

Abstract
Night vision requires signaling from rod photoreceptors to adjacent bipolar cells in the retina. Mutations in the genes NYX and GRM6, expressed in ON bipolar cells, lead to a disruption of the ON bipolar cell response. This dysfunction is present in patients with complete X-linked and autosomal-recessive congenital stationary night blindness (CSNB) and can be assessed by standard full-field electroretinography (ERG), showing severely reduced rod b-wave amplitude and slightly altered cone responses. Although many cases of complete CSNB (cCSNB) are caused by mutations in NYX and GRM6, in ∼60% of the patients the gene defect remains unknown. Animal models of human diseases are a good source for candidate genes, and we noted that a cCSNB phenotype present in homozygous Appaloosa horses is associated with downregulation of TRPM1. TRPM1, belonging to the family of transient receptor potential channels, is expressed in ON bipolar cells and therefore qualifies as an excellent candidate. Indeed, mutation analysis of 38 patients with CSNB identified ten unrelated cCSNB patients with 14 different mutations in this gene. The mutation spectrum comprises missense, splice-site, deletion, and nonsense mutations. We propose that the cCSNB phenotype in these patients is due to the absence of functional TRPM1 in retinal ON bipolar cells.
Main Text
Congenital stationary night blindness (CSNB) is a group of genetically and clinically heterogeneous retinal disorders. The genes involved in the different forms of CSNB encode proteins, which are confined to the phototransduction cascade or are important in retinal signaling from photoreceptors to adjacent bipolar cells.1 Most of the patients with mutations in these genes show a typical electrophysiological phenotype characterized by an electronegative waveform of the dark-adapted, bright-flash electroretinogram (ERG), in which the amplitude of the b-wave is smaller than that of the a-wave.2 This so-called Schubert-Bornschein type of ERG response allows the discrimination of two subtypes of CSNB: incomplete (ic) (CSNB2A [MIM 300071], CSNB2B [MIM 610427]) and complete (c) (CSNB1A [MIM 310500], CSNB1B [MIM 257270].3 The incomplete type is characterized by both a reduced rod b-wave and substantially reduced cone response, due to both ON and OFF bipolar cell dysfunction, whereas the complete type is associated with a drastically reduced rod b-wave response but largely normal cone b-wave amplitudes, due to ON bipolar cell dysfunction.4
In a considerable fraction of CSNB patients, mutations have been identified by direct sequencing of candidate genes or microarray analysis.5 However, from our CSNB cohort, the phenotype in ∼60% of the patients could not be associated with mutations in known genes, indicating that additional genes remain unidentified. Recently, a type of CSNB in Appaloosa horses has been described.6,7 Affected animals initially showed reduced vision in dim light conditions, which subsequently progressed to reduced vision even in normal light conditions in severely affected animals. No fundus abnormalities were present, but strabismus and nystagmus were described. Electrophysiological studies revealed a “negative ERG” resembling the human Schubert-Bornschein type of ERG response.8 Furthermore, the photopic flicker responses of affected horses seemed to be similar when compared with those of unaffected horses,6 suggesting a phenotype reminiscent of cCSNB. Association studies of the coat coloring in these horses revealed that this trait is directly linked with the CSNB phenotype. Gene expression analysis of genes linked to this disorder revealed that TRPM1 (MIM 603576), also known as melastatin (MLSN1), was significantly downregulated in the retina and skin of affected animals. Thus, it was proposed that TRPM1 is responsible for altering bipolar signaling as well as melanocyte function, causing both CSNB and the coat color phenotype in Appaloosa horses.9 Studies in mice lacking Trpm1 revealed a severely reduced b-wave in ERG recordings, similar to the Schubert-Bornschein type of ERG response.10 These findings support the hypothesis that this gene is important for night vision.
TRPM1 is a member of the transient receptor potential (TRP) channel family. These channels permit Ca2+ entry into hyperpolarized cells, producing intracellular responses linked to the phosphatidylinositol and protein kinase C signal transduction pathway.11 Because of the downregulation of TRPM1 in Appaloosa horses with CSNB, it was suggested that this gene may play a role in neural transmission in the human retina through changing cytosolic-free Ca2+ levels in the retina in ON bipolar cells. The mGluR6 protein (MIM 604096) of the ON bipolar cells is coupled to Gαo proteins (MIM 139311) and to TRPM1. TRPM1 might be the cation channel that is downstream of the Gαo protein in the ON bipolar cells.9
Altogether, the phenotype of Appaloosa horses, the downregulation of TRPM1 in affected animals, and its localization downstream of mGluR6 in ON bipolar cells rendered this gene a good candidate. Thus, we screened this gene in 38 clinically diagnosed CSNB patients from different centers in Europe and the United States (Belgium: Ghent; France: Paris, Montpellier, and Lille; Germany: Berlin, Freiburg, Giessen, and Tuebingen; Switzerland: Lausanne; United States: Philadelphia, PA). Prior to this study, most patients were excluded either for known mutations, by a CSNB genotyping microarray, or for known CSNB genes and additional candidate genes, by direct sequencing.5 Research procedures were conducted in accordance with institutional guidelines and the Declaration of Helsinki. Prior to genetic testing, informed consent was obtained from all patients and their family members. Ophthalmic examination included best corrected visual acuity, slit lamp examination, funduscopy, Goldmann kinetic perimetry, full-field ERG incorporating the ISCEV (International Society for Clinical Electrophysiology of Vision) standards,12 fundus autofluorescence, and optical coherence tomography (OCT) (extent of investigation depending on the referring center). Thirty fragments covering 27 exons of TRPM1 (RefSeq NM_002420.4, variant 70+TRPM113), two fragments corresponding to two recently identified exons (exon 1′ [variant 92+TRPM113] and exon 0 [variant 109+TRPM113]) of this gene (Figure 1), and the flanking intronic regions were directly sequenced from the PCR-amplified products (primers are listed in Table S1, available online) with the use of a sequencing mix (BigDye Terminator v1.1 Cycle Sequencing Kit, Applied Biosystems, Courtabœuf, France) and analyzed on an automated 48-capillary sequencer (ABI 3730 Genetic Analyzer, Applied Biosystems). The results were interpreted by a software application (SeqScape, Applied Biosystems).
Figure 1.
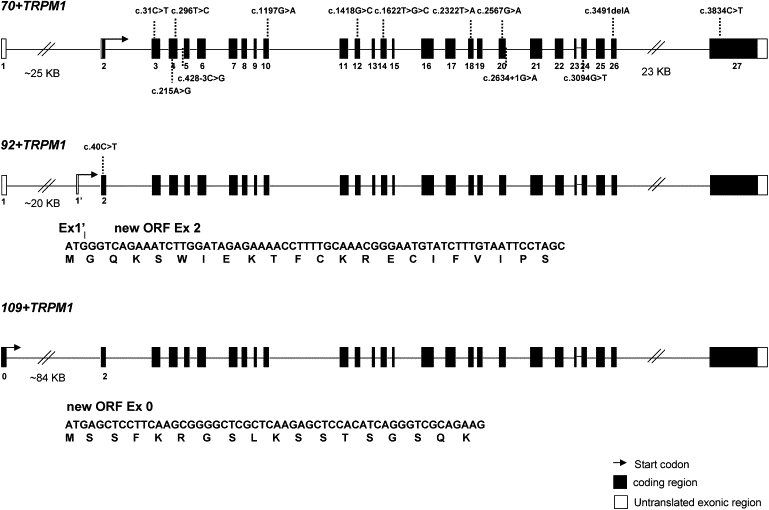
TRPM1 Isoforms and CSNB Mutations
Three isoforms of TRPM1 are presented: the 70+TRPM1 variant represents the previously published reference sequence (RefSeq NM_002420.4), whereas the 92+TRPM1 and 109+TRPM1 isoforms were only recently identified.13 In comparison to 70+TRPM1, the 92+TRPM1 isoform contains 22 additional amino acids, and the 109+TRPM1 isoform contains 39 additional amino acids. The new open reading frame of 92+TRPM1 is made up of the complete exon 2 with the initiation codon located in a new exon (exon 1′). The new ORF of 109+TRPM1 is made up of the complete exon 2 and a new exon (exon 0). Both new ORFs continue in the isoform 70+TRPM1, coding for 1625 and 1642 amino acids, respectively.
Analysis in TRPM1 revealed causative mutations in ten cCSNB patients (Figure 2: patient CIC00238 shown as an example of cCSNB) with a total of 14 different mutations (Figure 1 and Table 1). These comprise nonsense mutations, a deletion leading to a predicted premature stop codon, splice-site mutations, silent mutations, and missense mutations. None of these changes were found among control chromosomes (210–380 chromosomes). In those patients from whom family members could be investigated, the cCSNB phenotype cosegregated with the mutations and the genotypes were indicative for autosomal-recessive inheritance (Figures 3A and 3B: three patient examples). Five index patients (4497, 8214, CIC00612, 23625, and 758) showed compound heterozygous mutations (Table 1). Patients CIC00612 and 23625 both revealed a heterozygous p.Tyr72Cys substitution. From the origins of these patients, no close familial relationship was obvious. In three index patients (CIC00238, 691, and D0704708), an apparently homozygous mutation was found (Table 1). Homozygosity was proven for index patient D0704708 (Figure 3). Cosegregation analysis from family members of index patient CIC00238 revealed that another affected sister was apparently homozygous for the mutation, whereas the father was heterozygous. Two unaffected sisters and, interestingly, the mother did not show the mutation (Table 1). These findings indicated that the patient is most likely heterozygous for the missense mutation inherited from the father and would have a deletion in TRPM1 or a mutation in another gene, which would have been inherited from the mother. Four investigated SNPs (rs4779818 in intron 1, rs4779816 in exon 2, rs2241493 in exon 3, and rs2288242 in exon 18) were apparently homozygous in the patient and the parents. Therefore, the putative deletion could not be defined. Analyses of additional SNPs in genomic regions of TRPM1 or screening of candidate genes may enable us to localize the second mutation and will be investigated in the future. The parents of patient 691 were not available for genetic testing, and thus homozygosity could not be proven. For two patients, 14101 and 10731, only one heterozygous mutation was identified (Table 1). Again, the second mutation may be a large heterozygous deletion and thus not detectable by PCR-based sequencing. In addition, a mutation located in a second gene may disable signaling important for nocturnal vision. Three investigated SNPs (rs4779816 in exon 2, rs2241493 in exon 3, and rs3782599 in exon 4) were apparently homozygous in the patient and the parents, and thus the putative deletion could not be defined. Mutation analysis in patient 14101 on other known or candidate genes (NYX, AJ278865 [MIM 300278]; GRM6, NM_000843; CABP4, NM_145200 [MIM 608965]; CACNA2D4, NM_172364 [MIM 608171]; BHLHB4, BK000274 [MIM 609331]; CACN2B, NM_000724 [MIM 600003]; GNA01, NM_020988 and NM_138736; and TBC1D2, NM_018421 [MIM 609871]) did not reveal any mutation. Previous mutation analyses in the simplex case 10731 in known and candidate genes (NYX, CACNA1F, AJ006216 [MIM 300110], GRM6, CABP4, CACNA2D4, BHLHB4, CACN2B, GNA01, and TBC1D2) did not reveal any mutation. Thus, for both patients, the second mutation may be found in other regions of TRPM1, such as regulatory sequences or unidentified exons, or may represent a deletion in an as-yet-uninvestigated region of TRPM1. Alternatively, the second mutation may be found in a novel CSNB gene.
Figure 2.
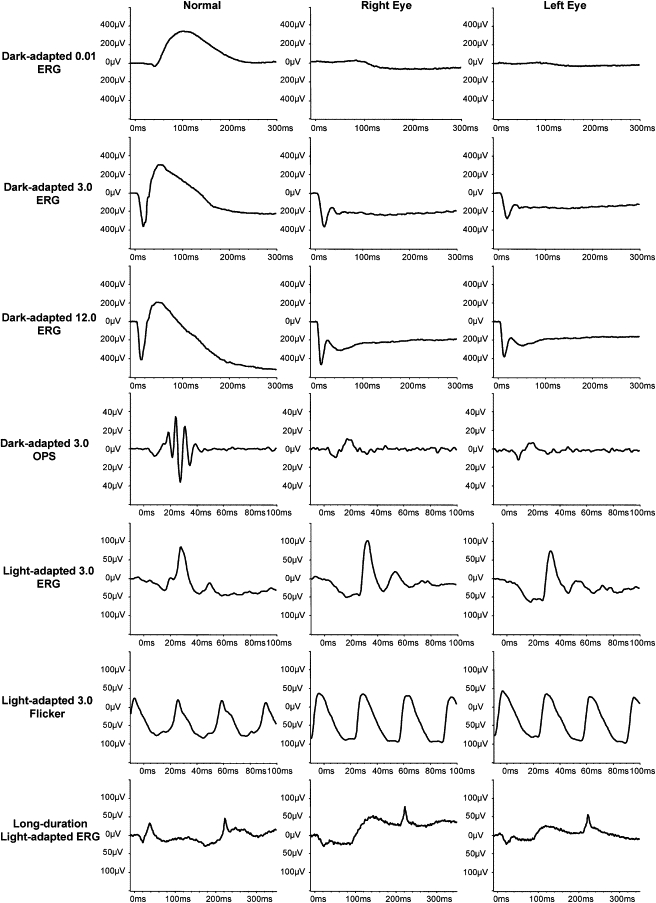
Electrophysiologic Description of Patient CIC00238 with cCSNB, as an Example
Full-field ERGs show typical ON bipolar pathway dysfunction: there are no detectable responses for the dark-adapted 0.01 ERG; dark-adapted 3.00 and 12.0 ERGs show an a-wave with a normal amplitude and implicit time but a severely reduced b-wave, leading to an electronegative waveform. Dark-adapted oscillatory potentials (OPs) are not detectable. Light-adapted 3.0 ERGs show normal amplitudes but implicit time shift for both the a-wave and the b-wave. The a-wave has a broadened trough, and there is a sharply rising b-wave with no OPs. Light-adapted 3.0 flicker ERGs show normal amplitudes but a broadened trough and a mildly delayed implicit time. These photopic ERG appearances are characteristic of selective dysfunction of the ON bipolar pathway with OFF bipolar pathway preservation.31 This is further confirmed with long-duration stimulations, which reveal a normal a-wave but a severely reduced ON-response b-wave and a preserved OFF-response d-wave.
Table 1.
Patients with Likely Pathogenic TRPM1 Mutations
| Index Patient, Location, Family Members | Ethnicity | Exon | Nucleotide Exchange | Allele State | Protein Effect | Control Alleles (Mut/WT) | Phenotype Index |
|---|---|---|---|---|---|---|---|
| CIC00238: Paris, France | Portuguese-French | 12 | c.1418G>C | hom? | p.Arg473Pro | 0/286 | cCSNB, myopia, nystagmus, strabismus |
| unaff. father CIC03424 | 12 | c.1418G>C | het | p.Arg473Pro | |||
| unaff. mother CIC03423 | - | no | - | - | |||
| unaff. sister CIC03421 | - | no | - | - | |||
| unaff. sister CIC03422 | - | no | - | - | |||
| aff. sister CIC03452 | 12 | c.1418G>C | hom? | p.Arg473Pro | cCSNB, myopia, nystagmus, strabismus | ||
| 4497a, II-1: Tuebingen, Germany | German | 3 | c.31C>T | het | p.Gln11X | 0/352 | cCSNB, nystagmus, myopia |
| 4 | c.296T>C | het | p.Leu99Pro | 0/224 | |||
| unaff. father 4608, I-1 | 4 | c.296T>C | het | p.Leu99Pro | |||
| unaff. mother 4610, I-2 | 3 | c.31C>T | het | p.Gln11X | |||
| unaff. sister 4600, II-2 | - | no | - | - | |||
| unaff. sister 4712, II-4 | 3 | c.31C>T | het | p.Gln11X | |||
| unaff. brother 4740, II-3 | 3 | c.31C>T | het | p.Gln11X | |||
| 691: Tuebingen, Germany | Turkish | 20 | c.2567G>A | hom? | p.Trp856X | 0/366 | cCSNB, myopia, nystagmus,strabismus |
|
8214: Tuebingen, Germany |
German |
10 | c.1197G>A | het | c.Pro399Pro/splice defect? | 0/350 | cCSNB, myopia, strabismus |
| 26 | c.3491delA | het | p.Gln1164ArgfsX31 | 0/266 | |||
| CIC00612: Paris, France | French | 4 | c.215A>G | het | p.Tyr72Cys | 0/210 | cCSNB, myopia, nystagmus, strabismus |
| 24 | c.3094G>T | het | p.Glu1032X | 0/370 | |||
| unaff. mother: CIC03359 | 24 | c.3094G>T | het | p.Glu1032X | |||
| unaff. brother: CIC03360 | - | no | - | - | |||
| 23625a, II-3: Lausanne, Switzerland | Italian | 4 | c.215A>G | het | p.Tyr72Cys | 0/210 | cCSNB, myopia |
| int4 | c.428-3C>G | het | splice defect | 0/298 | |||
| unaff. father 23628, I-1 | 4 | c.215A>G | het | p.Tyr72Cys | |||
| unaff. mother 23728, I-2 | int4 | c.428-3C>G | het | splice defect | |||
| unaff. brother CIC03365, II-1 | - | no | - | ||||
| unaff. brother CIC03364, II-2 | 4 | c.215A>G | het | p.Tyr72Cys | |||
|
758.01: Giessen, Germany |
German |
int20 | c.2634+1G>A | het | splice defect | 0/366 | cCSNB |
| 27 | c.3834C>T | het | p.Asn1278Asn | 0/304 | |||
| D0704708a, II-3: Ghent, Belgiumb | Flemish-Belgian | 2 | c.1-27C>T (70+TRPM1) or c.40C>T (92+TRPM1) | hom | 5′ UTR expression defect or p.Arg14Trp | 0/348 | cCSNB, strabismus, hypermetropia |
| unaff. father CIC03386, I-1 | 2 | ″ | het | 5′ UTR expression defect or p.Arg14Trp | |||
| unaff. mother D0704709, I-2 | 2 | ″ | het | 5′ UTR expression defect or p.Arg14Trp | |||
| unaff. sister CIC03389, II-1 | 2 | ″ | het | 5′ UTR expression defect or p.Arg14Trp | |||
| unaff. sister CIC03390, II-2 | 2 | ″ | het | 5′ UTR expression defect or p.Arg14Trp | |||
| unaff. brother CIC03391, II-3 | - | no | - | ||||
| 14101: Philadelphia, PA, USA | Austrian-Russian-Ashkenazi Jewish | 18 | c.2322T>A | het | p.Tyr774X | 0/380 | cCSNB, myopia |
| ? | ? | - | ? | ||||
| unaff. father 19037 | ? | ? | - | ? | |||
| unaff. mother 19038 | 18 | c.2322T>A | het | p.Tyr774X | |||
| 10731: Berlin, Germany | German | 14 | c.1622T>A | het | p.Met541Lys | 0/214 | cCSNB, myopia |
| ? | ? | - | ? | ||||
Index patients are presented in bold. Abbreviations are as follows: Mut, mutated; het, heterozygous; hom, homozygous; unaff., unaffected; aff., affected. CSNB mutations are annotated according to the recommendation of the Human Genome Variation Society, with nucleotide position +1 corresponding to the A of the translation-initiation codon ATG in the cDNA nomenclature RefSeq NM_002420.4, 70+TRPM1. For Exon 0 and Exon 1′, the respective A of the new translation initiation codon ATG was used.
See Figures 3A and 3B.
27533 Diagnostic: Zurich, Switzerland.
Figure 3.
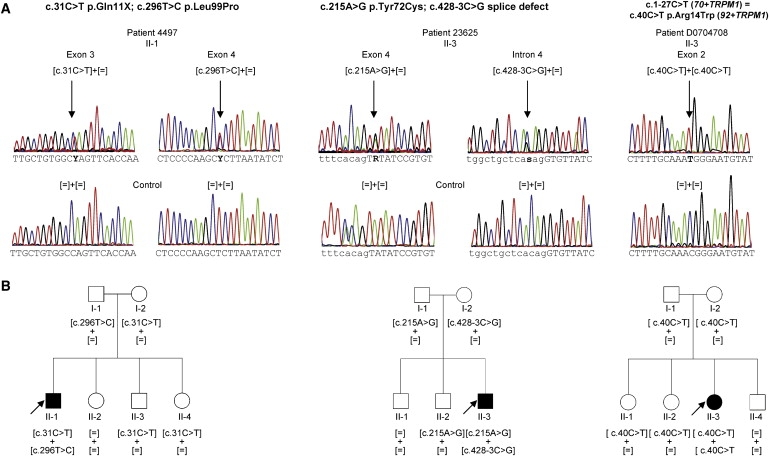
TRPM1 Mutations and Cosegregation Analysis in Families with CSNB
(A) Electropherograms of three index patients provided as an example, showing TRPM1 mutations, which are highlighted by an arrow. Exonic sequence is shown in capital letters. Intronic sequence is shown in lowercase letters.
(B) Corresponding pedigrees of selected cCSNB patients with TRPM1 mutations and cosegregation in available family members. Filled symbols represent affected individuals, and unfilled symbols represent unaffected persons. Squares indicate males, and circles indicate females. Arrows reflect the index patients.
One other patient (13830) with icCSNB was compound heterozygous for two missense changes: c.1195C>A, causing a p.Pro399Thr substitution in exon 10, and c.3483G>C, leading to a p.Gln1161His substitution in exon 26, respectively (Table 2). However, the c.1195C>A change was found in eight of 350 control chromosomes and the c.3483G>C in two of 266. Thus, both variants are most likely non-disease-causing variants. This is also consistent with the fact that TRPM1 mutations in our study specifically lead to cCSNB and not to icCSNB. Another variant (c.4123G>T) in exon 27, leading to a p.Glu1375X, was detected in three patients but turned out to be a SNP (rs378489), which was detected in 20 of 320 control chromosomes. Other presumably non-disease-causing variants were detected and are summarized in Table 3.
Table 2.
Likely Non-Disease-Causing TRPM1 Variants Identified in Patients with CSNB
| Exon | Nucleotide Exchange | Allele State | Protein Effect | Control Alleles (Mut/WT) | Conclusion |
|---|---|---|---|---|---|
| 0 | c.16C>T | het or hom | p.Arg6Trp | frequent in patients and controls | new, but T occurs also in Platypus; thus, SNP |
| 10 | c.1195C>A | het | p.Pro399Thr | 8/350 | SNP |
| 26 | c.3483G>C | het | p.Gln1161His | 2/266 | SNP |
| 27 | c.4123G>T | het | p.Glu1375X | 20/320 | SNP |
| c.4264C>T | het or hom | p.Arg1422Trp | frequent in patients and controls | new, but 2/334 alleles showed exchange; thus, SNP |
Abbreviations are as follows: Mut, mutated; het, heterozygous; hom, homozygous. CSNB mutations are annotated according to the recommendation of the Human Genome Variation Society, with nucleotide position +1 corresponding to the A of the translation-initiation codon ATG in the cDNA nomenclature RefSeq NM_002420.4, 70+TRPM1. For Exon 0 and Exon 1′, the respective A of the new translation initiation codon ATG was used.
Table 3.
Benign TRPM1 Variants Identified in CSNB Patients
| Exon | Nucleotide Exchange | Protein Effect | SNP ID |
|---|---|---|---|
| 2 | c.2T>C | p.Met1Thr | rs4779816 |
| 3 | c.95G>A | p.Ser32Asn | rs2241493 |
| 11 | c.1239G>A | p.Thr413Thr | rs1035705 |
| 16 | c.1813G>A | p.Val605Met | rs17815774 |
| 18 | c.2307T>C | p.Tyr769Tyr | rs12913672 |
| c.2340T>C | p.Asn780Asn | rs2288242 | |
| 19 | c.2475C>T | p.Asn825Asn | rs12911350 |
| 27 | c.3686A>C | p.Asn1229Thr | rs17227996 |
| c.4135C>A | p.Pro1379Thr | rs61734298 | |
| c.4139G>A | p.Val1395Ile | rs3784588 | |
| c.4494T>A | p.His1483Gln | rs12898290 |
CSNB mutations are annotated according to the recommendation of the Human Genome Variation Society, with nucleotide position +1 corresponding to the A of the translation-initiation codon ATG in the cDNA nomenclature RefSeq NM_002420.4, 70+TRPM1.
The most likely pathogenic mutations identified herein were predicted to localize at different sites of the TRP channel. Five missense mutations, which were found in evolutionarily conserved residues (Figure 4), one silent mutation, and one nonsense mutation were predicted to localize in the N-terminal intracellular part of TRPM1 (Figure 5), the function of which is not yet understood.11 All missense mutations were predicted by homology-based programs (SIFT amd Polyphen, data not shown) to be pathogenic. Another silent mutation was identified in the C terminus of TRPM1. For all of these, in addition to the splice-site mutations, splicing could be influenced because different splicing proteins were predicted to bind to the mutated variants in comparison to the control (ESEfinder, data not shown). In addition, mislocalization of the mutated proteins or channel-gating defects could be the underlying pathogenic mechanisms leading to cCSNB. In total, five different mutations, predicted to lead to premature-termination codons in different locations of the protein, were identified. We assume that the corresponding mutant mRNAs of these alleles would probably be subjected to nonsense-mediated decay or produce a short nonfunctional form of TRPM1. Previous studies showed that a shorter, alternatively spliced N-terminal form of TRPM1 devoid of any putative transmembrane segments (amino acids ∼1–500) can directly interact and suppress the activity of the full-length form by preventing its translocation to the plasma membrane and thus inhibiting Ca2+ entry into the cell.14 It was suggested that under normal conditions, this mechanism regulates the exact amount of molecules necessary for proper channel function.
Figure 4.

Evolutionary Conservation of the Altered Amino Acid Residues in Other Orthologs
Multiple amino acid sequence alignments show evolutionary conservation of mutated residues (depicted in green). Amino acid substitutions are highlighted in red. The position of the respective amino acids is shown in black numbers.
Figure 5.
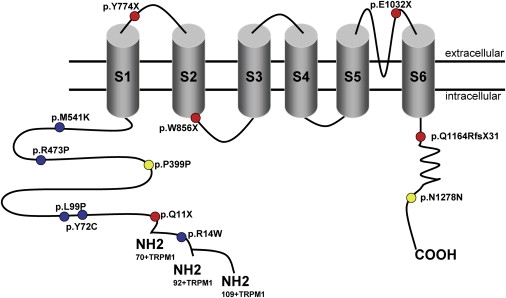
Localization of TRPM1 Mutations with Respect to Predicted Channel Domains
The specific domains for the TRPM1 channel were estimated by the use of different publications and prediction programs14,32 (UniProtKB-Swiss-Prot).
Currently, there are two genes implicated in complete CSNB: NYX and GRM6.15–18 They code for the proteins nyctalopin and mGluR6, respectively, which localize postsynaptically to the photoreceptors in the retina in ON bipolar cells.19 Whereas the function of nyctalopin is not yet understood, mGluR6 was shown to be important for the glutamate uptake released from the photoreceptors (Figure 6). The most obvious phenotypical feature of patients with cCSNB is a defect of the ON response, resulting in an electronegative combined rod-cone ERG, based on a severely reduced b-wave.2 In the dark, glutamate is released from photoreceptors, binds to mGluR6, and activates the Gαo1 subunit of a heterotrimeric G protein. This in turn leads by an unidentified mechanism to the closure of an as-yet-unknown cation channel (Figure 6).1,20–25 Upon light exposure, photoreceptor glutamate release decreases and the ON response is initiated with the shutting down of the G protein cascade. Subsequently, the cation channel opens, leading to ON bipolar cell depolarization, giving rise to the b-wave. Mutations in GRM6 lead to the loss of mGluR6 at the cell surface. Modulation of glutamate released from the photoreceptors cannot be correctly sensed by the bipolar cells, resulting in the failure of depolarization and thus a severely reduced b-wave.26 Recent findings in Appaloosa horses with CSNB and a specific coat patterning caused by low expression of a TRP channel, Trpm1, suggested that this specific channel is specifically linked to the depolarization of the ON bipolar cells during light exposure. However, no direct sequencing of the Trpm1 gene was performed in the horse, and thus the loss of ON bipolar cell function could rather be due to a secondary effect than to the mutated Trpm1. Nevertheless, mice lacking this channel showed the same ocular phenotype with a severely reduced b-wave.10 Together these findings indicated that mutations in this gene could be responsible for CSNB in patients, and indeed, our study presented herein revealed 14 different mutations in TRPM1 in ten different families with autosomal-recessive cCSNB.
Figure 6.
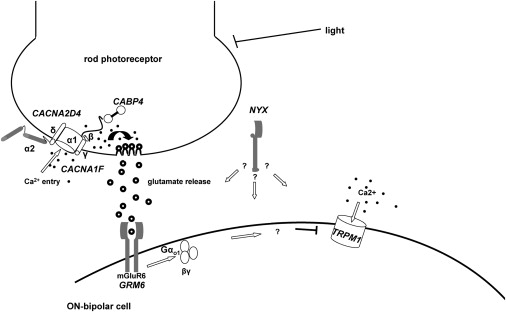
Schematic Drawing of Proteins Involved in Signal Transmission from Photoreceptors to Adjacent Bipolar Cells, the Disruption of Which Leads to CSNB
Arrows indicate the course of the signal transmission. In darkness, Ca2+ ions enter the rod photoreceptors, which results in glutamate release from the photoreceptors. Activated glutamate receptor activates Gαo1 (arrow), which then closes the TRPM1 channel by an unknown mechanism, indicated by a question mark, and thus ON bipolar cells are hyperpolarized. The exact role of NYX, encoding nyctalopin in this signal transduction cascade, remains to be solved in the future (indicated by a question mark).
Patients carrying mutations in TRPM1 reveal a similar ocular phenotype. All showed cCSNB with selective dysfunction of the ON bipolar pathway and OFF bipolar pathway preservation. Most of them revealed at least one of the following additional ocular abnormalities: myopia, nystagmus, or strabismus. These clinical observations are in accordance with the phenotype observed in the night-blind Appaloosa horses also showing nystagmus and strabismus. Although Bellone et al. showed downregulation of Trpm1 in these horses,9 it will be interesting to see which mutations in Trpm1 lead to its downregulation. Because of the fact that the ocular phenotype was similar in all patients and because of the presence of a large fraction of nonsense and splice-site mutations, we hypothesize that this form of autosomal-recessive cCSNB is due to a lack of TRPM1 mRNA or functional TRPM1 protein on the surface, rather than to functional alterations in the biophysical properties of this channel. The study of animal models carrying the identified mutations and investigation of transcript in the retina are needed for verification of this hypothesis.
To date, three genes have been associated with icCSNB (CACNA1F, CABP4, and CACNA2D4),27–30 and with the findings now reported here, there are also three genes associated with cCSNB (NYX, GRM6, and TRPM1)15–18 (Figure 7). With respect to the other autosomal-recessive CSNB genes identified so far, TRPM1 seems to be the most frequently mutated gene.
Figure 7.
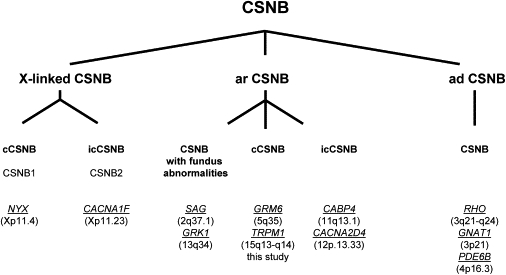
Genes Underlying CSNB
Different forms of human CSNB are classified according to their mode of inheritance, phenotype, and mutated genes. Abbreviations are as follows: cCSNB, complete CSNB; icCSNB, incomplete CSNB; ar, autosomal recessive; ad, autosomal dominant. Genes are indicated in italics and underlined. Chromosomal location is given between brackets. The phenotype of patients with mutations in icCSNB is more variable and can even lead to progressive cone or cone-rod dystrophy.1
Acknowledgments
The authors are grateful to the families described in this study, to Dominique Santiard-Baron and Christine Chaumeil for their help in DNA collection, and to the clinical staff. They thank Anne Friedrich, as well, for investigation of possibilities for the modeling of TRPM1 on a three-dimensional basis. The project was financially supported by Agence Nationale de la Recherche (to S.S.B), Foundation Voir et Entendre and BQR (Bonus Qualité Recherche), Université Pierre et Marie Curie6 (to C.Z.), Foundation Fighting Blindness (FFB) (to I.A., grant no. CD-CL-0808-0466-CHNO; and the CIC503 recognized as an FFB center, grant no. C-CMM-0907-0428-INSERM04), EU FP7, Integrated Project “EVI-GENORET” (LSHG-CT-2005-512036), the Swiss National Science Foundation (to F.L.M. and D.F.S., grant no. 320030_127558), Research Foundation Flanders (FWO) (to B.P.L., grant no. G.0043.06N), and the Deutsche Forschungsgemeinschaft (to S.K., B.W., and E.Z.; grant no. KFO134-KO2176/2-1 and KFO134-ZR1/17-2).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
ESEfinder, http://rulai.csh2.edu/tools/ESE
GenCards, PolyPhen (Polymorphism Phenotyping), http://tux.embl-heidelberg.de/ramensky/
National Center for Biotechnology Information (NCBI), http://ncbi.nlm.nih.gov/
Online Mendelian Inheritance in Man http (OMIM), http://ncbi.nlm.nih.gov//Omim/
SIFT (Sorting Intolerant From Tolerant), http://blocks.fhcrc.org/sift/SIFT.html
University of California-Santa Cruz (UCSC) Human Genome Browser http://genome.ucsc.edu/
UniProtKB-Swiss-Prot, http://www.uniprot.org
References
- 1.Zeitz C. Molecular genetics and protein function involved in nocturnal vision. Expert Rev Ophthalmol. 2007;2:467–485. [Google Scholar]
- 2.Schubert G., Bornschein H. Ophthalmologica. 1952;123:396–413. doi: 10.1159/000301211. [DOI] [PubMed] [Google Scholar]
- 3.Miyake Y., Yagasaki K., Horiguchi M., Kawase Y., Kanda T. Congenital stationary night blindness with negative electroretinogram. A new classification. Arch. Ophthalmol. 1986;104:1013–1020. doi: 10.1001/archopht.1986.01050190071042. [DOI] [PubMed] [Google Scholar]
- 4.Audo I., Robson A.G., Holder G.E., Moore A.T. The negative ERG: clinical phenotypes and disease mechanisms of inner retinal dysfunction. Surv. Ophthalmol. 2008;53:16–40. doi: 10.1016/j.survophthal.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 5.Zeitz C., Labs S., Lorenz B., Forster U., Ueksti J., Kroes H.Y., De Baere E., Leroy B.P., Cremers F.P., Wittmer M. Genotyping microarray for CSNB-associated genes. Invest Ophthalmol Vis Sci. 2009 doi: 10.1167/iovs.09-3548. Published online July 2, 2009. [DOI] [PubMed] [Google Scholar]
- 6.Sandmeyer L.S., Breaux C.B., Archer S., Grahn B.H. Clinical and electroretinographic characteristics of congenital stationary night blindness in the Appaloosa and the association with the leopard complex. Vet. Ophthalmol. 2007;10:368–375. doi: 10.1111/j.1463-5224.2007.00572.x. [DOI] [PubMed] [Google Scholar]
- 7.Witzel D.A., Smith E.L., Wilson R.D., Aguirre G.D. Congenital stationary night blindness: an animal model. Invest. Ophthalmol. Vis. Sci. 1978;17:788–795. [PubMed] [Google Scholar]
- 8.Sandmeyer L.S., Grahn B.H., Breaux C.B. Diagnostic ophthalmology. Congenital stationary night blindness (CSNB) Can Vet J. 2006;47:1131–1133. [PMC free article] [PubMed] [Google Scholar]
- 9.Bellone R.R., Brooks S.A., Sandmeyer L., Murphy B.A., Forsyth G., Archer S., Bailey E., Grahn B. Differential gene expression of TRPM1, the potential cause of congenital stationary night blindness and coat spotting patterns (LP) in the Appaloosa horse (Equus caballus) Genetics. 2008;179:1861–1870. doi: 10.1534/genetics.108.088807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shen Y., Heimel J.A., Kamermans M., Peachey N.S., Gregg R.G., Nawy S. A transient receptor potential-like channel mediates synaptic transmission in rod bipolar cells. J. Neurosci. 2009;29:6088–6093. doi: 10.1523/JNEUROSCI.0132-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clapham D.E., Runnels L.W., Strubing C. The TRP ion channel family. Nat. Rev. Neurosci. 2001;2:387–396. doi: 10.1038/35077544. [DOI] [PubMed] [Google Scholar]
- 12.Marmor M.F., Fulton A.B., Holder G.E., Miyake Y., Brigell M., Bach M. ISCEV Standard for full-field clinical electroretinography (2008 update) Doc. Ophthalmol. 2009;118:69–77. doi: 10.1007/s10633-008-9155-4. [DOI] [PubMed] [Google Scholar]
- 13.Oancea E., Vriens J., Brauchi S., Jun J., Splawski I., Clapham D.E. TRPM1 forms ion channels associated with melanin content in melanocytes. Sci Signal. 2009;2:ra21. doi: 10.1126/scisignal.2000146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu X.Z., Moebius F., Gill D.L., Montell C. Regulation of melastatin, a TRP-related protein, through interaction with a cytoplasmic isoform. Proc. Natl. Acad. Sci. USA. 2001;98:10692–10697. doi: 10.1073/pnas.191360198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zeitz C., van Genderen M., Neidhardt J., Luhmann U.F., Hoeben F., Forster U., Wycisk K., Matyas G., Hoyng C.B., Riemslag F. Mutations in GRM6 cause autosomal recessive congenital stationary night blindness with a distinctive scotopic 15-Hz flicker electroretinogram. Invest. Ophthalmol. Vis. Sci. 2005;46:4328–4335. doi: 10.1167/iovs.05-0526. [DOI] [PubMed] [Google Scholar]
- 16.Bech-Hansen N.T., Naylor M.J., Maybaum T.A., Sparkes R.L., Koop B., Birch D.G., Bergen A.A., Prinsen C.F., Polomeno R.C., Gal A. Mutations in NYX, encoding the leucine-rich proteoglycan nyctalopin, cause X-linked complete congenital stationary night blindness. Nat. Genet. 2000;26:319–323. doi: 10.1038/81619. [DOI] [PubMed] [Google Scholar]
- 17.Pusch C.M., Zeitz C., Brandau O., Pesch K., Achatz H., Feil S., Scharfe C., Maurer J., Jacobi F.K., Pinckers A. The complete form of X-linked congenital stationary night blindness is caused by mutations in a gene encoding a leucine-rich repeat protein. Nat. Genet. 2000;26:324–327. doi: 10.1038/81627. [DOI] [PubMed] [Google Scholar]
- 18.Dryja T.P., McGee T.L., Berson E.L., Fishman G.A., Sandberg M.A., Alexander K.R., Derlacki D.J., Rajagopalan A.S. Night blindness and abnormal cone electroretinogram ON responses in patients with mutations in the GRM6 gene encoding mGluR6. Proc. Natl. Acad. Sci. USA. 2005;102:4884–4889. doi: 10.1073/pnas.0501233102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morgans C.W., Ren G., Akileswaran L. Localization of nyctalopin in the mammalian retina. Eur. J. Neurosci. 2006;23:1163–1171. doi: 10.1111/j.1460-9568.2006.04647.x. [DOI] [PubMed] [Google Scholar]
- 20.Nomura A., Shigemoto R., Nakamura Y., Okamoto N., Mizuno N., Nakanishi S. Developmentally regulated postsynaptic localization of a metabotropic glutamate receptor in rat rod bipolar cells. Cell. 1994;77:361–369. doi: 10.1016/0092-8674(94)90151-1. [DOI] [PubMed] [Google Scholar]
- 21.Vardi N., Morigiwa K. ON cone bipolar cells in rat express the metabotropic receptor mGluR6. Vis. Neurosci. 1997;14:789–794. doi: 10.1017/s0952523800012736. [DOI] [PubMed] [Google Scholar]
- 22.Vardi N., Duvoisin R., Wu G., Sterling P. Localization of mGluR6 to dendrites of ON bipolar cells in primate retina. J. Comp. Neurol. 2000;423:402–412. doi: 10.1002/1096-9861(20000731)423:3<402::aid-cne4>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 23.Nawy S. The metabotropic receptor mGluR6 may signal through G(o), but not phosphodiesterase, in retinal bipolar cells. J. Neurosci. 1999;19:2938–2944. doi: 10.1523/JNEUROSCI.19-08-02938.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dhingra A., Lyubarsky A., Jiang M., Pugh E.N., Jr., Birnbaumer L., Sterling P., Vardi N. The light response of ON bipolar neurons requires G[alpha]o. J. Neurosci. 2000;20:9053–9058. doi: 10.1523/JNEUROSCI.20-24-09053.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dhingra A., Jiang M., Wang T.L., Lyubarsky A., Savchenko A., Bar-Yehuda T., Sterling P., Birnbaumer L., Vardi N. Light response of retinal ON bipolar cells requires a specific splice variant of Galpha(o) J. Neurosci. 2002;22:4878–4884. doi: 10.1523/JNEUROSCI.22-12-04878.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zeitz C., Forster U., Neidhardt J., Feil S., Kalin S., Leifert D., Flor P.J., Berger W. Night blindness-associated mutations in the ligand-binding, cysteine-rich, and intracellular domains of the metabotropic glutamate receptor 6 abolish protein trafficking. Hum. Mutat. 2007;28:771–780. doi: 10.1002/humu.20499. [DOI] [PubMed] [Google Scholar]
- 27.Bech-Hansen N.T., Naylor M.J., Maybaum T.A., Pearce W.G., Koop B., Fishman G.A., Mets M., Musarella M.A., Boycott K.M. Loss-of-function mutations in a calcium-channel alpha1-subunit gene in Xp11.23 cause incomplete X-linked congenital stationary night blindness. Nat. Genet. 1998;19:264–267. doi: 10.1038/947. [DOI] [PubMed] [Google Scholar]
- 28.Strom T.M., Nyakatura G., Apfelstedt-Sylla E., Hellebrand H., Lorenz B., Weber B.H., Wutz K., Gutwillinger N., Ruther K., Drescher B. An L-type calcium-channel gene mutated in incomplete X-linked congenital stationary night blindness. Nat. Genet. 1998;19:260–263. doi: 10.1038/940. [DOI] [PubMed] [Google Scholar]
- 29.Zeitz C., Kloeckener-Gruissem B., Forster U., Gebhart M., Magyar I., Mátyás G., Striessnig J., Berger B. Mutations in the calcium-binding protein 4 (CABP4) cause autosomal recessive night blindness. Invest. Ophthalmol. Vis. Sci. 2007;49 E-Abstract 6085. [Google Scholar]
- 30.Wycisk K.A., Zeitz C., Feil S., Wittmer M., Forster U., Neidhardt J., Wissinger B., Zrenner E., Wilke R., Kohl S. Mutation in the auxiliary calcium-channel subunit CACNA2D4 causes autosomal recessive cone dystrophy. Am. J. Hum. Genet. 2006;79:973–977. doi: 10.1086/508944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miyake Y., Horiguchi M., Terasaki H., Kondo M. Scotopic threshold response in complete and incomplete types of congenital stationary night blindness. Invest. Ophthalmol. Vis. Sci. 1994;35:3770–3775. [PubMed] [Google Scholar]
- 32.Fang D., Setaluri V. Expression and Up-regulation of alternatively spliced transcripts of melastatin, a melanoma metastasis-related gene, in human melanoma cells. Biochem. Biophys. Res. Commun. 2000;279:53–61. doi: 10.1006/bbrc.2000.3894. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.