary is located at or around CpG pair 66. We investigated the capacity of this transition sequence to bind nuclear proteins from human cells. Nuclear extracts from human HCT116 cells were incubated with a 630 bp DIG-labeled DNA fragment containing the region of the methylation boundary (red bar in Figure 1C). In some experiments, the 630 bp segment was premethylated with M.Sss I. Protein-DNA complexes were visualized by electrophoresis on native polyacrylamide gels. Several bands of shifted DNA, presumably DNA-protein complexes, were observed (Figure 5B, lane c, arrow and asterisks). The specificity of these complexes was assessed in the following competition experiments by the use of unlabeled 630 bp DNA fragments as competitors in > 2000-fold excess:
-
(1)
The unlabeled transition DNA fragment (Figure 5B, lane d) competes all shifted DNA-protein complexes (seven independent experiments).
-
(2)
The unlabeled 595 bp DraI-RsaI fragment from the β-lactamase (bla) gene (MIM 608440), of plasmid pcDNA3.1 (+) (Figure 5B, lane f), was used as a nonspecific competitor and competes only the slowly migrating bands (indicated by asterisks) but not the complex close to the 1033 bp size marker (Figure 5B, lane f).
-
(3)
The unlabeled M.Sss I-premethylated transition DNA fragment (Figure 5B, lane e) competes with probe binding to all complexes, much like the unmethylated fragment. In three independent experiments, competition by the M.Sss I-premethylated probe generated a DNA band of slightly higher molecular mass than that of the unmethylated probe. This control was performed because the transition fragment is methylated from CpG pair 66 upstream in DNA from all non-FRAXA human DNA sources.
Figure 5.
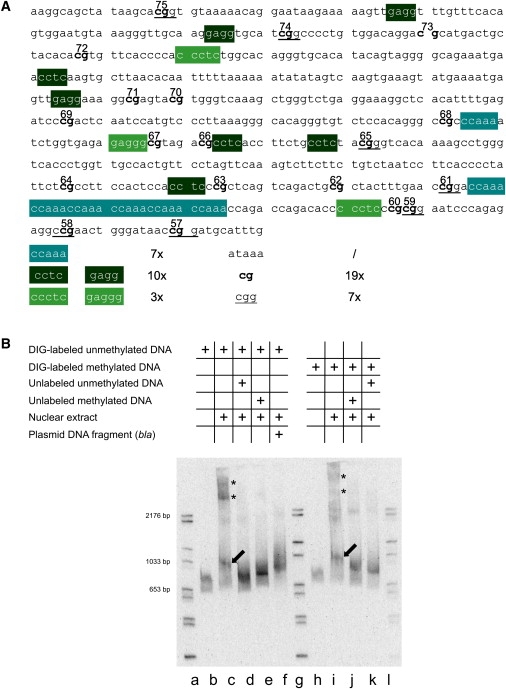
Specific Binding of Nuclear Proteins from Human Cells to DNA Sequences in the Boundary Region
(A) Nucleotide sequence in the transition zone between fully methylated and unmethylated DNA in the FMR1 5′-upstream region. Several DNA motifs and a 5′-(CCAAA)6-3′ repeat are indicated.
(B) Nuclear extracts from human HCT116 cells (2.64 μg of protein) were incubated with the DIG-labeled 630 bp DNA fragment from the boundary region in the FMR1 5′-upstream genome segment as indicated. The reaction products were analyzed by electrophoretic mobility shift assays. Lanes a, g, and l: Marker DNA lanes with DIG-labeled DNA fragments of sizes as marked. Lane b: DIG-labeled 630 bp fragment incubated without the addition of nuclear extracts; lane c shows the same after incubation with nuclear extracts. Lane d: conditions as in lane c, except that a > 2000-fold excess of an unlabeled 630 bp fragment was added as specific competitor. Lane e: conditions as in lane c, except that a > 2000-fold excess of an unlabeled M.Sss I-premethylated 630 bp DNA fragment from the transition region was used for competition. Lane f: conditions as in lane d, except that the competitor was a 595 bp DraI-RsaI fragment from the bla gene of E. coli plasmid pcDNA3.1 (+). In lanes h–k, an M.Sss I-premethylated 630 bp DNA fragment from the transition region was used for binding or competition experiments. Competition experiments were performed as indicated in the graph. Electrophoretic mobility was toward the bottom. After electrophoresis in a precast 6% polyacrylamide gel, the DNA bands were visualized as described in Material and Methods.
In (B), the presumably unspecific complexes are designated by asterisks (∗), the specific ones by arrows.
The M.Sss I-premethylated and DIG-labeled transition fragment was also shifted upon binding to proteins from nuclear extracts (Figure 5B, lane i). The nuclear protein binding to the M.Sss I-premethylated DIG-labeled transition fragment was competed by an excess of both unlabeled methylated (Figure 5B, lane j) and unmethylated (Figure 5B, lane k) transition DNA fragments.
On the basis of the competition patterns, we conclude that nuclear proteins bind specifically to sequences in the transition DNA fragment in both its unmethylated and M.Sss I-premethylated forms. We cannot exclude the possibility that there is a slight difference in the binding of nuclear proteins to the unmethylated as compared to the methylated boundary fragment. The slowly migrating DNA complexes (marked by asterisks in Figure 5B, lanes c and i), which are competed by the bla gene fragment, appear to be nonspecific or less sequence-specific.
Discussion
DNA-Methylation Boundary
In a human genome segment of about 5500 bp in the 5′-upstream segment of the FMR1 gene, we have identified a distinct methylation boundary 650 to 800 nucleotides and 65 to 70 CpG pairs upstream of the CGG repeat in the human FMR1 gene. This boundary is defined by an upstream genome segment of about 4900 bp with completely methylated CpG dinucleotides and a shorter downstream 670 bp stretch that is devoid of CpG methylation. The methylation boundary is often preceded on its downstream side by a short genome segment that is characterized by a methylation mosaic, with some of the chromosomes methylated and others unmethylated, in a fuzzy transition zone (Figures 1 and 2, Figure S2). This less sharply structured region might be characteristic of chromatin “breathing” in the transition zone, with some of the molecules fully methylated and others unmethylated.
The methylation boundary region is conserved in DNA from both male and female human probands, regardless of age, in human cell lines and in the mouse genome. In samples from females, the 5′-upstream and promoter regions of the FMR1 gene contain DNA sequences from both X chromosomes. DNA from one X chromosome carries a more heavily methylated DNA segment with loss of the methylation boundary; DNA from the other X allele, with its downstream region mainly unmethylated, carries a methylation boundary very similar, if not identical, to that of male X chromosomes. This interpretation is supported by the close 1:1 ratio of methylated to unmethylated molecules in the region between CpG dinucleotides 20 to 69 or 70 in female DNA (Figures 2C and 2D). The methylation border was present at the same location in DNA from both fetal and adult tissues (Figure S2).
Occasionally, the occurrence of 5-mC nucleotides in dinucleotide combinations other than CpGs have been reported.36,37 In the present data, 5-mC has not been detected in non-CpG dinucleotide combinations in about 700 bp of the 5′-upstream segment of the human FMR1 gene in HCT116 and GM07072 (FRAXA individual) DNAs.
In unrelated promoters in the human genome previously analyzed in our laboratory (RET [MIM 164761], CGGBP1 [MIM 603363], genes of the erythrocyte membrane, e.g., MIM 605331), such a methylation boundary has not been observed at a comparable distance from the site of transcriptional initiation.38–40 However, at the 5′- end of a CpG island of the glutathione S-transferase (GSTP1) gene (MIM 134660), a 5′-(ATAAA)19–24-3′ repeat defines a distinct border between a methylated and an unmethylated domain in several different human tissues that express the GSTP1 gene. This methylation boundary is located at CpG site 44.41 In prostate cancer (MIM 176807), this boundary is lost, the promoter fully methylated, and the gene silenced.41 In the upstream methylation boundary of the FMR1 gene, a 5′-(ATAAA)-3′ repeat is not present. In the FMR1 segment between CpG dinucleotides 60 and 61, downstream of the actual boundary, a 5′-(CCAAA)6-3′ repeat is located (Figure 5A).
Methylation Boundary at Equivalent Site in the Mouse Genome
Bisulfite sequencing of the 5′-upstream segment in the mouse genome also reveals a methylation boundary at the site equivalent to that in the human genome (Figure 3). At the nucleotide sequence level, both genomes are only 46.7% identical in the 5′-upstream region of the FMR1 gene. Conservation of the methylation boundary across mammalian species, even in nucleotide sequences that differ by > 50%, underscores its potential structural and functional importance.
Loss of the Methylation Boundary in DNA from FRAXA Individuals
In FRAXA males, the boundary is completely lost, and almost all of the 88 CpG dinucleotides in the 2260 bp fragment extending down to the CGG repeat are methylated (Figures 4A–4C). In the roughly 3300-bp-long segment far upstream in the boundary encompassing CpG dinucleotides 89 to 104, the degree of DNA methylation seems to be lower, particularly in the fibroblast sample GMO5848. This finding might indicate an alteration in the structure of the methylation boundary in FRAXA individuals. CpG dinucleotides 89 to 104 in DNA from PBMC sample 14,451, however, are heavily methylated (Figure 4C). The methylation boundary in the FMR1 5′-upstream region possibly coincides with a specific chromatin structure that, when destabilized, allows methylation to spread downstream, which eventually culminates in the complete methylation of the FMR1 promoter, as well as FMR1 gene silencing, as shown for the GM07072 and GM05848 FRAXA fibroblasts.25
Isolated Unmethylated CpG Dinucleotides in De Novo Methylated DNA
In the DNA from FRAXA patients, even the completely de novo methylated promoter and 5′-upstream sequences of the FMR1 gene contain isolated unmethylated CpGs (Figure 4). In the region around CpG 30, some of these isolated unmethylated CpGs coincide with transcription factor binding sites (USF1 and USF2 or NRF1).28,42 Similar isolated unmethylated CpGs occur in the almost completely, also de novo, methylated adenovirus type 12 genome in a transformed hamster cell line that has been maintained in culture for several decades.32 Thus, mammalian and integrated viral genome segments of quite different derivations that are characterized by a high degree of de novo CpG methylation harbor one or several unmethylated CpG dinucleotides, which might be typical of de novo methylated DNA. Comparable isolated unmethylated CpGs are not apparent in de novo premethylated FMR1 DNA when the bacterial M.Sss I has been used (Figures S1E S1G, and S1H). Hence, these isolated unmethylated CpGs seem to arise only during de novo methylation in living cells and organisms, but not during in vitro methylation by bacterial M.Sss I, possibly because a specific chromatin structure and/or protein binding in intact mammalian chromosomes precludes de novo methylation at these specific sites.
Proteins Binding at the Boundary Sequence in the Human FMR1 5′- Upstream Region
We propose that the FMR1 region carrying the methylation boundary is characterized by a specific chromatin structure and serves to demarcate the human FMR1 and mouse Fmr1 promoter regions from interspersed 5′-upstream-located DNA sequences of as-yet-undetermined functions. This boundary region resembles an insulator element.43 The identification of specific DNA-protein complexes between the upstream FMR1 boundary sequence and nuclear proteins from human cells might provide a clue as to how this methylation boundary is maintained. It has been suggested that binding sites for putative transcription factors and for specific zinc finger proteins are overrepresented in border regions of methylation.44 The FMR1 upstream boundary region contains one AluI (5′-AGCT-3′) sequence and several additional motifs described in Figure 5A.
DNA in the transition zone from non-FRAXA individuals contains the methylated CpG dinucleotides 66 to 75 and the unmethylated CpGs 57 to 65. When the entire transition sequence of 630 bp was reacted with nuclear proteins, distinct DNA-protein complexes were observed (Figure 5B). DNA-protein complexes of nearly identical sizes were formed by the transition DNA fragment both in the unmethylated and the fully CpG M.Sss I-methylated modes. The specificity of these complexes was supported by the results of competition experiments. Both the unmethylated and the methylated DNA fragments competed the binding of nuclear proteins to very similar extents and eliminated complex formation with the labeled methylated and unmethylated probes, respectively (Figure 5B). A 595 bp fragment from the plasmid β-lactamase gene, chosen as unspecific competitor, eliminated the complexes of higher molecular mass, but not the complex migrating close to the 1033 bp size marker (Figure 5B, lane f). We therefore consider this latter complex the one with the highest degree of specificity. Alterations of chromatin structure at this site of transition between unmethylated and fully methylated genome segments, or the abrogation of a higher-order chromatin structure at this site, might be responsible for the loss of the methylation barrier and lead to the extension of de novo methylation into the FMR1 promoter that becomes silenced in response.25 The less-complete methylation profile in the far 5′-upstream region in fibroblasts of one of the analyzed FRAXA cases (Figure 4B) might be an indication of this destabilization.
Supplemental Data
Supplemental Data include two figures and two tables and can be found with this article online at http://www.cell.com/AJHG/.
Supplemental Data
Web Resources
The URL for data presentend herein is as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Acknowledgments
We are indebted to the Institute for Virology of Erlangen University Medical School (EUMS) for their hospitality extended to W.D.'s Senior Research Group. We thank Cornelia Kraus, Institute of Human Genetics, EUMS, for PBMC DNA (14,451) from a FRAXA individual and the Department of Pediatrics, EUMS, for providing the human foreskin fibroblasts. This project was supported by the Fritz Thyssen Foundation and by amaxa/LONZA GmbH, both in Köln, Germany (to W.D.), and by U.S. National Institutes of Health grant GM52948 (to E.F.). The project was initiated during W.D.'s sabbatical stay (Deutscher Akademischer Austauschdienst) in E.F.'s laboratory at Vanderbilt University, Nashville, TN, USA.
References
- 1.Jones P.A., Takai D. The role of DNA methylation in mammalian epigenetics. Science. 2001;293:1068–1070. doi: 10.1126/science.1063852. [DOI] [PubMed] [Google Scholar]
- 2.Razin A., Kantor B. DNA methylation in epigenetic control of gene expression. Prog. Mol. Subcell. Biol. 2005;38:151–167. doi: 10.1007/3-540-27310-7_6. [DOI] [PubMed] [Google Scholar]
- 3.Sutter D., Doerfler W. Methylation of integrated adenovirus type 12 DNA sequences in transformed cells is inversely correlated with viral gene expression. Proc. Natl. Acad. Sci. USA. 1980;77:253–256. doi: 10.1073/pnas.77.1.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vardimon L., Neumann R., Kuhlmann I., Sutter D., Doerfler W. DNA methylation and viral gene expression in adenovirus-transformed and -infected cells. Nucleic Acids Res. 1980;8:2461–2473. doi: 10.1093/nar/8.11.2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Razin A., Riggs A.D. DNA methylation and gene function. Science. 1980;210:604–610. doi: 10.1126/science.6254144. [DOI] [PubMed] [Google Scholar]
- 6.Doerfler W. DNA methylation and gene activity. Annu. Rev. Biochem. 1983;52:93–124. doi: 10.1146/annurev.bi.52.070183.000521. [DOI] [PubMed] [Google Scholar]
- 7.Allfrey V.G., Mirsky A.E. Structural modifications of histones and their possible role in the regulation of RNA synthesis. Science. 1964;144:559. doi: 10.1126/science.144.3618.559. [DOI] [PubMed] [Google Scholar]
- 8.Jones P.L., Wolffe A.P. Relationships between chromatin organization and DNA methylation in determining gene expression. Semin. Cancer Biol. 1999;9:339–347. doi: 10.1006/scbi.1999.0134. [DOI] [PubMed] [Google Scholar]
- 9.Bernstein B.E., Meissner A., Lander E.S. The mammalian epigenome. Cell. 2007;128:669–681. doi: 10.1016/j.cell.2007.01.033. [DOI] [PubMed] [Google Scholar]
- 10.Kochanek S., Toth M., Dehmel A., Renz D., Doerfler W. Interindividual concordance of methylation profiles in human genes for tumor necrosis factors alpha and beta. Proc. Natl. Acad. Sci. USA. 1990;87:8830–8834. doi: 10.1073/pnas.87.22.8830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Doerfler W. In pursuit of the first recognized epigenetic signal - DNA methylation: a 1976 to 2008 synopsis. Epigenetics. 2008;3:125–133. doi: 10.4161/epi.3.3.6249. [DOI] [PubMed] [Google Scholar]
- 12.O'Donnell W.T., Warren S.T. A decade of molecular studies of fragile X syndrome. Annu. Rev. Neurosci. 2002;25:315–338. doi: 10.1146/annurev.neuro.25.112701.142909. [DOI] [PubMed] [Google Scholar]
- 13.Terracciano A., Chiurazzi P., Neri G. Fragile X syndrome. Am. J. Med. Genet. C. Semin. Med. Genet. 2005;137C:32–37. doi: 10.1002/ajmg.c.30062. [DOI] [PubMed] [Google Scholar]
- 14.Pieretti M., Zhang F.P., Fu Y.H., Warren S.T., Oostra B.A., Caskey C.T., Nelson D.L. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell. 1991;66:817–822. doi: 10.1016/0092-8674(91)90125-i. [DOI] [PubMed] [Google Scholar]
- 15.Fu Y.H., Kuhl D.P., Pizzuti A., Pieretti M., Sutcliffe J.S., Richards S., Verkerk A.J., Holden J.J., Fenwick R.G., Warren S.T. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell. 1991;67:1047–1058. doi: 10.1016/0092-8674(91)90283-5. [DOI] [PubMed] [Google Scholar]
- 16.Ashley C.T., Warren S.T. Trinucleotide repeat expansion and human disease. Annu. Rev. Genet. 1995;29:703–728. doi: 10.1146/annurev.ge.29.120195.003415. [DOI] [PubMed] [Google Scholar]
- 17.Verkerk A.J., Pieretti M., Sutcliffe J.S., Fu Y.H., Kuhl D.P., Pizzuti A., Reiner O., Richards S., Victoria M.F., Zhang F.P. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–914. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- 18.Oberlé I., Rousseau F., Heitz D., Kretz C., Devys D., Hanauer A., Boué J., Bertheas M., Mandel J. Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science. 1991;252:1097–1102. doi: 10.1126/science.252.5009.1097. [DOI] [PubMed] [Google Scholar]
- 19.Hansen R.S., Gartler S.M., Scott C.R., Chen S.H., Laird C.D. Methylation analysis of CGG sites in the CpG island of the human FMR1 gene. Hum. Mol. Genet. 1992;1:571–578. doi: 10.1093/hmg/1.8.571. [DOI] [PubMed] [Google Scholar]
- 20.Smeets H.J., Smits A.P., Verheij C.E., Theelen J.P., Willemsen R., van de Burgt I., Hoogeveen A.T., Oosterwijk J.C., Oostra B.A. Normal phenotype in two brothers with a full FMR1 mutation. Hum. Mol. Genet. 1995;4:2103–2108. doi: 10.1093/hmg/4.11.2103. [DOI] [PubMed] [Google Scholar]
- 21.Tabolacci E., Moscato U., Zalfa F., Bagni C., Chiurazzi P., Neri G. Epigenetic analysis reveals a euchromatic configuration in the FMR1 unmethylated full mutations. Eur. J. Hum. Genet. 2008;16:1487–1498. doi: 10.1038/ejhg.2008.130. [DOI] [PubMed] [Google Scholar]
- 22.Cleary J.D., Nichol K., Wang Y.H., Pearson C.E. Evidence of cis-acting factors in replication-mediated trinucleotide repeat instability in primate cells. Nat. Genet. 2002;31:37–46. doi: 10.1038/ng870. [DOI] [PubMed] [Google Scholar]
- 23.Nichol Edamura K., Leonard M.R., Pearson C.E. Role of replication and CpG methylation in fragile X syndrome CGG deletions in primate cells. Am. J. Hum. Genet. 2005;76:302–311. doi: 10.1086/427928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brylawski B.P., Chastain P.D., Cohen S.M., Cordeiro-Stone M., Kaufman D.G. Mapping of an origin of DNA replication in the promoter of fragile X gene FMR1. Exp. Mol. Pathol. 2007;82:190–196. doi: 10.1016/j.yexmp.2006.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gray S.J., Gerhardt J., Doerfler W., Small L.E., Fanning E. An origin of DNA replication in the promoter region of the human fragile X mental retardation (FMR1) gene. Mol. Cell. Biol. 2007;27:426–437. doi: 10.1128/MCB.01382-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lucas I., Palakodeti A., Jiang Y., Young D.J., Jiang N., Fernald A.A., Le Beau M.M. High-throughput mapping of origins of replication in human cells. EMBO Rep. 2007;8:770–777. doi: 10.1038/sj.embor.7401026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Genç B., Müller-Hartmann H., Zeschnigk M., Deissler H., Schmitz B., Majewski F., von Gontard A., Doerfler W. Methylation mosaicism of 5′-(CGG)n-3′ repeats in fragile X, premutation and normal individuals. Nucleic Acids Res. 2000;28:2141–2152. doi: 10.1093/nar/28.10.2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pietrobono R., Pomponi M.G., Tabolacci E., Oostra B., Chiurazzi P., Neri G. Quantitative analysis of DNA demethylation and transcriptional reactivation of the FMR1 gene in fragile X cells treated with 5-azadeoxycytidine. Nucleic Acids Res. 2002;30:3278–3285. doi: 10.1093/nar/gkf434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johns M.B., Paulus-Thomas J.E. Purification of human genomic DNA from whole blood using sodium perchlorate in place of phenol. Anal. Biochem. 1989;180:276–278. doi: 10.1016/0003-2697(89)90430-2. [DOI] [PubMed] [Google Scholar]
- 30.Frommer M., McDonald L.E., Millar D.S., Collis C.M., Watt F., Grigg G.W., Molloy P.L., Paul C.L. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. USA. 1992;89:1827–1831. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clark S.J., Harrison J., Paul C.L., Frommer M. High sensitivity mapping of methylated cytosines. Nucleic Acids Res. 1994;22:2990–2997. doi: 10.1093/nar/22.15.2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hochstein N., Muiznieks I., Mangel L., Brondke H., Doerfler W. Epigenetic status of an adenovirus type 12 transgenome upon long-term cultivation in hamster cells. J. Virol. 2007;81:5349–5361. doi: 10.1128/JVI.02624-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eichler E.E., Holden J.J., Popovich B.W., Reiss A.L., Snow K., Thibodeau S.N., Richards C.S., Ward P.A., Nelson D.L. Length of uninterrupted CGG repeats determines instability in the FMR1 gene. Nat. Genet. 1994;8:88–94. doi: 10.1038/ng0994-88. [DOI] [PubMed] [Google Scholar]
- 34.Hwu W.L., Lee Y.M., Lee S.C., Wang T.R. In vitro DNA methylation inhibits FMR-1 promoter. Biochem. Biophys. Res. Commun. 1993;193:324–329. doi: 10.1006/bbrc.1993.1627. [DOI] [PubMed] [Google Scholar]
- 35.Drouin R., Angers M., Dallaire N., Rose T.M., Khandjian E.W., Rousseau F. Structural and functional characterization of the human FMR1 promoter reveals similarities with the hnRNP-A2 promoter region. Hum. Mol. Genet. 1997;6:2051–2060. doi: 10.1093/hmg/6.12.2051. [DOI] [PubMed] [Google Scholar]
- 36.Toth M., Muller U., Doerfler W. Establishment of de novo DNA methylation patterns. Transcription factor binding and deoxycytidine methylation at CpG and non-CpG sequences in an integrated adenovirus promoter. J. Mol. Biol. 1990;214:673–683. doi: 10.1016/0022-2836(90)90285-T. [DOI] [PubMed] [Google Scholar]
- 37.Woodcock D.M., Crowther P.J., Jefferson S., Diver W.P. Methylation at dinucleotides other than CpG: implications for human maintenance methylation. Gene. 1988;74:151–152. doi: 10.1016/0378-1119(88)90273-9. [DOI] [PubMed] [Google Scholar]
- 38.Munnes M., Patrone G., Schmitz B., Romeo G., Doerfler W. A 5′-CG-3′-rich region in the promoter of the transcriptionally frequently silenced RET protooncogene lacks methylated cytidine residues. Oncogene. 1998;17:2573–2583. doi: 10.1038/sj.onc.1202165. [DOI] [PubMed] [Google Scholar]
- 39.Naumann F., Remus R., Schmitz B., Doerfler W. Gene structure and expression of the 5′-(CGG)n-3′-binding protein (CGGBP1) Genomics. 2004;83:106–118. doi: 10.1016/s0888-7543(03)00212-x. [DOI] [PubMed] [Google Scholar]
- 40.Remus R., Kanzaki A., Yawata A., Nakanishi H., Wada H., Sugihara T., Zeschnigk M., Zuther I., Schmitz B., Naumann F. DNA methylation in promoter regions of red cell membrane protein genes in healthy individuals and patients with hereditary membrane disorders. Int. J. Hematol. 2005;81:385–395. doi: 10.1532/ijh97.04171. [DOI] [PubMed] [Google Scholar]
- 41.Millar D.S., Paul C.L., Molloy P.L., Clark S.J. A distinct sequence (ATAAA)n separates methylated and unmethylated domains at the 5′-end of the GSTP1 CpG island. J. Biol. Chem. 2000;275:24893–24899. doi: 10.1074/jbc.M906538199. [DOI] [PubMed] [Google Scholar]
- 42.Schwemmle S., de Graaf E., Deissler H., Gläser D., Wöhrle D., Kemmerknecht I., Just W., Oostra B.A., Doerfler W., Vogel W., Steinbach P. Characterization of FMR1 promoter elements by in vivo-footprinting analysis. Am. J. Hum. Genet. 1997;60:1354–1362. doi: 10.1086/515456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wallace J.A., Felsenfeld G. We gather together: insulators and genome organization. Curr. Opin. Genet. Dev. 2007;17:400–407. doi: 10.1016/j.gde.2007.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fan S., Fang F., Zhang X., Zhang M.Q. Putative zinc finger protein binding sites are over-represented in the boundaries of methylation-resistant CpG islands in the human genome. PLoS. ONE. 2007;2:e1184. doi: 10.1371/journal.pone.0001184. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.