

Abstract
Desbuquois dysplasia is a severe condition characterized by short stature, joint laxity, scoliosis, and advanced carpal ossification with a delta phalanx. Studying nine Desbuquois families, we identified seven distinct mutations in the Calcium-Activated Nucleotidase 1 gene (CANT1), which encodes a soluble UDP-preferring nucleotidase belonging to the apyrase family. Among the seven mutations, four were nonsense mutations (Del 5′ UTR and exon 1, p.P245RfsX3, p.S303AfsX20, and p.W125X), and three were missense mutations (p.R300C, p.R300H, and p.P299L) responsible for the change of conserved amino acids located in the seventh nucleotidase conserved region (NRC). The arginine substitution at position 300 was identified in five out of nine families. The specific function of CANT1 is as yet unknown, but its substrates are involved in several major signaling functions, including Ca2+ release, through activation of pyrimidinergic signaling. Importantly, using RT-PCR analysis, we observed a specific expression in chondrocytes. We also found electron-dense material within distended rough endoplasmic reticulum in the fibroblasts of Desbuquois patients. Our findings demonstrate the specific involvement of a nucleotidase in the endochondral ossification process.
Main Text
Desbuquois dysplasia (DBQD) is an autosomal-recessive chondrodysplasia belonging to the multiple dislocation group1 and characterized by severe prenatal and postnatal growth retardation (<−5 SD), joint laxity, short extremities, and progressive scoliosis. The main radiological features are short long bones with metaphyseal splay, a “swedish key” appearance of the proximal femur (exaggerated trochanter), and advanced carpal and tarsal bone age with a delta phalanx (Figure 1).2,3 We have previously distinguished two forms of Desbuquois dysplasia on the basis of the presence (type 1) or absence (type 2) of characteristic hand anomalies4, and we have subsequently mapped the Desbuquois type 1 gene to a 1.65 Mb interval on chromosome 17q25.5
Figure 1.
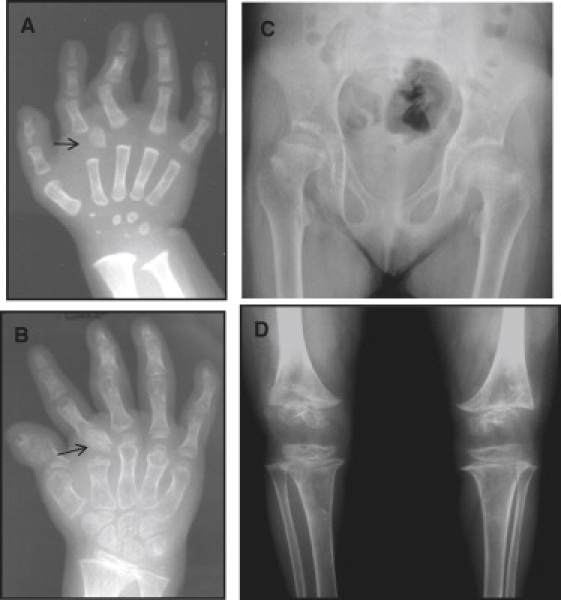
Radiological Manifestations of Desbuquois Dysplasia
(A and B) Hand X-rays at 1 and 3 years of age. Note the advanced carpal ossification and the delta phalanx (arrow).
(C) Hip at 3 years of age. Note the Swedish key appearance of the proximal femur.
(D) Knee at 3 years of age. Note the metaphyseal irregularities.
We report here the identification of calcium-activated nucleotidase 1 (CANT1) mutations in nine families with Desbuquois type 1. This gene encodes a soluble nucleotidase that preferentially hydrolyzes UDP followed by GDP and UTP but whose specific function is as yet unknown.6,7 Interestingly, we also found a distended rough endoplasmic reticulum (ER) in patient fibroblasts compared to controls.
Criteria for inclusion in the study were severe prenatal and postnatal growth retardation, joint laxity, short long bones, a Swedish key appearance of the proximal femur, and advanced carpal bone age with delta phalanx (Figure 1). A total of ten children who had Desbuquois dysplasia type 1 and belonged to nine families were included in the study. Eight families were consanguineous and originated from Turkey, Sri Lanka, Iran, United Arab Emirates, Morocco, or France. Table 1 summarizes the major clinical findings for these families. Blood samples were obtained with written consent in accordance with the French ethical standards regarding human subjects.
Table 1.
Clinical Manifestations in the Nine Families with Desbuquois Syndrome Type 1
| Family | Origin | Sex | Age | Birth Length | Height (SD) | Other Anomalies | Orthopedic Surgery | Scoliosis/Lordosis | Walking difficulties | Joint dislocation |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Sri Lanka | F | Death: day 2 Heart failure | 34 cm (35 WG) | −10 SD | Atrial septal defect | - | - | - | Hip, knee, elbow |
| 2 | Turkey | M | Terminated pregnancy 34 WG | 33 cm (34 WG) | 33 cm | Polyhydramnios | - | - | - | Hip, knee |
| 3 | Turkey | M | 18 months | 34 cm | −12 SD | Delay in verbal speech | Yes, knees | - | Not ambulatory sits without support | Hip, knee |
| 4 | Turkey | F | 20 months | ? | −4SD | - | Yes, knees | Thoracic scoliosis | Yes | Left elbow and both knees |
| 5 | Iran | M | 4 years | ? | −9 SD | - | - | Not ambulatory without any support | Hip and Knee | |
| 6 | France | M | 27 years | 43 cm (term) | −8SD | Glaucoma | Yes, knees | Lordosis | Yes | Hip, knee and fingers |
| 7 | United Arab Emirates | M | 4 years on report (17 years now) | ? | < −4SD | Mental retardation | Yes, hips and knees | Lordosis | Yes | Hip and knee, But limitation of elbow |
| 8 | Morocco | M | Death: 3 months Cardio-respiratory failure | 37 cm | < −6SD | - | - | - | - | Elbow, hip and right knee |
| 9 | Brazil Case 1 | M | Neonatal death | 36 cm (37 WG) | 36 cm | Omphalocele, Pulmonary hypoplasia, Ventricular septal defect, Hydronephrosis | - | - | - | Multiple dislocation |
| Case 2 | M | Neonatal death | 35 cm (39 WG) | 35 cm | Pulmonary hypoplasia, Coarctation of aorta | - | - | - | Multiple dislocation |
WG: weeks of gestation. ?: unknown. -: no clinical manifestation.
Using the human genome resources (Ensembl and UCSC Browser), we selected several candidate genes among the 14 genes located in our region of interest of 1.65 Mb on chromosome 17q25. These genes included the STAT induced inhibitor-3 gene (SSI-3), the phosphatidylglycerophosphate synthase gene (PGS1), the Pleckstrin homology Sec 7 and coiled/coil domains 1 gene (PSCD1), the human tissue inhibitor of metalloproteinases 2 gene (TIMP2), the C1q and tumor necrosis factor-related protein 1 gene (C1QTNF1), and the lectin galactoside-binding soluble 3 binding protein gene (LGALS3BP). After having excluded these six genes by direct sequencing, we considered the calcium-activated nucleotidase 1 gene (CANT1) as a candidate. CANT1, a member of the apyrase family, is a soluble nucleotidase that preferentially hydrolyzes UDP followed by GDP and UTP.6,7 Its exact function in humans remains unclear.8
CANT1 is composed of five coding exons and has three transcripts (Figure 2). The first transcript encodes a protein of 401 amino acids, characterized by eight nucleotidase conserved regions (NRC).9 By direct sequencing, we identified three missense and four nonsense mutations (including a large deletion of 2703 bp encompassing the 5′ UTR and exon 1) in a total of nine Desbuquois type 1 families (Table 2). The mutations were located throughout the gene (Figure 2). They cosegregated with the disease and were not identified in 210 control chromosomes. The missense mutations (p.R300H, p.R300C, and p.P299L) were responsible for the change of conserved amino acids located in NCR7. The substitution of arginine at position 300 was identified at the homozygote state in five out of nine families.
Figure 2.
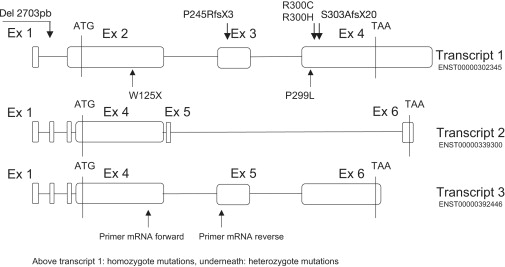
CANT1 Mutations Identified in Desbuquois Patients
Position of the primers used for the RT-PCR.
Table 2.
CANT1 Mutations Identified in Nine Families with Desbuquois Syndrome Type 1
| Family | Ethnic Origin | Consanguinity | Number of Affected Children | Nucleotide Change | Amino Acid Change | Location (Ref: transcript 1) |
|---|---|---|---|---|---|---|
| 1 | Sri lanka | Yes | 1 | del 2703 bp | _ | 5′UTR and Ex1 |
| 2 | Turkey | Yes | 1 | c.734 delC | p.P245RfsX3 | Ex3 |
| 3 | Turkey | Yes | 1 | c.898C > T | p.R300C | Ex4 |
| 4 | Turkey | Yes | 1 | c.898C > T | p.R300C | Ex4 |
| 5 | Iran | Yes | 1 | c.898C > T | p.R300C | Ex4 |
| 6 | France | Yes | 1 | c.899G > A | p.R300H | Ex4 |
| 7 | United Arab Emirates | Yes | 1 | c.899G > A | p.R300H | Ex4 |
| 8 | Morocco | Yes | 1 | c.907-911insGCGCC | p.S303AfsX20 | Ex4 |
| 9 | Brazil | No | 2 | c.374G > A | p.W125X | Ex2 |
| c.896C > T | p.P299L | Ex4 |
To further understand the function of CANT1 in humans, we studied its expression pattern by RT-PCR analysis and were able to observe a specific signal in control lymphocytes, cultured skin fibroblasts, and chondrocytes but not in osteoblasts. A specific CANT1 mRNA signal was still detected in Desbuquois dysplasia patients' fibroblasts and lymphocytes, which carried homozygous p.P245RfsX3 and p.R300H mutations. Yet, CANT1 mRNA was not detectable in patient 1, who presented a large homozygous deletion encompassing the 5′ UTR region and the noncoding exon 1 (Figure 3).
Figure 3.
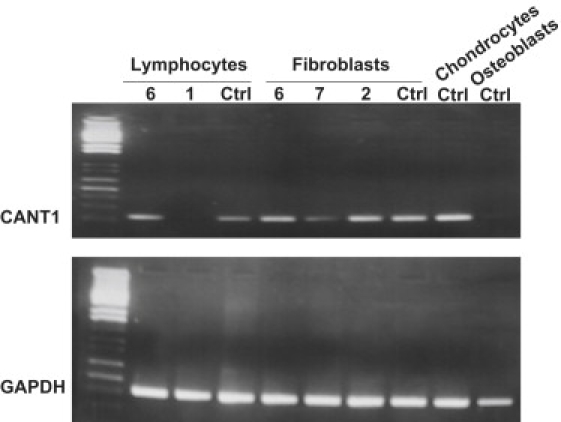
CANT1 mRNA Expression by RT-PCR in Desbuquois Patient Lymphocytes and Fibroblasts and in Wild-Type Chondrocytes and Osteoblasts
The presence of inclusion bodies within distended rough endoplasmic reticulum (RER) in Desbuquois patient chondrocytes10 prompted us to further study the cultured skin fibroblasts of our patients. Ultrastructural analysis of cultured fibroblasts from three Desbuquois patients demonstrated the presence of dilated RER cisternae containing proteinaceous material (Figure 4), comparable to what has been reported in chondrocytes. Importantly, dilatation of RER was not observed in three control cultured fibroblasts from the same age and same passage.
Figure 4.
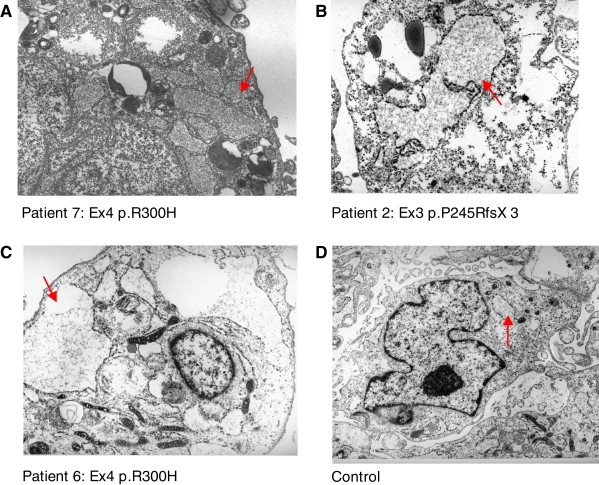
Transmission Electron Microscopy in Fibroblasts from Three Desbuquois Dysplasia Patients
(A–C) RER cisternae appeared markedly dilated in the vast majority of cells, in which there was an accumulation of slightly electron-dense, fibrillar, or finely granular proteinaceous material (arrows). Interestingly, there is no evidence of ribosome detachment, commonly observed in severe oxidative stress.
(D) Cultured fibroblasts from healthy individuals failed to display significant enlargement of RER (arrowhead). (staining was with uranyl acetate and lead citrate; original magnification ×15,000).
Here, we report seven distinct CANT1 mutations in nine families with Desbuquois dysplasia type 1. These mutations include four nonsense and three missense mutations. All missense mutations are located in the NCR7-encoding region, which is highly conserved among related apyrases, and two of them (identified in five out of nine families) substitute arginine at position 300.9 This amino acid belongs to a pentad of alternating positively and negatively charged residues (Asp 114, Lys 394, Glu 365, Arg 300, and Glu 284) that comprise a network of four salt bridges involved in the catalytic site of CANT1.11 Direct mutagenesis of Arg 300 has been shown to disrupt the electrostatic interactions in the salt bridge and result in decreased enzyme activity without altering calcium binding or producing a conformational change.11
All children presented with similar skeletal manifestations. However, an early death due to cardio-respiratory failure was observed in children with nonsense mutations. Among the whole series, extra-skeletal manifestations included heart defects (three out of ten), mental retardation (two), glaucoma (one), and hydronephrosis (one out of ten).
The specific function of CANT1 in humans, as well as its cellular localization, is unknown.
Desbuquois syndrome shares phenotypic features, namely advanced carpal bone maturation, with Diastrophic dysplasia (DTD), and it also shares features, namely congenital joint dislocations, with recessive Larsen syndrome (CHST3 deficiency). Both DTD and CHST3 deficiency involve a defect in sulfation, the final step of proteoglycan synthesis. CANT1 deficiency might interfere with the availability of UDP-sugars needed for proteoglycan synthesis. However, to date, the involvement of CANT1 in proteoglycan synthesis has not been demonstrated.12
CANT1 substrates (UDP > GDP > UTP) are involved in several major signaling functions, notably in calcium release, through activation of pyrimidinergic signaling.8,13,14 Indeed, the binding of pyrimidinergic nucleotides (UTP/UDP) to P2Y receptors generates inositol 1,4,5-triphosphate (IP3) through their coupling to phospholipase C.8,14 IP3 binding to the IP3 receptor at the surface of the endoplasmic reticulum (ER) allows rapid release of calcium from the intracellular stores.13 However, the link between CANT1 mutations and RER distension (observed in chondrocytes and fibroblasts of Desbuquois dysplasia patients) is unclear but may be related to impaired ER function. Accordingly, deletion of APY-1, the Caenorhabditis elegans homolog of CANT1, sensitized worms to ER stress and induced defects in pharynx and muscle organization, leading to a reduced lifespan.15
With the identification of CANT1 mutations in Desbuquois dysplasia, we demonstrate for the first time, to our knowledge, the key role of a nucleotidase in the endochondral ossification process. It is hoped that future studies will lead to further understanding of the specific function of CANT1 in the endochondral ossification process.
Acknowledgments
We thank the patients and their families for their participation in this study. C. Huber and M. Fradin are supported by the MD-PhD program of the Fondation pour la Recherche Médicale (FRM). M. Chami was supported by an INSERM young researcher contract. B. Oulès was supported by the MD-PhD program of the École de l'INSERM Liliane Bettencourt.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Ensembl genome browser, http://www.ensembl.org/Homo_sapiens/Info/Index
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omin/
UCSC genome browser, http://www.genome.ucsc.edu/
References
- 1.Superti-Furga A., Unger S. Nosology and classification of genetic skeletal disorders: 2006 revision. Am. J. Med. Genet. A. 2007;143:1–18. doi: 10.1002/ajmg.a.31483. [DOI] [PubMed] [Google Scholar]
- 2.Desbuquois G., Grenier B., Michel J., Rossignol C. Nanisme chondrodystrophique avec ossification anarchique et polymalformations chez deux sœurs. Arch. Fr. Pediatr. 1966;23:573–587. [Google Scholar]
- 3.Faivre L., Cormier-Daire V., Young I., Bracq H., Finidori G., Padovani J.P., Odent S., Lachman R., Munnich A., Maroteaux P., Le Merrer M. Long-term outcome in Desbuquois dysplasia: A follow-up in four adult patients. Am. J. Med. Genet. 2004;124A:54–59. doi: 10.1002/ajmg.a.20441. [DOI] [PubMed] [Google Scholar]
- 4.Faivre L., Cormier-Daire V., Eliott A.M., Field F., Munnich A., Maroteaux P., Le Merrer M., Lachman R. Desbuquois dysplasia, a reevaluation with abnormal and “normal” hands: radiographic manifestations. Am. J. Med. Genet. 2004;124A:48–53. doi: 10.1002/ajmg.a.20440. [DOI] [PubMed] [Google Scholar]
- 5.Faivre L., Le Merrer M., Al-Gazali L.I., Ausems M.G., Bitoun P., Bacq D., Maroteaux P., Munnich A., Cormier-Daire V. Homozygosity mapping of a Desbuquois dysplasia locus to chromosome 17q25.3. J. Med. Genet. 2003;40:282–284. doi: 10.1136/jmg.40.4.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murphy D.M., Ivanenkov V.V., Kirley T.L. Bacterial expression and characterization of a novel, soluble, calcium-binding, and calcium-activated human nucleotidase. Biochemistry. 2003;42:2412–2421. doi: 10.1021/bi026763b. [DOI] [PubMed] [Google Scholar]
- 7.Smith T.M., Hicks-Berger C.A., Kim S., Kirley T.L. Cloning, expression, and characterization of a soluble calcium-activated nucleotidase, a human enzyme belonging to a new family of extracellular nucleotidases. Arch. Biochem. Biophys. 2002;406:105–115. doi: 10.1016/s0003-9861(02)00420-4. [DOI] [PubMed] [Google Scholar]
- 8.Lecca D., Ceruti S. Uracil nucleotides: from metabolic intermediates to neuroprotection and neuroinflammation. Biochem. Pharmacol. 2008;75:1869–1881. doi: 10.1016/j.bcp.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 9.Yang M., Kirley T.L. Site-directed mutagenesis of human soluble calcium-activated nucleotidase 1 (hSCAN-1): identification of residues essential for enzyme activity and the Ca(2+)-induced conformational change. Biochemistry. 2004;43:9185–9194. doi: 10.1021/bi049565o. [DOI] [PubMed] [Google Scholar]
- 10.Shohat M., Lachman R., Gruber H.E., Hsia Y.E., Golbus M.S., Witt D.R., Bodell A., Bryke C.R., Hogge W.A., Rimoin D.L. Desbuquois syndrome: Clinical, radiographic, and morphologic characterization. Am. J. Med. Genet. 1994;52:9–18. doi: 10.1002/ajmg.1320520104. [DOI] [PubMed] [Google Scholar]
- 11.Dai J., Liu J., Deng Y., Smith T.M., Lu M. Structure and protein design of a human platelet function inhibitor. Cell. 2004;116:649–659. doi: 10.1016/s0092-8674(04)00172-2. Erratum in Cell 117, 413. [DOI] [PubMed] [Google Scholar]
- 12.Hermanns P., Unger S., Rossi A., Perez-Aytes A., Cortina H., Bonafé L., Boccone L., Setzu V., Dutoit M., Sangiorgi L. Congenital joint dislocations caused by carbohydrate sulfotransferase 3 deficiency in recessive Larsen syndrome and humero-spinal dysostosis. Am. J. Hum. Genet. 2008;82:1368–1374. doi: 10.1016/j.ajhg.2008.05.006. Erratum in Am. J. Hum. Genet. 83, 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clapham D.E. Calcium signaling. Cell. 2007;131:1047–1058. doi: 10.1016/j.cell.2007.11.028. [DOI] [PubMed] [Google Scholar]
- 14.Abbracchio M.P., Burnstock G., Boeynaems J.M., Barnard E.A., Boyer J.L., Kennedy C., Knight G.E., Fumagalli M., Gachet C., Jacobson K.A., Weisman G.A. International Union of Pharmacology LVIII: update on the P2Y G protein-coupled nucleotide receptors: From molecular mechanisms and pathophysiology to therapy. Pharmacol. Rev. 2006;58:281–341. doi: 10.1124/pr.58.3.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Uccelletti D., Pascoli A., Farina F., Alberti A., Mancini P., Hirschberg C.B., Palleschi A.C. APY-1, a novel Caenorhabditis elegans apyrase involved in unfolded protein response signalling and stress responses. Mol. Biol. Cell. 2008;19:1337–1345. doi: 10.1091/mbc.E07-06-0547. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.