

Abstract
Complete congenital stationary night blindness (cCSNB) is associated with loss of function of rod and cone ON bipolar cells in the mammalian retina. In humans, mutations in NYX and GRM6 have been shown to cause the condition. Through the analysis of a consanguineous family and screening of nine additional pedigrees, we have identified three families with recessive mutations in the gene TRPM1 encoding transient receptor potential cation channel, subfamily M, member 1, also known as melastatin. A number of other variants of unknown significance were found. All patients had myopia, reduced central vision, nystagmus, and electroretinographic evidence of ON bipolar cell dysfunction. None had abnormalities of skin pigmentation, although other skin conditions were reported. RNA derived from human retina and skin was analyzed and alternate 5′ exons were determined. The most 5′ exon is likely to harbor an initiation codon, and the protein sequence is highly conserved across vertebrate species. These findings suggest an important role of this specific cation channel for the normal function of ON bipolar cells in the human retina.
Main Text
Congenital stationary night blindness (CSNB) is a group of genetically determined, nondegenerative disorders of the retina associated with lifelong deficient vision in the dark and often nystagmus and myopia. Two common subgroups have been characterized, despite an essentially normal fundus appearance, on the basis of the electroretinographic (ERG) and psychophysical findings:1 complete (cCSNB) and incomplete (icCSNB). Both subtypes show a characteristic “negative” ERG in response to a bright white flash in the dark-adapted eye, such that the b-wave is of lower amplitude than a normal a-wave. The complete form shows an undetectable rod-specific ERG to a dim light under dark adaptation, characteristic changes in the standard photopic ERGs, and a characteristic loss of the ON b wave, derived from rod and cone ON bipolar cells, with a normal OFF response, derived from cone OFF bipolar cells, on long-duration photopic stimulation.2 To date, mutations in NYX (MIM ∗300278), on chromosome X,3 and GRM6 (MIM ∗604096), on chromosome 5,4 have been shown to cause cCSNB. GRM6 encodes the metabotropic glutamate receptor 6 (mGluR6) situated on the dendrites of rod and cone ON bipolar cells.5 It mediates the sign inversion that occurs at the first synapse, such that glutamate release in the dark by photoreceptors causes hyperpolarization of the ON bipolar cell membrane.6 The function of the NYX protein product remains unknown.7–10 The nature of the channel or channels, as well as the components of the signaling pathway, that respond to mGluR6 signaling in ON bipolar cells also remains uncharacterized.11,12 GRM6 mutations have not been detected in all cases of autosomal-recessive cCSNB, suggesting that further autosomal genes remain to be identified. Recently, reduced expression of the gene encoding transient receptor potential cation channel, subfamily M, member 1 (TRPM1 [MIM ∗603576]) has been discovered in the skin and retina of horses homozygous for the Appaloosa trait locus, characterised by night blindness, a cCSNB phenotype on electroretinography, and skin depigmentation,13 although the genomic DNA variant causing the reduced expression is yet to be determined. Binding of the TRPM1 channel protein with nyctalopin has recently been shown,14 and those findings suggest this channel as a candidate for effecting the membrane voltage change in ON bipolar cells in response to glutamate. Moreover, ON bipolar cell transduction currents recently determined in mouse retina strongly suggest that a transient receptor potential (TRP) channel mediates the light-induced depolarization of this cell type.15
All patients involved in this study provided informed consent as part of a research project approved by the local research ethics committee, and all investigation was conducted in accordance with the principles of the Declaration of Helsinki. We investigated a consanguineous family (family 1, Figure 1) of South Asian ethnicity, in which the proband, V:1, a two-year-old girl, was diagnosed with CSNB at 11 months of age. She was noticed to have nystagmus and convergent squint at the age of 5 months. There are multiple consanguineous marriages within the family; her mother and a maternal uncle also had a nonprogressive night blindness. Cycloplegic refraction of the proband at 13 months was −14.00/−1.00×170 in the right eye (OD) and −13.00/−1.00×20 in the left eye (OS). Fundoscopy showed pale optic discs in both eyes and signs of pallor of the fundal reflex, consistent with myopia. The father and paternal grandmother had a normal ocular examination. Her mother, 28 years old, presented with nonprogressive nyctalopia from an early age, as well as convergent squint. She had bilateral high myopia (−23D spherical equivalent in each eye). Corrected visual acuities were 0.26 logMAR (logarithm of the minimal angle of resolution) for the right and 0.32 logMAR for the left eye. Fundus examination showed myopic fundi (Figure 2A). A second daughter, who developed nystagmus within the first year of life, has not been examined formally. The affected mother, elder daughter, and maternal uncle were all said to have dry skin and required application of emollients but no further dermatological treatments. Electrophysiology was performed on the mother, and the results are shown in the top trace of Figure 3. The features were typical of cCSNB, suggesting ON- pathway dysfunction in rods and all cone types.
Figure 1.
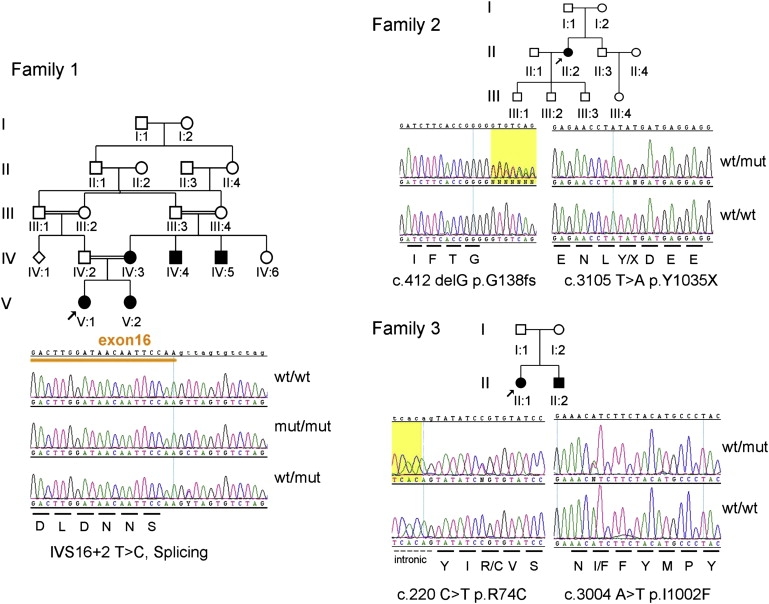
Pedigrees from Evaluated Families
Electropherograms of DNA sequence surrounding likely disease-causing variants are shown beneath each pedigree.
Figure 2.
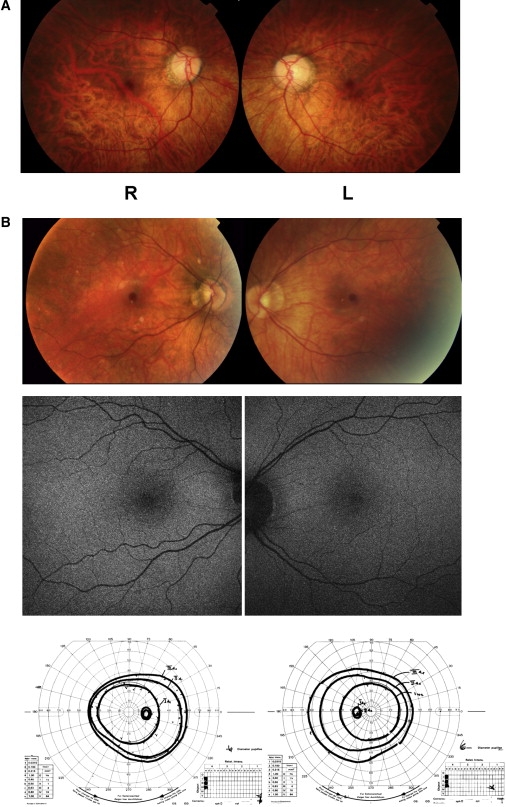
Color Fundus Photographs
(A) Color fundus photographs of patient IV:3 in family 1.
(B) Color fundus photographs, fundus autofluorescence, and Goldmann kinetic perimetry of patient II:2 in family 2. Goldmann perimetry isopters for targets III4a, II4a, and I4a are shown.
Figure 3.
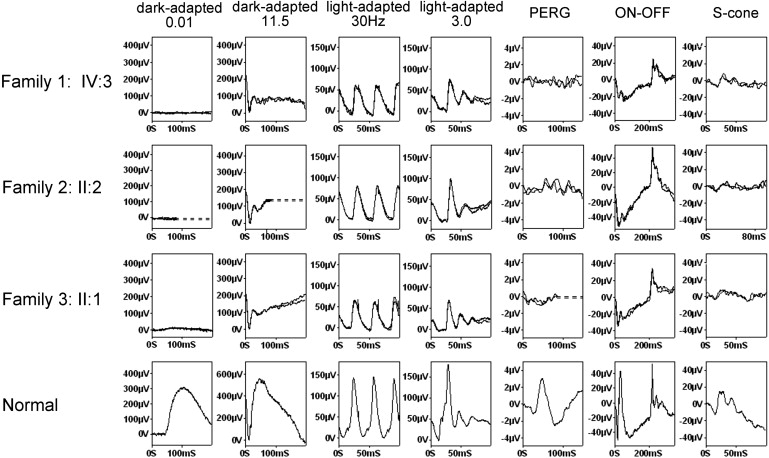
International-Standard Full-Field ERGs and Pattern ERGs from One Eye of Three Affected Individuals and One Control Subject
Top to bottom: patient IV:3 in family 1, patient II:2 in family 2, patient II:1 in family 3, control subject. Columns 1 and 2 show dark-adapted ERGs for flash intensities of 0.01 and 11.5 cd.s/m2 (columns 1 and 2). Columns 3 and 4 show light-adapted ERGs for a flash intensity of 3.0 cd.s/m2 presented at 30Hz (column 3) or 2Hz (column 4). Column 5 shows pattern ERGs (PERGs) recorded to an alternating checkerboard (check size 0.75 degrees, 4 reversals per s). Column 6 shows additional ON-OFF ERGs that use an orange stimulus (560 cd/m2, duration 200 ms) superimposed on a green background (150 cd/m2). Column 7 shows S-cone ERGs that use a blue stimulus (445 nm, 80 cd/m2) on an orange background (620 nm, 560 cd/m2). Dim-flash rod ERGs are undetectable (column 1). Bright-flash, dark-adapted ERGs are profoundly electronegative, with a b-wave of much lower amplitude than the borderline or minimally reduced a-wave. Photopic 30 Hz flicker ERGs show a slightly flattened trough and normal (patient II:1 in family 3) to marginally delayed peak times. Photopic single-flash ERGs show a broadened a-wave trough and a reduced-amplitude, sharply-rising b-wave lacking photopic oscillatory potentials. PERGs are undetectable, in keeping with macular involvement. Photopic ON-OFF ERGs show a profoundly reduced ON b-wave with relative sparing of the OFF response. S-cone ERGs show loss of the S-cone-specific component (the latter of the two peaks in the control subject), with some reduction in the amplitude of the L-M cone peak (the earlier of the two peaks in the normal). The findings therefore suggest ON pathway dysfunction in rods and all cone types.
DNA samples from the unaffected father and affected mother of family 1 were genotyped for 58,614 SNPs with the use of the Genechip Human Mapping 50k Xba Array (Affymetrix, Santa Clara, CA, USA). Samples were prepared with the use of the GeneChip Mapping 50K Xba Assay Kit as recommended by the manufacturer (Affymetrix). All chip data were imported into the Gene Chip Operating Software (GCOS) platform, and genotype data were extracted with the Gtype 4.0 SNP calling algorithm (Affymetrix). The pedigree was consistent with the propagation of a single mutant allele from a recent ancestor such that the unaffected father was heterozygous and the affected mother autozygous for this allele. A routine was written in Visual Basic within the Microsoft Excel program (Microsoft, Redmond, WA, USA) for the detection of genomic regions obeying this rule. The largest of four such regions, each greater than 1 Mb, consisted of 15.9 Mb comprising proximal chromosome 15q and included 280 contiguous SNPs. It was flanked by polymorphisms rs2090622 (proximal 15q) and rs10518928 (Figure 4). On interrogation of the Ensembl database, 106 distinct annotated Unigene clusters fell within the region, and each was inspected for its spectrum of tissue expression with the use of entries in the human dbEST database. One obvious candidate was a transient potential cation channel, subfamily M, member 1 (TRPM1) expressed in eye and skin. Primers were designed for amplification of the coding region and intron-exon boundaries of the 27 published exons (NM_002420.4) (Table S1, available online). Sanger sequencing with the use of fluorescently labeled di-deoxynucleotides (Big-Dye Terminator v3.1) on the ABI3730 platform was undertaken on genomic DNA (Applied Biosystems, Forster City, CA, USA) extracted from blood leukocytes of both parents. A homozygous variant was identified in the second nucleotide of the intron 16 donor site in the affected mother (IVS16+2T>C). The father was heterozygous for this variant. Such a base substitution would be expected to abrogate the canonical donor sequence and to affect efficient splicing of intron 16 of the gene. RNA was not available for a test of this assertion.
Figure 4.
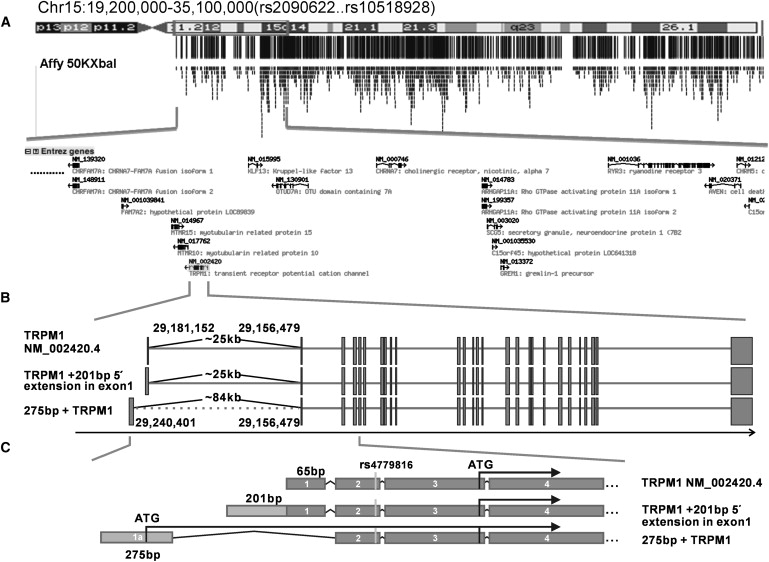
Region of Autozygosity Mapping and Analysis Of Gene Structure for the TRPM1 Gene
(A) Region of autozygosity for family 1 on chromosome 15q, of 15.9 Mb and 280 SNPs flanked by rs2090622 and rs10518928. A total of 106 Unigene clusters were found in the region. Only the partial region is shown in this figure.
(B) Results of RACE experiments for TRPM1 transcripts in human retina and skin. One transcript with a 201 bp 5′ extension of the published exon 1 was found in both skin and retinal tissue. An alternative and previously unreported 5′ exon of 275 bp, denoted here as exon 1a, was detected in retina approximately 84 Kb upstream of the published exon 2, spliced directly to exon 2 (omitting exon 1). Approximate physical distances are shown.
(C) The start codon of TRPM1 in the published sequence (NM_002420.4) is shown (rs4779816), with other likely translation start sites in the different transcripts.
Additional patients and families with a phenotype consistent with cCSNB, attending the retinal clinics at Moorfields Eye Hospital, were screened for mutations in GRM6 and, if male, NYX. We purposefully biased the cohort toward female patients and siblings, to enrich it for autosomal genes. Of 12 families screened for GRM6 mutations, three were found to have mutations. Only one family was consistent with X-linked segregation, and was therefore screened for NYX, which also showed no mutations. The nine familes that were negative were screened for mutations in TRPM1. Likely disease causing alleles were detected in two additional pedigrees (Figure 1, Table 1), both of white European descent. Also found were other variants whose pathological significance was unclear, which either failed to segregate with the disorder or were found on only one allele in an affected patient. All variants are listed in Table 1. The single affected person in family 2, a 36-year-old female, had onset of nystagmus in the first year of life and was diagnosed with CSNB at the age of 16 years. She had a pituitary gland enlargement without evidence of suprasellar extension or pituitary hormone dysfunction. She had received treatment for “eczema” in the past. Her best-corrected visual acuities were 1.00 logMAR OD and 0.84 logMAR OS. She was myopic (−15.50/−1.50×50 OD and −14.50/−1.00×90 OS) and had normal color vision and full visual fields on Goldmann kinetic perimetry. On examination, she had right convergent squint and pendular nystagmus, and myopic changes were evident on fundoscopy. Fundus autofluorescence imaging, performed with the Heidelberg Retinal Angiograph-II (Heidelberg Engineering, Germany), was normal (Figure 2B). Electrophysiology (second trace of Figure 3) was typical of cCSNB. The unaffected mother underwent electrophysiology for the determination of whether heterozygosity for TRPM1 mutation had a subclinical effect, and these results were entirely normal. In family 3, two siblings were affected with night blindness, and one, a 24-year-old female, was examined. She had nystagmus and a left divergent squint from birth and reported difficulties seeing at night and poorer vision in her left eye from an early age. She had been investigated for blistering of the skin and was thought to have an uncharacterized condition similar to epidermolysis bullosa simplex. She took regular thyroxine for hypothyroidism. Visual acuity at age 20 was 0.28 logMAR OD and 1.00 logMAR OS (which is thought to be amblyopic). She was myopic (−4.25D in each eye) and had a left divergent squint and nystagmus. Fundal examination showed bilateral tilted optic discs but was otherwise unremarkable Electrophysiology (third trace in Figure 3) was typical of cCSNB.
Table1.
Summary of DNA Variants
|
DNA Variants |
SIFTa |
PolyPhen |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Nucleotide | Protein | Type of Change | Family No. | Prediction | Tolerance Index | Predictionb | PSIC Score Differencec | Blosum62d | Frequency in Controlse |
| IVS16+2T>C | splicing | homozygous | 1 | N/A | N/A | N/A | N/A | N/A | 0/384 chromosomes |
| c.412 delG | p.G138fs | heterozygous | 2 | N/A | N/A | N/A | N/A | N/A | 0/384 |
| c.3105T>A | p.Y1035X | heterozygous | 2 | N/A | N/A | N/A | N/A | −4 | 0/384 |
| c.220C>T | p.R74C | heterozygous | 3 | intolerant | 0 | POD | 2.495 | −3 | 0/384 |
| c.3004A>T | p.I1002F | heterozygous | 3 | intolerant | 0 | POS | 1.799 | 0 | 0/384 |
| c.167A>G | p.Y56C | heterozygous | f | intolerant | 0 | POD | 2.544 | −2 | 0/384 |
| c.2162G>A | p.R721Q | heterozygous | f | intolerant | 0.01 | POS | 1.754 | 1 | 0/384 |
| c.2648A>G | p.E883G | heterozygous | f | intolerant | 0 | POD | 2.284 | −2 | 0/384 |
| c.2885A>C | p.M962T | heterozygous | g | intolerant | 0 | benign | 0 | −1 | 0/384 |
| c.4312A>G | p.R1438G | heterozygous | g | tolerant | 0.07 | POD | 2.006 | −2 | 0/384 |
| c.95G>A | p.S32N | tolerant | 0.49 | benign | 1.333 | 1 | rs2241493 | ||
| c.1239G>A | p.T413T | tolerant | 0.42 | N/A | N/A | 5 | rs1035705 | ||
| c.1813G>A | p.V605M | tolerant | 0.31 | benign | 1.289 | 1 | rs17815774 | ||
| c.2307T>C | p.Y769Y | tolerant | 0.12 | N/A | N/A | 7 | rs12913672 | ||
| c.2340T>C | p.N780N | tolerant | 0.18 | N/A | N/A | 6 | rs2288242 | ||
| c.4182G>A | p.V1395I | tolerant | 0.34 | benign | 0.127 | 3 | rs3784588 | ||
| c.2T>C | p.M1T | intolerant | 0.43 | benign | 0 | −1 | rs4779816–minor allele frequency 76/384 | ||
| c.16C>T | p.R6W | h | intolerant | 0 | POS | 1.8 | −3 | 6/32 chromosomes | |
| IVS14-64A>C | N/A | N/A | N/A | N/A | N/A | rs12907509 | |||
| IVS17+82G>T | N/A | N/A | N/A | N/A | N/A | rs12708421 | |||
| IVS23-24G>C | N/A | N/A | N/A | N/A | N/A | C allele in 8/28 chromosomes | |||
Unless otherwise specified, variant numbering is based on NM_002420.4
SIFT: Sorting Intolerant from Tolerant16—range 0–1, a lower score indicating intolerance to the substitution.
POD: probably damaging; POS: possibly damaging.
PSIC: Position-Specific Independent Counts. A high difference (range 0–4) indicates intolerance to the substitution.17
Blosum62: BLOcks of Amino Acid SUbstitution Matrix.26 Positive numbers indicate a substitution more likely to be tolerated evolutionarily, negative numbers indicate the opposite.
Controls were anonymized UK blood donors (ECACC). Unless otherwise specified, 192 samples were used.
Each were found in a simplex patient with cCSNB, in whom no other mutation was detected.
These two variants were found in cis, in a single female simplex patient. No other mutations were detected. Although the ERG showed a typical cCSNB phenotype, the night blindness was not congenital, having started in the third decade. A different molecular pathology is likely.
Numbering is based on TRPM1 exon 1a (see main text), registered as ss161641214 with dbSNP.
The affected proband of family 2 was found to have two likely disease-causing mutations in the TRPM1 gene (c.412delG [p.G138fs] and c.3105T>A [p.Y1035X]). Analysis of the mother showed that these variants were in trans. Both cause a premature termination codon, which would be predicted to cause the mRNA to succumb to nonsense-mediated decay. The proband of family 3 was found to harbor two missense variants not found in 192 unrelated individuals from the same ethnic group (c.220C>T [p.R74C] and c.3004A>T [p.I1002F]). It has so far not been possible to test segregation in other members of the family, nor to obtain RNA from the proband for amplification for the examination of cloned fragments. We cannot therefore be sure that these variants reside on different chromosomes. We employed the Sorting Intolerant From Tolerant (SIFT) and Polymorphism Phenotyping (PolyPhen) algorithms to assess these and other missense variants indentified in this study for likely pathogenicity, and the results are shown in Table 1. The SIFT algorithm deploys sequence homology to calculate a score, determining the evolutionary conservation status of the amino acid of interest and predicting whether its substitution will affect protein function. Substitutions at specific positions showing normalized probabilities less than the chosen cutoff value of 0.05 are predicted to be deleterious, and those greater than or equal to 0.05 are predicted to be tolerated. The higher the tolerance index, the less functional impact a particular amino acid substitution is likely to have.16 Polyphen evaluates the location of the substitution, the function of that region, and whether the substitution is likely to affect the three-dimensional structure of the protein. The prediction is based on the position-specific independent counts (PSIC) score derived from a composite of these calculations. PolyPhen scores greater than 2.0 indicate probable damage to protein function, scores of 1.5–2.0 indicate possible damage, and scores of lower than 1.5 indicate benign.17 Results are summarized in Table 1.
The published sequence (NM_002420.4) suggested an initiation methionine codon within exon 2, which was also listed as a single-nucleotide polymorphism (rs4779816). We tested this variant in 192 anonymized blood donors from the UK (European Collection of Cell Cultures, ECACC, Salisbury, UK) and found a minor allele prevalence of 0.199, which suggested strongly that another position in the transcript was used for initiation of protein translation. The next 3′ ATG codon in frame with the downstream sequence was located in exon 3. To investigate the full-length transcripts in human retina and skin RNA, we performed 5′ and 3′ RACE (rapid amplification of cDNA ends) experiments (Figure 5 and Table S2). cDNA products from the RACE reactions were analyzed on a 2% agarose gel and generated two distinct bands of approximately 500 bp and 200 bp. Bands were excised, subcloned into pCR4-TOPO vector (Invitrogen, Carlsbad, CA, USA), and sequenced in both directions. Clones derived from the 200 bp fragment included the published exon 1 sequence. Clones derived from the 500 bp fragments included a 201 bp 5′ extension of the currently published exon 1 of TRPM1 (Figure 4). This sequence did not extend the reading frame of the gene and contained three stop codons. Hence, this 5′ extension did not generate an alternative ATG start codon in frame with the downstream sequence. Secondly, a previously uncharacterized sequence at the 5′ end of some transcripts was found in a proportion of retinal clones with a similar molecular weight. Sequence analysis showed a 275-bp-long 5′ fragment contiguous with the sequence of exon 2 containing a 3′ extension of the open reading frame (Figure 4, accession number: GQ502181); with the use of the BLAT program, this exonic sequence was found to be situated approximately 84 Kbp upstream of exon 2 in the published human genomic DNA sequence. The published exon 1 was absent from these clones. ClustalW analysis of the TRPM1 homologs from diverse vertebrate genomes showed that this 5′ exon (hitherto referred to as exon 1a) was highly conserved (Figure S2). For confirmation of the presence of this splice variant, RT-PCR experiments were performed with the use of a forward primer complimentary to the 5′ end of exon 1a and two reverse primers from the sequence of TRPM1 exon 3–4 junction and exon 6. PCR products were readily visualized from the retinal tissue but were not found in same experiments for the skin tissue. Moreover, a band of the expected size was generated with the use of a 3′ primer complimentary to exon 27, confirming the presence of this exon 1a within the full-length transcript in human retina (∼4500 bp, data not shown). Additional RT-PCR experiments were performed with the 5′ primers with those complimentary to 3′ exons, in both skin and retinal RNA, and these experiments are summarized in Figure 5. Hence, it is likely that, at least in retina, the protein is longer at the amino end than is as yet appreciated. For the 3′ RACE experiment, it was mandatory to use an Oligo dT primer (GeneRacer Kit, Invitrogen) with a 24 nucleotide tail because a shorter primer appeared to bind internally to poly-A sequences. The resulting sequence analysis showed 3′-untranslated region sequences in both retina and skin that were identical to the published TRPM1 3′ sequence. The sequences found in this study were sequenced in all probands, and a common coding SNP was identified: p.R6W (dbSNP ss161641214, Table 1), but no further variants were found.
Figure 5.
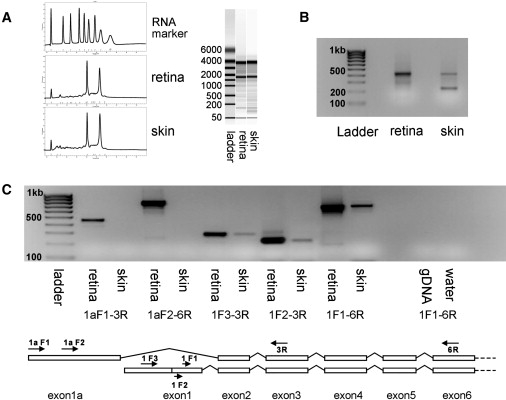
Experiments Examining TRPM1 RNA and 5′ RACE Experiments
(A) The integrity of RNA samples from human retina and skin were assessed with the Experion system (Bio-Rad, Hercules, CA, USA). The traces show similar peaks and ratios for 28S and 18S RNA molecules (vertical axis, fluorescence intensity; horizontal axis, migration time), suggesting low degradation and comparable quality from both tissues. The right panel shows the data as a computer-generated gel image.
(B) 2% agarose gel of DNA fragments generated from 5′ RACE experiments from the RNA assessed in Figure 5A (see main text and Table S2). With the use of a primer complimentary to a 5′ adapter oligonucleotide (Invitrogen, Carlsbad, CA, USA) and a gene-specific primer complimentary to exon 3, two main bands were produced, consistent with the published sequence (smaller band, 200 bp), as well as a mixture of the previously unreported 5′ exon 1a (∼475 bp) and as the 5' extension of exon 1 (475 bp), also previously unreported. The larger band was predominant in the retina, and the smaller was predominant in skin.
(C) After the generation of sequence from multiple clones of the 5′ RACE products (see main text), RT-PCR experiments were performed on skin and retina RNA and separated on 2% agarose, with the use of primers shown in the schematic below for determination of the presence of the various transcripts in the two tissues. The skin consistently failed to amplify for two different primers in exon 1a when used with reverse primers from exon 3 and 6. RT-PCR from comparable amounts of RNA gave less-dense bands than that in the retina for all other primer combinations. Lanes indicate the following: lane 0, ladder (subsequent lanes are paired reactions from retina and skin); lanes 1 and 2, 1aF and 3R; lanes 3 and 4, 1aF1 and 6R; lanes 5 and 6, 1F1 and 3R; lanes 7 and 8, 1F2 and 3R; lanes 9 and 10, 1F1 and 6R; lane 11, PCR with 1F1 and 6R from genomic DNA control; lane 12, same as 11 with water as control.
The finding of likely disease-causing mutations in TRPM1 in patients with electroretinographic evidence of loss of ON bipolar cell function (Figure 3), qualitatively identical to that seen in patients reported with GRM6 mutation,4 suggests an important role of this cation channel in the normal function of ON bipolar cells in the human retina. Transient receptor potential (TRP) channels are a family of membrane protein channels permeable to cations, with diverse functions. The first member of the family to be described followed the discovery of a mutant of Drosophila melanogaster with photoreceptor degeneration,18 and a mammalian homolog exists as TRPC1.19 To date, 28 mammalian TRP channels have been identified and can be divided into six subfamilies: TRPC, TRPV, TRPM, TRPP, TRPML, and TRPA.20–23 In the TRPM subfamily, TRPM1, the founding member was identified as an RNA sequence in a mouse melanoma cell line and was subsequently noticed to be expressed in melanomas with lower metastatic potential; the protein was subsequently termed melastatin.24 In the same year, Hunter et al. cloned a long transcript variant cDNA from a human retina cDNA library.25 Recently, TRPM1 has been described as the potential cause of CSNB and coat-spotting pattern in the Appaloosa horse.13 It is unclear whether the affected patients of the three families described here have skin manifestations of TRPM1 mutation. None had any suggestion of pigment disturbance such as that reported in the Appaloosa horse, each having a normal distribution of pigment in skin and hair and a tanning response no different to that of unaffected relatives. All probands reported dry or scaly skin, and one, the proband of family 3, required management by a specialist. Further clinical investigation is required if we are to know whether this represents a manifestation of TRPM1 dysfunction or absence in human skin. Also, a rare disorder, melanoma-associated retinopathy, is an accurate retinal phenocopy for the disorder described here and might suggest that an epitope of melastatin causes autoimmune targeting of retinal ON bipolar cells.
Intriguingly, in our cohort, there remained a handful of families with a cCSNB phenotype without mutations in NYX, GRM6, or TRPM1, suggesting that there may be additional genes that cause this disorder. The identification of such genes will provide further insights into the ON bipolar cell's signaling-pathway components that link the activation of metabotropic glutamate channels with the permeability state of TRPM1 membrane channels.
Acknowledgments
This study was supported by grants from the British Retinitis Pigmentosa Society, Foundation Fighting Blindness USA, Fight for Sight, and the National Institute for Health Research UK to the Biomedical Research Centre for Ophthalmology (Moorfields Eye Hospital [MEH] and the University College London [UCL] Institute of Ophthalmology). We are grateful to colleagues who referred patients to us at MEH and to Priya Banerjee and Jacob Raby of the UCL Genomics Microarray Facility for technical assistance. We acknowledge the cooperation and help provided by the family members in this study.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
An Organized View of the Transcriptome (Unigene), http://www.ncbi.nlm.nih.gov/sites/entrez?db=unigene
BLAST like Alignment tool (BLAT), http://genome.ucsc.edu/cgi-bin/hgBlat?command=start
Ensembl, http://www.ensembl.org/index.html
Expressed Sequence Tags database (dbEST), http://www.ncbi.nlm.nih.gov/dbEST/
Multiple sequence alignment program for DNA or proteins (ClustalW), http://www.ebi.ac.uk/Tools/clustalw2/index.html
NCBI SNP database (dbSNP), http://www.ncbi.nlm.nih.gov/projects/SNP/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/omim
Polymorphism Phenotyping (PolyPhen), http://genetics.bwh.harvard.edu/pph/index.html
Sorting Intolerant from Tolerant (SIFT), http://sift.jcvi.org/
Splice Site Prediction by Neural Network, http://www.fruitfly.org/seq_tools/splice.html
UCSC Genomic Bioinformatics, http://genome.ucsc.edu/index.html
Accession Numbers
The Genbank accession number for the TRPM1 exon 1a sequence reported in this paper is GQ502181.
The dbSNP database accession number for the SNP in exon 1a reported in this paper is ss161641214.
References
- 1.Miyake Y., Yagasaki K., Horiguchi M., Kawase Y., Kanda T. Congenital stationary night blindness with negative electroretinogram. A new classification. Arch. Ophthalmol. 1986;104:1013–1020. doi: 10.1001/archopht.1986.01050190071042. [DOI] [PubMed] [Google Scholar]
- 2.Miyake Y., Yagasaki K., Horiguchi M., Kawase Y. On- and off-responses in photopic electroretinogram in complete and incomplete types of congenital stationary night blindness. Jpn. J. Ophthalmol. 1987;31:81–87. [PubMed] [Google Scholar]
- 3.Bech-Hansen N.T., Naylor M.J., Maybaum T.A., Sparkes R.L., Koop B., Birch D.G., Bergen A.A., Prinsen C.F., Polomeno R.C., Gal A. Mutations in NYX, encoding the leucine-rich proteoglycan nyctalopin, cause X-linked complete congenital stationary night blindness. Nat. Genet. 2000;26:319–323. doi: 10.1038/81619. [DOI] [PubMed] [Google Scholar]
- 4.Dryja T.P., McGee T.L., Berson E.L., Fishman G.A., Sandberg M.A., Alexander K.R., Derlacki D.J., Rajagopalan A.S. Night blindness and abnormal cone electroretinogram ON responses in patients with mutations in the GRM6 gene encoding mGluR6. Proc. Natl. Acad. Sci. USA. 2005;102:4884–4889. doi: 10.1073/pnas.0501233102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vardi N., Duvoisin R., Wu G., Sterling P. Localization of mGluR6 to dendrites of ON bipolar cells in primate retina. J. Comp. Neurol. 2000;423:402–412. doi: 10.1002/1096-9861(20000731)423:3<402::aid-cne4>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 6.Slaughter M.M., Miller R.F. Characterization of an extended glutamate receptor of the on bipolar neuron in the vertebrate retina. J. Neurosci. 1985;5:224–233. doi: 10.1523/JNEUROSCI.05-01-00224.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pesch K., Zeitz C., Fries J.E., Munscher S., Pusch C.M., Kohler K., Berger W., Wissinger B. Isolation of the mouse nyctalopin gene nyx and expression studies in mouse and rat retina. Invest. Ophthalmol. Vis. Sci. 2003;44:2260–2266. doi: 10.1167/iovs.02-0115. [DOI] [PubMed] [Google Scholar]
- 8.Zeitz C., Scherthan H., Freier S., Feil S., Suckow V., Schweiger S., Berger W. NYX (nyctalopin on chromosome X), the gene mutated in congenital stationary night blindness, encodes a cell surface protein. Invest. Ophthalmol. Vis. Sci. 2003;44:4184–4191. doi: 10.1167/iovs.03-0251. [DOI] [PubMed] [Google Scholar]
- 9.O'Connor E., Eisenhaber B., Dalley J., Wang T., Missen C., Bulleid N., Bishop P.N., Trump D. Species specific membrane anchoring of nyctalopin, a small leucine-rich repeat protein. Hum. Mol. Genet. 2005;14:1877–1887. doi: 10.1093/hmg/ddi194. [DOI] [PubMed] [Google Scholar]
- 10.Bech-Hansen N.T., Cockfield J., Liu D., Logan C.C. Isolation and characterization of the leucine-rich proteoglycan nyctalopin gene (cNyx) from chick. Mamm. Genome. 2005;16:815–824. doi: 10.1007/s00335-005-0018-y. [DOI] [PubMed] [Google Scholar]
- 11.Dhingra A., Faurobert E., Dascal N., Sterling P., Vardi N. A retinal-specific regulator of G-protein signaling interacts with Galpha(o) and accelerates an expressed metabotropic glutamate receptor 6 cascade. J. Neurosci. 2004;24:5684–5693. doi: 10.1523/JNEUROSCI.0492-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nawy S. The metabotropic receptor mGluR6 may signal through G(o), but not phosphodiesterase, in retinal bipolar cells. J. Neurosci. 1999;19:2938–2944. doi: 10.1523/JNEUROSCI.19-08-02938.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bellone R.R., Brooks S.A., Sandmeyer L., Murphy B.A., Forsyth G., Archer S., Bailey E., Grahn B. Differential gene expression of TRPM1, the potential cause of congenital stationary night blindness and coat spotting patterns (LP) in the Appaloosa horse (Equus caballus) Genetics. 2008;179:1861–1870. doi: 10.1534/genetics.108.088807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bojang, P. Jr., Pearring, J.N., and Gregg, R.G. (2009) Biochem & Molecular Biology, University of Louisville, Louisville, KY. Nyctalopin Interacts with Transient Receptor Potential Channels in Yeast. ARVO 2009 Annual Meeting, FL, USA. Poster no. 5176.
- 15.Shen Y., Heimel J.A., Kamermans M., Peachey N.S., Gregg R.G., Nawy S. A transient receptor potential-like channel mediates synaptic transmission in rod bipolar cells. J. Neurosci. 2009;29:6088–6093. doi: 10.1523/JNEUROSCI.0132-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kumar P., Henikoff S., Ng P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protocols. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 17.Ramensky V., Bork P., Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30:3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cosens D.J., Manning A. Abnormal electroretinogram from a Drosophila mutant. Nature. 1969;224:285–287. doi: 10.1038/224285a0. [DOI] [PubMed] [Google Scholar]
- 19.Wes P.D., Chevesich J., Jeromin A., Rosenberg C., Stetten G., Montell C. TRPC1, a human homolog of a Drosophila store-operated channel. Proc. Natl. Acad. Sci. USA. 1995;92:9652–9656. doi: 10.1073/pnas.92.21.9652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ramsey I.S., Delling M., Clapham D.E. An introduction to TRP channels. Annu. Rev. Physiol. 2006;68:619–647. doi: 10.1146/annurev.physiol.68.040204.100431. [DOI] [PubMed] [Google Scholar]
- 21.Scott K., Zuker C. TRP, TRPL and trouble in photoreceptor cells. Curr. Opin. Neurobiol. 1998;8:383–388. doi: 10.1016/s0959-4388(98)80065-2. [DOI] [PubMed] [Google Scholar]
- 22.Clapham D.E., Runnels L.W., Strubing C. The trp ion channel family. Nat. Rev. Neurosci. 2001;2:387–396. doi: 10.1038/35077544. [DOI] [PubMed] [Google Scholar]
- 23.Clapham D.E. TRP channels as cellular sensors. Nature. 2003;426:517–524. doi: 10.1038/nature02196. [DOI] [PubMed] [Google Scholar]
- 24.Duncan L.M., Deeds J., Hunter J., Shao J., Holmgren L.M., Woolf E.A., Tepper R.I., Shyjan A.W. Down-regulation of the novel gene melastatin correlates with potential for melanoma metastasis. Cancer Res. 1998;58:1515–1520. [PubMed] [Google Scholar]
- 25.Hunter J.J., Shao J., Smutko J.S., Dussault B.J., Nagle D.L., Woolf E.A., Holmgren L.M., Moore K.J., Shyjan A.W. Chromosomal localization and genomic characterization of the mouse melastatin gene (Mlsn1) Genomics. 1998;54:116–123. doi: 10.1006/geno.1998.5549. [DOI] [PubMed] [Google Scholar]
- 26.Henikoff S., Henikoff J.G. Amino acid substituion matrices from protein blocks. Proc. Natl. Acad. Sci. USA. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.