

Abstract
Heterozygous copy-number variants and SNPs of CNTNAP2 and NRXN1, two distantly related members of the neurexin superfamily, have been repeatedly associated with a wide spectrum of neuropsychiatric disorders, such as developmental language disorders, autism spectrum disorders, epilepsy, and schizophrenia. We now identified homozygous and compound-heterozygous deletions and mutations via molecular karyotyping and mutational screening in CNTNAP2 and NRXN1 in four patients with severe mental retardation (MR) and variable features, such as autistic behavior, epilepsy, and breathing anomalies, phenotypically overlapping with Pitt-Hopkins syndrome. With a frequency of at least 1% in our cohort of 179 patients, recessive defects in CNTNAP2 appear to significantly contribute to severe MR. Whereas the established synaptic role of NRXN1 suggests that synaptic defects contribute to the associated neuropsychiatric disorders and to severe MR as reported here, evidence for a synaptic role of the CNTNAP2-encoded protein CASPR2 has so far been lacking. Using Drosophila as a model, we now show that, as known for fly Nrx-I, the CASPR2 ortholog Nrx-IV might also localize to synapses. Overexpression of either protein can reorganize synaptic morphology and induce increased density of active zones, the synaptic domains of neurotransmitter release. Moreover, both Nrx-I and Nrx-IV determine the level of the presynaptic active-zone protein bruchpilot, indicating a possible common molecular mechanism in Nrx-I and Nrx-IV mutant conditions. We therefore propose that an analogous shared synaptic mechanism contributes to the similar clinical phenotypes resulting from defects in human NRXN1 and CNTNAP2.
Introduction
The etiology of severe mental retardation (MR) is heterogeneous, and, despite a significant number of identified disease genes,1 the majority of cases, especially nonsyndromic cases, remain unsolved.2 Many of the currently known MR-related genes are involved in neurogenesis and neuronal migration, and awareness of the implication of synaptic organization and plasticity in MR has only recently begun to rise.3,4 In 2007, haploinsufficiency of the basic helix-loop-helix (bHLH) transcription factor 4 (TCF4) was identified as causative for Pitt-Hopkins syndrome (PTHS [MIM 610954]), a severe MR disorder with variable additional anomalies, such as breathing anomalies, epilepsy, and facial dysmorphism including a beaked nose and a wide mouth with a cupid's-bow-shaped upper lip.5,6 TCF4 belongs to the E-protein family of bHLH transcription factors, which bind as homo- and heterodimers to E-box consensus sequences in promoters of target genes.7 Like other E-proteins, TCF4 shows a broad expression pattern and a high expression in the CNS.8,9 After the identification of the underlying gene in 2007, approximately 50 patients have been reported,5,6,8–11 demonstrating the importance of a diagnostic test for the increased recognition and appreciation of a previously clinically underdiagnosed condition. Because of a similar severe degree of MR, commonly associated seizures, and microcephaly, PTHS has evolved as an important differential diagnosis to the two most common syndromic disorders in severe MR, Rett (MIM 312750) and Angelman (MIM 105830) syndromes.11 Because only 12% of patients referred to us with suspected PTHS showed mutations in TCF4 (Zweier et al.11 and unpublished data), the clinically relatively homogenous group of 179 TCF4-mutation-negative patients, including two sibling pairs, represented a suitable study cohort for searching for additional candidate genes for overlapping disorders.
Through molecular karyotyping and mutational analysis, we indeed identified recessive defects in two genes, CNTNAP2 and Neurexin I (NRXN1), in patients with a very similar severe MR disorder and variable additional symptoms, such as seizures and breathing anomalies, resembling Pitt-Hopkins syndrome. In light of the shared phenotype that characterizes our patients with recessive CNTNAP2 and NRXN1 defects, and on the basis of the theme of overlapping phenotypes being caused by genes that are linked with each other in molecular networks,12 we further aimed to address the hypothesis of a common molecular pathogenesis. We therefore utilized the fruit fly Drosophila melanogaster as a model and collected data that point to a common synaptic link between these two genes.
Subjects and Methods
Patients
Our study group consisted of 179 patients, including two sibling pairs, who were referred for TCF4 testing because of severe MR and variable additional features reminiscent of the PTHS spectrum, such as microcephaly, dysmorphic facial gestalt, or breathing anomalies. TCF4 mutational testing revealed normal results in all of these patients. Ethics approval for this study was obtained from the ethics committee of the Medical Faculty, University of Erlangen-Nuremberg, and informed consent was obtained from parents or guardians of the patients.
Molecular Karyotyping
Molecular karyotyping was performed in 48 patients with the Affymetrix 500 K SNP Array and in 12 patients with the Affymetrix 6.0 SNP Array, in accordance with the supplier's instructions. In the index patient of family 1, hybridization was performed with an Affymetrix GeneChip Mapping 500K SNP array, and the second affected patient and both parents were analyzed with the Affymetrix GeneChip Mapping 250K Nsp SNP array. Copy-number data were analyzed with the Nexus software (Biodiscovery) and the Affymetrix Genotyping Console 3.0.2 software. Molecular karyotyping in patients 2 and 3 was performed with the Affymetrix GeneChip Mapping 6.0 array platform, and copy-number data were analyzed with the Affymetrix software Genotyping Console 3.0.2. The identified copy-number variants (CNVs) were submitted to the Decipher database (patient 1a, 250902; patient 2, 250903; patient 3, 250904).
Mutational Screening
DNA samples from 177 patients, derived from peripheral-blood or lymphoblastoid cell lines, were screened for CNTNAP2 (NM_014141) and NRXN1 (NM004801) mutations by unidirectional direct sequencing of the coding exons 1–24 of CNTNAP2 and the coding exons 2–22 of NRXN1, including intronic flanking regions (ABI BigDye Terminator Sequencing Kit v.3; Applied Biosystems), with the use of an automated capillary sequencer (ABI 3730; Applied Biosystems). Mutations were confirmed with an independent PCR and bidirectional sequencing. Primer pairs can be found in Table S1, available online. For splice-site prediction, the online tools NNSPLICE 0.9 and HSF V2.3 were used.
FISH and MLPA
Fluorescence in situ hybridization (FISH) analysis was performed in family 1 with the directly Cy3-labeled bacterial artificial chromosome (BAC) clone RP4-558L10 on metaphase spreads, in accordance with standard protocols.
Probes for all coding exons of CNTNAP2 were designed and MLPA reaction was performed in accordance with the guidelines of MRC-Holland. The deletion in patient 3 was confirmed with MLPA with the use of a probe within exon 2 and a control probe within exon 12 of NRXN1. Probe sequences are listed in Table S2.
Analysis of Relationship
The relationship of individuals within family 1 was analyzed, with a four-generation family with known relationships used as background, with the Graphical Representation of Relationships (GRR) software.13 For GRR, we selected, from Affymetrix 250K arrays, 10,000 randomly distributed autosomal SNPs with a minimal minor allele frequency of 0.2 in Europeans. For each pair of individuals, GRR calculates over the 10,000 markers the identical-by-state (IBS) mean and standard deviation. The graphical plot of IBS mean versus IBS standard deviation facilitates distinguishing between relationships such as parents and offspring, siblings, half siblings, and cousins, as well as identical or unrelated individuals. Additionally, SNP genotypes around CNTNAP2 were analyzed in the family members.
Drosophila Genes and Lines
Drosophila orthologs of TCF4, NRXN1, and CASPR2 (daughterless [CG5102], Nrx-1 [CG7050], and Nrx-IV [CG6827]) were identified by the ENSEMBLE genome browser or by the reciprocal BLAST best-hit approach. Two RNAi lines, to Nrx-IV and daughterless, respectively, were obtained from the Vienna Drosophila Research Center (VDRC) and gave consistent phenotypes. VDRC lines no. 9039 (Nrx-IV) and no. 51297 (daughterless) were utilized for further analysis. RNA interference was induced with the UAS-Gal4 system. The w1118 line (VDRC no. 60000) was used as a control, representing the same genetic background as the RNAi lines. Flies were raised at 28°C for maximum efficiency of knockdown. The Nrx-I overexpression line pUAST-Nrx-I was obtained from Wei Xie from Nanjing, China. Gal4 driver lines and the inducible Nrx-IV overexpression line P(EP)Nrx-IVEP604 were obtained from the Bloomington stock center.
Quantitative Real-Time PCR
RNA extraction from 3×3 L3 larvae of each genotype was performed with the RNeasy Lipid Tissue Kit (QIAGEN) in accordance with the supplier's protocols. cDNA synthesis was performed with iScript (Biorad). Quantitative real-time PCR was performed with the Power SYBR Green PCR Master Mix on a 7500 Fast Real-Time PCR System (Applied Biosystems), and results were normalized to the endogenous control actin. Primer sequences can be found in Table S3.
Immunostaining and Data Acquisition
We harvested 10–18 hr embryos and fixed them with 3.7% PFA for 20–25 min. All primary antibodies—anti-elav (labels nuclei of all neurons), antibody 22c10 (sensory nervous system), antibody BP102 (axon tracts, central neuropile region), anti-fas II (motor- and central pioneer axons), and antibody nc82 (anti-bruchpilot, synaptic active zones) (all from the Developmental Studies Hybridoma Bank [DHSB])—were used in a 1:100 dilution. Late stage (16/17), nc-82-labeled embryos were assigned to one of three phenotypic categories: strong peripheral staining, moderate staining, and weak or residual staining, respectively. We performed statistical analysis of 69 wild-type (WT) embryos (w1118) and 73 Nrx-IV knockdown embryos from three independent experiments with a chi-square test and a Fisher's-exact test to obtain p values. The images for peripheral synaptic staining were obtained with a Zeiss Apotome.
Brains were dissected from L3 larvae and fixed for 30 min in 3.7% PFA. Pictures of WT and mutant brains were acquired with the use of the same microscope settings. Intensities of nc82 immunostainings were measured with Image J within two fields at two standardized positions in each CNS, one in the upper third and one in the lower third of the ventral nerve cord. The average of these two values was normalized to the average of controls for comparison of results from independent experiments. A total of 45 w1118 brains from six independent experiments, 11 elav-Gal4::Nrx-I brains from two independent experiments, 19 double-elav-Gal4::Nrx-I brains from three independent experiments, 12 elav-Gal4::Nrx-IV brains from two independent experiments, and 21 double-elav-Gal4::Nrx-IV brains from four independent experiments were measured. p values were obtained with a Wilcoxon test for two samples for comparison to the WT.
Type 1b neuromuscular junctions (NMJs) of muscle 4 were analyzed after dissection of L3 larvae and fixation in 3.7% PFA for 30 min. Costaining was performed with nc82 and DLG (both from DHSB) or HRP (Jackson Immuno Research) antibodies in a dilution of 1:500. NMJ pictures were stacked in ImageJ and processed in Adobe Photoshop. Numbers of active zones and branches were manually counted in an animated stack, and total synaptic area was determined by ImageJ. A total of 21 WT NMJs, 19 overexpression Nrx-I NMJs, and 20 overexpression Nrx-IV NMJs from at least two independent experiments were counted. For the evaluation of branches, 27 WT NMJs, 25 overexpression Nrx-I NMJs, and 20 overexpression Nrx-IV NMJs from three experiments were counted. We performed statistical evaluation with the Wilcoxon test for two samples, comparing each of the genotypes to the WT. The antibody against NrxIV was obtained from Christian Klämbt, Münster, Germany.14 Secondary antibodies for all stainings were either Alexa 568- or Alexa 488-labeled antibodies against mouse or rabbit (Molecular Probes). All data were acquired blind to the evaluated phenotype.
Results
Identification of Recessive Deletions and Mutations in CNTNAP2 and NRXN1
Molecular karyotyping led to the identification of a homozygous deletion of exons 2–9 within the CNTNAP2 gene on chromosome 7q35-q36.1 in a sibling pair of European origin (P1a and P1b), formerly published as possible clinical cases of Pitt-Hopkins syndrome.15 This deletion was confirmed by FISH analysis (Figure S2) and MLPA (data not shown) and is predicted to be in frame but result in the loss of several functional domains (Figure 1A, Figure 2A, and Figure S1). Consanguinity of the parents had been denied,15 and no indication for consanguinity was found by analysis of relationship with the use of the information of 10.000 SNPs. However, when the SNPs within and around CNTNAP2 were analyzed, they showed homozygosity in both children, indicating an allele of common ancestry. By subsequent mutational screening of CNTNAP2 in a larger cohort of 177 additional TCF4-mutation-negative patients, we identified a third patient of European origin (P2) with compound heterozygosity for the splice mutation IVS10-1G>T and a partial in-frame deletion of exons 5–8, identified by molecular karyotyping (Figure 1B and Figure 2A) and confirmed with MLPA. The splice-site mutation resulted in lack of recognition of the splice acceptor site by two splice-site-prediction programs and is therefore predicted to result in loss of exon 10, leading to a frame shift, and the deletion is predicted to result in the loss of two laminin G domains. The splice-site mutation was not found in 384 control chromosomes, and no CNTNAP2 deletion was found in 667 molecularly karyotyped control individuals. In both families, the parents were heterozygous carriers of one of the respective defects.
Figure 1.
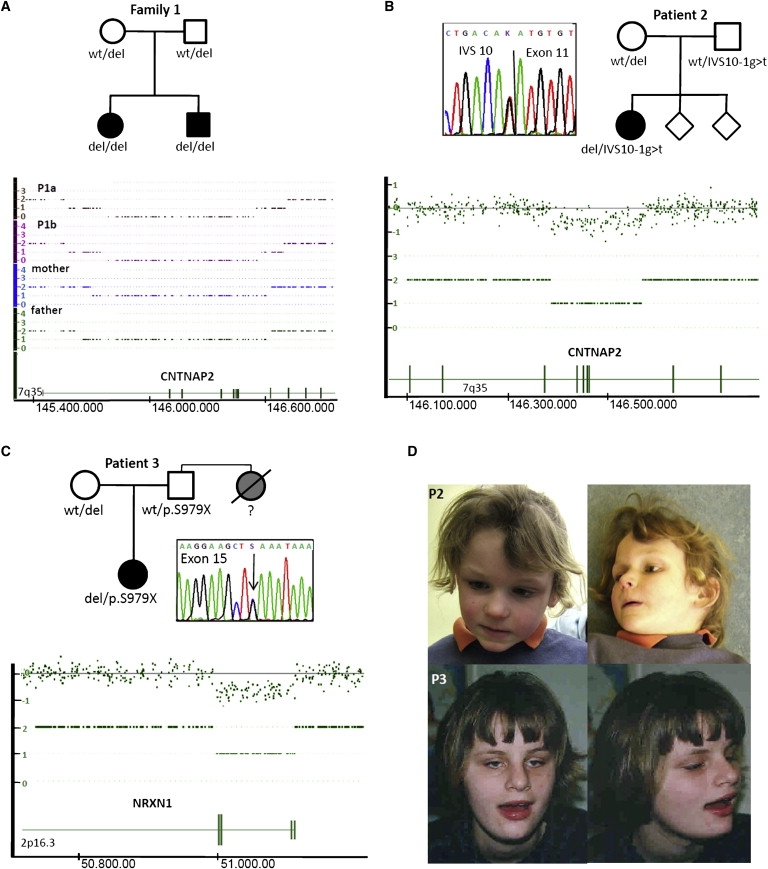
Pedigrees and Results of Molecular Karyotyping
(A) Pedigree of family 1, with two affected children and homozygous deletion of CNTNAP2 affecting exons 2–9. Both parents are heterozygous carriers of the deletion. Results are from molecular karyotyping with Affymetrix 250K SNP arrays and analysis with the Genotyping Console 3.0.2 software (Affymetrix). The deletion-flanking SNPs in the 500K array of P1a are SNP_A-1991616 (145,562,641 Mb; UCSC Human Genome Browser version 18 [hg18]) and SNP_A-1991672 (146,730,410 Mb; hg18), with a maximal deletion size of 1,167,269 bp and a minimal size of 1,146,016 bp (Nexus software).
(B) Pedigree of P2, with one affected child. The patient harbors an in-frame 180 kb deletion affecting CNTNAP2 exons 5–8 and a splice-site mutation in the splice donor site of exon 11. Results of molecular karyotyping data from the Affymetrix 6.0 SNP array were analyzed with the Genotyping Console 3.0.2 software, showing a deletion from CN_1217185 (146,387,354 Mb; hg18) to SNP_A-4269862 (146,566,863 Mb; hg18). See Figure S1 for SNP copy-number profiles.
(C) Pedigree of P3, with a compound-heterozygous deletion of NRXN1 exons 1–4 and a stop mutation in exon 15. Results of molecular karyotyping data from the Affymetrix 6.0 SNP array were analyzed with the Genotyping Console 3.0.2 (Affymetrix), showing a 113 kb deletion between CN_864223 (51,001,003 Mb; hg18) and CN_864269 (51,113,677 Mb; hg18).
(D) Clinical pictures of P2, with a compound-heterozygous deletion and mutation in CNTNAP2 and of patient 3, with a compound-heterozygous deletion and mutation in NRXN1. Apart from a wide mouth in patient 3, no specific dysmorphisms are noted.
Figure 2.
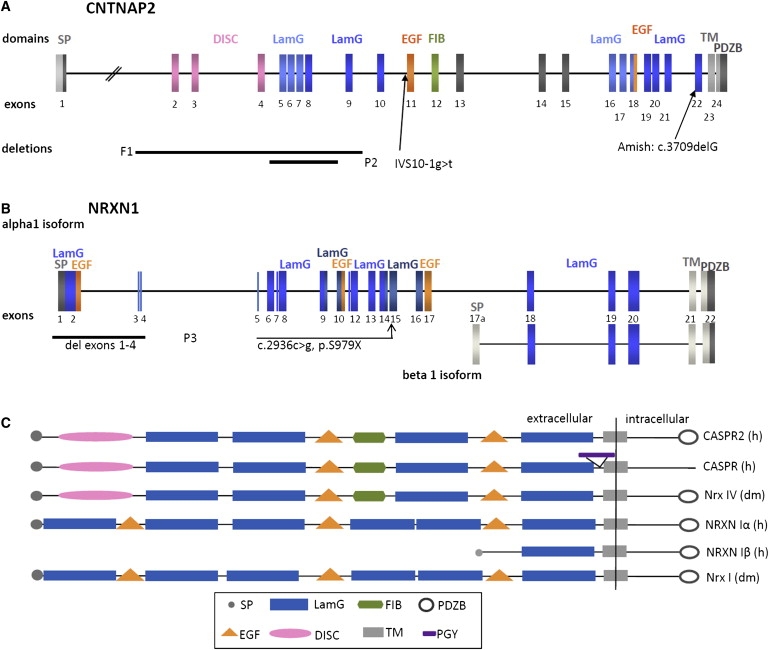
Structure of CNTNAP2 and NRXN1
(A) Schematic drawing of the genomic structure of CNTNAP2 with color coding for domain-coding exons and localization of mutations and deletions. Black bars represent deletions. Abbreviations are as follows: SP, signal peptide; DISC, discoidin-like domain; LamG, laminin-G domain; EGF, epidermal growth factor-like domain; FIB, fibrinogen-like domain; TM, transmembran region; PDZPB, PDZ-domain-binding site ; F1, family 1; P2, patient 2; Amish, homozygous mutation in the Amish population, published by Strauss et al.36
(B) Schematic drawing of the genomic structure of α-NRXN1 and β-NRXN1 with color coding for domain-coding exons and localization of the mutation and deletion in patient 3, the deletion being represented by a black bar. Abbreviations are as follows: SP, signal peptide; LamG, laminin-G domain; EGF, epidermal growth factor-like domain; TM, transmembrane region; PDZPB, PDZ-domain-binding site.
(C) Schematic drawing of the domain structure of neurexins, CASPR2, and CASPR in humans and Drosophila. In contrast to CASPR, CASPR2 contains a PDZ-domain-binding site but lacks a PGY repeat region, rich in proline, glycine, and tyrosine residues. Both neurexin I and CASPR2/Nrx-IV contain PDZ-domain-binding sites at their intracellular C terminus but differ in the presence of discoidin-like and fibrinogen-like domains and in the order of laminin-G domains.
In another European patient of our cohort who had a very similar phenotype (P3), we identified a heterozygous 180 kb deletion within the NRXN1 gene on chromosome 2p16.3, spanning exons 1–4, including the start codon. This deletion was inherited from the healthy mother, but no deletions affecting the coding region of NRXN1 were found in 667 molecularly karyotyped healthy controls. Subsequent sequencing of NRXN1 in this patient revealed a stop mutation in exon 15 on the second allele, which was inherited from the healthy father (Figure 1C and Figure 2B). Both mutations are predicted to result in loss of the so-called alpha-isoform of NRXN1, one of two NRXN1 isoforms that are transcribed from alternative promoters. The presumably remaining shorter beta-isoform (Figures 2B and 2C) appears not to be sufficient to ensure normal function, which is in accordance with findings in alpha-neurexin knockout mice.16 Mutational screening of NRXN1 in our study cohort did not reveal any additional defects.
Clinical Characterization
As far as data are available, birth measurements of all patients (P1a, P1b, P2, and P3) were normal. Further growth development was also normal, apart from short stature in the siblings from family 1 and additional microcephaly in one of them. All four patients with recessive defects in CNTNAP2 or NRXN1 showed severe MR with lack of speech or with speech limited to single words (P1b), whereas motor milestones were normal or only mildly delayed, with a walking age of 2 years in P3. Episodes of hyperbreathing occurred in all patients, and seizures with an age at onset between 4 months and 30 months were observed in P1a, P1b, and P2. Additional variable anomalies were cerebellar hypoplasia, autistic behavior, and stereotypic movements. Apart from a wide mouth with thick lips in P1a and P1b and a wide mouth in P3, no specific facial dysmorphisms were noted (Figure 1D). Parents of all patients were healthy, and the deceased sister of the father of P3 was said to have had epilepsy and mild MR. P1a and P1b have been described in detail by Orrico et al.,15 and an overview of clinical details of all patients is shown in Table 1. Lack of NRXN1 and CNTNAP2 expression in blood or fibroblasts (Bakkaloglu et al.17 and data not shown) precluded functional studies on human material.
Table 1.
Phenotype in Patients with CNTNAP2 and NRXN1 Mutations
|
Siblings |
|||||
|---|---|---|---|---|---|
| Patients | P1a | P1b | P2 | Amish36 (N = 9) | P3 |
| Mutations | CNTNAP2 deletion of exons 2–9, homozygous | CNTNAP2 deletion of exons 2–9, homozygous | CNTNAP2 deletion of exons 5–8 + IVS10-1G>T | CNTNAP2 c.3709delG, homozygous | NRXN1 deletion of exons 1–4 + p.S979X |
| Age | 20 yrs | 15 yrs | 11 yrs | 1–10 yrs | 18 yrs |
| Sex | f | m | f | not reported | f |
| Parents | healthy | healthy | healthy | not reported | healthy |
| Birth weight Length OFC |
3700 g 51 cm 34.5 cm |
not known | 3700 g at term |
not reported | 3450 g normal |
| Height | <P3 | <P3 | normal | P4–P57 | P50–P75 |
| Weight | P10 | P5–P10 | P50 | not reported | P50–P75 |
| OFC | <P3 | P75 | P75 | P18–P99 | P25 |
| MR | severe | severe | severe | all | severe |
| Age of walking | normal | normal | not known | 16–30 mo | 2 yrs |
| Speech | none | single words | none | yes, but regression | none |
| Age of seizure onset | 22 mo | 25–30 mo | 4–8 mo | 14–20 mo | none |
| MRI results | cerebellar hypoplasia | normal | not known | dysplasia in 43% | normal |
| Hyperbreathing | yes | yes | yes | not reported | yes |
| Stereotypies | none | not noted | tooth grind., rep. hand movements | yes | |
| Autistic behaviour | not noted | not noted | yes | 67% | yes |
| Developmental regression | not noted | not noted | considered normal until 8 months | yes, with onset of seizures | normal the first years |
| Constipation | not noted | not noted | no | not reported | yes |
| Decreased deep tendon reflexes | not known | not known | not known | 89% | UE: decreased LE: normal |
| Other | broad mouth, thick lips | broad mouth, thick lips | dry skin | broad mouth, strabismus, protruding tongue, excessive drooling, abnormal sleep-wake cycles, hypermotoric behavior |
|
| Normal testinga | FRAXA, UBE3A, MECP2, TCF4 |
FRAXA,UBE3A, MECP2 |
UBE3A, CDKL5, MECP2, TCF4 | array CGH:bUBE3A, MECP2, ZEB2, TCF4 | |
Patients P1a and P1b are siblings from family 1, clinically described by Orrico et al.;15 Abbreviations are as follows: f, female; m, male; hom, homozygous; het, heterozygous; MR, mental retardation; OFC, occipito-frontal circumference; rep. hand movements, repetitive hand movements; tooth grind., tooth grinding; UE, upper extremities; LE, lower extremities.
Previous genetic testing with reported normal results.
Array-based comparative-genome hybridization via an Agilent 244K oligonucleotide array (Agilent).
Analysis of CNTNAP2 and NRXN1 Orthologs Nrx-IV and Nrx-I in Drosophila
Although a synaptic role for NRXN1 is known, this has not yet been established for CNTNAP2. However, the high similarity of clinical phenotypes caused by defects in the two genes suggested a potential common molecular contribution. To address this, as well as a further possible connection with TCF4, we utilized Drosophila as a model organism. All three genes, TCF4, NRXN1, and CNTNAP2, are highly conserved in evolution and have orthologs in Drosophila.
We initially hypothesized that the TCF4 ortholog daughterless might regulate Nrx-I and Nrx-IV as transcriptional targets. Knockdown of daughterless to 60% of WT levels by the use of two different ubiquitous driver lines (promoter-Gal4 lines that regulate inducible RNAi alleles; see Subjects and Methods) resulted in pupal lethality, confirming the importance of daughterless for fly development and viability.18 However, expression of Nrx-I and Nrx-IV in L3 larvae of these genotypes was not significantly changed (Figure S3).
Drosophila Nrx-IV and Nrx-1 Both Determine the Level of the Synaptic Protein Bruchpilot
Knockdown of Nrx-IV, either ubiquitously with the use of actin-Gal4 drivers or specifically in neurons with the use of an elav-Gal4 driver, resulted in late embryonic lethality, with animals completing embryogenesis but failing to hatch. Together with an only recently reported expression of Nrx-IV in neurons,14,19 this suggests a crucial role in this cell type. To evaluate the cause for lethality upon neuronal knockdown, we performed immunostainings on embryos with a series of neuronal markers, labeling all neuronal nuclei, the sensory nervous system, main central axon tracts, and motor- and central pioneer axons, respectively. None of these showed abnormal position or morphology (Figure S4 and data not shown). However, we noted an overall diminished staining intensity of the presynaptic protein bruchpilot (nc82) in Nrx-IV knockdown embryos (Figure 3A). Quantification revealed that the fraction of embryos with weak or residual staining on peripheral motor synapses was significantly increased to 33% in Nrx-IV knockdown embryos, compared to 9% in WT embryos, whereas the fraction of strongly stained embryos was decreased to 25% in Nrx-IV knockdown embryos, compared to 55% in the WT (Figure 3B).
Figure 3.
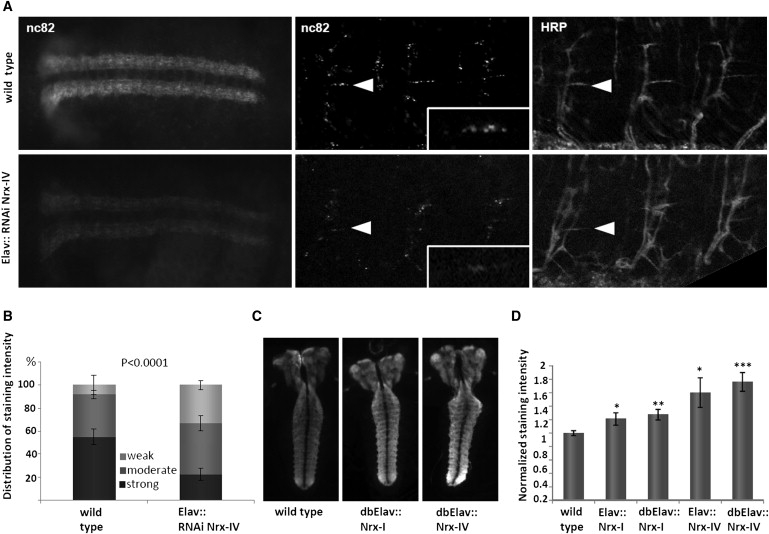
The Presynaptic Protein Bruchpilot Is Misregulated in Nrx-IV Knockdown Embryos and Larval Brains with Neuronal Overexpression of Nrx-I and Nrx-IV
(A and B) For quantitative evaluation, embryos have been assigned to one of three categories of bruchpilot (nc82) intensities: strong, moderate, and weak peripheral staining. (A) Images of strong and moderate central (ventral nerve cord, left panel) and peripheral (middle panel) presynaptic bruchpilot staining, representing the major fraction in WT and Nrx-IV knockdown embryos, respectively (UAS-Nrx-IVRNAi/+; elav-Gal4/+ [Elav::RNAi Nrx-IV]). Note that peripheral and central synaptic staining of bruchpilot in the Nrx-IV knockdown embryos is diminished. Costaining with an anti-HRP marker, staining neuronal membranes, ensured the same focal plane in mutant and WT embryos. Arrowheads point to a specific identifiable synapse (the muscle 6/7 synapse). Bruchpilot staining of this synapse is depicted in insets in the middle panels. (B) Quantitative analysis of nc82 (anti-bruchpilot, synaptic active zones) labeling. Diagram shows mean with standard deviation.
(C) Representative pictures of bruchpilot staining in brains with neuronal overexpression of Nrx-I or Nrx-IV as a result of the following genotypes: WT, UAS-Nrx-I/ elav-Gal4; elav-Gal4/+ (dbElav:: Nrx-I) and elav-Gal4/+; UAS-Nrx-IV/ elav-Gal4 (dbElav:: Nrx-IV). Bruchpilot immunoreactivity is increased in both mutant conditions.
(D) Quantitative assessment of bruchpilot immunoreactivity in brains of genotypes shown in (C). The diagram shows the mean of normalized intensity with the standard error of the mean. p values in (B) and (D) are related to the WT, respectively. Single asterisk, p < 0.05; double asterisk, p < 0.01; triple asterisk, p < 0.0001.
Interestingly, bruchpilot is known to colocalize with Nrx-I at presynaptic active zones, the domains of neurotransmitter release,20 and reduced levels of bruchpilot immunoreactivity have been reported in larval brains of Nrx-I mutants.21 We studied bruchpilot levels after neuronal overexpression of either Nrx-I or Nrx-IV and found a significant dosage-dependent increase of bruchpilot-staining intensity in larval brains. Upon overexpression with one copy of a panneuronal elav-Gal4 driver, bruchpilot intensity was increased 1.2- and 1.3-fold, and introduction of a second copy (dbElav) resulted in a 1.6- and 1.8-fold increase (Figures 3C and 3D).
Drosophila Nrx-IV Is Present at Synapses and Can, Like Nrx-I, Reorganize Them
For further analyses at the subsynaptic level, we utilized Drosophila larval NMJs, giant synapses that share a series of features with central excitatory synapses in the mammalian brain and represent an established model for the study of synaptic development and plasticity.22 Staining of these synapses with a recently characterized specific antibody14 for Nrx-IV detected the presence of Nrx-IV at synaptic terminals. Nrx-IV localizes in a pattern of subsynaptic foci that overlap active zones (Figure 4A), resembling the pattern previously reported for Nrx-I.20
Figure 4.
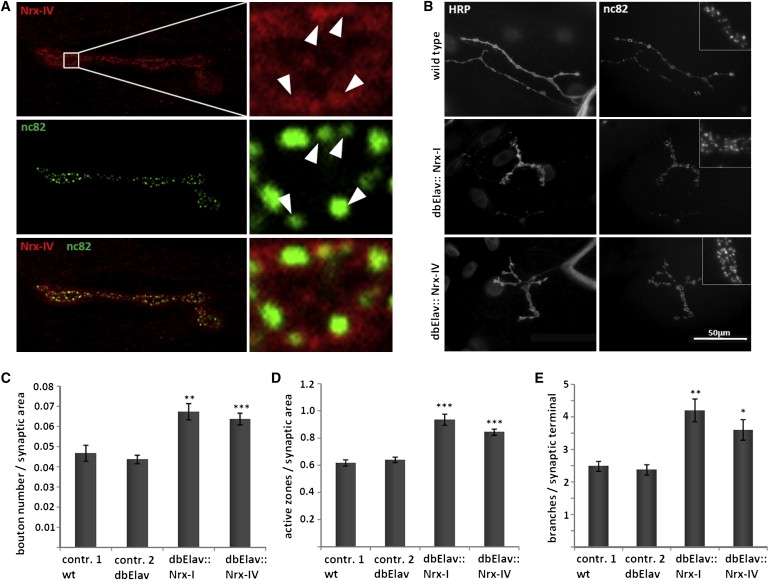
Nrx-IV Localizes to Synapses and Its Overexpression Reorganizes Synapse Architecture
(A) Presence of endogenous Nrx-IV at type 1b neuromuscular junctions (NMJs) of muscle 4, overlapping with active zones stained with nc82. White arrowheads point to Nrx-IV, labeling overlapping active zones.
(B) Synaptic terminal with increased bouton number and increased number of synaptic branches upon Nrx-I or Nrx-IV overexpression, stained with an anti-HRP marker and the nc82 antibody. The enlarged section depicts the increased density of active zones.
(C) Increased bouton number per μm2 NMJ area in UAS-Nrx-I/ elav-Gal4; elav-Gal4/+ (dbElav:: Nrx-I) and elav-Gal4/+; UAS-Nrx-IV/ elav-Gal4 (dbElav:: Nrx-IV) versus control w1118 animals (contr. 1) and dbElav control animals (contr. 2).
(D) Quantitative analysis of increased active-zone density (number per μm2 NMJ area) in neuronal Nrx-I and Nrx-IV overexpression.
(E) Increased number of synaptic branches in Nrx-I and Nrx-IV overexpression. Error bars indicate standard error of the mean. p values are related to the WT (control 1). Single asterisk, p < 0.008; double asterisk, p < 0.0003; triple asterisk, p < 0.0001.
Previous studies in Nrx-I null mutants revealed a decreased number of synaptic boutons in NMJs, whereas overexpression of Nrx-I resulted in an increased bouton number.20 Our results, measuring total synaptic area in Nrx-I-overexpression animals, show that this goes along with a morphological reorganization into smaller boutons, as illustrated by an increased number of boutons per μm2 NMJ area (Figures 4B and 4C). Strikingly, overexpression of Nrx-IV is capable of inducing the same morphological changes (Figures 4B and 4C). Furthermore, overexpression of either Nrx-I or Nrx-IV resulted in a highly significant increase in density of active zones (Figures 4B and 4D) and in a significant increase in branching of synaptic terminals (Figures 4B and 4E).
Discussion
Recessive Defects in CNTNAP2 and NRXN1 Cause Severe MR
We report here on homozygous and compound-heterozygous deletions and mutations in NRXN1 and CNTNAP2 that cause severe MR with additional features such as epilepsy, autistic behavior, and breathing anomalies. Heterozygous CNVs or SNPs in both genes have recently been extensively reported in association with autism-spectrum disorder (ASD) (AUTS15, [MIM 612100]), epilepsy, or schizophrenia.17,23–33 Additionally, for CNTNAP2, an association with Gilles de la Tourette syndrome and obsessive compulsive disorder34 was reported to be due to a disruption of the gene but was not confirmed in another family.35 Furthermore, a homozygous stop mutation in CNTNAP2 in Old Order Amish children was implicated in a distinct disorder, CDFE syndrome (MIM 610042), which is characterized by cortical dysplasia and early onset, intractable focal epilepsy leading to language regression, and behavioral and mental deterioration;36 as well as periventricular leukomalacia and hepatomegaly in an additional patient.37 Histological examination of temporal-lobe specimens of these patients showed evidence of abnormal neuronal migration and structure, as well as a possibly altered expression of Kv1.1 and Nav1.2 channels, therefore providing a possible explanation for the cortical dysplasia and epilepsy phenotypes.36
Taken together, published data and the present study indicate that heterozygous defects or variants in both NRXN1 and CNTNAP2 can represent susceptibility factors for variable cognitive, neurological, and psychiatric disorders, whereas biallelic defects result in a fully penetrant, severe neurodevelopmental disease such as that observed in our patients, thus representing different ends of the clinical spectrum caused by either monoallelic or biallelic defects in these two genes. This is in accordance with a report by Zahir et al.,38 who described a patient with a heterozygous de novo NRXN1 deletion affecting the same exons as those in our patient 3 but without detectable mutation on the second allele. This heterozygous deletion was associated with vertebral anomalies, behavioral problems, and only mild cognitive abnormalities. Of note, in our families, none of the heterozygous parents had a history of autism, epilepsy, or schizophrenia, and none of other family members are known to have these disorders. However, the deceased sister of the father of patient 3 was said to have had epilepsy and mild MR. Whether or not this relates to the mutation in the father's family remains elusive.
A common feature in the Amish patients with a homozygous stop mutation and in our patients with homozygous deletions or compound-heterozygous deletion and mutation in CNTNAP2 is the early onset of seizures. In contrast, none of our patients showed regression of speech development. Another difference is episodic hyperbreathing, present in our patients but not reported in the Amish patients. Cortical dysplasia, occurring in some of the Amish patients, was not observed in our patients; however, the number of patients examined with MRI is too low to allow any definite conclusions to be drawn. The phenotypic differences between our patients and the reported Amish patients may be explained by the loss of C-terminal transmembrane and cytoplasmic domains in the Amish mutation36 and by the loss of N-terminal extracellular domains in our patients (Figure 2 and Table 1). However, clinical bias cannot be excluded.
Resemblance among the patients in this study who have recessive defects in CNTNAP2 or NRXN1 is high, with the exception of patient 3, who has NRXN1 defects but no seizures. It might even be speculated that the epilepsy, observed in patients with recessive CNTNAP2 defects but not in our patient with the compound-heterozygous NRXN1 defect, might be associated to the previously observed neuronal-migration anomalies,36 whereas the remaining common symptoms such as severe MR and autistic behavior might be caused by overlapping synaptic anomalies. All of the reported patients were originally referred for TCF4 analysis as a result of phenotypic resemblance to Pitt-Hopkins syndrome with regard to facial aspects, the severity of MR, and breathing anomalies. Nevertheless, phenotypical differences were noticeable. In contrast to patients with Pitt-Hopkins syndrome, who present an equally severe delay or lack of both motor and speech development, patients with CNTNAP2 or NRXN1 defects, also severely impaired in speech development, present with normal or only mildly to moderately delayed motor milestones. These findings are in line with the previously reported specific involvement of CNTNAP2 in language development.26
Because of the similar phenotypes caused by defects in both NRXN1 and CNTNAP2 and the resemblance to Pitt-Hopkins syndrome, caused by haploinsufficiency of TCF4, we investigated an as-yet-unappreciated common molecular basis contributing to these disorders, using the fruitfly Drosophila as a model. The Drosophila TCF4 ortholog daughterless belongs to the achaete-scute complex, which encodes members of the bHLH class of transcriptional factors with a well-established proneural function.18 Our initial hypothesis, that daughterless might regulate Nrx-I and Nrx-IV as transcriptional targets, could not be confirmed by our analysis of their expression levels in the daughterless knockdown condition (Figure S3). However, given that the data were obtained from knockdown animals, not null animals, this excludes neither transcriptional regulation that is undetectable by our assay nor other nontranscriptional interactions.
CNTNAP2 and NRXN1 Drosophila Orthologs Nrx-IV and Nrx-I Might Be Involved in a Common Synaptic Mechanism
Our identification of a fully penetrant and severe MR phenotype due to recessive defects in NRXN1 is in agreement with NRXN1's important role in synaptic function, as previously suggested on the basis of its heterozygous alterations that are associated with neuropsychiatric disorders and work with animal models.39 NRXN1 belongs to the evolutionarily conserved family of neurexins, presynaptic transmembrane proteins. Each of the three vertebrate neurexins has two promoters, generating longer alpha-neurexins and shorter beta-neurexins. In addition, extensive alternative splicing occurs, generating a large number of variants, which may mediate target recognition and synaptic specificity.40 Alpha-neurexins also play a role in neurotransmitter release by coupling Ca2+ channels to synaptic-vesicle exocytosis.41 In contrast to the three neurexins in mammals, there is only one Drosophila neurexin, termed Nrx-I or DNRX.20,21 Previously, defects in several genes involved in synaptic pathways and complexes have been reported to be causative for or associated with MR or ASD, among them neurexin interactors. The extracellular region of neurexins binds to postsynaptic neuroligins, a family of proteins associated with autism and MR,42 which in turn interact with the postsynaptic protein SHANK3, also associated with ASD.43 The binding of neurexins to neuroligins connects pre- and postsynaptic neurons and mediates signaling across the synapse,39 and the NRXN-NLGN-SHANK pathway is supposed to be crucial during synaptogenesis, given that mutations within this pathway lead to abnormal synaptogenesis and excess of inhibitory currents.44 The intracellular domains of neurexins interact with the synaptic vesicle protein synaptotagmin and with PDZ-domain proteins such as CASK,20,40 in which X-linked recessive mutations were only recently identified to be causative for MR and brain malformations.45
In contrast, a synaptic role of CNTNAP2 has not yet been established. CNTNAP2 encodes CASPR2, a protein related to neurexins. However, on the basis of its additional motifs, different domain organization,40 and phylogenetic analyses,20 only a distant relationship was assumed.46 Caspr2 regulates neuron-glia contact in vertebrates and has been shown to colocalize with Shaker-like K+ channels in the juxtaparanodal areas of Ranvier nodes in myelinated axons of both the central and peripheral nervous system.47,48 Until recently, its fly ortholog Nrx-IV was reported to be almost exclusively expressed in glial cells49 and to regulate glia-glia contact in insects via septate junctions that are important for maintaining an intact blood-brain barrier.46 A detected decrease in evoked neurotransmitter release and an occasional failure to form synapses in Nrx-IV null mutants was interpreted as a consequence secondary to glial dysfunction.49 However, recently, a neuronal Nrx-IV isoform was also identified.14,19 Despite the knowledge of neuronal expression of vertebrate Caspr2,50 so far only a report on detection of the protein in fractionated rat synaptic plasma membranes pointed to a possible synaptic presence.17 Our findings now support a synaptic localization of Nrx-IV (Figure 4).
Previous studies at Drosophila neuromuscular junctions had reported a decreased number of synaptic boutons in Nrx-I null mutants and an increase upon overexpression of Nrx-I.20 Our observation that the latter goes in hand with an increased active-zone density and the finding that Nrx-IV overexpression is causing the very same phenotypes (Figure 4) suggest a crucial and possibly common role for both proteins in the morphological organization of synapses. Moreover, published data for Nrx-I mutants21 and our analysis of Nrx-IV knockdown and overexpression conditions of Nrx-I and Nrx-IV suggest an important role of both proteins in bidirectional regulation of bruchpilot levels. Bruchpilot is a presynaptic protein crucial for the structure of active zones, the subsynaptic domains of neurotransmitter release, and is present at most if not all synapses of Drosophila. It shows high sequence and functional homology to the vertebrate family of ELKS/CAST proteins,51 corresponding to ERC1 and ERC2 in humans with similar reported functions. The mechanism by which Nrx-I and Nrx-IV determine bruchpilot levels remains the subject of future study. Given that all three contain C-terminal PDZ-domain binding sites, which are necessary for the formation of large complexes with active-zone proteins,51 one reasonable hypothesis is that the three proteins might assemble into a synaptic complex or macromolecular network.
In conclusion, we have identified here autosomal-recessive defects in CNTNAP2 and NRXN1 in patients with severe MR and variable additional features overlapping with Pitt-Hopkins syndrome. With a frequency of at least 1% in this cohort, mutations in CNTNAP2 in particular appear to significantly contribute to severe MR. Using the fruitfly as a model organism, we observed that not only Nrx-I but also Nrx-IV, previously unrecognized as a synaptic protein in vertebrates and in Drosophila, is present at synapses and regulates levels of the active-zone protein bruchpilot. It is therefore tempting to hypothesize that misregulation of the human bruchpilot ortholog might underlie a common synaptic pathomechanism contributing to the similar phenotypes observed in patients with defects in CNTNAP2 and NRXN1.
Acknowledgments
We thank the patients and families for participation in this study and Christine Zeck-Papp, Michaela Kirsch, and Daniela Schweitzer for excellent technical assistance. We are grateful to Wei Xie (Genetics Research Center, Southeast University Medical School, Nanjing, China) and to Christian Klämbt (Institute for Neurobiology, Münster, Germany) for providing the pUAST-Nrx-I flies and the Nrx-IV antibody, respectively. We thank H. Brunner and H. van Bokhoven for critical reading of the manuscript. This work was supported by a grant from the Deutsche Forschungsgemeinschaft (DFG) to C.Z.; by a grant from the German Mental Retardation Network (MRNET) funded by the German Federal Ministry of Education and Research (BMBF) as a part of the National Genome Research Network (NGFNplus), to A.Re., A.Ra., and A.S.; and by a VIDI grant from the Netherlands Organisation for Scientific Research (NWO) to A.S.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Basic Local Alignment Search Tool (BLAST), http://blast.ncbi.nlm.nih.gov/Blast.cgi
Database of Drosophila Genes and Genomes, http://flybase.org
Decipher database, http://decipher.sanger.ac.uk/application/
Graphic Representation of Relationships, http://www.sph.umich.edu/csg/abecasis/GRR/index.html
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Splice site prediction, http://www.fruitfly.org/seq_tools/splice.html and http://www.umd.be/HSF/
University of California, Santa Clara (UCSC) Genome Browser, http://genome.ucsc.edu
References
- 1.Inlow J.K., Restifo L.L. Molecular and comparative genetics of mental retardation. Genetics. 2004;166:835–881. doi: 10.1534/genetics.166.2.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rauch A., Hoyer J., Guth S., Zweier C., Kraus C., Becker C., Zenker M., Huffmeier U., Thiel C., Ruschendorf F. Diagnostic yield of various genetic approaches in patients with unexplained developmental delay or mental retardation. Am. J. Med. Genet. A. 2006;140:2063–2074. doi: 10.1002/ajmg.a.31416. [DOI] [PubMed] [Google Scholar]
- 3.Humeau Y., Gambino F., Chelly J., Vitale N. X-linked mental retardation: focus on synaptic function and plasticity. J. Neurochem. 2009;109:1–14. doi: 10.1111/j.1471-4159.2009.05881.x. [DOI] [PubMed] [Google Scholar]
- 4.Vaillend C., Poirier R., Laroche S. Genes, plasticity and mental retardation. Behav. Brain Res. 2008;192:88–105. doi: 10.1016/j.bbr.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 5.Amiel J., Rio M., de Pontual. L., Redon R., Malan V., Boddaert N., Plouin P., Carter N.P., Lyonnet S., Munnich A. Mutations in TCF4, encoding a class I basic helix-loop-helix transcription factor, are responsible for Pitt-Hopkins syndrome, a severe epileptic encephalopathy associated with autonomic dysfunction. Am. J. Hum. Genet. 2007;80:988–993. doi: 10.1086/515582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zweier C., Peippo M.M., Hoyer J., Sousa S., Bottani A., Clayton-Smith J., Reardon W., Saraiva J., Cabral A., Gohring I. Haploinsufficiency of TCF4 causes syndromal mental retardation with intermittent hyperventilation (Pitt-Hopkins syndrome) Am. J. Hum. Genet. 2007;80:994–1001. doi: 10.1086/515583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murre C., McCaw P.S., Vaessin H., Caudy M., Jan L.Y., Jan Y.N., Cabrera C.V., Buskin J.N., Hauschka S.D., Lassar A.B. Interactions between heterologous helix-loop-helix proteins generate complexes that bind specifically to a common DNA sequence. Cell. 1989;58:537–544. doi: 10.1016/0092-8674(89)90434-0. [DOI] [PubMed] [Google Scholar]
- 8.Brockschmidt A., Todt U., Ryu S., Hoischen A., Landwehr C., Birnbaum S., Frenck W., Radlwimmer B., Lichter P., Engels H. Severe mental retardation with breathing abnormalities (Pitt-Hopkins syndrome) is caused by haploinsufficiency of the neuronal bHLH transcription factor TCF4. Hum. Mol. Genet. 2007;16:1488–1494. doi: 10.1093/hmg/ddm099. [DOI] [PubMed] [Google Scholar]
- 9.de Pontual L., Mathieu Y., Golzio C., Rio M., Malan V., Boddaert N., Soufflet C., Picard C., Durandy A., Dobbie A. Mutational, functional, and expression studies of the TCF4 gene in Pitt-Hopkins syndrome. Hum.Mutat. 2009;30:669–676. doi: 10.1002/humu.20935. [DOI] [PubMed] [Google Scholar]
- 10.Giurgea I., Missirian C., Cacciagli P., Whalen S., Fredriksen T., Gaillon T., Rankin J., Mathieu-Dramard M., Morin G., Martin-Coignard D. TCF4 deletions in Pitt-Hopkins Syndrome. Hum. Mutat. 2008;29:E242–E251. doi: 10.1002/humu.20859. [DOI] [PubMed] [Google Scholar]
- 11.Zweier C., Sticht H., Bijlsma E.K., Clayton-Smith J., Boonen S.E., Fryer A., Greally M.T., Hoffmann L., den Hollander N.S., Jongmans M. Further delineation of Pitt-Hopkins syndrome: phenotypic and genotypic description of 16 novel patients. J. Med. Genet. 2008;45:738–744. doi: 10.1136/jmg.2008.060129. [DOI] [PubMed] [Google Scholar]
- 12.Oti M., Brunner H.G. The modular nature of genetic diseases. Clin. Genet. 2007;71:1–11. doi: 10.1111/j.1399-0004.2006.00708.x. [DOI] [PubMed] [Google Scholar]
- 13.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. GRR: graphical representation of relationship errors. Bioinformatics. 2001;17:742–743. doi: 10.1093/bioinformatics/17.8.742. [DOI] [PubMed] [Google Scholar]
- 14.Stork T., Thomas S., Rodrigues F., Silies M., Naffin E., Wenderdel S., Klambt C. Drosophila Neurexin IV stabilizes neuron-glia interactions at the CNS midline by binding to Wrapper. Development. 2009;136:1251–1261. doi: 10.1242/dev.032847. [DOI] [PubMed] [Google Scholar]
- 15.Orrico A., Galli L., Zappella M., Lam C.W., Bonifacio S., Torricelli F., Hayek G. Possible case of Pitt-Hopkins syndrome in sibs. Am. J. Med. Genet. 2001;103:157–159. doi: 10.1002/ajmg.1523. [DOI] [PubMed] [Google Scholar]
- 16.Zhang W., Rohlmann A., Sargsyan V., Aramuni G., Hammer R.E., Sudhof T.C., Missler M. Extracellular domains of alpha-neurexins participate in regulating synaptic transmission by selectively affecting N- and P/Q-type Ca2+ channels. J. Neurosci. 2005;25:4330–4342. doi: 10.1523/JNEUROSCI.0497-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bakkaloglu B., O'Roak B.J., Louvi A., Gupta A.R., Abelson J.F., Morgan T.M., Chawarska K., Klin A., Ercan-Sencicek A.G., Stillman A.A. 1). Molecular cytogenetic analysis and resequencing of contactin associated protein-like 2 in autism spectrum disorders. Am. J. Hum. Genet. 2008;82:165–173. doi: 10.1016/j.ajhg.2007.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Caudy M., Vassin H., Brand M., Tuma R., Jan L.Y., Jan Y.N. daughterless, a Drosophila gene essential for both neurogenesis and sex determination, has sequence similarities to myc and the achaete-scute complex. Cell. 1988;55:1061–1067. doi: 10.1016/0092-8674(88)90250-4. [DOI] [PubMed] [Google Scholar]
- 19.Wheeler S.R., Banerjee S., Blauth K., Rogers S.L., Bhat M.A., Crews S.T. Neurexin IV and Wrapper interactions mediate Drosophila midline glial migration and axonal ensheathment. Development. 2009;136:1147–1157. doi: 10.1242/dev.030254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li J., Ashley J., Budnik V., Bhat M.A. Crucial role of Drosophila neurexin in proper active zone apposition to postsynaptic densities, synaptic growth, and synaptic transmission. Neuron. 2007;55:741–755. doi: 10.1016/j.neuron.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zeng X., Sun M., Liu L., Chen F., Wei L., Xie W. Neurexin-1 is required for synapse formation and larvae associative learning in Drosophila. FEBS Lett. 2007;581:2509–2516. doi: 10.1016/j.febslet.2007.04.068. [DOI] [PubMed] [Google Scholar]
- 22.Koh Y.H., Gramates L.S., Budnik V. Drosophila larval neuromuscular junction: molecular components and mechanisms underlying synaptic plasticity. Microsc. Res. Tech. 2000;49:14–25. doi: 10.1002/(SICI)1097-0029(20000401)49:1<14::AID-JEMT3>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 23.Alarcon M., Abrahams B.S., Stone J.L., Duvall J.A., Perederiy J.V., Bomar J.M., Sebat J., Wigler M., Martin C.L., Ledbetter D.H. Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am. J. Hum. Genet. 2008;82:150–159. doi: 10.1016/j.ajhg.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arking D.E., Cutler D.J., Brune C.W., Teslovich T.M., West K., Ikeda M., Rea A., Guy M., Lin S., Cook E.H. A common genetic variant in the neurexin superfamily member CNTNAP2 increases familial risk of autism. Am. J. Hum. Genet. 2008;82:160–164. doi: 10.1016/j.ajhg.2007.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Friedman J.I., Vrijenhoek T., Markx S., Janssen I.M., van der Vliet W.A., Faas B.H., Knoers N.V., Cahn W., Kahn R.S., Edelmann L. CNTNAP2 gene dosage variation is associated with schizophrenia and epilepsy. Mol. Psychiatry. 2008;13:261–266. doi: 10.1038/sj.mp.4002049. [DOI] [PubMed] [Google Scholar]
- 26.Vernes S.C., Newbury D.F., Abrahams B.S., Winchester L., Nicod J., Groszer M., Alarcon M., Oliver P.L., Davies K.E., Geschwind D.H. A functional genetic link between distinct developmental language disorders. N. Engl. J. Med. 2008;359:2337–2345. doi: 10.1056/NEJMoa0802828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bucan M., Abrahams B.S., Wang K., Glessner J.T., Herman E.I., Sonnenblick L.I., Alvarez Retuerto A.I., Imielinski M., Hadley D., Bradfield J.P. Genome-wide analyses of exonic copy number variants in a family-based study point to novel autism susceptibility genes. PLoS. Genet. 2009;5:e1000536. doi: 10.1371/journal.pgen.1000536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feng J., Schroer R., Yan J., Song W., Yang C., Bockholt A., Cook E.H., Jr., Skinner C., Schwartz C.E., Sommer S.S. High frequency of neurexin 1beta signal peptide structural variants in patients with autism. Neurosci. Lett. 2006/11;409:10–13. doi: 10.1016/j.neulet.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 29.Glessner J.T., Wang K., Cai G., Korvatska O., Kim C.E., Wood S., Zhang H., Estes A., Brune C.W., Bradfield J.P. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459:569–573. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim H.G., Kishikawa S., Higgins A.W., Seong I.S., Donovan D.J., Shen Y., Lally E., Weiss L.A., Najm J., Kutsche K. Disruption of neurexin 1 associated with autism spectrum disorder. Am. J. Hum. Genet. 2008;82:199–207. doi: 10.1016/j.ajhg.2007.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marshall C.R., Noor A., Vincent J.B., Lionel A.C., Feuk L., Skaug J., Shago M., Moessner R., Pinto D., Ren Y. Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet. 2008;82:477–488. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rujescu D., Ingason A., Cichon S., Pietilainen O.P., Barnes M.R., Toulopoulou T., Picchioni M., Vassos E., Ettinger U., Bramon E. Disruption of the neurexin 1 gene is associated with schizophrenia. Hum. Mol. Genet. 2009;18:988–996. doi: 10.1093/hmg/ddn351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yan J., Noltner K., Feng J., Li W., Schroer R., Skinner C., Zeng W., Schwartz C.E., Sommer S.S. Neurexin 1alpha structural variants associated with autism. Neurosci. Lett. 2008;438:368–370. doi: 10.1016/j.neulet.2008.04.074. [DOI] [PubMed] [Google Scholar]
- 34.Verkerk A.J., Mathews C.A., Joosse M., Eussen B.H., Heutink P., Oostra B.A. CNTNAP2 is disrupted in a family with Gilles de la Tourette syndrome and obsessive compulsive disorder. Genomics. 2003;82:1–9. doi: 10.1016/s0888-7543(03)00097-1. [DOI] [PubMed] [Google Scholar]
- 35.Belloso J.M., Bache I., Guitart M., Caballin M.R., Halgren C., Kirchhoff M., Ropers H.H., Tommerup N., Tumer Z. Disruption of the CNTNAP2 gene in a t(7;15) translocation family without symptoms of Gilles de la Tourette syndrome. Eur. J. Hum. Genet. 2007;15:711–713. doi: 10.1038/sj.ejhg.5201824. [DOI] [PubMed] [Google Scholar]
- 36.Strauss K.A., Puffenberger E.G., Huentelman M.J., Gottlieb S., Dobrin S.E., Parod J.M., Stephan D.A., Morton D.H. Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2. N. Engl. J. Med. 2006;354:1370–1377. doi: 10.1056/NEJMoa052773. [DOI] [PubMed] [Google Scholar]
- 37.Jackman C., Horn N.D., Molleston J.P., Sokol D.K. Gene associated with seizures, autism, and hepatomegaly in an Amish girl. Pediatr. Neurol. 2009;40:310–313. doi: 10.1016/j.pediatrneurol.2008.10.013. [DOI] [PubMed] [Google Scholar]
- 38.Zahir F.R., Baross A., Delaney A.D., Eydoux P., Fernandes N.D., Pugh T., Marra M.A., Friedman J.M. A patient with vertebral, cognitive and behavioural abnormalities and a de novo deletion of NRXN1alpha. J. Med. Genet. 2008;45:239–243. doi: 10.1136/jmg.2007.054437. [DOI] [PubMed] [Google Scholar]
- 39.Sudhof T.C. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. 2008/10;455:903–911. doi: 10.1038/nature07456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Missler M., Sudhof T.C. Neurexins: three genes and 1001 products. Trends Genet. 1998;14:20–26. doi: 10.1016/S0168-9525(97)01324-3. [DOI] [PubMed] [Google Scholar]
- 41.Missler M., Zhang W., Rohlmann A., Kattenstroth G., Hammer R.E., Gottmann K., Sudhof T.C. Alpha-neurexins couple Ca2+ channels to synaptic vesicle exocytosis. Nature. 2003;423:939–948. doi: 10.1038/nature01755. [DOI] [PubMed] [Google Scholar]
- 42.Laumonnier F., Bonnet-Brilhault F., Gomot M., Blanc R., David A., Moizard M.P., Raynaud M., Ronce N., Lemonnier E., Calvas P. X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am. J. Hum. Genet. 2004;74:552–557. doi: 10.1086/382137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Durand C.M., Betancur C., Boeckers T.M., Bockmann J., Chaste P., Fauchereau F., Nygren G., Rastam M., Gillberg I.C., Anckarsater H. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 2007;39:25–27. doi: 10.1038/ng1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bourgeron T. A synaptic trek to autism. Curr. Opin. Neurobiol. 2009;19:231–234. doi: 10.1016/j.conb.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 45.Najm J., Horn D., Wimplinger I., Golden J.A., Chizhikov V.V., Sudi J., Christian S.L., Ullmann R., Kuechler A., Haas C.A. Mutations of CASK cause an X-linked brain malformation phenotype with microcephaly and hypoplasia of the brainstem and cerebellum. Nat. Genet. 2008;40:1065–1067. doi: 10.1038/ng.194. [DOI] [PubMed] [Google Scholar]
- 46.Bellen H.J., Lu Y., Beckstead R., Bhat M.A. Neurexin IV, caspr and paranodin–novel members of the neurexin family: encounters of axons and glia. Trends Neurosci. 1998;21:444–449. doi: 10.1016/s0166-2236(98)01267-3. [DOI] [PubMed] [Google Scholar]
- 47.Arroyo E.J., Xu T., Poliak S., Watson M., Peles E., Scherer S.S. Internodal specializations of myelinated axons in the central nervous system. Cell Tissue Res. 2001;305:53–66. doi: 10.1007/s004410100403. [DOI] [PubMed] [Google Scholar]
- 48.Poliak S., Salomon D., Elhanany H., Sabanay H., Kiernan B., Pevny L., Stewart C.L., Xu X., Chiu S.Y., Shrager P. Juxtaparanodal clustering of Shaker-like K+ channels in myelinated axons depends on Caspr2 and TAG-1. J. Cell Biol. 2003;162:1149–1160. doi: 10.1083/jcb.200305018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baumgartner S., Littleton J.T., Broadie K., Bhat M.A., Harbecke R., Lengyel J.A., Chiquet-Ehrismann R., Prokop A., Bellen H.J. A Drosophila neurexin is required for septate junction and blood-nerve barrier formation and function. Cell. 1996;87:1059–1068. doi: 10.1016/s0092-8674(00)81800-0. [DOI] [PubMed] [Google Scholar]
- 50.Poliak S., Gollan L., Martinez R., Custer A., Einheber S., Salzer J.L., Trimmer J.S., Shrager P., Peles E. Caspr2, a new member of the neurexin superfamily, is localized at the juxtaparanodes of myelinated axons and associates with K+ channels. Neuron. 1999;24:1037–1047. doi: 10.1016/s0896-6273(00)81049-1. [DOI] [PubMed] [Google Scholar]
- 51.Wagh D.A., Rasse T.M., Asan E., Hofbauer A., Schwenkert I., Durrbeck H., Buchner S., Dabauvalle M.C., Schmidt M., Qin G. Bruchpilot, a protein with homology to ELKS/CAST, is required for structural integrity and function of synaptic active zones in Drosophila. Neuron. 2006;49:833–844. doi: 10.1016/j.neuron.2006.02.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.