

Abstract
In genome-wide association studies, only a subset of all genomic variants are typed by current, high-throughput, SNP-genotyping platforms. However, many of the untyped variants can be well predicted from typed variants, with linkage disequilibrium (LD) information among typed and untyped variants available from an external reference panel such as HapMap. Incorporation of such external information can allow one to perform tests of association between untyped variants and phenotype, thereby making more efficient use of the available genotype data. When related individuals are included in case-control samples, the dependence among their genotypes must be properly addressed for valid association testing. In the context of testing untyped variants, an additional analytical challenge is that the dependence, across related individuals, of the partial information on untyped-SNP genotypes must also be assessed and incorporated into the analysis for valid inference. We address this challenge with ATRIUM, a method for case-control association testing with untyped SNPs, based on genome screen data in samples in which some individuals are related. ATRIUM uses LD information from an external reference panel to specify a one-degree-of-freedom test of association with an untyped SNP. It properly accounts for dependence in the partial information on untyped-SNP genotypes across related individuals. We demonstrate that ATRIUM is robust in that it maintains the nominal type I error rate even when the external reference panel is not well matched to the case-control sample. We apply the method to detect association between type 2 diabetes and variants on chromosome 10 in the Framingham SHARe data.
Introduction
With the rapid advances in high-throughput genotyping technology, genome-wide association studies have become a viable approach to elucidating the genetic basis of human complex disease. It is now affordable to analyze on the order of 105 to 106 markers throughout the genome. However, because the set of single-nucleotide polymorphisms (SNPs) assayed by the current genotyping platforms covers only a fraction of the total variation in the human genome, it is likely that many disease-susceptibility alleles are not directly genotyped. Therefore, it is of great interest to develop powerful statistical methods to detect association with untyped causal variants. To do this, one can use the linkage disequilibrium (LD) structure of the genome, together with data on typed variants, to detect association between untyped variants and phenotype.
In the context of unrelated individuals, several approaches have been developed, including imputation approaches,1–8 a likelihood-based method,9 testing of tag SNPs,10 and testing of haplotypes of tag SNPs,11 as well as TUNA12,13 and related approaches14 that contrast estimated allele frequencies for cases and controls, where the estimates are based on a linear combination of haplotype frequencies of tag SNPs. A recent extension of BEAGLE8 allows imputation of genotypes for parent-offspring pairs and trios as well as for unrelated individuals. This is an improvement over the approach of applying, to related individuals, imputation methods that were designed for unrelated individuals, because it allows one to use additional phase information from relatives and it avoids the introduction of Mendelian errors.
A key concern for case-control association testing with related individuals is that imputed genotypes are dependent among relatives, where the dependence among imputed genotypes differs from the ordinary dependence among genotypes and is affected by the type and amount of information available for each individual. This complex dependence among imputed genotypes would need to be taken into account in the analysis in order to construct a valid test. However, to our knowledge, the current generation of imputation methods gives information only on the marginal accuracy (e.g., marginal posterior probabilities and not joint posterior probabilities) of imputed genotypes, so these methods would not allow valid assessment of uncertainty in the general setting of case-control association testing with related individuals.
A few methods of case-control association testing that allow arbitrary combinations of related and unrelated individuals have been developed, including methods to detect association with a typed marker15–17 and methods to detect haplotype association.18–20 In this article, we propose the Association Test with Related Individuals for Untyped Markers, or ATRIUM. ATRIUM is a one-degree-of-freedom (1-df) association test based on genotype data from multiple typed SNPs that are in LD with the untyped SNP, where information on the joint distribution of typed and untyped SNPs is obtained from an external reference panel, and where the sample can include family members as well as unrelated individuals. ATRIUM properly accounts for dependence in the partial information on untyped SNP genotypes across related individuals. Because we condition the analysis on the external LD information, ATRIUM still properly controls type I error even when the reference panel is not well matched to the case-control sample. Through simulation studies, we also investigate issues that may affect the power of ATRIUM, including limited size of the reference panel and mismatch between the reference panel and the case-control sample. We compare the power of ATRIUM with that of some existing approaches for testing in this context. We apply the new method to test for association of untyped SNPs with type 2 diabetes (MIM 125853) in the Framingham SHARe data set.
Material and Methods
Suppose a particular untyped genetic variant, U, plays an important role in the disease or binary trait of interest, and suppose that this leads to an association between U and case-control status. The idea behind ATRIUM is that, if we consider an appropriate set of typed markers, , known to be in strong LD with U based on external information, then the primary association of U with case-control status will induce a secondary association of haplotypes of ℳ with case-control status. Rather than applying a generic test of association between haplotypes of ℳ and case-control status, we can improve power to detect association attributable to the untyped variant U by using the information, available from an external reference such as HapMap, on the joint distribution of alleles of U and haplotypes of ℳ, to construct a 1-df test that has power in the direction that corresponds to association of U with the trait. By “has power in the direction that corresponds to association of U with the trait,” we mean that we test for the specific pattern of change in frequencies of haplotypes of ℳ between cases and controls that would be expected if the haplotype frequency change were driven by association between the trait and U. The major methodological challenges in developing ATRIUM are due to (1) allowing samples that contain related individuals and (2) the fact that genotypes, not haplotypes, are observed. The haplotype information used by ATRIUM for each sampled individual is based on the individual's own genotypes as well as genotype information on the individual's parents when available. The uncertainty in haplotype information and the dependence of haplotype information across related individuals are directly taken into account in the analysis through the use of the IQLS method.20 The result is a test of association with an untyped SNP, based on genotype data on associated markers ℳ with missing data allowed, where the test is applicable in samples that contain related individuals, assuming that the individuals are outbred and that pedigree information is available.
We first briefly outline the method; more detailed development is given in the following paragraphs. To construct the ATRIUM test, we start with the MQLS association model,17 which was developed for testing case-control association with a typed SNP in samples that contain related individuals. The MQLS model for the association of the untyped SNP U with the trait implies a model for association of the haplotypes of ℳ with the trait. This allows us to form a 1-df, complete-data, quasi-likelihood score test for case-control association with the untyped SNP in related individuals, based on haplotype data from ℳ. To extend the test to the more realistic situation in which genotype, not haplotype, information is available, we use the IQLS framework,20 which provides a general approach to quasi-likelihood inference in the presence of both dependent and missing data. Application of the IQLS framework allows us to test for case-control association with U by forming the 1-df, incomplete-data, ATRIUM test based on genotype data on associated markers ℳ in related individuals.
Mean Model for Untyped SNP
Suppose we have a case-control sample of n outbred individuals, some of whom may be related with relationships specified by known pedigrees. We arbitrarily label the two alleles of the untyped SNP U as “0” and “1,” and we let U be the unobserved vector of genotypes at U, where we define U = (U1, …, Un)T by , 1 ≤ i ≤ n. We let A be the observed phenotype vector, where we define A = (A1, …, An)T by Ai = 1 if individual i is affected, − K/(1 − K) if i is unaffected, and 0 if i's phenotype is unknown. Here 0 < K < 1 is a constant that represents an external estimate of the population prevalence of the trait. (The prevalence estimate is permitted to be very rough; the MQLS test is, in fact, valid for arbitrary fixed K.) The MQLS model specifies that
| (Equation 1) |
Here p represents the population frequency of allele 1 at U, which is treated as an unknown nuisance parameter; ϕij represents the kinship coefficient between individuals i and j, which is assumed known; Φ is the n × n kinship matrix having (i, j)th element 2ϕij; and γ is the unknown parameter of interest representing the strength and direction of association between the phenotype and the alleles of U. This model incorporates the enrichment effect, which specifies, for example, that affected individuals with affected relatives are more likely to have alleles predisposing to the trait than are affected individuals without affected relatives. The MQLS score test based on this model was previously shown17 to have high power to detect case-control association with a typed SNP, for a variety of multilocus trait models, in samples containing related individuals.
Mean Model for Haplotypes at Typed Markers in LD with Untyped SNP
For the untyped SNP U, a set of typed SNPs ℳ is chosen based on some multilocus measure of association,14,21–23 so that haplotypes of ℳ are highly informative about alleles of U. Suppose there are H + 1 possible haplotypes of ℳ, where for 1 ≤ j ≤ H + 1; we assume 0 < hj < 1 is the population frequency of the jth haplotype, with . The haplotype frequency vector h = (h1, …, hH)T is treated as an unknown nuisance parameter in the analysis. We let Y = (Y11, …, Y1H, …, Yn1, …, YnH)T, where , 1 ≤ i ≤ n, 1 ≤ j ≤ H + 1. If we assume that any association between haplotypes of ℳ and the trait is a secondary association that is attributable to the direct effect of U on the trait, then we can derive a mean model for haplotypes of ℳ based on Equation 1. More precisely, we assume that given the allele at U on a particular chromosome, the haplotype at ℳ on that chromosome is conditionally independent of the phenotype information. Then we find (Appendix A) that
| (Equation 2) |
where p1|j is defined to be the conditional probability that a chromosome has allele 1 at U given that it has the jth haplotype at ℳ. Thus, the association effect for the jth haplotype is γhj(p1|j − p)/[p(1 – p)], where γ is the association effect for allele 1 of U, and hj(p1|j − p)/[p(1 − p)] is the slope of the regression line for the simple linear regression of the haplotype indicator Yj = (Y1j, …, Ynj)T on the allele indicator U. If we reparameterize the model in terms of a new association parameter r = γ/[p(1 − p)] and apply the identity , then we obtain the equivalent mean model
| (Equation 3) |
The genotype data for the case-control sample give direct information on h but give no information at all on the p1|j's. Therefore we replace each p1|j by an estimate, , which is obtained from the reference panel and is taken to be
| (Equation 4) |
provided , where denotes an estimate of haplotype frequency hj in the reference panel, and denotes an estimate, from the reference panel, of the frequency of the haplotype having allele 1 at U and haplotype j at ℳ. Then we define ηj by
| (Equation 5) |
which can be treated in the analysis as a fixed function of the unknown haplotype frequency h. (When , we set , which, when plugged into Equation (5), leads to ηj = 0.) Combining Equations 5 and 3, we finally obtain
| (Equation 6) |
which is the mean model for haplotypes of ℳ, based on the information, from the reference panel, on LD between alleles of U and haplotypes of ℳ. Alternatively, we could use a logistic version (Appendix B) of the model in Equation 6, and we would still obtain the same quasi-likelihood score test as in the next subsection. We note that the role of the reference panel information is solely to determine the direction in which to perform a 1-df test, so the accuracy of the reference panel information will affect only the power, not the validity, of the score tests we propose in the next two subsections.
ATRIUM When Haplotypes Are Observed
When the haplotypes at ℳ are observed, ATRIUM is a 1-df, quasi-likelihood score test based on the mean model in Equation 6. Our null hypothesis is H0:r = 0, and our alternative hypothesis is HA:r ≠ 0. The null hypothesis represents no association of haplotypes of ℳ with the trait, whereas the alternative hypothesis is formulated to detect the specific kind of association that would be expected if the haplotype association were driven by the effects of the untyped SNP U.
To form ATRIUM in the case of observed haplotypes, we require the null conditional covariance matrix of Y, which can be written as
| (Equation 7) |
where B is the H × H matrix having (j, k)th element Bjk = hj(1 − hj)/2 if j = k and − hjhk/2 if j ≠ k. We can also think of B as being the correlation matrix of the H vector (Yi1, …, YiH) for any individual i. Similarly, note that Φ (defined in the previous subsection) is the correlation matrix of the n vector Yj = (Y1j, …, Ynj)T for any haplotype j. The Kronecker product ⊗ is defined in Appendix C. ATRIUM for the case when haplotypes are observed is the quasi-likelihood score test based on the model of Equations 6 and 7, with the resulting ATRIUM test statistic given by
| (Equation 8) |
where 1 is a vector with every entry equal to 1, and is the BLUE24 of hj under the null hypothesis of no association. In the special case when the individuals are unrelated, the term [ATΦA − (AT1)2/(1TΦ−11)] in the denominator reduces to , where is the sample average of A. Under regularity conditions, the ATRIUM test statistic T is asymptotically χ21 distributed under the null hypothesis of no association. This asymptotic null distribution holds regardless of whether the reference panel provides biased estimates of the p1|j, as might happen, for instance, when the reference panel and case-control sample are drawn from different populations. Thus, validity of our test does not depend on choice of reference panel.
While accuracy of does not affect validity of the test, it can affect power. When is exactly equal to the true p1|j, which one could think of as the case of an infinitely large, perfectly matched reference panel, there is an optimality result for our test, namely, that it is asymptotically locally most powerful among tests based on Y (under regularity conditions and assuming that Equations 3 and 7 hold). Thus, compared to other association tests between haplotypes of ℳ and the trait, ATRIUM should have increased power to detect case-control association with the untyped SNP U.
ATRIUM When Unphased Genotypes Are Observed
For the ith sampled individual, 1 ≤ i ≤ n, let Gi denote the observed, unphased genotype data on the associated markers ℳ, where missing genotypes are allowed, and let G = (G1, …, Gn). Note that G provides only partial information about the haplotype indicator vector Y. To obtain the ATRIUM test statistic in this setting, we use the IQLS method,20 which provides a quasi-likelihood score test that can be used with missing and dependent data. Instead of being based on Y, which is now only partially observed, ATRIUM is the quasi-likelihood score test based on a vector, Z, of conditional expectations of elements of Y, which incorporates partial haplotype information. We define Z = (Z11, …, Z1H, …, Zn1, …, ZnH)T with Zij = E(Yij|Gi, Gmi, Gfi, A), where Gmi and Gfi denote the observed, unphased genotype data for the mother and father of individual i, respectively, where these may be missing. Throughout the analysis, we condition on the observed pattern of missing genotypes, and we assume that the pattern of missingness is not informative about the underlying haplotypes.
We have E(Z|A) = E(Y|A), so the mean model of Equation 6 still applies to Z. As before, we are interested in testing the null hypothesis of no association between the trait and the untyped SNP U, H0:r = 0 versus HA:r ≠ 0. Note that explicit computation of Z under the alternative hypothesis would require additional assumptions about the genetic model. Fortunately, the IQLS method allows one to perform the quasi-likelihood score test without having to actually compute Z under the alternative model. We define Ω = Var0(Z|A) = Var0(Z) to be the conditional covariance matrix of Z under the null hypothesis, and we let and , where with μij given by Equation 6 and where E0(·) denotes expectation under the null hypothesis. Additional details on how Ω, Fr, and Fh are obtained can be found in Appendix D. Then the ATRIUM test statistic has the form
| (Equation 9) |
where the entire right-hand side is evaluated at . Here is the IQL estimator of h when r = 0, which is the solution to the IQLS equation , when r = 0. This equation is easily solved numerically by an iterative algorithm. The ATRIUM test statistic asymptotically follows a χ21 distribution under the null hypothesis of no association, under regularity conditions.20
Connections with Previous Methods for Unrelated Individuals
The WHAP method14 has been developed for the special case of complete haplotype information on the set of markers ℳ in a sample consisting of equal numbers of unrelated cases and controls, where no distinction is made between unaffected controls and unphenotyped (i.e., general population) controls. The optimal-weight WHAP test statistic is obtained as a special case of our complete-data ATRIUM test statistic in Equation 8, under the assumptions that the sampled individuals are unrelated and that the controls are all of one type, either all unaffected or all unphenotyped.
The TUNA method12,13 uses a similar idea to test for association with an untyped SNP in a sample of unrelated cases and controls, when unphased genotype data are available on the set of markers ℳ. Where ATRIUM uses a score test, the TUNA software13 uses a Wald test based on different null and alternative hypotheses than those of ATRIUM. Nonetheless, in the special case of complete haplotype data on unrelated individuals, where all controls are of the same type, the TUNA and ATRIUM test statistics are identical except for the choice of variance estimator.
Comparison to Other Approaches for Related Individuals
In the next section, we perform simulation studies to compare the power of ATRIUM to that of (1) the single-SNP MQLS association test17 with the SNP among that has highest r2 with the untyped SNP; (2) the full-degree-of-freedom IQLS haplotype association test20 applied to haplotypes of ℳ; and (3) a 1-df haplotype association test for deviation in the direction of the single haplotype of ℳ that has highest r2 with the untyped SNP, where test (3) is novel, to our knowledge. We now give a brief overview of tests (2) and (3).
The full-degree-of-freedom IQLS haplotype association test20 is similar to ATRIUM in that it is a quasi-likelihood score test based on the vector Z, but it uses a different mean model given by
| (Equation 10) |
where is the parameter of interest, which measures trait-haplotype association. The full-degree-of-freedom IQLS test allows one to test versus . Under regularity conditions, the asymptotic null distribution of the IQLS test is χ2H.
In addition, we propose a 1-df haplotype association test that, to our knowledge, is novel. Let α denote the haplotype of ℳ that has highest r2 with the untyped SNP. We modify the model of Equation 10 to incorporate the constraint , for j ≠ α. The resulting model has a one-dimensional parameter of interest, γα, which represents association between the trait and haplotype α. The constraint specifies that conditional on a haplotype being not of type α, its type is independent of the phenotype information. Then we perform the quasi-likelihood score test of the null hypothesis H0:γα = 0 versus HA:γα ≠ 0, which is a test of association between the trait and haplotype α. This test is equivalent to the ATRIUM test with the setting , and , for j ≠ α. In other words, we are effectively assuming that the untyped SNP is in perfect LD with haplotype α.
Results
Simulation Studies
We perform simulation studies to explore the validity and power of ATRIUM. We consider a scenario in which a case-control sample is genotyped for the Illumina 300K SNP set. The European (CEU) HapMap sample is taken to be a well-matched reference panel for the population from which the case-control sample is drawn. (Note that although we use the CEU HapMap sample to choose tag SNPs, we actually simulate a reference panel for each replicate of our simulation studies, with the simulated reference panel used for calculation of .) We refer to any SNP in HapMap that is not in the Illumina 300K set as an “untyped” SNP. For each untyped SNP on chromosome 1, we use the TUNA software to find a set of four Illumina 300K tag SNPs that maximizes the MD information measure23 within a 400 kb window, based on the 60 HapMap CEU parents' data. (MD can be viewed as a multilocus extension of r2. It can be interpreted as the asymptotic ratio of sample sizes needed to obtain the same power when the SNP is typed versus when it is untyped and a particular set of tag SNPs is used.23) From each set of four tag SNPs, we then remove SNPs that do not provide significant information on the untyped SNP, based on the adjusted MD measure.13 We randomly choose five of the untyped SNPs on chromosome 1, from among those with MD ≥ 0.4, to be used in the simulations. Tables 1–5 list the joint distributions, estimated from the CEU HapMap sample, for each of these five untyped SNPs with their Illumina 300K tag SNPs. In the simulations, we assume that the distributions in Tables 1–5 represent the true joint distributions, of the corresponding SNP sets, in the population from which the case-control sample is drawn.
Table 1.
Haplotype Frequencies, Estimated from the CEU HapMap Sample, for SNP rs10797373, denoted by U, and Three Tag SNPs on the Illumina 300K Set
| Haplotype | U | Frequency | ||||||
|---|---|---|---|---|---|---|---|---|
| H1 | 0 | 0 | 1 | 1 | 0.158 | |||
| H2 | 0 | 1 | 1 | 1 | 0.042 | |||
| H3 | 1 | 0 | 0 | 1 | 0.092 | |||
| H4 | 1 | 0 | 1 | 0 | 0.075 | |||
| H5 | 1 | 1 | 0 | 1 | 0.067 | |||
| H6 | 1 | 1 | 1 | 0 | 0.567 |
Table 2.
Haplotype Frequencies, Estimated from the CEU HapMap Sample, for SNP rs10907174, Denoted by U, and Three Tag SNPs on the Illumina 300K Set
| Haplotype | U | Frequency | ||||||
|---|---|---|---|---|---|---|---|---|
| H1 | 0 | 0 | 1 | 0 | 0.017 | |||
| H2 | 0 | 1 | 0 | 0 | 0.708 | |||
| H3 | 1 | 0 | 1 | 1 | 0.083 | |||
| H4 | 1 | 1 | 0 | 0 | 0.017 | |||
| H5 | 1 | 1 | 0 | 1 | 0.125 | |||
| H6 | 1 | 1 | 1 | 0 | 0.033 | |||
| H7 | 1 | 1 | 1 | 1 | 0.017 |
Table 3.
Haplotype Frequencies, Estimated from the CEU HapMap Sample, for SNP rs2794347, Denoted by U, and Three Tag SNPs on the Illumina 300K Set
| Haplotype | U | Frequency | ||||||
|---|---|---|---|---|---|---|---|---|
| H1 | 0 | 0 | 0 | 0 | 0.050 | |||
| H2 | 0 | 0 | 0 | 1 | 0.008 | |||
| H3 | 0 | 1 | 0 | 0 | 0.675 | |||
| H4 | 0 | 1 | 0 | 1 | 0.067 | |||
| H5 | 0 | 1 | 1 | 0 | 0.017 | |||
| H6 | 0 | 1 | 1 | 1 | 0.042 | |||
| H7 | 1 | 0 | 0 | 0 | 0.008 | |||
| H8 | 1 | 0 | 0 | 1 | 0.075 | |||
| H9 | 1 | 1 | 0 | 0 | 0.050 | |||
| H10 | 1 | 1 | 0 | 1 | 0.008 |
Table 4.
Haplotype Frequencies, Estimated from the CEU HapMap Sample, for SNP rs12031614, Denoted by U, and Three Tag SNPs on the Illumina 300K Set
| Haplotype | U | Frequency | ||||||
|---|---|---|---|---|---|---|---|---|
| H1 | 0 | 0 | 0 | 1 | 0.025 | |||
| H2 | 0 | 0 | 1 | 1 | 0.208 | |||
| H3 | 0 | 1 | 0 | 0 | 0.108 | |||
| H4 | 0 | 1 | 0 | 1 | 0.133 | |||
| H5 | 1 | 0 | 0 | 0 | 0.008 | |||
| H6 | 1 | 0 | 0 | 1 | 0.033 | |||
| H7 | 1 | 0 | 1 | 0 | 0.050 | |||
| H8 | 1 | 1 | 0 | 0 | 0.433 |
Table 5.
Haplotype Frequencies, Estimated from the CEU HapMap Sample, for SNP rs10910097, Denoted by U, and Four Tag SNPs on the Illumina 300K Set
| Haplotype | U | Frequency | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| H1 | 0 | 0 | 0 | 0 | 0 | 0.025 | ||||
| H2 | 0 | 0 | 0 | 0 | 1 | 0.050 | ||||
| H3 | 0 | 0 | 0 | 1 | 1 | 0.050 | ||||
| H4 | 0 | 0 | 1 | 0 | 0 | 0.250 | ||||
| H5 | 0 | 0 | 1 | 1 | 0 | 0.075 | ||||
| H6 | 0 | 1 | 0 | 0 | 0 | 0.067 | ||||
| H7 | 0 | 1 | 0 | 1 | 0 | 0.383 | ||||
| H8 | 1 | 0 | 0 | 0 | 1 | 0.083 | ||||
| H9 | 1 | 1 | 0 | 0 | 1 | 0.017 |
We simulate the case-control data based on a trait model with two unlinked causal SNPs, both acting dominantly, with epistasis between them. The minor allele frequencies (MAFs) of SNP 1 and SNP 2 are denoted by p1 and p2, respectively. In addition to the two allele frequencies, there are two penetrance parameters, f1 and f2 (f1 > f2), with penetrance f1 for individuals who have at least one copy of the minor allele at SNP 1 and at least one copy of the minor allele at SNP 2 and penetrance f2 for all other individuals. We consider five different parameter settings, which are listed as Models a–e in Table 6. For each model, we set SNP 2 to be one of the untyped SNPs in Tables 1–5, and its MAF, p2, is determined accordingly. Table 6 gives the trait model parameters; the resulting population prevalence, K; and the sibling risk ratio, λs = Ks/K, where Ks is the prevalence conditioned on having an affected sibling. We sample 90 outbred, three-generation, 16-person pedigrees, of which 30 pedigrees have four affected individuals, 30 have five affected individuals, and 30 have six affected individuals. In each sampled pedigree, phenotypes for all 16 individuals are observed. The individual's genotypes are observed if and only if at least 30% of the individual's siblings, parents, and offspring are affected. This is similar to the study design in a previous report.17
Table 6.
Parameter Settings for Simulation Models
| Model | SNP 2 |
Minor Allele Frequencies |
Penetrance Parameters |
K | λs | ||
|---|---|---|---|---|---|---|---|
| p1 | p2 | f1 | f2 | ||||
| a | rs10797373 | 0.15 | 0.358 | 0.15 | 0.05 | 0.066 | 1.118 |
| b | rs10907174 | 0.30 | 0.225 | 0.22 | 0.09 | 0.116 | 1.075 |
| c | rs2794347 | 0.40 | 0.200 | 0.40 | 0.20 | 0.246 | 1.045 |
| d | rs12031614 | 0.50 | 0.400 | 0.18 | 0.07 | 0.123 | 1.077 |
| e | rs10910097 | 0.35 | 0.200 | 0.25 | 0.10 | 0.131 | 1.081 |
An important feature of the ATRIUM analysis is that it is conditional on the value of obtained from the reference panel. This results in a theoretical robustness property for type I error of the test, namely, ATRIUM will be valid, in the sense of having the correct type I error, even when is a biased or inaccurate estimate of p1|j. We verify this in our simulations, and we also use simulations to assess the impact of the reference panel on power. Specifically, we consider the effect on power of (1) bias in introduced by a mismatched reference panel and (2) variability in because of small sample size of the reference panel. To assess (1), we compare results from simulations based on three different types of reference panel: well-matched, in which the reference panel consists of phased genotype data on 60 unrelated individuals simulated based on the CEU HapMap sample; mismatched, in which the reference panel consists of phased genotype data on 90 unrelated individuals simulated based on the Asian (JPT+CHB) HapMap sample; and extremely mismatched, in which the reference panel consists of phased genotype data on 60 unrelated individuals simulated based on the African (YRI) HapMap sample. To assess (2), we compare results from simulations in which the true value of p1|j is plugged in for (“perfect panel”) to those in which is estimated from the well-matched reference panel of 60 unrelated individuals described above. In our simulations, every replicate of the case-control sample has its own simulated reference panel (except “perfect panel” in which true values are used instead of a reference panel).
For the assessment of type I error, association is tested with an untyped SNP that is unlinked and unassociated with any causal variant, where phenotype is simulated according to Model b. We compare the proportion of simulations in which the statistic exceeds the (1 − α)th quantile of the χ12 distribution to the nominal type I error level α, for α = 0.01 and 0.05. Table 7 gives the empirical type I error of the ATRIUM test, based on 10,000 replicates, where is obtained from a reference panel that mimics the HapMap CEU, YRI, or JPT+CHB sample. We find that even for an extremely mismatched (YRI) reference panel, type I error is not significantly different from the nominal level when α = 0.05 or 0.01. These results verify that the type I error of the ATRIUM test for an untyped SNP is robust to the choice of reference sample.
Table 7.
Power and Type I Error of ATRIUM, When the Reference Panel Is Well Matched to the Sample or Mismatched, Based on 5,000 Simulated Replicates for Power or 10,000 Simulated Replicates for Type I Error
| Reference Panel |
Empirical Type I Error (SE) |
Estimated Power (SE) with Significance Level of 0.05 |
|||||
|---|---|---|---|---|---|---|---|
| α = 0.05 | α = 0.01 | Model a | Model b | Model c | Model d | Model e | |
| CEU (match) | 0.050 (0.002) | 0.009 (0.0010) | 0.915 (0.004) | 0.957 (0.003) | 0.868 (0.005) | 0.859 (0.005) | 0.985 (0.002) |
| JPT+CHB (mismatch) | 0.049 (0.002) | 0.011 (0.0010) | 0.911 (0.004) | 0.944 (0.003) | 0.447 (0.007) | 0.782 (0.006) | 0.932 (0.004) |
| YRI (extreme mismatch) | 0.051 (0.002) | 0.010 (0.0010) | 0.838 (0.005) | 0.750 (0.006) | 0.355 (0.007) | 0.432 (0.007) | 0.930 (0.004) |
To assess power for each model, we mask the genotypes at causal SNPs 1 and 2 and perform the tests based only on the tag SNPs for SNP 2, which are given in Tables 1–5 for Models a–e, respectively. These tag SNPs form the set ℳ described in Material and Methods. Table 7 illustrates the power of ATRIUM for the three different types of reference panel. Under Models a, b, and e, power remains high even when the mismatched JPT+CHB reference panel is used, and power is reasonable even for the extremely mismatched YRI reference panel. However, in Models c and d, power is compromised with a mismatched reference panel. This results from the fact that the joint haplotype distribution of SNP 2 with its tag SNPs differs dramatically across the three populations (where this distribution for the CEU sample is given in Tables 3 and 4).
Table 8 compares the power of ATRIUM, with a well-matched reference panel of 60 unrelated individuals, to the power of three other tests that are valid in samples containing related individuals: (1) the single-SNP MQLS association test with the SNP in ℳ that has the highest r2 with the untyped SNP; (2) the full-degree-of-freedom haplotype test applied to the haplotypes of ℳ; and (3) the 1-df haplotype association test for deviation in the direction of the single haplotype of ℳ that has the highest r2 with the untyped SNP. In every setting, the ATRIUM test outperforms the other three, verifying that in samples containing related individuals, the strategy of using reference panel information to select an optimal direction for testing association with an untyped SNP improves power over other approaches. To assess the possible effects on power of variability in as a result of small sample size of the reference panel, we also compare the power of ATRIUM with the well-matched reference panel of 60 unrelated individuals to ATRIUM with the true value of p1|j plugged in for (“perfect panel”). Based on these simulations, there appears to be little loss of power between the unattainable perfect panel and the well-matched reference panel.
Table 8.
Power of ATRIUM Compared to Single-SNP and Haplotype Association Tests, Based on 5,000 Simulated Replicates, α = 0.05, CEU Reference Panel
| Test |
Estimated Power (SE) |
||||
|---|---|---|---|---|---|
| Model a | Model b | Model c | Model d | Model e | |
| SNP | 0.555 (0.007) | 0.946 (0.003) | 0.530 (0.007) | 0.749 (0.006) | 0.793 (0.006) |
| HAP (full degree of freedom) | 0.723 (0.006) | 0.856 (0.005) | 0.713 (0.006) | 0.650 (0.007) | 0.899 (0.004) |
| HAP (1 degree of freedom) | 0.849 (0.005) | 0.935 (0.003) | 0.723 (0.006) | 0.832 (0.005) | 0.741 (0.006) |
| ATRIUM | 0.915 (0.004) | 0.957 (0.003) | 0.868 (0.005) | 0.859 (0.005) | 0.985 (0.002) |
| ATRIUM (perfect panel)a | 0.915 (0.004) | 0.960 (0.003) | 0.887 (0.005) | 0.867 (0.005) | 0.987 (0.002) |
ATRIUM (perfect panel) is ATRIUM with true p1|j plugged in for .
Analysis of Type 2 Diabetes in the Framingham SHARe Data
The Framingham Heart Study (FHS)25 is a multicohort, longitudinal study of risk factors for cardiovascular disease. The FHS sample consists of unrelated individuals as well as individuals from multigenerational pedigrees. For individuals in Cohort 1 (original Framingham cohort), we use the data from exams 1–27 to determine type 2 diabetes status, which is coded as follows: individuals with at least one exam with (nonfasting) blood glucose (BG) level ≥ 200 mg/dl or who were under treatment for diabetes, where the measurement or treatment occurred between the ages of 35 and 75 years, are classified as affected. We classify as unaffected those who satisfy all of the following conditions: (1) ≥ 70 years at the time of the last exam for which BG is available, (2) BG < 200 mg/dl for all exams for which it is available, and (3) not taking any treatment by the time of the last exam. We classify as unknown phenotype those who were < 70 years at the time of the last exam for which BG is available and who satisfy both conditions (2) and (3). For individuals in Cohort 2 (offspring cohort), we use the data from exams 1–7, and for individuals in Cohort 3 (generation three cohort), we use the data from exam 1 to determine type 2 diabetes status, which is coded as follows for Cohorts 2 and 3: individuals with at least one exam with fasting plasma glucose (FPG) ≥ 126 mg/dl or who were under treatment for diabetes, where the measurement or treatment occurred between the ages of 35 and 75 years, are classified as affected. We classify as unaffected those who satisfy all of the following conditions: (1) ≥ 70 years at the time of the last exam for which FPG is available, (2) FPG < 126 mg/dl for all exams for which it is available, and (3) not taking any treatment by the time of the last exam. We classify as unknown phenotype those who were < 70 years at the time of the last exam for which FPG is available and who satisfy both conditions (2) and (3). Note that for exams 1 and 2 for Cohort 2, instead of the FPG ≥ 126 mg/dl criterion, we use the hand-curated diabetes mellitus (DM) status, which is based on detailed chart review and is available for exams 1 and 2 of Cohort 2 in Framingham SHARe. This study had approval from dbGaP and from the University of Chicago Institutional Review Board.
Among the FHS individuals who are genotyped on the Affymetrix 500K array, and who are coded as affected, unaffected, or unknown phenotype based on the above criteria, we include only those who satisfy the following quality-control conditions: (1) completeness > 96%, where completeness is the proportion of all markers on the Affymetrix 500K array for which a given individual has genotypes called, and (2) diagonal of empirical kinship matrix < 1.05. We also use the off-diagonals of the empirical kinship matrix to exclude an additional 298 individuals with kinship values that are not consistent with the pedigree information. Based on the above criteria, a total of 7,678 individuals are retained in the analysis, with 576 affected, 1,254 unaffected, and 5,848 unknown phenotype, of which 793 are original cohort, 3,142 are offspring cohort, and 3,743 are third generation.
We perform case-control association testing of type 2 diabetes with both typed and untyped SNPs on chromosome 10 in the Framingham SHARe data. The typed SNPs in the analysis consist of the 21,777 SNPs on chromosome 10 that are on the Affymetrix 500K array, pass the Affymetrix 500K quality-control tests, and meet the following additional criteria: (1) call rate ≥ 96%, (2) Mendelian error rate ≤ 0.02, and (3) MAF ≥ 0.01. The untyped SNPs in the analysis are the 60,344 SNPs on chromosome 10 in HapMap that meet the following criteria: (1) are not among the 21,777 typed SNPs and (2) are tagged by at least two typed SNPs, where the tagging is done by applying TUNA to the CEU HapMap samples, in a similar fashion to what is described in detail in subsection Simulation Studies (with the Illumina 300K array replaced by the Affymetrix 500K array and with chromosome 1 replaced by chromosome 10). We use MQLS to test for association with each typed SNP and ATRIUM to test for association with each untyped SNP. The prevalence of diagnosed and undiagnosed diabetes among people aged 60 years or older in the United States is reported to be ∼23%, with type 2 diabetes accounting for about 90%–95% of all diagnosed cases of diabetes.26 Therefore, we set K = 0.2 in the MQLS and ATRIUM analyses.
Table 9 gives the results for SNPs for which the corresponding ATRIUM or MQLS test has a p value < 8.0 × 10−5. The first SNP in Table 9 (rs2904802) is in an intron of the gene encoding aldo-keto reductase family 1, member C1 (AKR1C1 [MIM 600449]), located at 10p15.1. The next four SNPs in Table 9 (rs7901695, rs4506565, rs7903146, and rs4132670) are intronic SNPs for the gene encoding transcription factor 7-like 2 (TCF7L2 [MIM 602228]), located at 10q25.2. Recently, five separate type 2 diabetes genome-wide association studies27–31 have identified association between TCF7L2 and type 2 diabetes. One study32 has shown that the rs7903146 T allele is associated with hepatic insulin resistance and diminished glucose-stimulated plasma insulin secretion. The sixth SNP in Table 9 (rs859510) is 78.5 kb from the gene encoding G protein-coupled receptor 26 (GPR26 [MIM 604847]), located at 10q26.13. This SNP lies in a previously identified,33 6.3 Mb linkage region, for hemoglobin AIc (HbA1c), a diabetes-related quantitative glucose trait, in the FHS based on the Affymetrix 100K array. The last three SNPs in Table 9 (rs4615933, rs6482989, and rs9418739) are intronic SNPs for the gene encoding dedicator of cytokinesis 1 (DOCK1 [MIM 601403]), located at 10q26.2.
Table 9.
Type 2 Diabetes Association Results in Framingham SHARe, for Typed and Untyped SNPs on Chromosome 10
| Region | Gene | SNP | Position (nucleotides) | Typed | # of Tag SNPs | MD | Test | p Value |
|---|---|---|---|---|---|---|---|---|
| 10p15.1 | AKR1C1 | rs2904802 | 4,999,364 | no | 4 | 0.44 | ATRIUM | 4.8e−5 |
| 10q25.2 | TCF7L2 | rs7901695 | 114,744,078 | yes | – | – | MQLS | 4.9e−6 |
| 10q25.2 | TCF7L2 | rs4506565 | 114,746,031 | yes | – | – | MQLS | 3.1e−6 |
| 10q25.2 | TCF7L2 | rs7903146 | 114,748,339 | no | 3 | 0.94 | ATRIUM | 1.9e−5 |
| 10q25.2 | TCF7L2 | rs4132670 | 114,757,761 | yes | – | – | MQLS | 2.4e−5 |
| 10q26.13 | GPR26a | rs859510 | 125,337,377 | no | 4 | 1.00 | ATRIUM | 7.8e−5 |
| 10q26.2 | DOCK1 | rs4615933 | 128,716,002 | no | 4 | 0.65 | ATRIUM | 3.4e−5 |
| 10q26.2 | DOCK1 | rs6482989 | 128,753,959 | no | 3 | 0.68 | ATRIUM | 1.6e−5 |
| 10q26.2 | DOCK1 | rs9418739 | 128,758,976 | yes | – | – | MQLS | 3.4e−5 |
rs859510 is 78.5 kb upstream of the gene GPR26.
Previous studies have found the region 10q25-q26 to be syntenic to several quantitative trait loci for both weight and type 2 diabetes in rats.34–36 10q25-q26 also contains a 6.3 Mb region showing evidence for linkage to HbA1c in FHS.33 Figure 1 shows our FHS type 2 diabetes association results for the 10q25-q26 region, where we plot −log 10(p value) for each of 12,653 untyped HapMap SNPs, based on ATRIUM, and 4,550 typed Affymetrix 500K SNPs, based on MQLS. Overall, the untyped SNPs are well represented among those SNPs having small p values, taking into account the fact that although they represent about 75% of the tested SNPs, the tests for untyped SNPs are generally expected to have lower power because of lower information content. In the previously identified linkage region (119.5–125.8 Mb), the untyped SNPs provide stronger evidence of association than do the typed SNPs.
Figure 1.
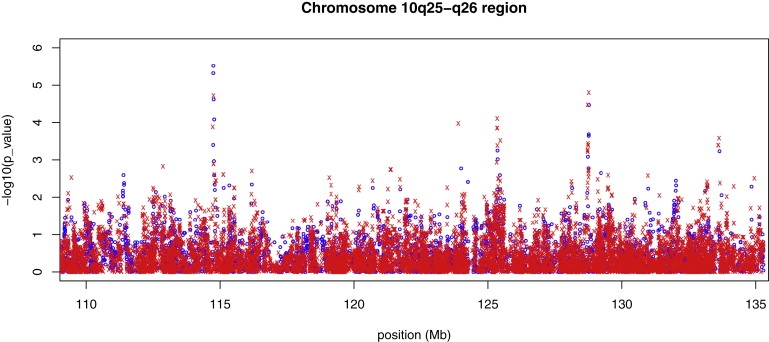
Association Results for Type 2 Diabetes in Framingham SHARe for the Region 10q25-q26
−log10(p value) is plotted against chromosomal location for 4,550 typed SNPs (blue circles) and 12,653 untyped SNPs (red x).
Assessment of Computation Time
The computational burden of untyped SNP analysis is much greater in samples containing related individuals than it is in samples of unrelated individuals. This is because the dependence, across related individuals, of the partial information on untyped-SNP genotypes must be assessed and incorporated into the analysis. We note that this dependence differs from the ordinary dependence among genotypes and is affected by the type and amount of information available, on the untyped-SNP genotype, for each individual. Thus, the computationally costly part of ATRIUM is the calculation, at each iteration, of the covariance matrix Ω.
The Framingham SHARe data we analyze contain some extremely large pedigrees. For instance, there is one family with 317 genotyped individuals who are included in our analysis, and there are four other families that each have more than 90 genotyped individuals included in our analysis. Accordingly, we did two different assessments of the computation time needed to test the 60,344 untyped SNPs of chromosome 10: (1) computation with extremely large pedigrees, where we include 7,678 individuals from 1,147 families with the number of genotyped individuals per family ranging from 1 to 317, with five families having 90 or more genotyped individuals; and (2) computation with moderate-size pedigrees, where we include 4,926 individuals from 1,084 families in which there are no more than 20 genotyped individuals per family. With extremely large pedigrees, the analysis took 17.5 hr on an Intel 2.6 GHz Mac laptop with 4 GB RAM when the haplotype information for an individual is based only on that individual's genotype (i.e., when Zij is taken to be Zij = E(Yij|Gi, A)), and it took 45 hr when we included parental genotype data when considering an individual's haplotype information (i.e., when Zij is taken to be Zij = E(Yij|Gi, Gmi, Gfi, A)). With moderate-size pedigrees, it took 4.5 hr to do the analysis when haplotype information for an individual is based only on that individual's genotype, and it took 12 hr to do the analysis when we included parental genotype data when considering an individual's haplotype information. We have not optimized the code, so it is likely that these times could be greatly improved.
Discussion
We propose the ATRIUM method for testing association with untyped genetic variants in samples containing general combinations of related and unrelated individuals. An important feature of ATRIUM is that it properly accounts for dependence in the partial information on untyped-SNP genotypes across related individuals, which is crucial for construction of a valid test. ATRIUM is potentially useful in a wide range of study designs, including extremely large pedigrees as well as samples that combine families and unrelated individuals. ATRIUM uses information from an external reference panel, such as HapMap, to select an optimal direction for testing association with an untyped SNP, based on genotype data from typed SNPs. ATRIUM allows both phased and unphased genotype data for both the case-control sample and the reference panel. We demonstrate, both theoretically and through simulation, that the validity of ATRIUM is robust to mismatch between the reference panel and the case-control sample, though power is highest when the reference panel is reasonably well-matched to the case-control sample. We also find that small sample size of the reference panel results in little loss of power. We further demonstrate, both theoretically and through simulation, that ATRIUM provides higher power to detect association with untyped SNPs than do other single-SNP and haplotype tests based on typed SNPs.
We apply ATRIUM to the Framingham SHARe data to test for association between type 2 diabetes and SNPs on chromosome 10 that are in HapMap but are untyped on the Affymetrix 500K array in the Framingham sample. We replicate association between type 2 diabetes and intronic SNPs of the TCF7L2 gene, where we obtain p values for association in the range of 3.1e−6 to 2.4e−5 for three typed and one untyped intronic SNP of TCF7L2. We also obtain p values < 8e−5 for SNPs in or near three other genes on chromosome 10, including one typed and two untyped intronic SNPs for the DOCK1 gene. In a previously identified linkage region for a diabetes-related phenotype,33 the untyped SNPs, analyzed with ATRIUM, provide stronger evidence for association than do the typed SNPs.
A key challenge that arises in association analysis with samples of related individuals is that specification of an alternative model is vastly more problematic with related than with unrelated individuals. This is because the background effects of environmental factors and multiple loci, other than the particular variant being tested, can create very different patterns of dependence of phenotype among related individuals under different modeling assumptions. This makes it particularly challenging to develop appropriate likelihood-based or Bayesian analyses in this context. The use of a score-function or quasi-score-function approach, as in ATRIUM, avoids this problem. A close connection between the MQLS and IQLS tests and the retrospective likelihood score test has previously been shown.20 A key point is that the retrospective likelihood score, MQLS, IQLS, and ATRIUM tests can be formed without specifying the joint distribution of phenotypes among related individuals under the null or alternative hypothesis, whereas likelihood-based and Bayesian approaches would, in principle, require this to be specified.
Ideally, one should be able to improve the power of ATRIUM by making use of one or more of the available hidden Markov model (HMM) imputation approaches1–8 for the modeling and computation of the conditional probabilities that are needed from the reference database. A difficulty is that the calculation of the required covariance matrix Ω becomes increasingly onerous as the number of possible haplotypes of typed SNPs used to predict the untyped SNP increases. An analogous problem arises if imputation methods are used directly on the case-control sample with related individuals, because it would be necessary to compute and take into account the joint posterior distribution, across related individuals, of the untyped SNP genotypes. To our knowledge, current imputation methods do not provide these joint posterior probabilities for related individuals.
When comparing p values across SNPs, particularly when there are both typed and untyped SNPs being tested, it is useful to keep in mind that the p value does not make any adjustment for the differing power of the tests. This is particularly relevant when typed and untyped SNP p values are compared, because power to detect association with an untyped SNP will be reduced to the extent that the untyped SNP is not well characterized by haplotypes of typed SNPs. The extent of information on the untyped SNP, relative to the information that would be available if the SNP were typed, can be assessed by the MD measure,23 which can be helpful in interpreting the resulting p values.
When the sample size of the reference panel is small, it can arise that the case-control sample contains a haplotype (call it haplotype j), for the typed SNPs, that has an estimated frequency of zero in the reference panel. Thus, the reference panel lacks information on LD between haplotype j and the untyped SNP. As described in subsection, Mean Model for Haplotypes at Typed Markers in LD with Untyped SNP, we solve this problem by treating haplotype j as independent of the untyped SNP. An alternative approach12 would be to group haplotype j with the closest haplotype (call it haplotype k), among those having nonzero estimated frequency in the reference panel, and then set . This approach assumes that the untyped SNP has the same conditional distribution given haplotype j as given haplotype k. In most cases, one would expect little difference between the two approaches, but if haplotype j were greatly enriched in the case-control sample, the results of the two approaches might be somewhat different. This problem could be resolved with larger reference panels.
Appendix A
We derive the mean model of Equation 2 for haplotypes of ℳ from the mean model of Equation 1 for alleles of U. Let Xi = (ai, xi) be a random variable representing a randomly chosen combined haplotype from individual i, where ai is the allele at U and xi is the haplotype at ℳ. Then our assumption is that P(xi = j|ai = m, A) = pj|m, for m = 0, 1, j = 1, …, H + 1, i.e., that given the allele at U, the haplotype at ℳ is conditionally independent of the phenotype information. Note that E(Ui|A) = P(ai = 1|A) and E(Yij|A) = P(xi = j|A). Then we have E(Yij|A) = E(Ui|A)P(xi = j|ai = 1, A) + [1 − E(Ui|A)]P(xi = j|ai = 0, A) = E(Ui|A)pj|1 + [1 − E(Ui|A)]pj|0, and the result follows, where we use the fact that pj|1p + pj|0(1 − p) = hj and that pj|1 − pj|0 = hj(p1|j − p)/[p(1 − p)].
Appendix B
Instead of Equation 6, we could use the logistic model
| (Equation 11) |
where all quantities have the same definitions as in Equation 6. The quasi-likelihood score test for H0:r = 0 versus HA:r ≠ 0 based on Equation 11 is identical to that based on Equation 6, where this test statistic is given in Equation (8). Conceptually, the advantage of the logistic model (11) over the linear model (6) is that in the logistic model, r can be any real number, whereas in the linear model, r is constrained in a rather complicated way relative to h = (h1, …, hH)T to ensure that 0 ≤ E(Yij|A) ≤ 1.
Appendix C
Given the n × m matrix A with (i, j)th element aij and the p × q matrix B with (i, j)th element bij, their Kronecker product, denoted by A ⊗ B, is the np × mq matrix with block structure
Appendix D
We assume that the markers in the set ℳ are tightly linked, and that, under the null hypothesis, both Hardy-Weinberg equilibrium and Mendelian inheritance hold. Let Z0 denote Z evaluated under the null hypothesis, r = 0. Then we have Zij0 = E0(Yij|Gi, Gmi, Gfi), which can be explicitly computed, as a function of h and (Gi, Gmi, Gfi), for an outbred, parent-offspring trio, where we allow some genotypes to be missing. When we plug in for h, we obtain Z evaluated at , which is needed for Equation 9. We can obtain by finding the joint conditional distribution of (Gi, Gmi, Gfi, Gj, Gmj, Gfj) for each pair of sampled individuals (i, j), given the pattern of missing genotypes, where this distribution is explicitly computed as a function of h, the pedigree information, and the pattern of missingness. To obtain Fh, note that , where 1n is an n-vector with all entries equal to 1 and IH is the H × H identity matrix. We can explicitly obtain ∂Z0/∂h from Z0, and the null expectation is obtained from the joint conditional distribution of (Gi, Gmi, Gfi), given the pattern of missing genotypes, for each sampled individual i. Finally, consider , where . Note that knowledge of Z0 is not sufficient to obtain E0(∂Z/∂r|A). At the same time, it is not necessary to fully specify Z under the alternative model either. Instead, we need the first-order term of the power series expansion for Z around r = 0. This first-order term is the same for any two-allele disease model for the untyped SNP, and we use it to obtain Fr. Note that this is the same assumption used by Thornton and McPeek (2007)17 to obtain the MQLS mean model, so we do not need to impose any additional assumptions to obtain Fr.
Web Resources
The URLs for data presented herein are as follows:
EntrezGene, http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene
ATRIUM source code, http://www.stat.uchicago.edu/∼mcpeek/software/index.html
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Acknowledgments
We thank Mark Abney and William Wen for discussion, critical comments, and help with software implementation; Dan Nicolae, Matthew Stephens, and Peter McCullagh for discussion and critical comments; and two anonymous reviewers for critical comments. This study was supported by National Institutes of Health grant R01 HG001645. The Framingham Heart Study (FHS) and the Framingham SHARe project are conducted and supported by the National Heart, Lung, and Blood Institute (NHLBI) in collaboration with Boston University. The Framingham SHARe data used for the analyses described in this manuscript were obtained through dbGaP (phs000007.v6.p3). This work was not prepared in collaboration with investigators of the FHS and does not necessarily reflect the opinions or views of the FHS, Boston University, or NHLBI.
References
- 1.Stephens M., Scheet P. Accounting for decay of linkage disequilibrium in haplotype inference and missing-data imputation. Am. J. Hum. Genet. 2005;76:449–462. doi: 10.1086/428594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scheet P., Stephens M. A fast and flexible statistical method for large-scale population genotype data: Application to inferring missing genotypes and haplotypic phase. Am. J. Hum. Genet. 2006;78:629–644. doi: 10.1086/502802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marchini J., Howie B., Myers S., McVean G., Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat. Genet. 2007;39:906–913. doi: 10.1038/ng2088. [DOI] [PubMed] [Google Scholar]
- 4.Servin B., Stephens M. Imputation-based analysis of association studies: Candidate regions and quantitative traits. PLoS Genet. 2007;3:e114. doi: 10.1371/journal.pgen.0030114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Browning S.R., Browning B.L. Rapid and accurate haplotype phasing and missing data inference for whole genome association studies using localized haplotype clustering. Am. J. Hum. Genet. 2007;81:1084–1097. doi: 10.1086/521987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Willer C.J., Sanna S., Jackson A.U., Scuteri A., Bonnycastle L.L., Clarke R., Heath S.C., Timpson N.J., Najjar S.S., Stringham H.M. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat. Genet. 2008;40:161–169. doi: 10.1038/ng.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guan Y., Stephens M. Practical issues in imputation-based association mapping. PLoS Genet. 2008;4:e1000279. doi: 10.1371/journal.pgen.1000279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Browning B.L., Browning S.R. A Unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am. J. Hum. Genet. 2009;84:210–223. doi: 10.1016/j.ajhg.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin D.Y., Hu Y., Huang B.E. Simple and efficient analysis of disease association with missing genotype data. Am. J. Hum. Genet. 2008;82:444–452. doi: 10.1016/j.ajhg.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson G.C., Esposito L., Barratt B.J., Smith A.N., Heward J., DiGenova G., Ueda H., Cordell H.J., Eaves I.A., Dudbridge F. Haplotype tagging for the identification of common disease genes. Nat. Genet. 2001;29:233–237. doi: 10.1038/ng1001-233. [DOI] [PubMed] [Google Scholar]
- 11.de Bakker P.I.W., Yelensky R., Pe'er I., Gabriel S.B., Daly M.J., Altshuler D. Efficiency and power in genetic association studies. Nat. Genet. 2005;37:1217–1223. doi: 10.1038/ng1669. [DOI] [PubMed] [Google Scholar]
- 12.Nicolae D.L. Testing untyped alleles (TUNA) – applications to genome-wide association studies. Genet. Epidemiol. 2006;30:718–727. doi: 10.1002/gepi.20182. [DOI] [PubMed] [Google Scholar]
- 13.Wen X., Nicolae D.L. Association studies for untyped markers with TUNA. Bioinformatics. 2008;24:435–437. doi: 10.1093/bioinformatics/btm603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zaitlen N., Kang H.M., Eskin E., Halperin E. Leveraging the HapMap correlation structure in association studies. Am. J. Hum. Genet. 2007;80:683–691. doi: 10.1086/513109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Slager S.L., Schaid D.J. Evaluation of candidate genes in case-control studies: A statistical method to account for related subjects. Am. J. Hum. Genet. 2001;68:1457–1462. doi: 10.1086/320608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bourgain C., Hoffjan S., Nicolae R., Newman D., Steiner L., Walker K., Reynolds R., Ober C., McPeek M.S. Novel case-control test in a founder population identifies P-selectin as an atopy-susceptibility locus. Am. J. Hum. Genet. 2003;73:612–626. doi: 10.1086/378208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thornton T., McPeek M.S. Case-control association testing with related individuals: A more powerful quasi-likelihood score test. Am. J. Hum. Genet. 2007;81:321–337. doi: 10.1086/519497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Browning S.R., Briley J.D., Briley L.P., Chandra G., Charnecki J.H., Ehm M.G., Johansson K.A., Jones B.J., Karter A.J., Yarnall D.P., Wagner M.J. Case-control single-marker and haplotypic association analysis of pedigree data. Genet. Epidemiol. 2005;28:110–122. doi: 10.1002/gepi.20051. [DOI] [PubMed] [Google Scholar]
- 19.Zhang J., Schneider D., Ober C., McPeek M.S. Multilocus linkage disequilibrium mapping by the decay of haplotype sharing with samples of related individuals. Genet. Epidemiol. 2005;29:128–140. doi: 10.1002/gepi.20081. [DOI] [PubMed] [Google Scholar]
- 20.Wang Z., McPeek M.S. An incomplete-data quasi-likelihood approach to haplotype-based genetic association studies on related individuals. J. Am. Stat. Assoc. 2009;104:1251–1260. doi: 10.1198/jasa.2009.tm08507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chapman J.M., Cooper J.D., Todd J.A., Clayton D.G. Detecting disease associations due to linkage disequilibrium using haplotype tags: A class of tests and the determinants of statistical power. Hum. Hered. 2003;56:18–31. doi: 10.1159/000073729. [DOI] [PubMed] [Google Scholar]
- 22.Stram D.O. Tag SNP selection for association studies. Genet. Epidemiol. 2004;27:365–374. doi: 10.1002/gepi.20028. [DOI] [PubMed] [Google Scholar]
- 23.Nicolae D.L. Quantifying the amount of missing information in genetic association studies. Genet. Epidemiol. 2006;30:703–717. doi: 10.1002/gepi.20181. [DOI] [PubMed] [Google Scholar]
- 24.McPeek M.S., Wu X., Ober C. Best linear unbiased allele-frequency estimation in complex pedigrees. Biometrics. 2004;60:359–367. doi: 10.1111/j.0006-341X.2004.00180.x. [DOI] [PubMed] [Google Scholar]
- 25.Splansky G.L., Corey D., Yang Q., Atwood L.D., Cupples L.A., Benjamin E.J., D'Agostino R.B., Fox C.S., Larson M.G., Murabito J.M. The third generation cohort of the National Heart, Lung, and Blood Institute's Framingham Heart Study: Design, recruitment, and initial examination. Am. J. Epidemiol. 2007;165:1328–1335. doi: 10.1093/aje/kwm021. [DOI] [PubMed] [Google Scholar]
- 26.National Institute of Diabetes and Digestive and Kidney Diseases . U.S. Department of Health and Human Services, National Institutes of Health; Bethesda, MD: 2008. National Diabetes Statistics, 2007 Fact Sheet. [Google Scholar]
- 27.Sladek R., Rocheleau G., Rung J., Dina C., Shen L., Serre D., Boutin P., Vincent D., Belisle A., Hadjadj S. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature. 2007;445:881–885. doi: 10.1038/nature05616. [DOI] [PubMed] [Google Scholar]
- 28.Saxena R., Voight B.F., Lyssenko V., Burtt N.P., de Bakker P.I.W., Chen H., Roix J.J., Kathiresan S., Hirschhorn J.N., Daly M.J. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316:1331–1336. doi: 10.1126/science.1142358. [DOI] [PubMed] [Google Scholar]
- 29.Zeggini E., Weedon M.N., Lindgren C.M., Frayling T.M., Elliott K.S., Lango H., Timpson N.J., Perry J.R.B., Rayner N.W., Freathy R.M. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science. 2007;316:1336–1341. doi: 10.1126/science.1142364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scott L.J., Mohlke K.L., Bonnycastle L.L., Willer C.J., Li Y., Duren W.L., Erdos M.R., Stringham H.M., Chines P.S., Jackson A.U. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341–1345. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Steinthorsdottir V., Thorleifsson G., Reynisdottir I., Benediktsson R., Jonsdottir T., Walters G.B., Styrkarsdottir U., Gretarsdottir S., Emilsson V., Ghosh S. A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nat. Genet. 2007;39:770–775. doi: 10.1038/ng2043. [DOI] [PubMed] [Google Scholar]
- 32.Wegner L., Hussain M.S., Pilgaard K., Hansen T., Pedersen O., Vaag A., Poulsen P. Impact of TCF7L2 rs7903146 on insulin secretion and action in young and elderly Danish twins. J. Clin. Endocrinol. Metab. 2008;93:4013–4019. doi: 10.1210/jc.2008-0855. [DOI] [PubMed] [Google Scholar]
- 33.Meigs J.B., Manning A.K., Fox C.S., Florez J.C., Liu C., Cupples L.A., Dupuis J. Genome-wide association with diabetes-related traits in the Framingham Heart Study. BMC Med. Genet. 2007;8(Suppl 1):S16. doi: 10.1186/1471-2350-8-S1-S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kato N., Hyne G., Bihoreau M.T., Gauguier D., Lathrop G.M., Rapp J.P. Complete genome searches for quantitative trait loci controlling blood pressure and related traits in four segregating populations derived from Dahl hypertensive rats. Mamm. Genome. 1999;10:259–265. doi: 10.1007/s003359900983. [DOI] [PubMed] [Google Scholar]
- 35.Galli J., Li L.S., Glaser A., Ostenson C.G., Jiao H., Fakhrai-Rad H., Jacob H.J., Lander E.S., Luthman H. Genetic analysis of non-insulin dependent diabetes mellitus in the GK rat. Nat. Genet. 1996;12:31–37. doi: 10.1038/ng0196-31. [DOI] [PubMed] [Google Scholar]
- 36.Klöting I., Kovács P., van den Brandt J. Sex-specific and sex independent quantitative trait loci for facets of the metabolic syndrome in WOKW rats. Biochem. Biophys. Res. Commun. 2001;284:150–156. doi: 10.1006/bbrc.2001.4932. [DOI] [PubMed] [Google Scholar]