

Abstract
Blood lipids are important cardiovascular disease (CVD) risk factors with both genetic and environmental determinants. The Whitehall II study (n = 5592) was genotyped with the gene-centric HumanCVD BeadChip (Illumina). We identified 195 SNPs in 16 genes/regions associated with 3 major lipid fractions and 2 apolipoprotein components at p < 10−5, with the associations being broadly concordant with prior genome-wide analysis. SNPs associated with LDL cholesterol and apolipoprotein B were located in LDLR, PCSK9, APOB, CELSR2, HMGCR, CETP, the TOMM40-APOE-C1-C2-C4 cluster, and the APOA5-A4-C3-A1 cluster; SNPs associated with HDL cholesterol and apolipoprotein AI were in CETP, LPL, LIPC, APOA5-A4-C3-A1, and ABCA1; and SNPs associated with triglycerides in GCKR, BAZ1B, MLXIPL, LPL, and APOA5-A4-C3-A1. For 48 SNPs in previously unreported loci that were significant at p < 10−4 in Whitehall II, in silico analysis including the British Women's Heart and Health Study, BRIGHT, ASCOT, and NORDIL studies (total n > 12,500) revealed previously unreported associations of SH2B3 (p < 2.2 × 10−6), BMPR2 (p < 2.3 × 10−7), BCL3/PVRL2 (flanking APOE; p < 4.4 × 10−8), and SMARCA4 (flanking LDLR; p < 2.5 × 10−7) with LDL cholesterol. Common alleles in these genes explained 6.1%–14.7% of the variance in the five lipid-related traits, and individuals at opposite tails of the additive allele score exhibited substantial differences in trait levels (e.g., >1 mmol/L in LDL cholesterol [∼1 SD of the trait distribution]). These data suggest that multiple common alleles of small effect can make important contributions to individual differences in blood lipids potentially relevant to the assessment of CVD risk. These genes provide further insights into lipid metabolism and the likely effects of modifying the encoded targets therapeutically.
Introduction
LDL cholesterol (LDL-C), HDL cholesterol (HDL-C), and triglycerides (TG) are correlated phenotypes inherited as complex traits. Their measurements are used clinically to evaluate risk of future cardiovascular disease (CVD)3,4 and to guide prescription of statin drugs for primary prevention. Whereas LDL-C and apolipoprotein (apo) B, which are lowered by statins, have been shown to play a causal role in atherosclerosis,5 the causal relevance of the other lipid-related traits is currently uncertain. ApoAI is the major protein moiety of HDL-C, and more recently it has been suggested that the ratio of apoB/apoAI, representing the balance between proatherogenic apoB-containing particles and the antiatherogenic apoAI-containing particles, is among the strongest predictor of CVD risk.6
To date, genome-wide association studies (GWAS) have been successful in identifying 22 genes/regions associated with blood lipids, although they contribute only 3%–5% of the total variance of LDL-C, HDL-C, and TG,7 but less information is available on the genetic determinants of apoB and apoAI. Many of these GWAS discoveries reconfirm previously validated lipid-related genes, though substantial new insight into pathways relevant to lipid metabolism has been provided by identification of hitherto unsuspected genes.7 Intervals of association discovered in GWAS studies have often been large and may contain more than one annotated gene, and so the identity, mechanism, and number of causal sites remains uncertain for the most part. SNP arrays utilized for GWAS have been designed primarily for broad coverage of the genome, and are therefore not ideal tools for identification of causal variants or resolution of independent signals within these intervals of association. Studies with dense SNP coverage may uncover additional independently associated variants at known loci, and also at previously unknown loci not well covered by GWAS studies, both of which may contribute to some of the remaining unexplained variance.
For many genes identified by whole-genome analysis, such as the association of CELSR2 (MIM 604265) locus with LDL-C,8,9 replication efforts have rightly focused on a subset of the most strongly associating SNPs, rather than on capturing the full genetic architecture of the region. It has been proposed that a gene-centric SNP array that includes SNP content from many of the genes or regions identified by GWAS, as well as additional candidate genes, could help refine association signals in a gene or region to help delineate independently associated variants that better mark or represent causal sites.
The Human CVD Beadchip (Cardiochip) was developed in part for this purpose, and a small multiethnic study examining the association of lipid traits with the same array has been published recently, but reported only four gene-lipid trait associations.10 We have used this gene-centric SNP array (incorporating content from GWAS and candidate gene studies, animal models of hyperlipidaemia and atherosclerosis, and prior biological knowledge)11 in a large prospective observational study of middle-aged civil servants (Whitehall II; WH-II).12
The design of the Cardiochip included three levels of coverage. Group 1 loci (genes and regions with a high likelihood of functional significance, including established mediators of vascular disease, loci derived from GWAS, and those shown to be associated with phenotypes of interest) had average coverage of 36 SNPs per locus, a higher density of coverage than for the SNP arrays used for the majority of GWAS reported to date. Group 2 loci (defined as candidate loci that are potentially involved in phenotypes of interest or established loci that required very large numbers of tagging SNPs) had average coverage of 16.3 SNPs/locus, similar to that of the most widely used whole-genome SNP arrays, whereas Group 3 loci comprised rarer nonsynonymous SNPs and those with established functionality with MAF > 0.01.11
Our aim was to identify genes and regions associated with LDL-C and HDL-C and TG, and we extend these associations to their protein components, apoB and apoAI, in order to assess the extent to which associations may overlap across traits, and to identify a subset of independently associated variants at each region. To validate previously unreported associations, we undertook a pooled in silico analysis in the British Women's Heart and Health Study (BWHHS),13 the British Genetics of Hypertension (BRIGHT)14 study, the UK subgroup from the Anglo-Scandinavian Cardiac Outcomes Trial (ASCOT),1 and the NORDIL Study,2 and we performed further validation in the Northwick Park Heart Study II (NPHSII).15
Material and Methods
Whitehall II Study
The Whitehall II study (WH-II) recruited 10,308 participants (70% men) between 1985 and 1989 from 20 London-based Civil service departments.16 Blood samples for DNA were collected in 2002–2004 from more than 6000 participants. Total cholesterol, HDL-C, and TG were measured within 72 hr in serum stored at 4°C via standard enzymatic colorimetric methods in the same laboratories with identical methodology for all subjects. LDL-C concentration was calculated with the Friedewald formula.17 ApoAI and apoB levels were determined by immunoturbidimetry.18 Details of the design, participants, lipid measurements, and genotyping of WH-II are presented in Table S1 (available online) together with summary details of the four other studies contributing to the replication phase of the analysis. The study was approved by the UCL Research Ethics Committee, and participants gave informed consent to each aspect of the study.
Genotyping and Quality Control
DNA from WH-II was extracted from 6156 individuals from whole blood samples (via magnetic bead technology; Medical Solutions, Nottingham, UK) and normalized to a concentration of 50 ng/μl. We used custom SNP arrays designed by the Institute of Translational Medicine and Therapeutics, the Broad Institute, and the National Heart Lung and Blood Institute supported Candidate-gene Association Resource Consortium (HumanCVD BeadChip; Illumina)11 on 5592 of these samples (this included 115 samples that were run as hidden duplicates). The initial genotype calls were generated with the cluster file provided by Illumina Beadstudio software. Call frequencies were below 98% for 1,798 out of the 49,094 SNPs. These were reviewed by eye and reclustered manually. SNPs that remained below 98% after reclustering were discarded.113 duplicate pairs had a concordance rate >99.5%, 1 pair had a concordance rate of 99.1%, and 1 pair had a concordance rate of 95%. Manual reclustering of some SNPs resulted in duplicate concordance rates of 100%. 5557 samples passed the prespecified sample call rate threshold of 80%. Removing 115 duplicates and 1 sample of ambiguous identity left 5441 samples of which 5067 were whites. Eight outliers from the principal component analysis were removed based on the genome-wide identity-by-state analysis implemented in PLINK (see Figure S1), leaving 5059 samples for further analysis. 48,032 SNPs with call rates greater than 0.98 were included in the final analysis marker set in 5059 individuals of white ancestry. The genomic inflation factors for all the analyses reported were close to 1, indicating negligible influence from population structure or genotyping errors.
Summary Demographics
DNA extraction methods, genotyping, and QC filters for the other participating studies are detailed in Table S1. Forty-eight SNPs identified in WH-II were exchanged with the prospective BWHHS, and three studies of hypertensive cohorts, BRIGHT, ASCOT, and NORDIL. These cohorts provided in silico results with the same analysis plan as performed in WH-II. Two of these SNPs (rs3184504 and rs1049817) were directly genotyped in NPHSII (n = 3052). All results were pooled by a fixed effect meta-analysis.
Statistical Analyses
Significance
We applied an additive genetic model in PLINK19 and used the corresponding p values to construct quantile-quantile and Manhattan plots. We adopted a conservative p value of 1 × 10−5 for declaration of significance. The significance threshold represented a trade-off between avoidance of false positive associations while taking into account the likely higher prior odds of association because of the nature of the array. The Bonferroni correction for the 50,000 SNPs is 10−6 for 50,000 independent tests with no prior information. Our cut-off was set to a more relaxed 10−5 because the prior odds that some of the SNPs tested would be associated with the phenotypes tested was higher than for a whole-genome array. Under the global null of no true associations, 10−4 would give an expected number of false positives of 5 for each independent phenotype. However, SNPs crossing a lower p value threshold of 1 × 10−4 were further analyzed in silico in BWHHS, BRIGHT, ASCOT, and NORDIL and the results pooled by a fixed effect meta-analysis of individual study beta coefficients, weighted by the inverse of the variance, via the metafor library in R. All results presented are from analyses adjusted for gender and age, the latter treated as a categorical variable with 5-year bands (39–44, 45–49, 50–54, 55–59, 60–65 in WH-II). Analyses were not adjusted for use of lipid-lowering drugs because the prevalence was generally low; of the 5059 individuals from WH-II, 39 (0.8%) were taking lipid-lowering medication at the time of lipid measurement. The proportion of subjects receiving lipid-lowering medication at the time of blood sampling varied between 0% and 12.6% in BWHHS, BRIGHT, ASCOT, and NORDIL (Table S1).
Independence of Association Signals
To assess the best genetic predictors at each locus, a variable selection algorithm was implemented as follows. SNPs were filtered according to whether they reached the p value cut-off of 10−5 in the univariate analyses and had a MAF > 0.001. The set of markers thus obtained was augmented for any missing genotype data with the software fastPHASE,20 and a stepwise selection scheme with the Akaike's Information Criterion (AIC)21 was implemented separately for each chromosome. The genetic model assumed an additive effect on the appropriate scale, adjusting for gender and age as in the univariate analyses.
Effect Size
We first assessed the proportion of the trait variance explained by SNPs both singly and in combination. The estimates for the proportion of the total trait variance explained (R2) were obtained by the regression of the SNP on the trait of interest without adjustment for covariates as well as for the covariates separately. To obtain the R2 for all loci showing significant association with the trait, we used SNPs selected from the stepwise regression (variable selection), and a linear model was fitted between age, gender, and the trait. The residuals of this model were then used to obtain the percentage of residual variance in the trait explained by all the SNPs characterizing a single gene or a gene cluster. We separately assessed the effect size as the beta coefficient of the linear regression of individual SNPs on trait level by using a per-allele model. Finally, to assess the joint effect of carriage of multiple independently associated SNPs, we developed a gene score and computed the odds of lying within the 10% tail of the risk factor distribution, based on the centile position in the frequency distribution of gene score values. The gene score, based on SNPs remaining in the model for the trait after variable selection, were recoded with the allele having the detrimental effect as 1 and the other as 0, and each individual was assigned a score of 0, 1, or 2 according to their genotype for each SNP. The simple unweighted version of the gene score was calculated by summing the scores across all relevant SNPs for each individual. The alternative approach of weighting each SNP score by the relevant beta coefficient yielded very similar findings (data not shown) and the simpler model is presented here.
Results
The anthropometric characteristics and lipid and lipoprotein profiles of the 5059 white men and women from WH-II after QC are presented in Table 1. A total of 195 SNPs with a MAF > 0.001 and Hardy-Weinberg p values > 0.0001 were found to be associated with one or more of LDL-C, HDL-C, TG, apoB, or apoAI at a prespecified significance level of p < 10−5. These SNPs encompassed 16 gene regions (Figure 1) and were robust to adjustment for age and gender (Table S2).
Table 1.
Lipid and Anthropometric Characteristics of the Whitehall-II Participants Taken at Phase 3
|
Trait |
Men n = 3720 |
Women n = 1338 |
||
|---|---|---|---|---|
| Mean | SD | Mean | SD | |
| Age (yrs) | 49.06 | 5.94 | 49.59 | 6.10 |
| BMI (kg/M2) | 25.03 | 3.08 | 25.31 | 4.70 |
| Hip (cm) | 96.88 | 6.14 | 97.03 | 9.53 |
| Waist (cm) | 87.15 | 9.16 | 74.41 | 11.59 |
| DBP (mmHg) | 80.72 | 8.92 | 76.14 | 9.31 |
| SBP (mmHg) | 121.52 | 12.77 | 116.64 | 13.65 |
| Chol (mmol/l) | 6.45 | 1.11 | 6.43 | 1.17 |
| LDL-C (mmol/l) | 4.43 | 0.99 | 4.199 | 1.07 |
| ApoB (g/L) | 1.30 | 0.29 | 1.19 | 0.30 |
| TG (mmol/l) | 1.55 | 1.20 | 1.13 | 0.69 |
| HDL-C (mmol/l) | 1.33 | 0.35 | 1.72 | 0.43 |
| ApoA1 (g/L) | 2.06 | 0.32 | 2.35 | 0.38 |
Abbreviations: SD, standard deviation; BMI, body mass index; DBP, diastolic blood pressure; SBP, systolic blood pressure; chol, cholesterol; LDL-C, low-density lipoprotein cholesterol; ApoB, apolipoprotein B; TG, triglycerides; HDL-C, high-density lipoprotein cholesterol; ApoAI, apolipoprotein AI.
Figure 1.
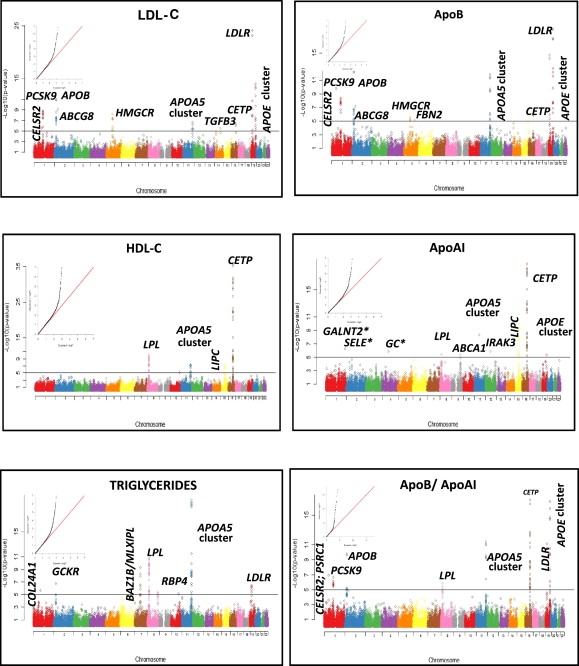
Manhattan Plots Showing the Associations of 48,032 SNPs by Chromosome for the 5 Lipid and Apolipoprotein Phenotypes versus −log10 p Value
The horizontal line indicates a p value threshold of 10−5. The Quantile/Quantile plots for the test statistics of the observed association p values plotted as a function of the expected SNP; association p values are inset for each trait. ∗SNPs with MAF < 0.001 but with p < 10−5.
Validation of Signals at Previously Reported Loci for Blood Lipids and Effects on ApoB and ApoAI
We identified 60 SNPs in 12 genes/clusters associated with LDL-C (Table S3A) and 53 SNPs in 9 genes/clusters associated with apoB (Table S3B). These included multiple strong associations for LDL-C and apoB, respectively, of SNPs in LDLR (MIM 606945) (rs17248720, p = 7.9 × 10−25 and p = 2.1 × 10−15), PCSK9 (MIM 607786) (rs11591147, p = 9.0 × 10−12 and p = 2.0 × 10−10), and APOB (MIM 107730) (rs563290, p = 1.3 × 10−11 and p = 7.0 × 10−13). We also identified multiple signals of association in a region of chromosome 19 that includes the TOMM40 (MIM 608061)-APOE (MIM 107741)-C1 (MIM 107710)-C2 (MIM 608083)-C4 (MIM 600745) gene cluster, and which, because of the extensive tagging in the Cardiochip, extended nearly 60 kb upstream to include SNPs in BCL3 (MIM 109560) and PVRL2 (MIM 600798) (Table S6). In addition, SNPs in LD with previously reported LDL-C-related SNPs in CELSR2 (rs12740374 and rs629301),22 HMGCR (MIM 142910) (rs12916),9,22 and CETP (MIM 118470) (rs3764261)23,24 also exhibited significant associations with both LDL-C and apoB. Highly significant associations with apoB (but not LDL-C) were observed for SNPs within the APOA5 (MIM 606368) cluster on chromosome 11, e.g., rs180327 (p = 8 × 10−7), rs2072560 (p = 1 × 10−12), and rs9804646 (p = 1 × 10−6). We also validated the association of ABCG8 with LDL-C (rs4299376; p = 9 × 10−10). This SNP is in complete LD (r2 = 1) with the previously reported ABCG8 (MIM 605460) rs654471322 and our results extend the association of ABCG8 to apoB (p = 8 × 10−8).
A total of 73 SNPs in 10 genes or clusters (Table S3C) were associated with TG levels, including SNPs in GCKR (MIM 600842) (rs1260326, p = 2 × 10−7),22,24,25 BAZ1B (MIM 605681)/MLXIPL (MIM 605678) (rs17145713, p = 6 × 10−5),26 and LPL (MIM 609708) (rs285, rs331, and rs3289; ranging from p = 3 × 10−6 to 2 × 10−11). There were also highly significant associations with the chr 11 APOA5 cluster (e.g., r651821, p = 9 × 10−21).
Five genes/regions were associated with both HDL-C (71 SNPs at p < 10−5) and apoAI (49 SNPs at p < 10−5) (Tables S3D and S3E). SNPs in CETP exhibited the most significant associations, including rs3764261 (p = 8 × 10−36) and rs17231506 (p = 1.4 × 10−19) for HDL-C and apoAI, respectively, as did SNPs in LPL (e.g., rs301, p = 9.0 × 10−11 and 4 × 10−6, respectively), as well as several SNPs in LIPC (MIM 151670) (e.g., rs261342, p = 6 × 10−8 and 3 × 10−11, respectively). The strongest determinants of apoB and apoAI, individually, were also the determinants of the apoB/apoAI ratio, analyzed as a separate phenotype.
New Association Signals
To conduct a sweep for previously unreported genes/regions, we undertook an in silico analysis of 48 SNPs associated with LDL-C, HDL-C, or TG with p values of ≤10−4 in WH-II in the BWHHS, BRIGHT, ASCOT, and NORDIL studies and pooled the effect estimates by meta-analysis. For two of these SNPs, rs3184504 and rs1049817, we also tested for replication in NPHSII. Ten SNPs including rs12467409 (BMPR2 [MIM 600799], chr 2), rs3184504 (SH2B3 [MIM 605093], chr 12), seven SNPs in BCL3/PVRL2 on chr 19 upstream of APOE/TOMM40, and rs1529729 (SMARCA4 [MIM 603254], upstream of LDLR, chr 19) were associated with LDL-C and yielded meta-analysis p value < 10−5 (Figure 2). Two SNPs associated with TG levels, rs2314028 in GMIP (MIM 609694) and a GTF3C2 (MIM 604883) rs1049817, yielded p values > 10−5 in meta-analyses.
Figure 2.
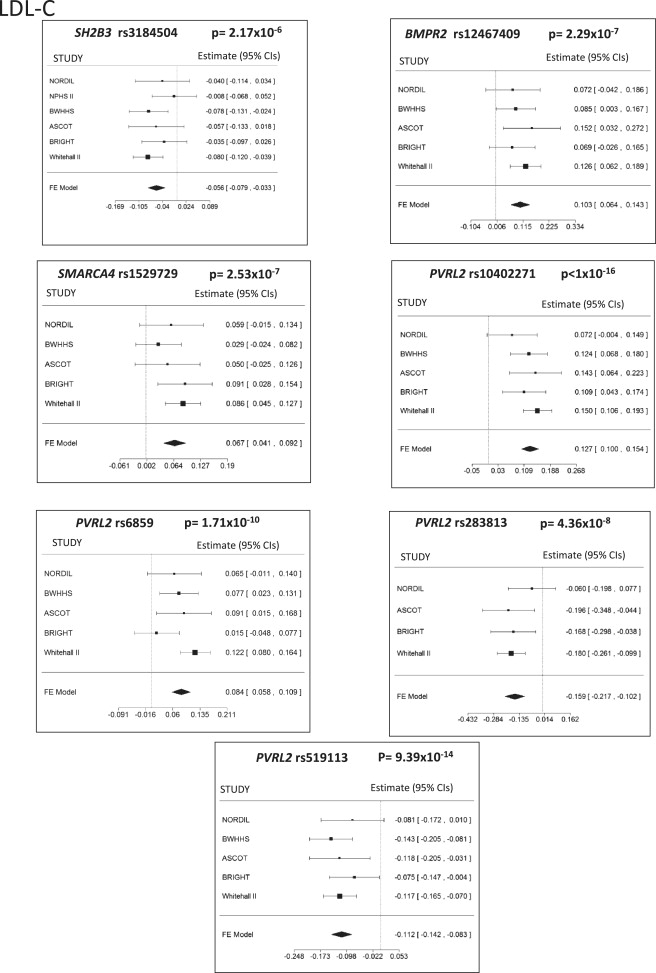
Forest Plots for Fixed Effects Meta-analyses for Previously Unreported Association Signals
Forest plots for fixed effects meta-analyses of beta-coefficients associated with LDL-C.
Refinement of Association Signals via Variable Selection
In the presence of multiple signals of association in regions of LD, we undertook a stepwise regression analysis by using the AIC to identify independently predictive SNPs for each associated gene or region (Figures 3–5; Figures S3A and S3B; and Table S6).
Figure 3.
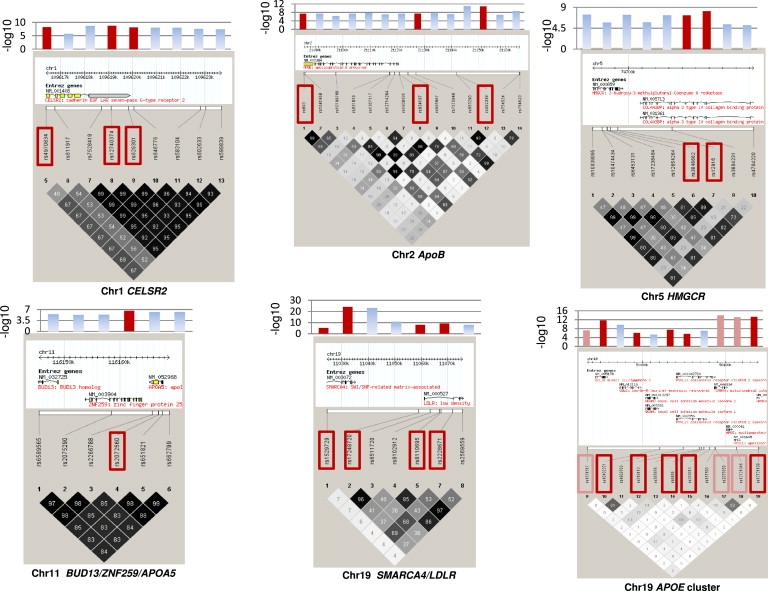
Haploview Plots of SNPs Showing Significant Associations with LDL-C before and after Variable Selection in Whitehall II
Haploview plots for LDL-C-associated SNPs significant by univariate analysis (at p < 10−5) and those retained after variable selection (red boxes). Bar charts show the −log10 p value for each SNP from the univariate analysis, with those in red corresponding to the values for SNPs retained after variable selection. For LDL-C and the chr 19 APOE cluster, orange boxes designate SNPs that were not retained after inclusion in the model of the APOE E2, E3, E4 variants that had been previously genotyped in WH-II.26
Figure 4.
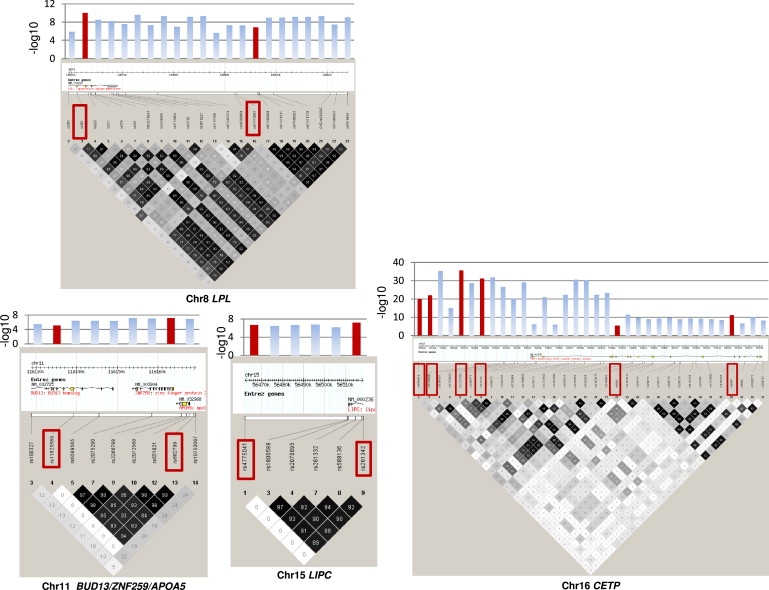
Haploview Plots of SNPs Showing Significant Associations with HDL-C before and after Variable Selection in Whitehall II
Haploview plots for HDL-C-associated SNPs significant by univariate analysis (at p < 10−5) and those retained after variable selection (red boxes). Bar charts show the −log10 p value for each SNP from the univariate analysis, with those in red corresponding to the values for SNPs retained after variable selection.
Figure 5.
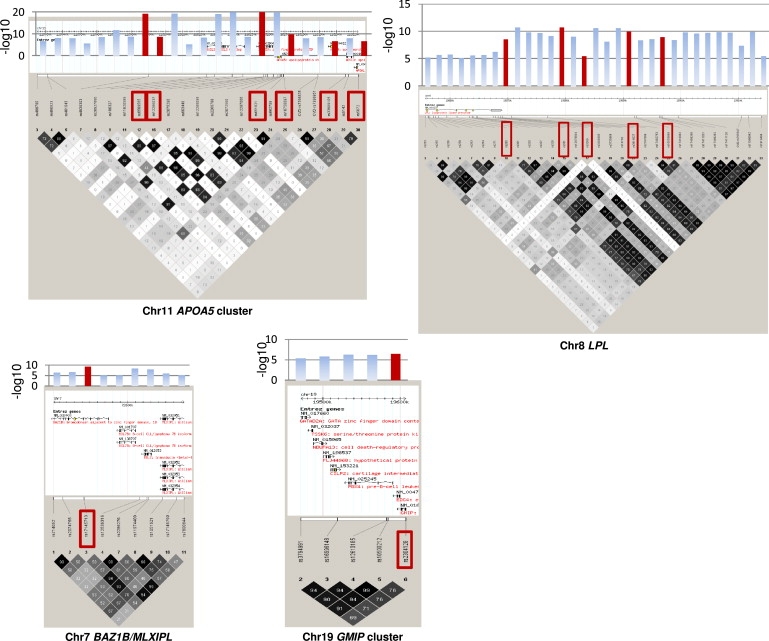
Haploview Plots of SNPs Showing Significant Associations with Triglycerides before and after Variable Selection in Whitehall II
Haploview plots for triglyceride-associated the SNPs significant by univariate analysis (at p < 10−5) and those retained after variable selection (red boxes). Bar charts show the −log10 p value for each SNP from the univariate analysis, with those in red corresponding to the values for SNPs retained after variable selection.
We detected additional predictors of association in the region of chr 19 that includes SNPs in BCL3, PVRL2, TOMM40, and APOE-C1-C2-C4, even after including the established functional cSNPs in APOE (rs429358 and rs7412) in the model (see Table S6 column “stepAIC”). Four of the PVRL2 SNPs exhibiting association with LDL-C in the meta-analyses were retained as independent predictors, as was BMPR2 rs12467409 on chr 2 (Figure 2). However, SMARCA4 rs1529729 (associated with LDL-C) located in close proximity to the LDLR on chr 19 and GTF3C2 rs1049817 (associated with TG), located close to GCKR, were not retained after variable selection.
Spectrum of Effects of Associating SNPs with Multiple Lipid and Lipoprotein Traits
The lipid-related traits we studied are intercorrelated (Figure S2), but the extent of overlap of the genetic associations across these phenotypes has not been extensively addressed. We summarize by means of a heat plot the spectrum of associations of the 195 SNPs that passed the p value threshold of 10−5 (Figure 6). Several distinct profiles of association emerged. For SNPs in PCSK9, CELSR2, APOB, LDLR, HMGCR, and the APOE gene cluster, the predominant associations were with both LDL-C and apoB but not with TG, HDL-C, or apoAI. By contrast, SNPs in CETP and LIPC were predominantly associated with HDL-C and apoAI but less so or not at all with LDL-C, apoB, or TG, whereas SNPs in LPL and APOA5 were associated most strongly with TG and HDL-C.
Figure 6.
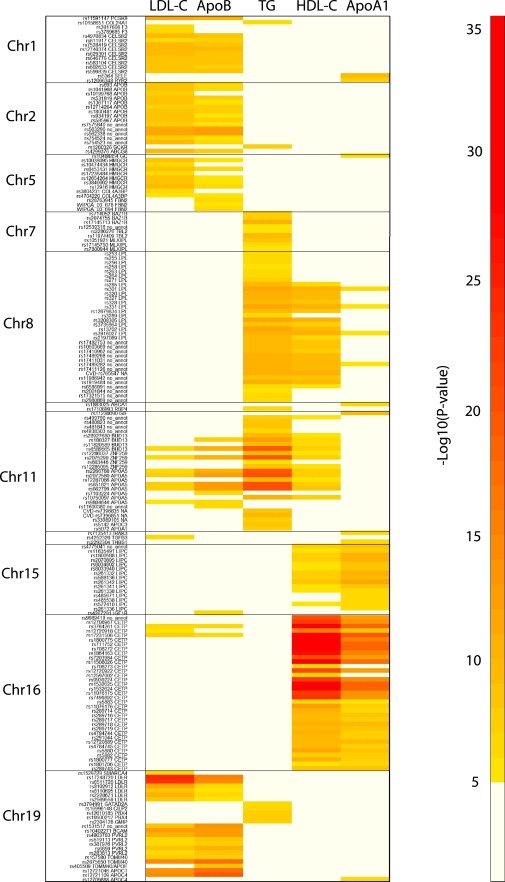
Heat Plot of the Associations of the 195 Significant SNPs across All the Lipid and Lipoprotein Variables Analyzed
Effect Size
The proportion of the variance explained by each single SNPs was typically <5%. However, the overall variance jointly explained by the common associating alleles was 14.6% for LDL-C, 14.7% for apoB, 9.5% for HDL-C, 6.1% for apoAI, and 7.8% for TG, which, in most cases, was greater than the variance explained by age and gender (Figures 7A–7C). When assessed in this way, SNPs in the APOE gene cluster, LDL-R, and APOB emerged as the major contributors to variance in both LDL cholesterol and apoB, and the CETP and LIPC genes as the major contributors to both HDL cholesterol and apoAI. SNPs in APOA5, LIPC, and BAZ1B/MLXIPL were the major genetic influences on TG levels.
Figure 7.
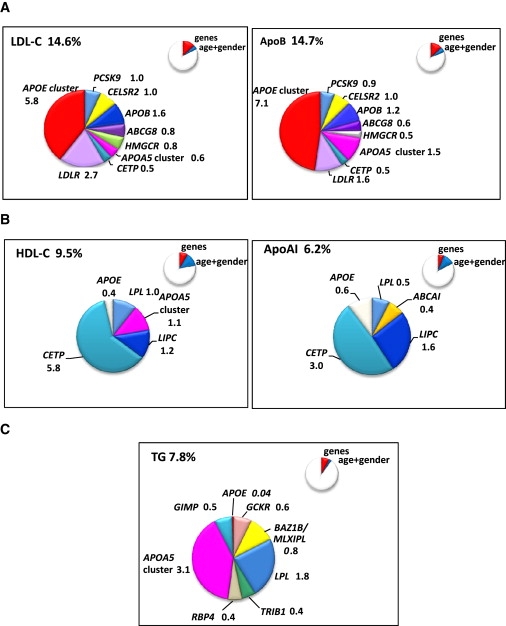
Pie Charts Showing the Contribution of SNPs to the Genetic Variance of Lipid, Lipoprotein, and Apolipoprotein Traits
Contribution of SNPs retained after variable selection to the genetic variance of the (A) LDL-C and ApoB, (B) HDL-C and ApoAI, and (C) triglycerides. Inset in each figure is shown as a proportion of the total variance of the trait compared to the variance of the trait explained by age and gender.
Assessment of effect size, based solely on the proportion of variance explained, can underemphasize the effect of variants that have a substantial effect on a trait, but which are rare in a population.
For LDL-C, the SNP having the largest individual effect was a low-frequency cSNP (rs11591147; L46R) in PCSK9 (MAF 0.016)9 with rare allele homozygotes having LDL-C levels around 32% (1.4 mmol/l) lower than common allele homozygotes (Table S6). For the LDLR SNP with the strongest effect, rs1724820, there was a 0.59 mmol/l mean difference in LDL-C between homozygous individuals whereas for LDLR rs2228671 (C27C, an association previously identified in GWAS27), there was a 0.24 mmol/l mean difference between homozygous subjects. Overall, genetic effects on apoB were proportionately similar to those seen with LDL (Table S6).
Common CETP SNPs had the largest individual effects for HDL and apoAI (0.1–0.2 mmol/l and 0.07–0.17 g/l, respectively, in comparison to homozygous subjects). Similar effect sizes were seen for SNPs in the APOA5 cluster with HDL-C, with rs651821 having the largest TG-raising effect of 0.5 mmol/l, reflecting a 20% difference in levels in rare versus common homozygotes. In this study, GCKR rs1260326 (P446L)22,24,25 was associated with a TG-raising effect of approximately 6% and the chr 7 locus BAZ1B/MLXIPL26 SNP rs17145713 was associated with an 8% TG-lowering effect.
We derived trait-specific gene scores comprising 21 SNPs for LDL-C, 19 for apoB, 15 for HDL-C, 10 for apoAI, 16 for TG, and 18 for apoB/apoAI. The gene score distribution and effect on trait levels are shown in Figures 8A and 8B for LDL-C and TG and Table S5 and Figures S4A–S4D for HDL-C, apoAI, apoB, and apoB/apoAI. Individuals in the highest 5% of the distribution of the respective traits had a higher predicted LDL-C (by 1.1 mmol/l, equivalent to approximately 1.1 standard deviations), a higher predicted apoB concentration (by 0.3 g/l [0.21 standard deviations]), and a higher predicted TG (by 0.61 mmol/l [0.17 standard deviations]) than those in the lowest 5% of the distribution. The corresponding differences were 0.30 mmol/l (0.12 standard deviations) for HDL-C and 0.25 g/l (0.11 standard deviations) for apoAI. The odds ratio for occupancy of the extreme 10% of the trait distribution at different cut points of the respective gene score distributions are shown in Figures 8A and 8B and Figures S4A–S4D. For example, individuals at or above the 95th centile of the gene score distribution of LDL-C-raising SNPs had a 17-fold higher risk of occupying the top 10% of the LDL-C distribution, compared with individuals below the 95th centile.
Figure 8.
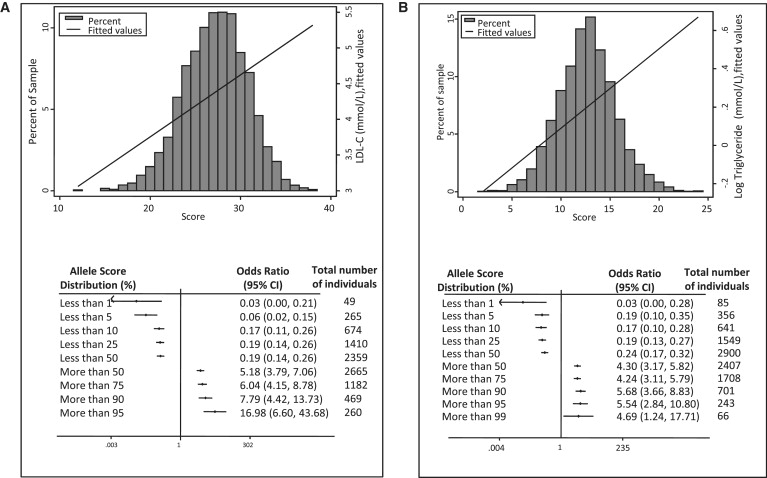
Frequency Distribution of the Gene Count Scores for LDL-C and Triglycerides
Frequency distribution of the gene count score for (A) LDL-C and (B) TG with the fitted line representing the effect of gene score on trait level, with increasing score. Below each gene score histogram is a plot of the odds ratio for being in either the top or bottom 10% of the trait distribution at different cut points of the respective gene score below. The medians (interquartile ranges) of the gene counts were 27 (25–30) for LDL-C and 13 (11–14) for TG.
Discussion
Loci Affecting LDL Cholesterol and ApoB
We identified an almost complete overlap of genes and regions influencing LDL-C and apoB concentrations, with similar influence on the proportion of the explained variance in the respective traits. The loci associated with LDL-C overlapped with those identified by prior GWAS. The locus having the greatest effect on LDL-C and apoB variance was the APOE cluster on chr 19, which had been identified to be associated with LDL-C in GWASs.22,23 Two SNPs, rs429358 and rs7412, together define the common APOE E2, E3, E4 genotypes, which are in essence haplotypes defined by the base changes altering residues 112 and 158 of the encoded protein, apoE. Although rs7412 is present on the chip, rs429358 failed the design and is not on the chip. Thus, with genotype data for only one SNP, these common APOE genotypes could not be determined. However, both SNPs had been typed previously in WH-II28 and when they were included in the regression model, three other SNPs in the APOE region were no longer retained as independent predictors. This suggests that the well-studied APOE variants that alter amino acid residues 112 and 158 of apoE, and which are known to affect receptor-binding and LDL-C catabolism (reviewed in 29), explain much of the effect.
The loci having the next largest effect are those encoding the LDL receptor and ApoB, proteins that play key roles in LDL-C catabolism, as the primary receptor, and the ligand for LDL clearance. Six loci have similar or smaller effects including PCSK9 and HMGCoA reductase (HMGCR), the rate-limiting enzyme in cholesterol biosynthesis, which is already the target for lipid-lowering statins. Variants in HMGCR were associated with effects on both LDL-C and apoB levels. The SNP, rs12916, which remained after variable selection, is in LD (r2 = 0.84) with, and likely to be marking, a recently identified functional variant, rs3846662, which in vitro resulted in 2.2-fold lower HMGCR expression and was associated with differences LDL-C levels.30 Concerning the SORT1/CELSR2/PSRC1 locus, data from liver expression studies in mice showed that Sort1 and Celsr2 were negatively correlated with LDL-C, in agreement with human findings, but Psrc1 expression was positively correlated to LDL-C.31 Further confirmation that SORT1/CELSR2 form a subnetwork comes from GO annotation that identifies them as being involved in the process of cell surface receptor-linked signal transduction, whereas PSRC1 is more likely to be involved in cell cycle pathways.31 A recent detailed study in more than 18,000 individuals of the SORT1 SNP, rs599839, confirmed its association with decreased LDL-C and lower risk of coronary heart disease and this was supported by expression array analysis and in vitro studies, demonstrating that the consequence of increased sortilin expression was to enhance LDL uptake into cells.32 Although not entirely excluding some cooperativity between SORT1 and CELSR2, this strongly suggests that SORT1 is involved in LDL-C determination.
All the above genes that showed association with LDL-C and, in the current study, with apoB levels have also been associated with coronary heart disease (CHD) risk.23 These findings further endorse the causal role of LDL-C in CHD and suggest additional therapeutic targets for LDL reduction.
Loci Affecting HDL-C and ApoAI
CETP, encoding cholesteryl ester transfer protein, exhibited the strongest associations with HDL-C and apoAI, confirming GWASs findings,22,23 with 6 of the 35 CETP SNPs being retained after variable selection, suggesting the existence of multiple functional SNPs in CETP. The intronic SNP rs708272 (TaqIB), not thought to be functional and the subject of many candidate gene studies,33 was strongly associated with both HDL-C and apoAI levels in univariate analyses, but was not retained after variable selection. SNP rs5880 (P390A), which is retained in the model, may affect CETP activity directly, as shown by the fact that it causes protein backbone strain.
Loci Affecting Triglycerides
The chromosome 11 gene cluster comprising BUD13/ZNF259/APOA5/APOA4/APOC3/APOA1 contained multiple SNPs associated with TG levels. The locus contributes 38% of the genetic variance of TG as well as explaining 11% of HDL-C, 4% of LDL-C, and 10% of apoB genetic variance. GCKR,9,24,25 BAZ1B/MLXIPL,34 and TRIB19,23 all recent GWAS hits, were replicated, and together contribute 22% of the explained genetic variance of TG.
However, several loci associated with these lipid traits that had been identified by GWAS studies did not reach our level of statistical significance and these are presented in Table S4.
Newly Identified Gene Regions
Of the 48 SNPs significant at ≤10−4 identified in WH-II, in regions not previously associated with these traits, SNPs from 4 regions retained association with LDL-C at ≤10−5 when examined by meta-analysis across the five studies and >12,500 individuals. The SNP rs3184504, a nonsynonymous variant in SH2B3 (R262W), previously shown to be associated with myocardial infarction and asthma risk, as well as with eosinophil count, type 1 diabetes (MIM 222100), celiac disease (MIM 212750), and more recently blood pressure,35–38 was associated with LDL-C in WH-II at p = 7 × 10−5, and at p = 2.17 × 10−6 in the pooled analysis of more than 15,000 subjects. It seems surprising this variant should not have been identified previously by pooled analyses of GWAS of LDL-C that have included >48,000 individuals in which this SNP was directly typed or imputed,22,24,27 because coverage of this region has been extensive on SNP platforms employed by prior GWAS (see Figure S7A). We observed evidence of a possible effect modification by gender, i.e., association in BWHHS (all women) and a null association in NPHS-II (all men). However, both the marginal association of this SNP with LDL-C, as well as the possibility of a gender interaction, will require further validation and replication. It cannot be excluded, at this point, that this is a chance finding, and it is essential that further studies are conducted to confirm or refute this association. Nevertheless, in the present analysis, the direction of effect and point estimates were consistent across studies. As outlined by Manolio,7 even in the three recent large GWAS for lipid genes,22,24,27 whereas 18 loci were common to all three studies, up to six loci were common to only two of the three studies, and between three and eight loci were unique to each study. Similarly, in the two recent GWAS meta-analyses for blood pressure loci,37,38 only three loci were common to both studies, while five additional loci were unique to each of the two meta-analyses. One explanation could be context-dependent effects that could contribute to the heterogeneity between studies. Alternatively, it could be an issue of power despite the large number of individuals genotyped.
SNPs in PVRL2 upstream of APOE, and in SMARCA4 upstream of LDLR, associated with LDL-C and retained in variable selection, also showed consistent association with LDL-C in the meta-analysis (Figure 2), suggesting a potential independent signal arising from these genes. The comparative coverage of these regions by GWAS and the Cardiochip is presented in Figures S5B and S5C. As with coverage of SH2B3, GWAS coverage of these loci were good so it is unclear why these regions have not been previously detected as affecting lipid traits.
The BMPR2 association with LDL-C was confirmed by the meta-analysis (Figure 2). This locus (see Table S5D for comparative coverage to GWAS chips) encodes bone morphogenetic protein receptor, type II. Mutations in this gene have been associated with primary pulmonary hypertension (MIM 178600),39 but links with LDL-C need further validation.
Patterns of Association across the Five Lipid, Lipoprotein, and Apolipoprotein Phenotypes
Although the panel of index SNPs were identified on the basis of association with a single lipid or lipoprotein phenotype, many SNPs were associated with more than one trait. One potential explanation for the joint signals of association could simply be the high degree of correlation between traits.40 Although this is a possibility for associations of a given SNP with LDL-C and its major apolipoprotein constituent apoB, and for HDL-C with apoAI, this seems an unlikely explanation for the joint association of some SNPs with several lipid traits. These distinct association patterns provide circumstantial evidence for distinct biological mechanisms that could be informative for the understanding of lipid and lipoprotein metabolism and on the likely consequences of modifying the target proteins pharmacologically.
Effect Size of Common Alleles
In the current analysis, independently associating SNPs at each locus jointly explained between 6% and 15% of the variance in these traits. It should be noted, however, that this will be an overestimate because of the potential bias introduced by selecting SNPs and estimating effect sizes within the same data set, so replication in an independent data set is required to validate this estimate. Additional variants (including common or rare SNPs and structural variants) or context-dependent genetic effects (e.g., gene-gene and gene-environment interactions) are likely to contribute in whole or part to the remaining unexplained variance. There is already a precedent for rare variants contributing to the variance in LDL41, HDL,42 and TG.43
However, variance is only one of a number of metrics of effect, and the gene score analysis provides evidence that carrying multiple common trait-modifying alleles can make an important contribution to the between-individual differences in trait levels. We found a >1.0 mmol/L difference in LDL-C between individuals at the 5% extremes of the distribution of the number of LDL-modifying alleles carried, an effect that is similar in size to that seen with the commonly used doses of statins in CHD prevention. This suggests that one in every 20 individuals in the population could be at substantially different risk of CHD by virtue of their complement of common LDL-modifying alleles, though this finding does not necessarily argue that typing of such panel of alleles would be useful in risk prediction. Moreover, the odds of being in the top 10% of the trait distribution increased sharply with the number of trait-modifying alleles carried, suggesting that occupancy of the upper ranks of the trait distribution of blood lipids is achieved in some individuals by carriage of a small number of rare alleles, e.g., in PCSK9 of large effect, and in others by carriage of a larger repertoire of common alleles of modest effect.
For the apoB/apoAI ratio, the odd ratio for individuals at or above the 95th centile of the gene score is 13-fold higher if they are in the top 10% of the apoB/apoAI than the rest. This is comparable to that for LDL-C and supports the argument that measurement of apoB and apoAI, expressed as the ratio defining pro- and antiatherogeneic particles, are as predictive and more reliable in their measurement, than LDL-C levels that are usually calculated.6
Relevance of the Current Findings for Future Research and Clinical Application
Whole-genome analysis has produced a surge in our understanding of the genetic contribution to interindividual differences in the blood lipids but we are still in the discovery phase and the challenge of identifying causal variants at each identified locus remains. Although denser genotyping at loci of interest can contribute to the fine mapping efforts, and our analysis indicates that multiple predictive SNPs (marking causal sites) might be a common feature of the genetic architecture of lipid phenotypes, additional efforts directed at resequencing individuals carrying the relevant haplotypes will be required.
The small individual contribution of common alleles to the variance in quantitative traits has led to concerns that the translational potential of recent GWAS findings may be limited. However, in the case of rarer SNPs that make a minor contribution to the proportion of the variance explained, the difference in trait levels per allele can be substantial. Moreover, as the gene score plots illustrate, differences in LDL cholesterol levels of a magnitude seen from the use of standard doses of statins5 can be observed in individuals at opposite extremes of a gene score distribution for common alleles.
Furthermore, because genotype is randomly allocated and invariant, genetic associations provide unique insight on causal pathways, and the findings can be considered as a type of natural randomized trial. It is notable, for example, that the effects of HMGCR and CETP SNPs identified here closely mimic the effects of HMGCR44 and CETP inhibitors45 on lipid levels seen in clinical trials (albeit with a smaller effect size), suggesting that genetic studies might be helpful in modeling the effects of potential lipid-lowering therapies during the process of drug development. Cholesterol lowering, whether by pharmacotherapeutic or lifestyle modification, has been suspected of increasing some adverse events, such as depression, with observational studies finding associations between low cholesterol and high depression.46 Genetic variants can be utilized in a Mendelian randomization framework47 and the availability of multiple independent related variants allows several of the key assumptions of this method to be interrogated. Taken together, these observations suggest that effect size per se is not the major indicator of potential translational utility of the recent genetic advances.
Appendix
BRIGHT Consortium Members and Their Affiliations
Toby Johnson,1 Stephen J. Newhouse,1 Mark Caulfield,1 Patricia B. Munroe,1 Philip J. Howard,1 Abiodun Onipinla,1 Anna Dominiczak,2 Nilesh J. Samani,3 Martin Farrall,4 John Connell,2 Morris Brown,5 Mark Lathrop6
1Clinical Pharmacology and Barts and the London Genome Centre, William Harvey Research Institute, Barts and the London School of Medicine, Queen Mary University of London, London, UK
2BHF Glasgow Cardiovascular Research Centre, Division of Cardiovascular and Medical Sciences, University of Glasgow, Western Infirmary, Glasgow, UK
3Department of Cardiovascular Sciences, University of Leicester, Glenfield Hospital, Leicester, UK
4Department of Cardiovascular Medicine, University of Oxford, Wellcome Trust Centre for Human Genetics, Oxford, UK
5Clinical Pharmacology and the Cambridge Institute of Medical Research, University of Cambridge, Addenbrooke's Hospital, Cambridge, UK
6Centre National de Genotypage, Evry, France
Supplemental Data
Supplemental Data include four figures and seven tables and can be found with this article online at http://www.cell.com/AJHG.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
SNPs3D, http://www.snps3d.org
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Acknowledgments
This work on WH-II was supported by the British Heart Foundation (BHF) PG/07/133/24260, RG/08/008, SP/07/007/23671, and a Senior Fellowship to A.D.H. (FS/2005/125). S.E.H., A.D., and N.J.S. are BHF Chairholders. M. Kumari's time on this manuscript was partially supported by the National Heart Lung and Blood Institute (NHLBI: HL36310). The WH-II study has been supported by grants from the Medical Research Council; British Heart Foundation; Health and Safety Executive; Department of Health; National Institute on Aging, NIH, US (AG13196); Agency for Health Care Policy Research (HS06516); and the John D. and Catherine T. MacArthur Foundation Research Networks on Successful Midlife Development and Socio-economic Status and Health. The British Women's Heart and Health Study (BWHHS) has been supported by funding from the BHF and the UK Department of Health. We thank all BWHHS participants and the BWHHS team for their contributions. HumanCVD BeadChip genotyping was funded by the BHF (PG/07/131/24254, PI T.R.G.). I.N.D. thanks the University of Bristol for set-up costs for Bristol Genetics Epidemiology Laboratory. Gemma Wiacek is thanked for sample handling. T.R.G. was a BHF Intermediate Fellow (FS/05/065/19497). The BRIGHT study is supported by the Medical Research Council of Great Britain (G9521010D) and the British Heart Foundation (PG/02/128). We would also like to thank Charles Mein, Richard Dobson, and the Barts Genome Centre staff. The BRIGHT study is extremely grateful to all the patients who participated in the study and the BRIGHT nursing team. A.D. is also supported by BHF grants (RG/07/005/23633 and SP/08/005/25115) and EU Ingenious HyperCare Consortium: Integrated Genomics, Clinical Research, and Care in Hypertension (LSHM-C7-2006-037093).
We thank all ASCOT trial participants, physicians, nurses, and practices in the participating countries for their important contribution to the study. In particular we thank Clare Muckian and David Toomey for their help in DNA extraction, storage, and handling. The ASCOT study and the collection of the ASCOT DNA repository were supported by Pfizer, New York, NY, USA. Funding for the ASCOT study was also provided by Servier Research Group, Paris, France, and Leo Laboratories, Copenhagen, Denmark. NPHSII was supported by MRC UK, US NIH (NHLBI 33014), and Du Pont Pharma, Wilmington, VA, USA. We thank Professor Thomas Hedner (Department of Clinical Pharmacology, Sahlgrenska Academy, Gotheburg, Sweden) and Professor Sverre Kjeldsen (Ullevaal University Hospital, University of Oslo, Oslo, Norway), who are investigators of the NORDIL study. Professor Kjeldsen is also an investigator of the ASCOT trial.
J.W. is an employee of GlaxoSmithKlein.
References
- 1.Sever P.S., Dahlof B., Poulter N.R., Wedel H., Beevers G., Caulfield M., Collins R., Kjeldsen S.E., McInnes G.T., Mehlsen J. Rationale, design, methods and baseline demography of participants of the Anglo-Scandinavian Cardiac Outcomes Trial. ASCOT investigators. J. Hypertens. 2001;19:1139–1147. doi: 10.1097/00004872-200106000-00020. [DOI] [PubMed] [Google Scholar]
- 2.Hansson L., Hedner T., Lund-Johansen P., Kjeldsen S.E., Lindholm L.H., Syvertsen J.O., Lanke J., de Faire U., Dahlof B., Karlberg B.E. Randomised trial of effects of calcium antagonists compared with diuretics and beta-blockers on cardiovascular morbidity and mortality in hypertension: The Nordic Diltiazem (NORDIL) study. Lancet. 2000;356:359–365. doi: 10.1016/s0140-6736(00)02526-5. [DOI] [PubMed] [Google Scholar]
- 3.Assmann G., Cullen P., Schulte H. Simple scoring scheme for calculating the risk of acute coronary events based on the 10-year follow-up of the prospective cardiovascular Munster (PROCAM) study. Circulation. 2002;105:310–315. doi: 10.1161/hc0302.102575. [DOI] [PubMed] [Google Scholar]
- 4.Wilson P.W., D'Agostino R.B., Levy D., Belanger A.M., Silbershatz H., Kannel W.B. Prediction of coronary heart disease using risk factor categories. Circulation. 1998;97:1837–1847. doi: 10.1161/01.cir.97.18.1837. [DOI] [PubMed] [Google Scholar]
- 5.Baigent C., Keech A., Kearney P.M., Blackwell L., Buck G., Pollicino C., Kirby A., Sourjina T., Peto R., Collins R. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366:1267–1278. doi: 10.1016/S0140-6736(05)67394-1. [DOI] [PubMed] [Google Scholar]
- 6.McQueen M.J., Hawken S., Wang X., Ounpuu S., Sniderman A., Probstfield J., Steyn K., Sanderson J.E., Hasani M., Volkova E. Lipids, lipoproteins, and apolipoproteins as risk markers of myocardial infarction in 52 countries (the INTERHEART study): A case-control study. Lancet. 2008;372:224–233. doi: 10.1016/S0140-6736(08)61076-4. [DOI] [PubMed] [Google Scholar]
- 7.Manolio T.A. Cohort studies and the genetics of complex disease. Nat. Genet. 2009;41:5–6. doi: 10.1038/ng0109-5. [DOI] [PubMed] [Google Scholar]
- 8.Sandhu M.S., Waterworth D.M., Debenham S.L., Wheeler E., Papadakis K., Zhao J.H., Song K., Yuan X., Johnson T., Ashford A. LDL-cholesterol concentrations: A genome -wide association study. Lancet. 2008;371:483–491. doi: 10.1016/S0140-6736(08)60208-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kathiresan S., Melander O., Guiducci C., Surti A., Burtt N.P., Rieder M.J., Cooper G.M., Roos C., Voight B.F., Havulinna A.S. Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat. Genet. 2008;40:189–197. doi: 10.1038/ng.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lanktree M.B., Anand S.S., Yusuf S., Hegele R.A., SHARE Investigators Replication of genetic associations with plasma lipoprotein traits in a multiethnic sample. J. Lipid Res. 2009;50:1487–1496. doi: 10.1194/jlr.P900008-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Keating B.J., Tischfield S., Murray S.S., Bhangale T., Price T.S., Glessner J.T., Galver L., Barrett J.C., Grant S.F., Farlow D.N. Concept, design and implementation of a cardiovascular gene-centric 50 k SNP array for large-scale genomic association studies. PLoS ONE. 2008;3:e3583. doi: 10.1371/journal.pone.0003583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marmot M.G., Smith G.D., Stansfeld S., Patel C., North F., Head J., White I., Brunner E., Feeney A. Health inequalities among British civil servants: The Whitehall II study. Lancet. 1991;337:1387–1393. doi: 10.1016/0140-6736(91)93068-k. [DOI] [PubMed] [Google Scholar]
- 13.Lawlor D.A., Bedford C., Taylor M., Ebrahim S. Geographical variation in cardiovascular disease, risk factors, and their control in older women. British Women's Heart and Health Study. J. Epidemiol. Community Health. 2003;57:134–140. doi: 10.1136/jech.57.2.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Caulfield M., Munroe P., Pembroke J., Samani N., Dominiczak A., Brown M., Benjamin N., Webster J., Ratcliffe P., O'Shea S. Genome-wide mapping of human loci for essential hypertension. Lancet. 2003;361:2118–2123. doi: 10.1016/S0140-6736(03)13722-1. [DOI] [PubMed] [Google Scholar]
- 15.Miller G.J., Bauer K.A., Barzegar S., Foley A.J., Mitchell J.P., Cooper J.A., Rosenberg R.D. The effects of quality and timing of venepuncture on markers of blood coagulation in healthy middle-aged men. Thromb. Haemost. 1995;73:82–86. [PubMed] [Google Scholar]
- 16.Marmot M., Brunner E. Cohort profile: The Whitehall II study. Int. J. Epidemiol. 2005;34:251–256. doi: 10.1093/ije/dyh372. [DOI] [PubMed] [Google Scholar]
- 17.Friedewald W.T., Levy R.I., Fredrickson D.S. Estimation of low density lipoprotein cholesterol without the use of the preparative ultracentrifuge. Clin. Chem. 1972;18:499–502. [PubMed] [Google Scholar]
- 18.Mount J.N., Kearney E.M., Rosseneu M., Slavin B.M. Immunoturbidimetric assays for serum apolipoproteins A1 and B using Cobas Biol. centrifugal analyser. J. Clin. Pathol. 1988;41:471–474. doi: 10.1136/jcp.41.4.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scheet P., Stephens M. A fast and flexible statistical model for large-scale population genotype data: applications to inferring missing genotypes and haplotypic phase. Am. J. Hum. Genet. 2006;78:629–644. doi: 10.1086/502802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Akaike H. A new look at the statistical model identification. IEEE Trans. Automat. Contr. 1974;19:716–723. [Google Scholar]
- 22.Kathiresan S., Willer C.J., Peloso G.M., Demissie S., Musunuru K., Schadt E.E., Kaplan L., Bennett D., Li Y., Tanaka T. Common variants at 30 loci contribute to polygenic dyslipidemia. Nat. Genet. 2009;41:56–65. doi: 10.1038/ng.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Willer C.J., Sanna S., Jackson A.U., Scuteri A., Bonnycastle L.L., Clarke R., Heath S.C., Timpson N.J., Najjar S.S., Stringham H.M. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat. Genet. 2008;40:161–169. doi: 10.1038/ng.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sabatti C., Service S.K., Hartikainen A.L., Pouta A., Ripatti S., Brodsky J., Jones C.G., Zaitlen N.A., Varilo T., Kaakinen M. Genome-wide association analysis of metabolic traits in a birth cohort from a founder population. Nat. Genet. 2009;41:35–46. doi: 10.1038/ng.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chasman D.I., Pare G., Zee R.Y.L., Parker A.N., Cook N.R., Buring J.E., Kwiatkowski D.J., Rose L.M., Smith J.D., Williams P.T. Genetic loci associated with plasma concentration of low-density lipoprotein cholesterol, high-density lipoprotein cholesterol, triglycerides, apolipoprotein A1, and apolipoprotein B among 6382 white women in genome-wide analysis with replication. Circ. Cardiovasc. Genet. 2008;1:21–30. doi: 10.1161/CIRCGENETICS.108.773168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kooner J.S., Chambers J.C., Aguilar-Salinas C.A., Hinds D.A., Hyde C.L., Warnes G.R., Gomez Perez F.J., Frazer K.A., Elliott P., Scott J. Genome-wide scan identifies variation in MLXIPL associated with plasma triglycerides. Nat. Genet. 2008;40:149–151. doi: 10.1038/ng.2007.61. [DOI] [PubMed] [Google Scholar]
- 27.Aulchenko Y.S., Ripatti S., Lindqvist I., Boomsma D., Heid I.M., Pramstaller P.P., Penninx B.W., Janssens A.C., Wilson J.F., Spector T. Loci influencing lipid levels and coronary heart disease risk in 16 European population cohorts. Nat. Genet. 2009;41:47–55. doi: 10.1038/ng.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Talmud P.J., Lewis S.J., Hawe E., Martin S., Acharya J., Marmot M.G., Humphries S.E., Brunner E.J. No APOEepsilon4 effect on coronary heart disease risk in a cohort with low smoking prevalence: the Whitehall II study. Atherosclerosis. 2004;177:105–112. doi: 10.1016/j.atherosclerosis.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 29.Mahley R.W., Weisgraber K.H., Huang Y. Apolipoprotein E: Structure determines function-from atherosclerosis to Alzheimer's disease to AIDS. J. Lipid Res. 2008;50:S183–S188. doi: 10.1194/jlr.R800069-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burkhardt R., Kenny E.E., Lowe J.K., Birkeland A., Josowitz R., Noel M., Salit J., Maller J.B., Pe'er I., Daly M.J. Common SNPs in HMGCR in micronesians and whites associated with LDL-cholesterol levels affect alternative splicing of exon13. Arterioscler. Thromb. Vasc. Biol. 2008;28:2078–2084. doi: 10.1161/ATVBAHA.108.172288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schadt E.E., Molony C., Chudin E., Hao K., Yang X., Lum P.Y., Kasarskis A., Zhang B., Wang S., Suver C. Mapping the genetic architecture of gene expression in human liver. PLoS Biol. 2008;6:e107. doi: 10.1371/journal.pbio.0060107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Linsel-Nitschke P., Heeren J., Aherrahrou Z., Bruse P., Gieger C., Illig T., Prokisch H., Heim K., Doering A., Peters A. Genetic variation at chromosome 1p13.3 affects sortilin mRNA expression, cellular LDL-uptake and serum LDL levels which translates to the risk of coronary artery disease. Atherosclerosis. 2009 doi: 10.1016/j.atherosclerosis.2009.06.034. in press. Published online July 8, 2009. [DOI] [PubMed] [Google Scholar]
- 33.Boekholdt S.M., Sacks F.M., Jukema J.W., Shepherd J., Freeman D.J., Mcmahon A.D., Cambien F., Nicaud V., de Grooth G.J., Talmud P.J. Cholesteryl ester transfer protein TaqIB variant, high-density lipoprotein cholesterol levels, cardiovascular risk, and efficacy of pravastatin treatment: individual patient meta-analysis of 13,677 subjects. Circulation. 2005;111:278–287. doi: 10.1161/01.CIR.0000153341.46271.40. [DOI] [PubMed] [Google Scholar]
- 34.Kooner J.S., Chambers J.C., Guilar-Salinas C.A., Hinds D.A., Hyde C.L., Warnes G.R., Gomez Perez F.J., Frazer K.A., Elliott P., Scott J. Genome-wide scan identifies variation in MLXIPL associated with plasma triglycerides. Nat. Genet. 2008;40:149–151. doi: 10.1038/ng.2007.61. [DOI] [PubMed] [Google Scholar]
- 35.Gudbjartsson D.F., Bjornsdottir U.S., Halapi E., Helgadottir A., Sulem P., Jonsdottir G.M., Thorleifsson G., Helgadottir H., Steinthorsdottir V., Stefansson H. Sequence variants affecting eosinophil numbers associate with asthma and myocardial infarction. Nat. Genet. 2009;41:342–347. doi: 10.1038/ng.323. [DOI] [PubMed] [Google Scholar]
- 36.Hunt K.A., Zhernakova A., Turner G., Heap G.A., Franke L., Bruinenberg M., Romanos J., Dinesen L.C., Ryan A.W., Panesar D. Newly identified genetic risk variants for celiac disease related to the immune response. Nat. Genet. 2008;40:395–402. doi: 10.1038/ng.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Newton-Cheh C., Johnson T., Gateva V., Tobin M.D., Bochud M., Coin L., Najjar S.S., Zhao J.H., Heath S.C., Eyheramendy S. Genome-wide association study identifies eight loci associated with blood pressure. Nat. Genet. 2009 doi: 10.1038/ng.361. in press. Published online May 10, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Levy D., Ehret G.B., Rice K., Verwoert G.C., Launer L.J., Dehghan A., Glazer N.L., Morrison A.C., Johnson A.D., Aspelund T. Genome-wide association study of blood pressure and hypertension. Nat. Genet. 2009 doi: 10.1038/ng.384. in press. Published online May 10, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lane K.B., Machado R.D., Pauciulo M.W., Thomson J.R., Phillips J.A., Loyd J.E., Nichols W.C., Trembath R.C. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. The International PPH Consortium. Nat. Genet. 2000;26:81–84. doi: 10.1038/79226. [DOI] [PubMed] [Google Scholar]
- 40.Ioannidis J.P., Thomas G., Daly M.J. Validating, augmenting and refining genome-wide association signals. Nat. Rev. Genet. 2009;10:318–329. doi: 10.1038/nrg2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cohen J.C., Boerwinkle E., Mosley T.H., Hobbs H.H. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N. Engl. J. Med. 2006;354:1264–1272. doi: 10.1056/NEJMoa054013. [DOI] [PubMed] [Google Scholar]
- 42.Cohen J.C., Kiss R.S., Pertsemlidis A., Marcel Y.L., McPherson R., Hobbs H.H. Multiple rare alleles contribute to low plasma levels of HDL cholesterol. Science. 2004;305:869–872. doi: 10.1126/science.1099870. [DOI] [PubMed] [Google Scholar]
- 43.Wang J., Cao H., Ban M.R., Kennedy B.A., Zhu S., Anand S., Yusuf S., Pollex R.L., Hegele R.A. Resequencing genomic DNA of patients with severe hypertriglyceridemia (MIM 144650) Arterioscler. Thromb. Vasc. Biol. 2007;27:2450–2455. doi: 10.1161/ATVBAHA.107.150680. [DOI] [PubMed] [Google Scholar]
- 44.Hunter D.J., Altshuler D., Rader D.J. From Darwin's finches to canaries in the coal mine—Mining the genome for new biology. N. Engl. J. Med. 2008;358:2760–2763. doi: 10.1056/NEJMp0804318. [DOI] [PubMed] [Google Scholar]
- 45.Sofat R., Hingorani A.D., Smeeth L., Humphries S.E., Talmud P.J., Cooper J.A., Shah T., Sandhu M.S., Ricketts S.L., Boekholdt S.M. Separating the mechanism-based and off-target actions of CETP inhibitors using CETP gene polymorphisms. Circulation. 2009 doi: 10.1161/CIRCULATIONAHA.109.865444. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Giltay E.J., van Reedt Dortland A.K., Nissinen A., Giampaoli S., van Veen T., Zitman F.G., Bots S., Kromhout D. Serum cholesterol, apolipoprotein E genotype and depressive symptoms in elderly European men: the FINE study. J. Affect. Disord. 2009;115:471–477. doi: 10.1016/j.jad.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 47.Davey Smith G., Ebrahim S. ‘Mendelian randomization’: Can genetic epidemiology contribute to understanding environmental determinants of disease? Int. J. Epidemiol. 2003;32:1–22. doi: 10.1093/ije/dyg070. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.