

Abstract
We report recessive mutations in the gene for the latent transforming growth factor-β binding protein 4 (LTBP4) in four unrelated patients with a human syndrome disrupting pulmonary, gastrointestinal, urinary, musculoskeletal, craniofacial, and dermal development. All patients had severe respiratory distress, with cystic and atelectatic changes in the lungs complicated by tracheomalacia and diaphragmatic hernia. Three of the four patients died of respiratory failure. Cardiovascular lesions were mild, limited to pulmonary artery stenosis and patent foramen ovale. Gastrointestinal malformations included diverticulosis, enlargement, tortuosity, and stenosis at various levels of the intestinal tract. The urinary tract was affected by diverticulosis and hydronephrosis. Joint laxity and low muscle tone contributed to musculoskeletal problems compounded by postnatal growth delay. Craniofacial features included microretrognathia, flat midface, receding forehead, and wide fontanelles. All patients had cutis laxa. Four of the five identified LTBP4 mutations led to premature termination of translation and destabilization of the LTBP4 mRNA. Impaired synthesis and lack of deposition of LTBP4 into the extracellular matrix (ECM) caused increased transforming growth factor-β (TGF-β) activity in cultured fibroblasts and defective elastic fiber assembly in all tissues affected by the disease. These molecular defects were associated with blocked alveolarization and airway collapse in the lung. Our results show that coupling of TGF-β signaling and ECM assembly is essential for proper development and is achieved in multiple human organ systems by multifunctional proteins such as LTBP4.
Introduction
The transforming growth factor betas (TGF-βs) compose a family of cytokines critical for regulating multiple cellular processes, including proliferation, differentiation, migration, and death.1 TGF-βs are generally secreted as a part of a large latent complex (LLC), consisting of the growth factor, its propeptide, and a latent TGF-β binding protein (LTBP), linked to the propeptide via two disulfide bonds.2 Release of TGF-β from the LLC, referred to as TGF-β activation because it is required for the binding of the cytokine to its receptors,3 occurs via proteolysis or integrin-mediated displacement. Within the LLC, the noncovalent association of the propeptide with TGF-ß confers latency, and the LTBP is thought to direct the latent TGF-ß and propeptide, the small latent complex (SLC), to specific sites in the extracellular matrix (ECM).
There are four known LTBP genes (LTBP1-4 [MIM 150390, 602091, 602090, 604710]), each yielding multiple LTBP isoforms by dual promoter use and alternative splicing.2 LTBP1 and LTBP3 bind all three isoforms of TGF-β, whereas LTBP4 binds only TGF-β1.4 LTBPs share structural homology with fibrillins and target the latent TGF-β complex to fibrillin microfibrils that are often associated with elastic fibers in the ECM.2 Thus, the LTBPs may serve as molecular modulators for the disposition of latent TGF-β and the assembly of other matrix proteins.
Elastic fibers consist of a scaffold of fibrillin microfibrils that surround elastin, a protein polymer of elastin precursors (tropoelastin) crosslinked by lysyl oxidase enzymes. Impaired elastic fiber formation leads to defective pulmonary, vascular, and connective tissue development. For example, deletions or point mutations in the elastin gene (ELN [MIM 130160]) lead to reduced amounts of elastin, causing supravalvular aortic stenosis either as a part of Williams-Beuren syndrome (WBS [MIM 194050]) or as an isolated familial disease (MIM 185500). Another class of mutations in ELN results in the synthesis and incorporation of mutant tropoelastin into the ECM, leading to disruption of the structure and function of elastic fibers in autosomal-dominant cutis laxa (ADCL [MIM 123700]).5–9
In addition to structural proteins and crosslinking enzymes, elastic fiber formation requires multiadhesive adaptor proteins, such as fibulin-410 and fibulin-5.11,12 Mutations in FBLN4 (EFEMP2 [MIM 604633])13,14 and FBLN5 (MIM 604580)15–17 cause autosomal-recessive cutis laxa type 1 (ARCL1 [MIM 219100]), associated with severe developmental emphysema and vascular malformations.
Recent human genetic and animal model studies have highlighted a close coupling of TGF-β signaling and elastic fiber integrity. Mutations in fibrillin-1 (FBN1 [MIM 134797]) cause Marfan syndrome (MFS [MIM 154700]),18 in which structural abnormalities in microfibrils lead to pathologically elevated TGF-β signaling.19 Conversely, in Loeys-Dietz syndrome (LDS [MIM 610168, 609192]), increased TGF-β signaling associated with TGF-β receptor mutations (TGFBR1 [MIM 190181] and TGFBR2 [MIM 190182]) leads to impaired elastic fiber deposition. Furthermore, in aortic tortuosity syndrome (ATS [MIM 208050]), SLC2A10 (MIM 606145) mutations lead to both elastic fiber disruption and altered TGF-β signaling.20
Here, we describe four patients with a constellation of lesions affecting the craniofacial, pulmonary, gastrointestinal, genitourinary, musculoskeletal, and integumentary systems and identify recessive mutations in LTBP4 as a cause. Furthermore, we show that elevated TGF-β signaling and disruption of the elastic fibers are key molecular characteristics of the disease process.
Subjects and Methods
The subjects of this study participated after giving their informed consent. This study was approved by the Human Research Protection Office (institutional review board) of the Washington University School of Medicine. Tissue samples were collected as skin biopsies or as autopsy material. Normal control tissue samples were provided by the Cooperative Human Tissue Network, which is funded by the National Cancer Institute. Other investigators may have received samples from the same subjects.
Patient 1
Patient 1 was born to second-cousin parents of Hispanic ancestry, at a gestational age of 38.7 wks, by a Cesarean section performed after breech presentation in an uneventful pregnancy. Length, weight, and head circumference percentiles at birth were 64th, 40th, and 90th, respectively. Physical examination showed lax, redundant, and inelastic skin, especially of the shoulder and neck region (Figure 1C) and the extremities (Figure 1D). The patient had hypertelorism (Figure 1A), microretrognathia, and dolichocephaly (Figure 1B). The fontanelles were large. The cheeks and the periorbital region were puffy (Figure 1A), the philtrum long, and the right earlobe folded. The patient had widely spaced first and second toes (Figures 1D and 1E) and bilateral transverse plantar creases (Figure 1E). He had a hoarse cry and a low muscle tone. Although initial serum ceruloplasmin level was low, repeat ceruloplasmin and copper tests performed several times showed results within normal limits. Hair shafts were normal; no abnormal twisting or angulation was observed.
Figure 1.
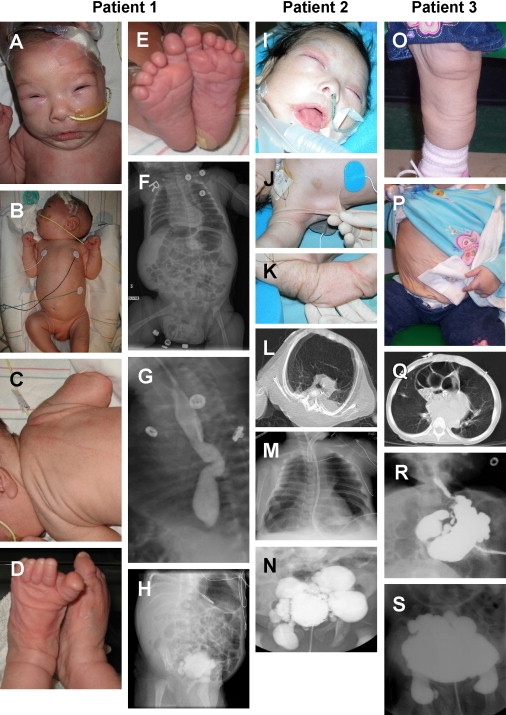
Clinical Manifestations of Patients with LTBP4 Mutations
(A–E) Characteristics of patient 1 at 1 mo of age included (A and B) flattened midface with a wide nasal bridge, long philtrum, micrognathia, swollen eyelids, receding forehead and dolichocephaly, (C) redundant skin on the trunk and (D) extremities, and (E) widely spaced first and second toes with transverse plantar creases.
(F) X-ray imaging demonstrated dilated intestines, laterally displaced liver, compressed chest, and inguinal hernia.
(G) Upper gastrointestinal imaging highlighted esophageal tortuosity.
(H) Contrast imaging of the bladder showed severe diverticulosis.
(I–K) Clinical pictures of patient 2 at 2 mo of age showed (I) craniofacial features similar to patient 1, as well as (J and K) cutis laxa.
(L) CT image at 3 mo showing hyperinflation of the anterior and atelectasis of the posterior lung.
(M) X-ray image showing bell-shaped chest, atelectasis of the right lung, and hyperinflation of the left lung.
(N) Severe bladder diverticulosis.
(O and P) Clinical photographs of Patient 3 taken at 3 yrs 1 mo illustrate cutis laxa with premature aged appearance throughout the body, including (O) the legs and (P) the abdomen. The patient also had a gastrostomy tube and tracheostomy.
(Q) Chest CT image of parasternal diaphragmatic hernia with cystic changes and atelectasis of the lung at 3 yrs 6 mo of age.
(R) Upper gastrointestinal contrast image showing stomach diverticula at 3 yrs 6 mo of age.
(S) Urinary tract contrast imaging showing bladder diverticula at 2 yrs 4 mo of age.
Respiratory and feeding difficulties prompted detailed clinical investigations within the first 2 mo of life, when the patient was in the neonatal intensive care unit. A gastrostomy tube was placed, but weight gain and growth remained slow. The length, weight, and head circumference percentiles at the age of 55 days were 37th, 36th, and 36th, respectively. Bronchoscopy results were normal, but chest X-ray showed occasional changes consistent with pneumonia.
Echocardiography showed moderate hypoplasia of the left pulmonary artery (pressure gradient 64 Hgmm, peak velocity 5.8 m/s), mild hypoplasia of the right pulmonary artery (pressure gradient 4 Hgmm), and mild right ventricular hypertrophy. Heart murmur consistent with peripheral pulmonary stenosis was also detected. Abdominal ultrasound showed normal caliber of the aorta and renal arteries.
Distension of the abdomen suggested gastrointestinal abnormalities. Subsequent X-ray images showed dilated intestinal structures (Figure 1F). Upper gastrointestinal contrast imaging demonstrated a dilated, tortuous esophagus (Figure 1G) but neither tracheoesophageal fistula nor vascular ring. Gastroesophageal reflux was absent by pH probe measurements. The second portion of the duodenum was also tortuous. A cystogram showed severe diverticulosis and thickened bladder wall (Figure 1H). Renal ultrasound revealed severe left hydronephrosis, renal cortical thinning, and dilated posterior urethra. He had both inguinal (Figure 1F) and umbilical hernias. After being discharged from the intensive care unit, the patient was hospitalized five times over a period of 6 mo for respiratory distress, pneumonia, and atelectasis. Mechanical ventilation was advised against because of the potential for barotrauma on the lung tissue. After his last hospitalization, at 8 mo, the patient was discharged to hospice care, and he died at home when he was 9 mo old. An autopsy was not performed.
Patient 2
Patient 2 was born at term to unaffected and unrelated parents of Mexican origin (Figure 2C). Birth length, weight, and head circumference were at the 75th–90th, >90th, and 75th–90th percentiles, respectively. He was noted at birth to have lax, redundant, and inelastic skin throughout the body (Figures 1J and 1K). He had a flattened midface with a receding forehead, wide nasal bridge, hypertelorism, long philtrum, micrognathia, and swollen eyelids (Figure 1I). His chest was bell-shaped (Figure 1M), with a prominent xiphoid. His ear canals were difficult to visualize as a result of his skin redundancy, and he failed the newborn hearing screen twice. He had hyperextensible small joints and decreased muscle tone. A wide space was also noted between his first and second toes, with a vertical crease on his soles starting between the first and second toes, extending over 2/3 of his soles. He was referred to pediatric dermatology for his skin condition at the age of 10 days and was diagnosed with cutis laxa. At that time, he was thriving without respiratory or feeding difficulties. At 20 days, he required surgery for pyloric stenosis. During this admission, a murmur was noted, and an echocardiogram was performed, showing normal anatomy with a patent foramen ovale. Because of the cutis laxa, a voiding cystourethrogram (VCUG), skeletal survey, and skull X-ray were performed. The VCUG demonstrated innumerable bladder diverticula of varying sizes (Figure 1N). Aside from bilateral hindfoot varus deformity, the skeletal survey was normal. The skull X-ray showed mandibular hypoplasia and decreased mineralization with widened cranial sutures. A hearing screen performed during this admission was normal. He demonstrated good oral intake and was discharged home.
Figure 2.
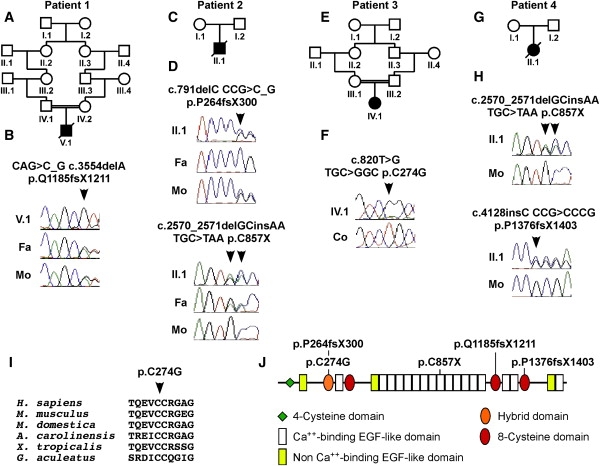
Mutations Found in LTBP4
(A) The parents of patient 1 were second cousins.
(B) DNA sequencing showed that patient 1 (V.1) was homozygous for mutation p.Q1185fsX1211, whereas his unaffected father (Fa) and mother (Mo) were both heterozygous.
(C) The parents of patient 2 were unaffected and unrelated.
(D) Patient 2 (II.1) was compound heterozygous for mutation p.P264fsX300 inherited from his mother (Mo) and mutation p.C857X inherited from his father (Fa).
(E) Patient 3 was an offspring of first-cousin parents.
(F) Homozygous mutation p.C274G (arrowhead) in patient 3 (IV.1) compared to normal sequence in a control individual (Co).
(G) The pedigree of patient 4.
(H) Patient 4 (II.1) was compound heterozygous for mutations p.C857X and p.P1376fsX1403. Neither of these mutations was found in his mother (Mo), suggesting that at least one of the mutations was de novo. No DNA sample was available from the father.
(I) The cysteine residue replaced by the mutation p.C274G found in patient 3 is highly conserved in all vertebrates.
(J) Schematic representation of the domain structure of LTBP4, showing the location of the mutations.
At 7 wks of age, he was readmitted to the hospital with tachypnea, hypoxia, and hypercapnia. On bronchoscopy, he was noted to have floppy upper airways, laryngomalacia, tracheomalacia, and bronchomalacia. His respiratory status worsened, and he required intubation and eventually tracheotomy. Chest computed tomography (CT) (Figure 1L) and X-ray (Figure 1M) showed hyperinflation with areas of atelectasis and cystic change. He showed poor weight gain, and both weight and length fell below the 3rd percentile when he was 2 mo of age, despite his being on high-calorie formula. A gastrostomy tube was placed for prolonged enteral feeding. Bilateral inguinal hernias were also observed, and additional imaging demonstrated tortuous vessels, including the internal jugular veins and inferior vena cava.
Despite positive and negative pressure ventilation, the patient continued to have difficulty with both oxygenation and ventilation. His respiratory function worsened, and he died at 4 mo. At autopsy, the right upper lobe of the lung was hyperinflated and extended into the left superior anterior hemithorax. The remainder of the lungs showed mixed areas of hyperinflation and atelectasis. The bronchial tree showed situs ambiguous with bilateral left-sided architecture. Multiple urinary bladder diverticula, bilateral congested renal cortices, mild left hydronephrosis, and bilateral adrenal hypoplasia were observed. The peritoneal cavity contained 510 ml ascites. The ascending colon had diverticula, and the ileocecal junction lacked the valve and was dilated. Osteopenia, thymic hypolasia and left sided accessory spleen (1 cm) were found.
Patient 3
Patient 3 was born to first-cousin parents (Figure 2E) of Palestinian ancestry, by spontaneous vaginal delivery. Birth length, weight, and head circumference were at the 10th–25th, 2nd, and 5th percentiles, respectively. The patient had respiratory distress at birth, and chest X-ray was suspicious for congenital diaphragmatic hernia. Exploratory laparotomy revealed a paraesophageal hernia, gastric strangulation, and the presence of a transverse colon in the chest. Reduction and repair of the defect were performed. Other neonatal findings included excessive skin laxity, bladder diverticula (Figure 1S), prolapse of the urethra and cervix, and pelvic floor herniation. Voiding cystourethrogram demonstrated spontaneous voiding without reflux or obstruction. The patient was discharged but was subsequently admitted at the age of 4 wks as a result of respiratory distress and was placed on an apnea monitor.
At 6 wks of age, the patient was intubated as a result of apnea, respiratory distress, and a right upper lobe infiltrate. A barium study showed evidence for gastresophageal reflux. Laparoscopic Nissen fundoplication and gastrostomy tube placement were performed. Because of ongoing respiratory problems, the patient eventually underwent tracheostomy. She receives bilevel positive airway pressure (BiPAP) ventilation and supplemental oxygen at night. A CT scan at 3.5 yrs showed Morgagni (parasternal) diaphragmatic hernia (Figure 1Q). Upper gastrointestinal contrast imaging showed severe diverticulosis of the stomach (Figure 1R). She did not start walking until after 3 yrs of age. At 7 yrs of age, she walks but fatigues easily and has difficulty jumping or running. She uses a speaking valve and can say about 70–80 words and form short, complete sentences. She is in kindergarten, can count to 17, and knows her letters, shapes, and colors. The physical exam is significant for generalized cutis laxa (Figures 1O and 1P), inverted V-shaped eyebrows, long philtrum, and a premature aged appearance.
Patient 4
Patient 4 was born to unaffected and unrelated parents (Figure 2G), at 38 wks gestation, by vertex vaginal delivery. Length, weight, and head circumference were at the >90th, 75th, and 40th percentiles, respectively. Apgar scores were 8 and 9. Respiratory status was initially normal, but oxygen saturations of 80%–90% prompted transfer to the neonatal intensive care unit. Mild respiratory distress ensued, necessitating oxygen, which was weaned over a 2–3 day period, consistent with transient tachypnea of the newborn.
Genetics was consulted on day 4 of life, for hypotonia, poor feeding, and skin laxity. The family history was noteworthy for several individuals on the maternal side with skin laxity at birth, which subsequently improved, resolving by adulthood. Therefore, a dominant form of cutis laxa was initially suspected. There was also a history of congenital heart disease, one maternal aunt dying from a “three-chambered heart” at 2 wks of age and a second aunt having heart disease that was surgically repaired. Two maternal aunts also had a history of large abdominal-wall defects with extrusion of abdominal contents. Despite a positive maternal family history, the mother did not carry any of the mutations found in patient 4 (Figure 2H). Thus, we concluded that the birth defects in maternal relatives were unrelated to the LTBP4 mutations found in the patient.
Physical examination revealed a mildly dysmorphic child with retrognathia, umbilical hernia, widely spaced first and second toes, and a unilateral transverse palmar crease. Loose skin that could be easily pulled 3–4 cm from the underlying surface was evident on the abdomen, legs, and thighs. Chromosomal analysis, methylation studies for Prader-Willi syndrome, echocardiogram, electrocardiogram, and magnetic resonance imaging of the brain were all normal.
Vomiting during the first 2 wks prompted an evaluation for obstruction, which revealed pyloric stenosis, leading to pyloromyotomy. The patient was discharged at 6 wks of age after the feeding problems resolved.
The patient was subsequently hospitalized because of respiratory distress at 3 mo of age and was found to have viral pneumonia, a right-sided Bochdalek diaphragmatic hernia, and rectal prolapse. During a complicated hospital course, which included a long stay in the intensive care unit, she had a variety of problems and procedures, including rhinovirus, parainfluenza pneumonia, pulmonary hypertension, and surgical repair of the diaphragmatic hernia, umbilical hernia, and rectal prolapse. A gastrostomy tube was placed for nutrition and medications. Given her tracheomalacia, a tracheostomy was recommended. However, because of her poor prognosis, the family opted for “do not resuscitate” status and hospice care, and she was discharged with ongoing oxygen supplementation through a nasal cannula.
Physical examination when the patient was 7 mo of age revealed a weight of 5.3 kg (< 3rd percentile) and a length of 64 cm (8th percentile). She exhibited markedly increased skin laxity, with redundant skin that could easily be stretched 4–5 cm from the surface hanging from her face and other areas of her body. Her abdomen was protuberant, but soft. The joints were mildly hyperextensible. The patient was stable until age 23 mo, when she died suddenly at home after an acute aspiration event.
Mutational Analysis
Peripheral blood or cultured skin fibroblasts were used as a source of DNA. Samples from the probands were subjected to mutational analysis of the entire coding region of LTBP4 and flanking noncoding sequences. Intron-derived primers for PCR amplification of all 35 LTBP4 exons were synthesized with the use of sequence information obtained from the reference sequence (GenBank accession number NT_011109). Intron and exon boundaries were deduced from information available in the database and by comparison with the LTBP4 cDNA isoforms (NM_001042544, NM_003573, NM_001042545). Primers were designed with the Vector NTI Advance program, version 9.1, on the basis of the annotated genomic sequence across each exon, including regions of at least 80–100 bp of flanking intervening sequence (primer sequences available upon request). PCR amplifications were performed in a 25 μl volume with 100 ng of genomic DNA, 25 μM of each primer, 200 μM dNTPs, 1 M betaine, optimized KTLA buffer pH 9 or 7.9, and 0.05 U of KlenTaq LA DNA polymerase (DNA polymerase facility, Washington University, WA, USA). Exon 6 was amplified with the use of platinum Pfx polymerase (Invitrogen, Carlsbad, CA, USA). Standard PCR cycle conditions were performed, and amplicons were visualized by agarose gel electrophoresis. PCR products were treated with 2 U each of exonuclease I and shrimp alkaline phosphatase (USB, Cleveland, OH, USA) for the removal of unincorporated dNTPs and excess primers. Purified PCR-amplification products were sequenced with BigDye Terminator Chemistry and were electrophoresed on ABI 3730XL (Applied Biosystems, Foster City, CA, USA). Sequence traces of both the sense and the antisense strands of the PCR-amplified fragments were compared with the reference gene sequences with Sequencher 4.5 software (GeneCodes, Ann Arbor, MI, USA). DNA sequencing of two independent amplification products confirmed mutations in each case. The inheritance of mutations was confirmed by sequence analysis of parental DNA, if available. The numbering of mutant DNA positions was based on NT_011109 (genomic DNA) and NM_003573 (cDNA).
RNA Expression Analysis
Fibroblasts were cultured to confluence at passages 2–8 in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 25 mM HEPES, L-glutamine, and antibiotics. Parallel cultures were incubated with 100 μg/ml cycloheximide for study of nonsense-mediated decay. Confluent cultures were used for extraction of RNA with TRI-reagent (Molecular Research Center, Cincinnati, OH, USA). Contaminating genomic DNA was removed with the Turbo DNA-free Kit (Ambion, Austin, TX, USA). First-strand cDNA was synthesized from total RNA samples (1 μg each) with the use of SuperScript III reverse transcriptase (Invitrogen). We performed reverse transcriptase-PCR (RT-PCR) by using the cDNA templates to amplify desired regions of LTBP4 for the determination of possible splicing changes caused by nonsense and frameshift mutations. After visualization on a 1.2% agarose gel, bands were purified with the QIAquick Gel Extraction Kit (QIAGEN, Valencia, CA, USA) and were subjected to direct sequencing. PCR primers were designed at least one exon away from the mutation in question. Quantitative PCR was performed with iQ SYBR Green Supermix and an iCycler iQ PCR System (Bio-Rad, Hercules, CA, USA) for the measurement of LTBP4 and TGFB1 (MIM 190180) expression. ACTB (β-actin [MIM 102630]) was used as a reference transcript (primer sequences available upon request). Experiments were performed in triplicate. “No RT” samples were used as a PCR control measure for gDNA contamination. We performed ΔCt analysis to assess relative RNA expression levels, by first calculating the difference between the threshold cycle number (Ct) value of the target gene (LTBP4, TGFB1) and the average Ct value of the corresponding ACTB sample. We estimated relative mRNA abundance by calculating 2−ΔCt.
Immunoblotting
Cell cultures were washed with serum-free media and incubated with fresh serum-free media for 24 hr. The conditioned media (CM) were collected, and protease inhibitors were added. The CM were clarified by centrifugation and stored at −80°C. Proteins in conditioned media samples were separated by SDS-PAGE with the use of 4%–20% gradient polyacrylamide mini gels and transferred to nitrocellulose membranes by electroblotting. Membranes were blocked in PBS/0.1% Tween-20 containing 5% nonfat milk for 1 hr at room temperature and then incubated with primary antibodies specific for LTBP-1 or LTBP-4 overnight at 4°C. The membranes were washed five times in PBS/0.1% Tween-20 and incubated in the appropriate secondary antibody for 1 hr at room temperature. After washing, the blots were developed by chemiluminescence with the use of enhanced chemiluminescence (ECL) (Thermo Scientific, Waltham, MA, USA) detection reagents.
Immunocytochemistry
For analysis of the expression of ECM proteins, cells were plated on coverslips in 6-well plates and grown for at least 2 wks past confluence. Postconfluent cultures were washed once with PBS, fixed with 100% ethanol at room temperature for 10 min, and processed for standard indirect immunofluorescence. Fixed cells were washed three times in PBS and were blocked with 1% goat serum in PBS for 1 hr at room temperature. Fibroblasts were stained for double immunofluorescence with the use of primary antibodies against LTBP1 (Ab39) or LTBP4 (pAb2101) together with mAb69, specific for FBN1. After three washes with PBS, the cells were incubated with secondary antibodies: anti-mouse Alexa Fluor 488 and anti-rabbit Alexa Fluor 594 (Invitrogen). Nuclei were stained with DAPI. The coverslips were mounted on glass slides with Prolong Gold anti-fading reagent (Invitrogen) and examined under the Axioskop 2 Mot Plus imaging microscope (Zeiss, Thornwood, NY, USA) with the use of a 40× objective. Images were acquired with an AxioCam MRm camera (Zeiss) and AxioVision 3.1 software (Zeiss).
TGF-β Activity Assay
Reporter mink lung epithelial cells (MLECs), which produce luciferase in response to TGF-β, were used as previously described.21 MLECs and test cells were each suspended at 5 × 105/mL in DMEM containing 2.5% FBS. MLECs were plated first at 100 μL/well (96-well plates), and 100 μl of test cell suspensions or DMEM supplemented with 2.5% FBS were added. TGF-β signaling was inhibited by addition of the ALK5 inhibitor, SB431542 (Sigma, St. Louis, MO, USA), to 5 mM final concentration. Cells were cocultured for 16–20 hr. Lysates were assayed for luciferase activity as described previously.21 Experiments were repeated three times, with similar results observed each time.
Histology
Skin tissue in paraffin blocks were cut to 5-μm-thick sections. Sections were deparaffinized in Histo-Clear (National Diagnostics, Atlanta, GA, USA) and rehydrated. All histological sections were stained with hematoxylin-eosin. For elastic fiber staining, sections were subjected to Hart's elastin stain. In brief, sections were immersed in 0.25% potassium permanganate solution for 5 min. Slides were cleared in 5% oxalic acid and soaked in resorcin-fuchsin solution overnight. After washing in water, sections were counterstained with tartrazine (yellow), dehydrated in ethanol, cleared in xylene, and mounted. Sections were viewed under bright-field optics with a BX60 microscope (Olympus, Center Valley, PA, USA).
Electron Microscopy
Biopsies were fixed in glutaraldehyde, stained sequentially with OsO4, tannic acid, and uranyl acetate, dehydrated, and embedded in Epon.22 Thin sections (60 nm) were cut, placed on formvar-coated grids, and counterstained with 7% methanolic uranyl acetate and lead citrate. Sections were viewed with a Tecnai 12 transmission electron microscope at 120 kV, and images were digitally captured.
Statistical Analysis
Group means and standard errors of means were calculated for continuous variables, including relative gene expression and TGF-β activity data. Unpaired t tests for samples with unequal variance were used for investigating whether pairwise differences of group means were significant (p < 0.05).
Results
LTBP4 Mutations
While performing comparative electron microscopic analysis of tissue samples from human patients with connective tissue diseases and from mice lacking the short form of Ltbp4 (Ltbp4S), we noted that these mice showed elastic fiber pathology that was strikingly similar to that of a patient (patient 1) who had cutis laxa with lethal pulmonary complications and gastrointestinal and urinary involvement. Moreover, Ltbp4S−/− mice also developed severe pulmonary and gastrointestinal disease,23,24 similar to patient 1. Therefore, we reasoned that patient 1 might have an alteration in LTBP4. We sequenced all exons of LTBP4, and we identified a homozygous mutation, p.Q1185fsX1211, in patient 1 (Figure 2B).
To match the phenotype of patient 1, we selected nine probands from a cohort of 100 probands with cutis laxa on the basis of the presence of severe pulmonary, gastrointestinal, and urinary involvement. Mutational analysis of LTBP4 and detailed clinical follow up remains incomplete in four of the nine patients. Thus, these four patients were removed from the study. Of the remaining five patients, three (patients 2–4) were positive and two were negative for LTBP4 mutations. Patients 1–4 had either homozygous or compound heterozygous mutations. Each homozygous patient was an offspring of a consanguineous union (Figures 2A and 2E). Patients 1 and 2 inherited one mutation from each parent. In contrast, the mother of patient 4 was negative for both of the mutations found in the patient, indicating that at least one of the mutations was de novo.
Four of the five identified mutations (Figure 2, Table 1) were predicted to lead to premature termination codons (p.P264fsX300, p.C857X, p.Q1185fsX1211, and p.P1376fsX1403), whereas one replaced a highly conserved cysteine in the hybrid domain of LTBP4 with glycine (p.C274G; Figures 2I and 2J). Interestingly, four of the five mutations were located in either a hybrid or an 8-cysteine domain, domains known to have long-range effects on fibrillin and LTBP conformation.25,26
Table 1.
LTBP4 Mutations
| Patient | Ethnicity | Exon | Domain | Status | Gene | cDNA | Protein |
|---|---|---|---|---|---|---|---|
| 1 | Hispanic | 28 | 8-CYS 2 | hom | g.29481delA | c.3554delA | p.Q1185fsX1211 |
| 2 | Mexican | 9 | HYB | het | g.12574 | c.791delC | P264fsX300 |
| 22 | EG 11 | het | g.25287_25288delGCinsAA | c.2570_2571delGCinsAA | p.C857X | ||
| 3 | Palestinian | 9 | HYB | hom | g.12603T>G | c.820T>G | p.C274G |
| 4 | Hispanic | 22 | EG 11 | het | g.20287_20288delGCinsAA | c.2570_2571delGCinsAA | p.C857X |
| 33 | 8-CYS 3 | het | g.33861insC | c.4128insC | p.P1376fsX1403 |
Abbreviations are as follows: EG, EGF-like domain; HYB, hybrid domain; 8-CYS, 8-cysteine domain; het, heterozygous; hom, homozygous.
Patients 2, 3, and 4 have undergone extensive mutational screening of known cutis laxa genes FBLN5 and FBLN4 (EFEMP2) and of candidate genes FBLN1 (MIM 135820), FBLN2 (MIM 135821), FBLN3 (EFEMP1 [MIM 601548]), SLC2A10 (GLUT10), LOX (MIM 153455), LOXL1 (MIM 153456), and LOXL2 (MIM 606663), all with negative results. FBLN5 mutations were also excluded in patient 1.
Clinical Features
The shared clinical characteristics among the patients positive for LTBP4 mutations are summarized in Figure 1 and Table 2. Craniofacial features included receding forehead, microretrognathia, wide fontanelles, periorbital swelling, hypertelorism, flat midface, and wide nasal bridge. Patients had severe infantile bronchopulmonary dysplasia with developmental emphysema, cystic, and atelectatic abnormalities, hypoplastic lungs, and susceptibility to pneumonia. Respiratory lesions were complicated by laryngomalacia, tracheomalacia, bronchomalacia, diaphragmatic hernia, and pulmonary hypertension with vascular stenoses at various levels of the pulmonary vessels. Three patients died of respiratory failure in infancy or in early childhood. Gastrointestinal anomalies included tortuosity, diverticulosis, and dilation of the digestive tract, pyloric stenosis, and umbilical hernias. Genitourinary malformations were severe, with bladder diverticulosis and hydronephrosis and inguinal hernias. Musculoskeletal abnormalities included joint laxity, muscle weakness, and widely spaced first and second toes with plantar creases. All patients had loose, redundant, and inelastic skin. Postnatal growth was impaired.
Table 2.
Clinical Features Associated with LTBP4 Mutations
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | |
|---|---|---|---|---|
| Skin | ||||
| Cutis laxa | + | + | + | + |
| Craniofacial | ||||
| Long philtrum | + | + | + | ? |
| Flat midface | + | + | + | ? |
| Receding forehead | + | + | ? | + |
| Periorbital swelling | + | + | ? | ? |
| Hypertelorism | + | + | ? | ? |
| Wide nasal bridge | + | + | ? | ? |
| Retrognathia, micrognathia, or mandibular hypoplasia | + | + | + | + |
| Wide sutures or fontanelles | + | + | ? | − |
| Pulmonary | ||||
| Tachypnea, respiratory distress | + | + | + | + |
| Tracheal tube | − | + | + | − (declined) |
| Lanryngo-, tracheo-, or bronchomalacia | − | + | ? | + |
| Emphysema or hypoplastic lung | + | + (1) | ? | + (1) |
| Diaphragmatic hernia or eventration | − | + (2) | 2× (3, 4) | + (5) |
| Pneumonia | + | − | − | + |
| Cardiovascular | ||||
| Pulmonary artery stenosis | + (6) | − | ? | + (7) |
| Patent foramen ovale | + | + | − | ? |
| Gastrointestinal | ||||
| Gastro-esophageal reflux | − | − | + | + |
| Diverticula | − | a. colon | gastric | ? |
| Gastric tube | + | + | + | + |
| Pyloric stenosis | − | + | − | + |
| Intestinal dilation, tortuosity | + (8) | + (9) | ? | − |
| Umbilical hernia | + | − | − | + |
| Rectal prolapse | − | − | + | + |
| Genitourinary | ||||
| Bladder diverticula | + | + | + | ? |
| Hydronephrosis | + (left) | + | − | ? |
| Inguinal hernia | + | bilateral | − | − |
| Musculoskeletal | ||||
| Joint laxity | + | + | + | + |
| Low muscle tone | + | + | ? | + |
| Widely spaced first and second toes | + | + | ? | + |
| Plantar crease | + | + | ? | − |
| Growth Delay | ||||
| Prenatal | − | − | + | − |
| Postnatal | + | + | + | + |
| Survival | ||||
| Age at death (mo) | 9 | 4 | − | 26 |
| Cause of death | resp | resp | − | resp |
| Lab | ||||
| Ceruloplasmin (mg/dL) | normal (10) | 13 (normal) | ? | ? |
| Copper (μg/L) | normal | 384 (low) | ? | normal |
| Chromosomes | ? | normal | normal | normal |
Data are indicated as follows: +, present; -, absent; ?, unknown; 2×, recurrent; a. colon, ascending colon; resp, respiratory; (1) hyperinflation, atelectasis, cystic change; (2) eventration; (3) hiatal hernia; (4) Morgagni hernia; (5) Bochdalek hernia; (6) left, right, and peripheral pulmonary stenoses, right ventricular hypertrophy; (7) pulmonary valve and infundibular stenosis, pulmonary hypertension; (8) tortuous duodenum; (9) dilated ileocecal junction, lacks valve; (10) initially low.
The two patients who were negative for LTBP4 mutations had dermal, pulmonary, gastrointestinal, and urinary lesions similar to those of mutation-positive individuals. However, one of the mutation-negative patients had severe enlargement of the aortic root, a lesion not observed among mutation-positive subjects.
Reduced Synthesis, Secretion, and Extracellular Deposition of LTBP4
We assessed the effect of mutations on LTBP4 biosynthesis by using skin fibroblasts from patients 1, 2, and 4, whose mutations were predicted to lead to premature termination codons. Fibroblasts were not available from patient 3, who was homozygous for missense mutation p.C274G. Quantitative RT-PCR analysis showed greatly reduced LTBP4 expression in each patient sample (5% of normal levels on average), suggesting that the premature termination mutations led to the destabilization of the mRNA via the nonsense-mediated decay pathway (Figure 3A). TGF-β1 mRNA levels were unaltered in the patient fibroblasts (Figure 3A).
Figure 3.
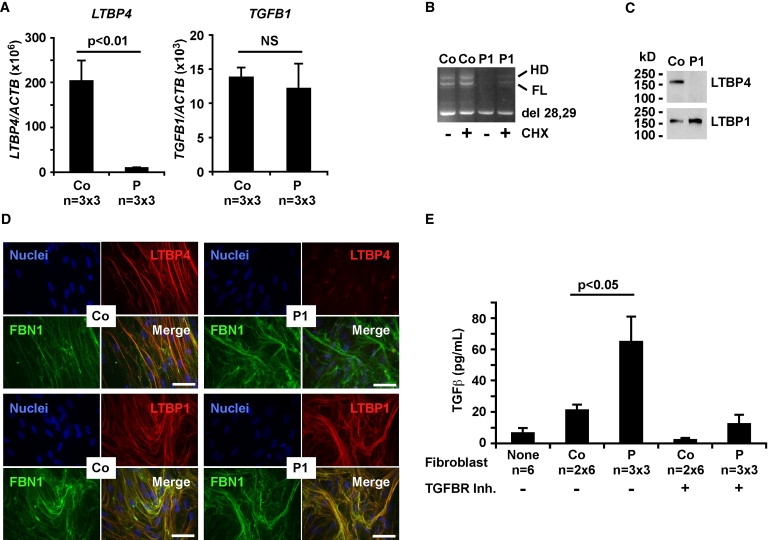
Functional Consequences of LTBP4 Mutations In Vitro
(A) Quantitative RT-PCR analysis showed severely reduced expression of LTBP4 in skin fibroblasts from three patients (1, 2, and 4) in comparison to three controls, triplicates each. Results were normalized to β-actin (ACTB) and expressed as group averages ± standard errors of means. The expression of TGF-β1 (TGFB1) was not altered significantly (NS) in patients.
(B) Patient 1 (P1) expressed low levels of full-length (FL) LTBP4 in comparison to control (Co) fibroblasts. Cycloheximide (CHX) treatment (+) partially rescued the expression of the FL isoform. Del 28,29 shows the exon 28, 29 skipping product, HD indicates a heteroduplex.
(C) Immunoblotting confirmed severely reduced LTBP4 levels in conditioned media of fibroblasts from patient 1 (P1). LTBP1 secretion was normal in both control (Co) and patient fibroblasts.
(D) Dual immunostaining in patient 1 (P1) shows an abundant fibrillin-1 (FBN1, green) network but very little staining for LTBP4 (red). Control cells (Co) show strong colocalization of FBN1 and LTBP4 (yellow, orange). LTBP1 (green) and FBN1 (red) were strongly colocalized in both patient and control cells. Nuclei were counterstained with DAPI (blue). Magnification bars represent 50 μm.
(E) Measurement of TGF-β signaling by coculturing of reporter cells with fibroblasts from patients 1, 2, and 4 (P, triplicates each) or two controls (Co, six replicates each). Negative controls (None, six replicates) were reporter cells only. The presence of TGF-β receptor inhibitor (TGFBR Inh) is shown by a + sign and absence by a − sign. Luminescence units were converted to TGF-β concentration with the use of a TGF-β1 standard curve. Significant difference is shown by a p value (t test) and error bars show standard errors of means.
Exons 28 and 29 encode the second 8-cysteine domain, which interacts with latent TGF-β1. However, in some tissues, these two exons can be spliced out, yielding an LTBP4 isoform unable to bind latent TGF-β127. This isoform (del 28,29) is abundantly expressed in skin fibroblasts. Because the mutation in patient 1, p.Q1185fsX1211, was located in exon 28 and was predicted to cause premature termination in exon 29, we studied the splicing of these exons in patient and control fibroblasts. The expression of the full-length (FL) LTBP4 isoform was significantly reduced in patient 1 in comparison to that observed in a control (Figure 3B). Treatment with cycloheximide, an inhibitor of nonsense-mediated decay, partially rescued the expression of FL LTBP4, providing evidence for mutation-induced destabilization of this isoform in patient 1 (Figure 3B). Consistent with reduced mRNA levels, conditioned media from patient 1 fibroblasts contained diminished amounts of LTBP4 but normal levels of LTBP1 (Figure 3C).
LTBP4 targets latent TGF-β to fibronectin and fibrillin-1 (FBN1)-containing microfibrils.28 Therefore, we examined the ability of patient fibroblasts to incorporate LTBP4 into the ECM. Whereas control fibroblasts showed strong colocalization of FBN1 and LTBP4, FBN1 microfibrils lacked LTBP4 in the ECM of cells from patient 1 (Figure 3D). In contrast, LTBP1 was efficiently incorporated into FBN1 microfibrils in both patient and control cells (Figure 3D). Given the significant reduction in LTBP4 expression at the mRNA level, we expect that incorporation of LTBP4 into the ECM is also severely impaired in patients 2 and 4.
Increased TGF-β Activation
To investigate the effect of LTBP4 mutations on latent TGF-β activation and signaling, we cocultured TGF-β reporter cells with fibroblasts from patients 1, 2, and 4 and from two controls. Patient cells produced significantly higher levels of active TGF-β than did control cells. TGF-β activity was greatly reduced by a chemical inhibitor (SB431542) of the TGF-β receptor I kinase (Figure 3E). However, TGF-β1 mRNA levels were normal in the patients (Figure 3A), suggesting that the release and/or storage, but not synthesis, of TGF-β were abnormal in LTBP4-deficient cells. Thus, loss of LTBP4 causes increased active TGF-β by altering TGF-β bioavailability in the ECM.
Abnormal Elastic Fiber Formation
Previous studies of Ltbp4S−/− mice23,24 revealed significant disruption of elastic fibers in multiple tissues (Figure 4M). Therefore, we investigated the distribution and morphology of elastic fibers and the overall histopathology of affected organs in our patients. In control skin (Figure 4A), elastic fibers were robust in both the deep and upper dermis. Conversely, in the skin of patients 1 (Figure 4B) and 2 (Figure 4C), the fine candelabra-like elastic fibers in the upper dermis were missing. Also, dermal elastic fibers were fragmented and weakly stained and had less-defined edges in patients than in controls. Skin tissue was not available from patients 3 and 4.
Figure 4.
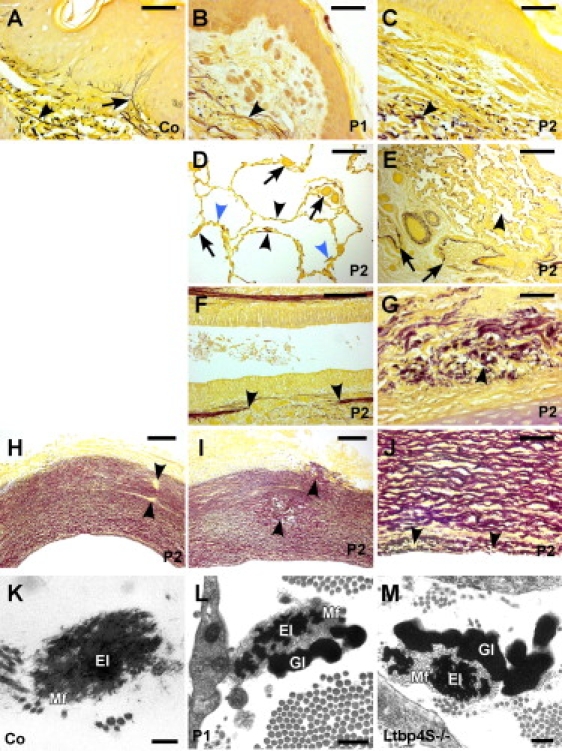
Impaired Elastic Fiber and Tissue Architecture in Patients with LTBP4 Mutations
(A) Hart's elastin stain of the skin from a 9-mo-old control (Co) shows robust, strongly stained horizontal elastic fibers in the deep dermis (arrowhead) and fine candelabra-like vertical fibers in the papillary dermis (arrow).
(B and C) Skin sections from (B) patient 1 (P1) and (C) patient 2 (P2) lack elastic fibers in the papillary dermis and have fragmented, diffuse fibers in the deep dermis.
(D) Lung sections from patient 2 showed dramatically enlarged (> 200 μm) saccular air spaces and rarefied elastic fibers confined mostly to the saccular wall (black arrowheads) or to arrested septal primordia (blue arrowheads). Capillaries were enlarged and had thickened walls (arrows).
(E) Other areas of the lung contained collapsed air spaces (arrowhead) and airways filled with proteinaceous exudates (arrows).
(F–J) Bronchi had (F) discontinuities (arrowheads) in the elastic sheet of the lamina propria and (G) fragmented elastic fibers in the submucosa. Aortic sections from patient 2 showed overall preserved vessel wall structure, but (H) discontinuities in elastic lamellae (arrowheads), (I) cystic degeneration of the media (arrowheads), and (J) fragmentation of the internal elastic lamina (arrowheads) were observed.
(K) Electron microscopy of dermal elastic fibers from a 7-mo-old control (Co) showed robust insoluble elastin core (El) surrounded by a fine layer of microfibrils (Mf).
(L) In patient 1 (2 mo old), dermal elastic fibers had a diminished elastin core (El), with abundant microfibrils (Mf) and globular elastin aggregates at the periphery of the fiber (Gl).
(M) Ultrastructural abnormalities of pulmonary elastic fibers in Ltbp4S−/− mice were identical to those of patient 1.
Magnification bars represent 20 μm (A–C, G, J); 200 μm (D, E, H, I); 100 μm (F); 0.5 μm (L); and 0.2 μm (K, M).
Histology of the lung revealed a severe septation defect in patient 2 (Figure 4D). The parenchyma had a saccular morphology, was devoid of alveoli, and showed extreme airspace enlargement. The elastic fibers were oriented parallel to the saccule walls, and occasional focal concentrations of elastic fibers were found at locations resembling failed alveolar septum formation. The capillaries were enlarged and had thickened walls. No inflammation was evident. Other areas contained collapsed parenchyma and airways (Figure 4E). In the lamina propria of the tracheal mucosa (Figure 4F) and submucosa (Figure 4G), the elastic fibers were discontinuous and fragmented, respectively. The tunica media of the aorta showed focal discontinuities (Figure 4H) and cystic changes (Figure 4I), and the internal elastic lamina was fragmented (Figure 4J). Lung, trachea, and aorta samples were not available from patients 1, 3, and 4.
Electron microscopy of dermal elastic fibers in control tissues showed typical elastic fibers, with a core of elastin surrounded by a peripheral mantle of microfibrils (Figure 4K). In contrast, elastic fibers in patient 1 revealed some elastin within the microfibril bundles but considerable elastin located external to the bundles in large, globular deposits (Figure 4L), an appearance identical to that of Ltbp4S−/− mice (Figure 4M). Skin samples fixed for electron microscopy were not available from patients 2, 3, and 4.
Discussion
Clinical and Diagnostic Considerations
To our knowledge, this is the first description of a syndrome characterized by craniofacial anomalies, severe pulmonary and visceral involvement, and skin laxity. Although some clinical features of this syndrome overlap with several cutis laxa syndromes, arterial tortuosity syndrome, Loeys-Dietz syndrome, and Marfan syndrome (MFS), the constellation of pulmonary, gastrointestinal, genitourinary, and dermal lesions is a consistent and unique feature of the patients reported here (Table 3). Because the multitude of specific lesions associated with this syndrome makes it difficult to devise a descriptive name, we propose the name Urban-Rifkin-Davis syndrome (URDS).
Table 3.
Clinical Characteristics in Comparison to Other Related Syndromes
| Present Study | ARCL1 (FBLN4) | ARCL1 (FBLN5) | ARCL2 | ADCL | ATS | LDS | MFS | CCA | |
|---|---|---|---|---|---|---|---|---|---|
| Skin laxity | +++ | +++ | +++ | +++ | +++ | +++ | + | − | − |
| Lethal BPD | +++ | ++ | ++ | − | − | − | − | − | − |
| GI malformations | +++ | − | − | − | − | − | − | − | − |
| GU malformations | +++ | − | − | − | − | − | − | − | − |
| Arterial tortuosity | − | ++ | ++ | − | − | +++ | +++ | − | − |
| Aortic aneurysm | − | ++ | − | − | + | + | +++ | +++ | + |
| Aortic stenosis | − | − | +++ | − | − | − | − | − | − |
| Pulmonary artery stenosis | ++ | ++ | + | − | + | ++ | ++ | − | − |
| Microretrognathia | +++ | − | − | − | − | +++ | ++ | − | − |
| Hypertelorism | ++ | − | − | − | − | + | +++ | − | − |
| Wide fontanelles | ++ | − | − | +++ | − | − | − | − | − |
| Microcephaly | − | − | − | +++ | − | − | − | − | − |
| Long philtrum | +++ | +++ | +++ | +++ | +++ | − | − | − | − |
| Cleft palate, bifid uvula | − | − | − | − | − | ++ | +++ | − | + |
| Joint laxity | ++ | +++ | − | +++ | + | +++ | +++ | +++ | − |
| Joint contractures | − | − | − | − | − | ++ | ++ | − | +++ |
| Arachnodactily | − | ++ | − | − | − | + | ++ | +++ | +++ |
| Developmental delay, MR | − | − | − | +++ | − | − | + | − | − |
| Postnatal growth delay | +++ | + | + | ++ | − | − | − | − | − |
Abbreviations are as follows: ARCL1, autosomal-recessive cutis laxa type 1; FBLN4, fibulin-4; FBLN5, fibulin-5; ARCL2, autosomal-recessive cutis laxa type 2; ADCL, autosomal-dominant cutis laxa; ATS, arterial tortuosity syndrome; BPD, bronchopulmonary dysplasia; LDS, Loeys-Dietz syndrome; MFS; Marfan syndrome; CCA, congenital contractural arachnodactily; GU, genito-urinary; MR, mental retardation.
Symbols are as follows: +++, typical; ++, common; +, present; −, absent.
Data are based on the following studies: ARCL1 (FBLN4),13,14 ARCL1 (FBLN5),15–17 ARCL2,33 ADCL,5–9 ATS,20 LDS,34 MFS,34 CCA.35
We recommend LTBP4 mutational screening in cutis laxa patients with severe pulmonary, gastrointestinal, and urinary abnormalities. On the basis of our current experience, we estimate the success rate of detecting LTBP4 mutations in this group of patients as approximately 67%. Expansion of mutational screening to regulatory regions of the LTBP4 gene, use of techniques that can detect deletions, and electron microscopic detection of characteristic dermal elastic fiber abnormalities prior to mutational analysis may further increase the success rate. The prevalence of LTBP4 mutations in patients ascertained on the basis of lethal alveolarization defects, severe diverticulosis or intestinal dilatation, tortuosity, or stenoses will need to be examined in future studies.
LTBP4 Function
Most LTBP4 mutations reported here were premature-termination mutations, leading to severely reduced mRNA abundance. Consistent with this finding, a reduced amount of secreted LTBP4 was found in the conditioned media of mutant fibroblasts. Thus, we conclude that reduced LTBP4 function is the primary cause of disease in this syndrome.
Ltbp4S−/− mice develop pulmonary septation defects, rectal prolapse, colorectal adenomas and carcinomas, and dilated cardiomyopathy, but they survive up to 6 months (mid-adult age) without major clinical symptoms.23,24 In contrast, patients with LTBP4 deficiency face high mortality and respiratory failure in infancy or early childhood. No cardiomyopathy or colorectal cancer was observed in the patients, but these conditions may require a longer time to develop. Patients had several lesions that have not been reported in Ltbp4S−/− mice, including craniofacial and musculoskeletal features; gastrointestinal stenoses, dilatation, and diverticulosis; bladder diverticulosis, and hydronephrosis. Overall, the milder phenotype of the Ltbp4S−/− mice may be related to residual expression of the long form of LTBP4 (Ltbp4L), not affected in this mouse model.23,24 Species-specific functions of LTBP4 or different amount of functional overlap with other LTBPs in different species may also contribute to differences in phenotypic outcomes of LTBP4 deficiency between humans and mice.
LTBP4 deficiency leads to altered production of active TGF-β and disorganization of elastic fibers. The intimate relationship between ECM integrity and TGF-β signaling has been highlighted by recent studies on MFS, which is caused by mutations in FBN1, encoding a major structural protein of microfibrils. Administration of neutralizing antibodies to TGF-β or treatment with the angiotensin II type 1 receptor antagonist, losartan, prevent the development of lung, aortic, and muscular abnormalities associated with MFS.19,29,30 Given the potential similarities in disease mechanisms between MFS and the LTBP4-deficiency syndrome described here, pharmacologic intervention for normalization of TGF-β signaling may be a treatment option.
LTBPs and Human Disease
The importance of LTBPs in human development has only begun to be understood. A new autosomal-recessive form of oligodontia, associated with short stature, increased bone density, and scoliosis, has been reported in a family with an LTBP3 mutation.31 Another recent report described recessive LTBP2 mutations in primary congenital glaucoma associated with extopia lentis and osteopenia.32 Thus, LTBP2 is essential for normal ocular development, whereas LTBP3 is required for tooth formation and bone remodeling. We now show that LTBP4 is necessary for the development of human visceral organs, including the lung, intestines, and urinary tract. Among the known LTBP-deficiency syndromes, mutations in LTBP4 result in the most severe human disease described to date.
Web Resources
The URL for data presented herein is as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/omim/
Acknowledgments
We thank the patients and their family members for participating in the study. We are also grateful to James Southern for providing pathological materials and to Barbara Mordue and Subhadra Ramanathan for clinical information and images. This study was funded in part by U.S. National Institutes of Health grants HL084922 (Z.U.), CA034282 (D.B.R.), and AR49698 (D.B.R.); March of Dimes grant 1-FY09-33 (Z.U.); Phillip Morris USA Inc. (D.B.R.), and Canadian Institutes of Health grant MOP86713 (E.C.D.). E.C.D. is a Canada Research Chair.
References
- 1.Wu M.Y., Hill C.S. Tgf-beta superfamily signaling in embryonic development and homeostasis. Dev. Cell. 2009;16:329–343. doi: 10.1016/j.devcel.2009.02.012. [DOI] [PubMed] [Google Scholar]
- 2.Rifkin D.B. Latent transforming growth factor-beta (TGF-beta) binding proteins: orchestrators of TGF-beta availability. J. Biol. Chem. 2005;280:7409–7412. doi: 10.1074/jbc.R400029200. [DOI] [PubMed] [Google Scholar]
- 3.Annes J.P., Munger J.S., Rifkin D.B. Making sense of latent TGFbeta activation. J. Cell Sci. 2003;116:217–224. doi: 10.1242/jcs.00229. [DOI] [PubMed] [Google Scholar]
- 4.Saharinen J., Keski-Oja J. Specific sequence motif of 8-Cys repeats of TGF-beta binding proteins, LTBPs, creates a hydrophobic interaction surface for binding of small latent TGF-beta. Mol. Biol. Cell. 2000;11:2691–2704. doi: 10.1091/mbc.11.8.2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rodriguez-Revenga L., Iranzo P., Badenas C., Puig S., Carrio A., Mila M. A novel elastin gene mutation resulting in an autosomal dominant form of cutis laxa. Arch. Dermatol. 2004;140:1135–1139. doi: 10.1001/archderm.140.9.1135. [DOI] [PubMed] [Google Scholar]
- 6.Szabo Z., Crepeau M.W., Mitchell A.L., Stephan M.J., Puntel R.A., Yin Loke K., Kirk R.C., Urban Z. Aortic aneurysmal disease and cutis laxa caused by defects in the elastin gene. J. Med. Genet. 2006;43:255–258. doi: 10.1136/jmg.2005.034157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tassabehji M., Metcalfe K., Hurst J., Ashcroft G.S., Kielty C., Wilmot C., Donnai D., Read A.P., Jones C.J. An elastin gene mutation producing abnormal tropoelastin and abnormal elastic fibres in a patient with autosomal dominant cutis laxa. Hum. Mol. Genet. 1998;7:1021–1028. doi: 10.1093/hmg/7.6.1021. [DOI] [PubMed] [Google Scholar]
- 8.Urban Z., Gao J., Pope F.M., Davis E.C. Autosomal dominant cutis laxa with severe lung disease: synthesis and matrix deposition of mutant tropoelastin. J. Invest. Dermatol. 2005;124:1193–1199. doi: 10.1111/j.0022-202X.2005.23758.x. [DOI] [PubMed] [Google Scholar]
- 9.Zhang M.C., He L., Giro M., Yong S.L., Tiller G.E., Davidson J.M. Cutis laxa arising from frameshift mutations in exon 30 of the elastin gene (ELN) J. Biol. Chem. 1999;274:981–986. doi: 10.1074/jbc.274.2.981. [DOI] [PubMed] [Google Scholar]
- 10.McLaughlin P.J., Chen Q., Horiguchi M., Starcher B.C., Stanton J.B., Broekelmann T.J., Marmorstein A.D., McKay B., Mecham R., Nakamura T. Targeted disruption of fibulin-4 abolishes elastogenesis and causes perinatal lethality in mice. Mol. Cell. Biol. 2006;26:1700–1709. doi: 10.1128/MCB.26.5.1700-1709.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nakamura T., Lozano P.R., Ikeda Y., Iwanaga Y., Hinek A., Minamisawa S., Cheng C.F., Kobuke K., Dalton N., Takada Y. Fibulin-5/DANCE is essential for elastogenesis in vivo. Nature. 2002;415:171–175. doi: 10.1038/415171a. [DOI] [PubMed] [Google Scholar]
- 12.Yanagisawa H., Davis E.C., Starcher B.C., Ouchi T., Yanagisawa M., Richardson J.A., Olson E.N. Fibulin-5 is an elastin-binding protein essential for elastic fibre development in vivo. Nature. 2002;415:168–171. doi: 10.1038/415168a. [DOI] [PubMed] [Google Scholar]
- 13.Dasouki M., Markova D., Garola R., Sasaki T., Charbonneau N.L., Sakai L.Y., Chu M.L. Compound heterozygous mutations in fibulin-4 causing neonatal lethal pulmonary artery occlusion, aortic aneurysm, arachnodactyly, and mild cutis laxa. Am. J. Med. Genet. A. 2007;143A:2635–2641. doi: 10.1002/ajmg.a.31980. [DOI] [PubMed] [Google Scholar]
- 14.Hucthagowder V., Sausgruber N., Kim K.H., Angle B., Marmorstein L.Y., Urban Z. Fibulin-4: a novel gene for an autosomal recessive cutis laxa syndrome. Am. J. Hum. Genet. 2006;78:1075–1080. doi: 10.1086/504304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Claus S., Fischer J., Megarbane H., Megarbane A., Jobard F., Debret R., Peyrol S., Saker S., Devillers M., Sommer P. A p.C217R mutation in fibulin-5 from cutis laxa patients is associated with incomplete extracellular matrix formation in a skin equivalent model. J. Invest. Dermatol. 2008;128:1442–1450. doi: 10.1038/sj.jid.5701211. [DOI] [PubMed] [Google Scholar]
- 16.Elahi E., Kalhor R., Banihosseini S.S., Torabi N., Pour-Jafari H., Houshmand M., Amini S.S., Ramezani A., Loeys B. Homozygous missense mutation in fibulin-5 in an Iranian autosomal recessive cutis laxa pedigree and associated haplotype. J. Invest. Dermatol. 2006;126:1506–1509. doi: 10.1038/sj.jid.5700247. [DOI] [PubMed] [Google Scholar]
- 17.Loeys B., Van Maldergem L., Mortier G., Coucke P., Gerniers S., Naeyaert J.M., De Paepe A. Homozygosity for a missense mutation in fibulin-5 (FBLN5) results in a severe form of cutis laxa. Hum. Mol. Genet. 2002;11:2113–2118. doi: 10.1093/hmg/11.18.2113. [DOI] [PubMed] [Google Scholar]
- 18.Dietz H.C., Cutting G.R., Pyeritz R.E., Maslen C.L., Sakai L.Y., Corson G.M., Puffenberger E.G., Hamosh A., Nanthakumar E.J., Curristin S.M. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352:337–339. doi: 10.1038/352337a0. [DOI] [PubMed] [Google Scholar]
- 19.Neptune E.R., Frischmeyer P.A., Arking D.E., Myers L., Bunton T.E., Gayraud B., Ramirez F., Sakai L.Y., Dietz H.C. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat. Genet. 2003;33:407–411. doi: 10.1038/ng1116. [DOI] [PubMed] [Google Scholar]
- 20.Coucke P.J., Willaert A., Wessels M.W., Callewaert B., Zoppi N., De Backer J., Fox J.E., Mancini G.M., Kambouris M., Gardella R. Mutations in the facilitative glucose transporter GLUT10 alter angiogenesis and cause arterial tortuosity syndrome. Nat. Genet. 2006;38:452–457. doi: 10.1038/ng1764. [DOI] [PubMed] [Google Scholar]
- 21.Abe M., Harpel J.G., Metz C.N., Nunes I., Loskutoff D.J., Rifkin D.B. An assay for transforming growth factor-beta using cells transfected with a plasminogen activator inhibitor-1 promoter-luciferase construct. Anal. Biochem. 1994;216:276–284. doi: 10.1006/abio.1994.1042. [DOI] [PubMed] [Google Scholar]
- 22.Davis E.C. Smooth muscle cell to elastic lamina connections in developing mouse aorta. Role in aortic medial organization. Lab. Invest. 1993;68:89–99. [PubMed] [Google Scholar]
- 23.Dabovic B., Chen Y., Choi J., Vassallo M., Dietz H.C., Ramirez F., von Melchner H., Davis E.C., Rifkin D.B. Dual functions for LTBP in lung development: LTBP-4 independently modulates elastogenesis and TGF-beta activity. J. Cell. Physiol. 2009;219:14–22. doi: 10.1002/jcp.21643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sterner-Kock A., Thorey I.S., Koli K., Wempe F., Otte J., Bangsow T., Kuhlmeier K., Kirchner T., Jin S., Keski-Oja J. Disruption of the gene encoding the latent transforming growth factor-beta binding protein 4 (LTBP-4) causes abnormal lung development, cardiomyopathy, and colorectal cancer. Genes Dev. 2002;16:2264–2273. doi: 10.1101/gad.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jensen S.A., Iqbal S., Lowe E.D., Redfield C., Handford P.A. Structure and interdomain interactions of a hybrid domain: a disulphide-rich module of the fibrillin/LTBP superfamily of matrix proteins. Structure. 2009;17:759–768. doi: 10.1016/j.str.2009.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lack J., O'Leary J.M., Knott V., Yuan X., Rifkin D.B., Handford P.A., Downing A.K. Solution structure of the third TB domain from LTBP1 provides insight into assembly of the large latent complex that sequesters latent TGF-beta. J. Mol. Biol. 2003;334:281–291. doi: 10.1016/j.jmb.2003.09.053. [DOI] [PubMed] [Google Scholar]
- 27.Koli K., Saharinen J., Karkkainen M., Keski-Oja J. Novel non-TGF-beta-binding splice variant of LTBP-4 in human cells and tissues provides means to decrease TGF-beta deposition. J. Cell Sci. 2001;114:2869–2878. doi: 10.1242/jcs.114.15.2869. [DOI] [PubMed] [Google Scholar]
- 28.Kantola A.K., Keski-Oja J., Koli K. Fibronectin and heparin binding domains of latent TGF-beta binding protein (LTBP)-4 mediate matrix targeting and cell adhesion. Exp. Cell Res. 2008;314:2488–2500. doi: 10.1016/j.yexcr.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 29.Brooke B.S., Habashi J.P., Judge D.P., Patel N., Loeys B., Dietz H.C. Angiotensin II blockade and aortic-root dilation in Marfan's syndrome. N. Engl. J. Med. 2008;358:2787–2795. doi: 10.1056/NEJMoa0706585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cohn R.D., van Erp C., Habashi J.P., Soleimani A.A., Klein E.C., Lisi M.T., Gamradt M., ap Rhys C.M., Holm T.M., Loeys B.L. Angiotensin II type 1 receptor blockade attenuates TGF-beta-induced failure of muscle regeneration in multiple myopathic states. Nat. Med. 2007;13:204–210. doi: 10.1038/nm1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Noor A., Windpassinger C., Vitcu I., Orlic M., Rafiq M.A., Khalid M., Malik M.N., Ayub M., Alman B., Vincent J.B. Oligodontia is caused by mutation in LTBP3, the gene encoding latent TGF-beta binding protein 3. Am. J. Hum. Genet. 2009;84:519–523. doi: 10.1016/j.ajhg.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ali M., McKibbin M., Booth A., Parry D.A., Jain P., Riazuddin S.A., Hejtmancik J.F., Khan S.N., Firasat S., Shires M. Null mutations in LTBP2 cause primary congenital glaucoma. Am. J. Hum. Genet. 2009;84:664–671. doi: 10.1016/j.ajhg.2009.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hucthagowder V., Morava E., Kornak U., Lefeber D.J., Fischer B., Dimopoulou A., Aldinger A., Choi J., Davis E.C., Abuelo D.N. Loss-of-function mutations in ATP6V0A2 impair vesicular trafficking, tropoelastin secretion and cell survival. Hum. Mol. Genet. 2009;18:2149–2165. doi: 10.1093/hmg/ddp148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Loeys B.L., Chen J., Neptune E.R., Judge D.P., Podowski M., Holm T., Meyers J., Leitch C.C., Katsanis N., Sharifi N. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat. Genet. 2005;37:275–281. doi: 10.1038/ng1511. [DOI] [PubMed] [Google Scholar]
- 35.Callewaert B.L., Loeys B.L., Ficcadenti A., Vermeer S., Landgren M., Kroes H.Y., Yaron Y., Pope M., Foulds N., Boute O. Comprehensive clinical and molecular assessment of 32 probands with congenital contractural arachnodactyly: report of 14 novel mutations and review of the literature. Hum. Mutat. 2009;30:334–341. doi: 10.1002/humu.20854. [DOI] [PubMed] [Google Scholar]