

Abstract
Blood cells participate in vital physiological processes, and their numbers are tightly regulated so that homeostasis is maintained. Disruption of key regulatory mechanisms underlies many blood-related Mendelian diseases but also contributes to more common disorders, including atherosclerosis. We searched for quantitative trait loci (QTL) for hematology traits through a whole-genome association study, because these could provide new insights into both hemopoeitic and disease mechanisms. We tested 1.8 million variants for association with 13 hematology traits measured in 6015 individuals from the Australian and Dutch populations. These traits included hemoglobin composition, platelet counts, and red blood cell and white blood cell indices. We identified three regions of strong association that, to our knowledge, have not been previously reported in the literature. The first was located in an intergenic region of chromosome 9q31 near LPAR1, explaining 1.5% of the variation in monocyte counts (best SNP rs7023923, p = 8.9 × 10−14). The second locus was located on chromosome 6p21 and associated with mean cell erythrocyte volume (rs12661667, p = 1.2 × 10−9, 0.7% variance explained) in a region that spanned five genes, including CCND3, a member of the D-cyclin gene family that is involved in hematopoietic stem cell expansion. The third region was also associated with erythrocyte volume and was located in an intergenic region on chromosome 6q24 (rs592423, p = 5.3 × 10−9, 0.6% variance explained). All three loci replicated in an independent panel of 1543 individuals (p values = 0.001, 9.9 × 10−5, and 7 × 10−5, respectively). The identification of these QTL provides new opportunities for furthering our understanding of the mechanisms regulating hemopoietic cell fate.
Main Text
Blood cells play an integral role in vital physiological processes, from gas transport to blood clotting and various aspects of immunity. Their production, sequestration, and apoptosis is under complex regulatory control, which can be compromised in rare clinical conditions such as hemophilia (MIM 306900) but also, to some extent, in more common disorders, including asthma (MIM 600907)1 and atherosclerosis (MIM 209010).2 Therefore, important insights into the etiology of these conditions can be gained from a detailed understanding of the molecular mechanisms regulating blood cell numbers. Given the large variation in blood cell numbers between individuals and their documented high heritability,3 we sought to map new quantitative trait loci (QTL) for a variety of hematology traits through a whole-genome association study and reasoned that these could provide new insights into the mechanisms regulating hemopoietic cell fate.
Thirteen hematology traits were measured in the peripheral blood of up to 6015 individuals from the Australian and Dutch general populations, ascertained and clinically tested as described in detail elsewhere.3–6 The traits measured included total blood hemoglobin (Hb), red blood cell count (RBC), mean cell erythrocyte volume (MCV), platelet count (PLT), and white blood cell count (WBC). From these values, the derived traits hematocrit (HT), mean cell hemoglobin (MCH), and mean cell hemoglobin concentration (MCHC) were calculated through the appropriate formulae. A five-part differential provided estimates for neutrophil (NEUT), monocyte (MONO), eosinophil (EOS), basophil (BASO), and lymphocyte (LYMPH) counts (Table S1, available online). For each trait, outlier observations (six standard deviations [SD] above the mean) were excluded from analysis and an inverse normal transformation was applied so that normality was ensured. For the Australian cohort, phenotypes were measured in some individuals at three time points (ages 12, 14, and 16) and for these the average across all available data was computed. Trait heritabilities ranged from 35% to 94%, with low to moderate cross-trait phenotypic correlations (Table S2).
Genotyping was performed with Illumina (Australian cohort) and Perlegen (Dutch cohort) chips, with 529,721 and 435,291 single-nucleotide polymorphisms (SNP), respectively, passing quality control (QC) (Table S3). To facilitate the combined analysis of both cohorts, we inferred unmeasured HapMap SNPs in each panel separately by using the MACH program (see Web Resources). In brief, we first compared data from a set of 350 randomly chosen individuals to the phased haplotype data from the CEU HapMap samples (phase I+II, release22, build 36) to generate recombination and error maps. We then used these to impute unmeasured SNPs for each group by using the same reference panel. After imputation, data for 2.5 million Hapmap SNPs were available for the Australian and Dutch cohorts. Of these, in each cohort we dropped SNPs with an imputation score < 0.3 (indicating low imputation confidence; ∼2% of SNPs), a minor allele frequency (MAF) < 0.01, or a Hardy-Weinberg (HW) equilibrium test p < 10−6 (3%). Next, we merged data from both cohorts and further excluded SNPs if they were imputed with high confidence in only one cohort (3%), failed the MAF or HW thresholds in the overall sample (1%), or had significant allele frequency differences between the two cohorts (p < 0.001, 19%). After these exclusions, there remained 1,872,254 SNPs available for analysis. Simulations showed that this data set was well powered for the detection of a QTL that explained ∼0.8% of the phenotypic variance (Figure S1). This study was performed with the approval of the appropriate ethics committee and with informed written consent from all participants.
Individual SNPs were tested for association under an additive model with the use of the family-based SCORE test implemented in Merlin7 after adjustment for the significant effects of age, sex, and time of day of blood collection (including a square component). Similar results were obtained for our top regions of association with the more computationally intensive variance-components likelihood-ratio test, which accounts for identity-by-descent sharing when modeling the correlation between relatives (not shown). The genomic inflation factor ranged between 1.00 and 1.02 (Figure S2), indicating that potential technical or population-stratification artifacts had a negligible impact on the results.
Genome-wide association study (GWAS) results for the combined analysis of the Australian and Dutch cohorts are shown in Figure S3 and are available for download (see Web Resources). Six loci exceeded our significance threshold of p = 5.5 × 10−9, which was adjusted to account for the effective number of independent traits tested (n = 9, p = 5 × 10−8 / 9 = 5.5 × 10−9).
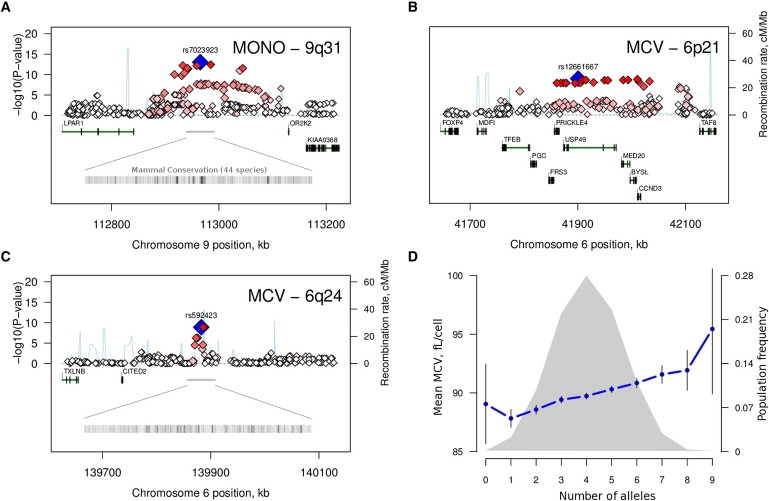
Of the six loci reaching experiment-wide significance, three have not previously been reported as associating with hematology traits (Table 1). The first of these was located 124 kb upstream of the lysophosphatidic acid receptor 1 gene (LPAR1 [MIM 602282]) on chromosome 9q31 (Figure 1A), with multiple SNPs associated with monocyte counts (best SNP: rs7023923; p = 8.9 × 10−14, explaining 1.5% of the trait variance). This locus did not influence the variation of other blood cell levels (not shown).
Table 1.
Loci Associated with Variation in Monocyte Count and Mean Erythrocyte Cell Volume
| Locus | Trait | SNP, Allelea | Panel | Na | Allele Frequency | Effectb | SE | h2 (%)c | Association p Value | Heterogeneity p Valued |
|---|---|---|---|---|---|---|---|---|---|---|
| 9q31 | Monocytes | rs7023923, C | GWAS | 4225 | 0.50 | −0.17 | 0.02 | 1.5 | 8.9 × 10−14 | 0.691 |
| Replication | 1517 | 0.48 | −0.12 | 0.04 | 0.8 | 0.001 | 0.104 | |||
| 6p21 | MCV | rs12661667, C | GWAS | 5946 | 0.73 | 0.13 | 0.02 | 0.7 | 1.2 × 10−9 | 0.078 |
| Replication | 1542 | 0.75 | 0.17 | 0.05 | 1.2 | 9.9 × 10−5 | 0.245 | |||
| 6q24 | MCV | rs592423, C | GWAS | 5938 | 0.53 | −0.11 | 0.02 | 0.6 | 5.3 × 10−9 | 0.305 |
| Replication | 1405 | 0.56 | −0.15 | 0.04 | 0.9 | 7.2 × 10−5 | 0.361 | |||
Abbreviations are as follows: MCV, mean cell volume; N, sample size; SE, standard error.
Sample size corresponds to the number of phenotyped and genotyped individuals. For the Australian cohort, genotype data from 1457 parents who were not phenotyped were also included in the analysis.
Effect corresponds to standard-deviation units for the transformed phenotype.
h2 represents the proportion of phenotypic variance explained by the SNP.
Results for the test of heterogeneity of effect size between the two samples that form the GWAS panel (Australian [n = 2538] and Dutch [n = 3477] cohorts) or between the two samples that form the replication panel (Busselton [n = 1294] and GenomeEUtwin [n = 249] cohorts). A significant p value indicates significant evidence for heterogeneity.
Figure 1.
Main Association Results
(A–C) Regional association plots for three previously unpublished loci associated with peripheral-blood monocyte counts (A) and mean cell erythrocyte volume (MCV; B and C). The most-associated SNP for each region is shown in blue, and the color of the remaining markers reflects the linkage disequilibrium (r2) with the top SNP in each panel (increasing red hue associated with increasing r2). The recombination rate (second y axis) is plotted in light blue and is based on the CEU HapMap population. Exons for each gene are represented by vertical bars, based on all isoforms available from the March 2006 UCSC Genome Browser assembly.
(D) Mean MCV volume (main y axis) as a function of the number of high-MCV alleles at five confirmed MCV loci: 6p21 (rs12661667), 6q24 (rs592423), HBS1L/MYB (rs11154792), TMPRSS6 (rs4820268), and HFE (rs1408272). The gray curve represents the population frequency (second y axis) of the ten observed cumulative allele categories. Vertical bars correspond to the 95% confidence interval around the mean.
The second locus was located on chromosome 6p21 and associated with mean cell erythrocyte volume (rs12661667; p = 1.2 × 10−9, 0.7% variance explained), a region that spanned 133 kb and included five genes (Figure 1B), the most notable of these a member of the D-cyclin gene family (CCND3 [MIM 123834]), which is thought to be critical for expansion of hematopoietic stem cells.8 The third region was also associated with erythrocyte volume and was located in an intergenic region on chromosome 6q24 (rs592423; p = 5.3 × 10−9, 0.6% variance explained [Figure 1C]). The 6p21 and 6q24 loci also accounted for a significant proportion of the variation in mean cell hemoglobin levels (0.5% for both; p = 1.3 × 10−5 and p = 4.0 × 10−5, respectively). There was no evidence for significant heterogeneity of effects between the Australian and Dutch cohorts for these three regions (Table 1).
We first sought to confirm the three associations that had not been previously published and that reached genome-wide significance in our discovery sample in an independent replication panel (n = 1543), also ascertained from the general population. Two studies contributed data for replication: the longitudinal Busselton Health Study9,10 and the GenomEUtwin Study.11 The Busselton Health Study consists of cross-sectional,whole-population health surveys conducted at intervals of 3 yrs in adults residing in Busselton, Western Australia. For this study, data were available for 1294 adults (57% females: mean age 53, range 17–91) tested in 1994 and 1995 and genotyped on the Illumina Human 610-Quad BeadChip. The GenomEUtwin Study consists of an unselected population-based cohort of twins genotyped with the Illumina HumanHap300-Duo BeadChip; phenotypes were available for 249 unrelated monozygotic females (mean age 33, range 17–69). SNPs that were not directly genotyped in either study were imputed with high confidence (imputation score > 0.95) with the MACH (Busselton, rs592423 and rs7023923) or PLINK (GenomEUtwin, all three variants) program.
The three loci replicated convincingly (p = 0.001 for monocytes and rs7023923, p = 9.9 × 10−5 for MCV and rs12661667, p = 7.2 × 10−5 for MCV and rs592423), with the same direction of effect observed in the replication and GWAS samples (Table 1). These results thus confirm that genetic variants in these three regions significantly influence variation in monocyte counts (9q31) and erythrocyte volume (6p21 and 6q24). Given the extent of linkage disequilibrium (LD) and/or the lack of obvious candidate genes for all three, additional studies will be required for identification of the underlying causal variants and associated genes. The degree of sequence conservation across mammals suggests that for 9q31, and to some extent also for 6q24, new exonic or regulatory regions may be located near the association signal (Figures 1A and 1C).
In addition to these three regions, an additional three loci that have previously been reported to associate with hematology traits were identified with genome-wide significance: namely, the HBS1L (MIM 612450)/MYB (MIM 189990) locus,12,13 TMPRSS6 (MIM 609862), and HFE (MIM 235200).14 Our results for these loci are consistent with previous findings (Table S4).
Regions with less-stringent evidence for association, which we did not attempt to replicate, are listed in Table S5. Most notable were the associations between hematocrit and the hexokinase 1 gene (HK1; rs10159477, p = 5.8 × 10−9), consistent with recent reports;15,16 between neutrophil count and the colony-stimulating factor 3 gene (CSF3 [MIM 138970]; rs2227322, p = 9.2 × 10−7), which stimulates the proliferation and differentiation of granulocyte progenitor cells;17 and between lymphocyte counts and the major histocompatibility complex (MHC) locus (rs3099844, p = 4.7 × 10−8).
For most traits tested, the combined effect of all identified QTL to date explains only a small proportion of the total phenotypic variance. For example, for MCV, which we estimated to be > 80% heritable (Table S2), the five loci that now have confirmed effects for this trait explained only ∼6% of the between-individual variation in our replication panel, with largely additive effects within and across loci (Figure 1D). We addressed three potential factors that could partly explain this discrepancy. First, we tested whether the remaining unexplained genetic variance could to some degree be attributed to heterogeneous effects between populations. We performed a genome-wide comparison of genetic effects between the Australian and Dutch cohorts but found that these were largely comparable (Figure S4).
We also investigated the possibility that multiple independent variants with weak effects may exist within a gene, which our main single-locus analysis would have been underpowered to detect. We applied a gene-based approach implemented in PLINK18 to test the overall evidence for association with 17,062 genes, while accounting for the size, number of SNPs, and LD structure for each gene, but we found no additional loci associated with any of the traits tested (Table S6).
Finally, we used a multivariate approach19 to search for loci with weak but potentially pleiotropic effects on multiple traits that could also have been missed by the main analysis. We identified 12 regions that were not detected (p < 10−5) in the univariate analysis, but these did not have experiment-wide significance (Table S7). Of note was the association of a nonsynonymous SNP in the transcription factor 19 gene (TCF19 [MIM 600912]; rs7750641) with lymphocyte count, mean cell hemoglobin, white blood cell count, hematocrit count, and eosinophil count (p = 2.3 × 10−7). Thus, consistent with recent results for most complex traits and diseases,20 we conclude that the genetic variants that remain to be identified for the hematology traits that we tested are not well tagged by the current SNP sets and/or have effect sizes that require larger cohorts to be identified.
In summary, our analyses identified a QTL with specific regulatory effects on peripheral-blood monocyte numbers and two loci that influence erythrocyte volume. Monocytes represent 10% of all leukocytes and develop from the common myeloid progenitor in the bone marrow through several commitment steps regulated by growth and transcription factors.21 Once they emigrate from the bone marrow to the peripheral blood in response to proinflammatory cytokines, monocytes assume their innate immune functions, including the removal of apoptotic cells and toxic compounds.22 Through these, they also play a key role in healing after myocardial infarction23 and contribute to the development of inflammatory diseases, including atherosclerosis.2 The currently accepted model of monocyte development is far from complete,21 so the identification of a confirmed genetic variant for monocyte numbers in the gene-poor 9q31 region provides new opportunities for furthering our understanding of monocyte biology and potentially new avenues for the control of inflammatory diseases.
The two variants identified for erythrocyte volume bring the total number of confirmed genetic loci for this trait up to five (HBS1L/MYB, TMPRSS6, HFE, 6p21, and 6q24). The size of red blood cells determines not only oxygen carrying capacity but also diffusibility and flexibility of movement through capillaries. It is therefore under tight control, with increased size associated with increased mortality risk, namely due to cardiovascular disease and lower respiratory tract disease.24 It is therefore paramount to further characterize the molecular mechanisms underlying the genetic associations reported in our study, as well as to investigate the extent to which the five known QTL interact with known environmental determinants of cell size, including iron, folate, vitamin B12, and alcohol.
Acknowledgments
QIMR. We thank the Brisbane twins and their families for their participation, as well as Dixie Statham, Ann Eldridge, Marlene Grace; Lisa Bowdler, Steven Crooks; David Smyth, Harry Beeby; and Peter Visscher. Funding: Australian National Health and Medical Research Council (NHMRC, grants 241944, 339462, 389927, 389875, 389891, 389892, 389938, 443036, 442915, 442981, 496739, 552485, 552498), Australian Research Council (A7960034, A79906588, A79801419, DP0212016, DP0343921). Busselton Health Study. We thank the people of the Busselton community for their participation. We also acknowledge the support for the 1994 and 1995 follow-up study from Healthway, Western Australia (WA). The Busselton population studies are supported by The Great Wine Estates of the Margaret River region of WA. The authors acknowledge the assistance of the WA DNA Bank (NHMRC Enabling Facility) with DNA samples and the support of the WA Genetic Epidemiology Resource (NHMRC Enabling Facility). NTR and NESDA studies. Funding: The Netherlands Society for Scientific Research (NWO 904-61- 090, 904-61-193, 480-04-004, 400-05-717; SPI 56-464-14192), Center for Medical Systems Biology (NWO Genomics), Geestkracht program of ZonMW (10-000-1002), matching funds from institutes involved in NESDA (GGZ Buitenamstel- Geestgronden, Rivierduinen, University Medical Center Groningen, GGZ Lentis, GGZ Friesland, GGZ Drenthe), Centre for Neurogenomics and Cognitive Research VU University (CNCR-VU), European Science Foundation (EU/QLRT-2001-01254), the National Institute of Mental Health (NIMH) (R01 MH059160), and matching funds from participating institutes in NTR. Genotyping was funded by the Genetic Association Information Network (GAIN), and analysis was supported by grants from GAIN and the NIMH (MH081802).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Genome-wide association study results, http://genepi.qimr.edu.au/staff/manuelF/gwas_results/main.html
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
References
- 1.Kay A.B. The role of eosinophils in the pathogenesis of asthma. Trends Mol. Med. 2005;11:148–152. doi: 10.1016/j.molmed.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 2.Mestas J., Ley K. Monocyte-endothelial cell interactions in the development of atherosclerosis. Trends Cardiovasc. Med. 2008;18:228–232. doi: 10.1016/j.tcm.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Evans D.M., Frazer I.H., Martin N.G. Genetic and environmental causes of variation in basal levels of blood cells. Twin Res. 1999;2:250–257. doi: 10.1375/136905299320565735. [DOI] [PubMed] [Google Scholar]
- 4.Aitken J.F., Green A.C., MacLennan R., Youl P., Martin N.G. The Queensland Familial Melanoma Project: study design and characteristics of participants. Melanoma Res. 1996;6:155–165. doi: 10.1097/00008390-199604000-00011. [DOI] [PubMed] [Google Scholar]
- 5.Boomsma D.I., Willemsen G., Sullivan P.F., Heutink P., Meijer P., Sondervan D., Kluft C., Smit G., Nolen W.A., Zitman F.G. Genome-wide association of major depression: description of samples for the GAIN Major Depressive Disorder Study: NTR and NESDA biobank projects. Eur. J. Hum. Genet. 2008;16:335–342. doi: 10.1038/sj.ejhg.5201979. [DOI] [PubMed] [Google Scholar]
- 6.Sullivan P.F., de Geus E.J., Willemsen G., James M.R., Smit J.H., Zandbelt T., Arolt V., Baune B.T., Blackwood D., Cichon S. Genome-wide association for major depressive disorder: a possible role for the presynaptic protein piccolo. Mol. Psychiatry. 2009;14:359–375. doi: 10.1038/mp.2008.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen W.M., Abecasis G.R. Family-based association tests for genomewide association scans. Am. J. Hum. Genet. 2007;81:913–926. doi: 10.1086/521580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kozar K., Ciemerych M.A., Rebel V.I., Shigematsu H., Zagozdzon A., Sicinska E., Geng Y., Yu Q., Bhattacharya S., Bronson R.T. Mouse development and cell proliferation in the absence of D-cyclins. Cell. 2004;118:477–491. doi: 10.1016/j.cell.2004.07.025. [DOI] [PubMed] [Google Scholar]
- 9.James A.L., Knuiman M.W., Divitini M.L., Musk A.W., Ryan G., Bartholomew H.C. Associations between white blood cell count, lung function, respiratory illness and mortality: the Busselton Health Study. Eur. Respir. J. 1999;13:1115–1119. doi: 10.1034/j.1399-3003.1999.13e29.x. [DOI] [PubMed] [Google Scholar]
- 10.James A.L., Palmer L.J., Kicic E., Maxwell P.S., Lagan S.E., Ryan G.F., Musk A.W. Decline in lung function in the Busselton Health Study: the effects of asthma and cigarette smoking. Am. J. Respir. Crit. Care Med. 2005;171:109–114. doi: 10.1164/rccm.200402-230OC. [DOI] [PubMed] [Google Scholar]
- 11.Vink J.M., Smit A.B., de Geus E.J., Sullivan P., Willemsen G., Hottenga J.J., Smit J.H., Hoogendijk W.J., Zitman F.G., Peltonen L. Genome-wide association study of smoking initiation and current smoking. Am. J. Hum. Genet. 2009;84:367–379. doi: 10.1016/j.ajhg.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thein S.L., Menzel S., Peng X., Best S., Jiang J., Close J., Silver N., Gerovasilli A., Ping C., Yamaguchi M. Intergenic variants of HBS1L-MYB are responsible for a major quantitative trait locus on chromosome 6q23 influencing fetal hemoglobin levels in adults. Proc. Natl. Acad. Sci. USA. 2007;104:11346–11351. doi: 10.1073/pnas.0611393104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Menzel S., Jiang J., Silver N., Gallagher J., Cunningham J., Surdulescu G., Lathrop M., Farrall M., Spector T.D., Thein S.L. The HBS1L-MYB intergenic region on chromosome 6q23.3 influences erythrocyte, platelet, and monocyte counts in humans. Blood. 2007;110:3624–3626. doi: 10.1182/blood-2007-05-093419. [DOI] [PubMed] [Google Scholar]
- 14.Benyamin B., Ferreira M.A., Willemsen G., Gordon S., Middelberg R.P., McEvoy B.P., Hottenga J.J., Henders A.K., Campbell M.J., Wallace L. Common variants in TMPRSS6 are associated with iron status and erythrocyte volume. Nat. Genet. 2009 doi: 10.1038/ng.456. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paré G., Chasman D.I., Parker A.N., Nathan D.M., Miletich J.P., Zee R.Y., Ridker P.M. Novel association of HK1 with glycated hemoglobin in a non-diabetic population: a genome-wide evaluation of 14,618 participants in the Women's Genome Health Study. PloS Genet. 2008;4:e1000312. doi: 10.1371/journal.pgen.1000312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bonnefond A., Vaxillaire M., Labrune Y., Lecoeur C., Chèvre J.C., Bouatia-Naji N., Cauchi S., Balkau B., Marre M., Tichet J. A genetic variant in HK1 is associated with pro-anemic state and HbA1c but not other glycemic control related traits. Diabetes. 2009 doi: 10.2337/db09-0652. Published online August 3, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Metcalf D. The granulocyte-macrophage colony-stimulating factors. Science. 1985;229:16–22. doi: 10.1126/science.2990035. [DOI] [PubMed] [Google Scholar]
- 18.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ferreira M.A., Purcell S.M. A multivariate test of association. Bioinformatics. 2009;25:132–133. doi: 10.1093/bioinformatics/btn563. [DOI] [PubMed] [Google Scholar]
- 20.Goldstein D.B. Common genetic variation and human traits. N. Engl. J. Med. 2009;360:1696–1698. doi: 10.1056/NEJMp0806284. [DOI] [PubMed] [Google Scholar]
- 21.Auffray C., Sieweke M.H., Geissmann F. Blood monocytes: development, heterogeneity, and relationship with dendritic cells. Annu. Rev. Immunol. 2009;27:669–692. doi: 10.1146/annurev.immunol.021908.132557. [DOI] [PubMed] [Google Scholar]
- 22.Williams M.J. Drosophila hemopoiesis and cellular immunity. J. Immunol. 2007;178:4711–4716. doi: 10.4049/jimmunol.178.8.4711. [DOI] [PubMed] [Google Scholar]
- 23.Nahrendorf M., Swirski F.K., Aikawa E., Stangenberg L., Wurdinger T., Figueiredo J.L., Libby P., Weissleder R., Pittet M.J. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 2007;204:3037–3047. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perlstein T.S., Weuve J., Pfeffer M.A., Beckman J.A. Red blood cell distribution width and mortality risk in a community-based prospective cohort. Arch. Intern. Med. 2009;169:588–594. doi: 10.1001/archinternmed.2009.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.