

Abstract
Bestrophin-1 is preferentially expressed at the basolateral membrane of the retinal pigmented epithelium (RPE) of the retina. Mutations in the BEST1 gene cause the retinal dystrophies vitelliform macular dystrophy, autosomal-dominant vitreochoroidopathy, and autosomal-recessive bestrophinopathy. Here, we describe four missense mutations in bestrophin-1, three that we believe are previously unreported, in patients diagnosed with autosomal-dominant and -recessive forms of retinitis pigmentosa (RP). The physiological function of bestrophin-1 remains poorly understood although its heterologous expression induces a Cl−-specific current. We tested the effect of RP-causing variants on Cl− channel activity and cellular localization of bestrophin-1. Two (p.L140V and p.I205T) produced significantly decreased chloride-selective whole-cell currents in comparison to those of wild-type protein. In a model system of a polarized epithelium, two of three mutations (p.L140V and p.D228N) caused mislocalization of bestrophin-1 from the basolateral membrane to the cytoplasm. Mutations in bestrophin-1 are increasingly recognized as an important cause of inherited retinal dystrophy.
Introduction
Human bestrophin-1 (MIM 607854) is one of a family of four transmembrane proteins and is primarily expressed in the retinal pigment epithelium (RPE).1–4 The family was originally defined from over 20 related sequences in Caenorhabditis elegans,5 and orthologs exist down to bacteria.6 The conservation of their transmembrane domains suggest that bestrophins perform a common critical function.6 Considerable evidence shows that bestrophins act as chloride channels: exogenous expression in various cell lines induces a new Cl− current,1 the paralogues have different biophysical properties,5 and endogenous Cl− currents can be modified by knockdown of bestrophin-1 expression.7 However there is also evidence that human bestrophin-1 can regulate voltage-gated Ca2+ channels in an RPE cell line.8 This raises the possibility that bestrophin-1 may be a channel and a channel regulator.9 Such a dual function has been reported for the cystic fibrosis transmembrane conductance regulator (CFTR [MIM 602421]) which functions as a chloride channel10 and regulates transport proteins, e.g., chloride-coupled bicarbonate transporters11 and the epithelial sodium channel.12
Given the localization of bestrophin-1 to the RPE, it has been suggested that it is responsible for generating, or at least modulating, the light peak of the electrooculogram (EOG).1,13 The light peak is thought to be due to the activation of a Cl− conductance at the basolateral membrane of the RPE,14 and it is used in the clinical assessment of RPE health and function. Abnormal EOGs are characteristic of the bestrophinopathies, the first of which was vitelliform macular dystrophy (VMD [MIM 153700], also known as Best disease), in which yellow material accumulates under the retina and RPE in the area of the macula so that it resembles an egg yolk (vitelliform).6 We have described the molecular cause of other retinopathies caused by BEST1 mutations. In autosomal-dominant vitreoretinochoroidopathy (ADVIRC [MIM 193220]), missense mutations cause the expression of an isoform lacking an internal exon in addition to the full-length missense isoform.15 The condition had been clinically described previously, and as the name suggests, it consists of retinal and vitreal findings.16 Subsequently, we described the previously unreported phenotype of autosomal-recessive bestrophinopathy (ARB [MIM 611809]), a condition that represents the human bestrophin-1 null phenotype in one family with the characteristic phenotype and homozygous nonsense mutations.17 In addition to having an abnormal EOG, patients also have abnormal electroretinograms (ERG) and a retinopathy that includes an abnormal RPE, frequently with scattered punctate flecks. A recently described condition, canine multifocal retinopathy, is likely to be the naturally occurring equivalent of ARB in dogs.18
In this study, we report four missense mutations in BEST1, three previously unreported, that have been identified in patients diagnosed with retinitis pigmentosa (RP) in five unrelated families. The phenotypic heterogeneity associated with BEST1 is becoming increasingly complicated, with five separate ocular diseases now being ascribed to mutations in it (VMD, adult-onset VMD, ADVIRC, ARB, and autosomal-dominant RP [adRP]). Although the incidence of each of these bestrophinopathies is individually rare, the increasing number of phenotypes associated with BEST1 mutations suggests that these mutations are an increasingly important cause of genetic ocular disease. To our knowledge, this is the first report of mutations in an RPE gene as a cause of adRP.
Subjects and Methods
Patient Details
The study was approved by research ethics committees at Manchester Royal Eye Hospital. Informed consent was obtained from all participants prior to the investigations. Clinical examination of selected members of the affected families included visual acuity testing, fundus examination and photography, and autofluorescence imaging. Electroretinogram testing on families 1, 2, and 5 was performed in accordance with the standards of the International Society for Clinical Electrophysiology of Vision (ISCEV). Electrooculogram testing was not performed on any of the families because of the extinguished or greatly reduced ERGs (where recorded).
Blood was taken from all family members by venesection, and genomic DNA was extracted via standard procedures.
SNP Microarray and Linkage Analysis of Family 1
Whole-Genome Amplification
Whole-genome amplification was used for those samples that did not contain the minimum amount of DNA required for microarray analysis. Amplification reactions were set up with the use of the REPLI-g Mini Kit (QIAGEN, Crawley, West Sussex, UK), in accordance with the manufacturer's instructions. Samples were incubated at 30°C for 16 hr.
SNP Microarray and Linkage Analysis
SNP genotyping was carried out with the use of Affymetrix Genechip 250K Nsp Arrays (Affymetrix, High Wycombe, Buckinghamshire, UK). DNA was prepared in accordance with the manufacturer's instructions and hybridized onto the array at 49°C for 16–18 hr. Staining of the arrays was carried out with the Affymetrix Fluidics Station 450, followed by scanning performed with the Affymetrix Genechip Scanner 3000 7G system.
Genotypes were called with the Bayesian Robust Linear Model with Mahalanobis (BRLMM) algorithm within the GTYPE 3.1 software provided by Affymetrix, with a modified call threshold of 0.1. Files for linkage analysis with Merlin19 were produced with the use of the SNP Export Tool within the GTYPE 3.1 software.
Parametric linkage analysis was carried out with the Merlin software package. So that accuracy would be increased, files were filtered for the removal of unlikely genotypes with the use of the error-detection facility within Merlin prior to linkage analysis. Parametric linkage analysis was carried out with the use of a fully penetrant dominant model with a disease-allele frequency of 0.01% and a phenocopy rate of 0.01%. Merlin was also used for generating haplotypes, which were then viewed with Haploview.20
DNA Sequencing
All ten coding exons and flanking intronic boundaries were sequenced in each of the five family probands with the use of methods and primers as previously described.17 The relevant exon containing the sequence alteration was sequenced in other family members as indicated in the Results section and in Figure 1. The absence of the three previously unreported mutations (c.418c>g, c.614t>c, and c.682g>a) in 210 ethnically matched control chromosomes (white of European descent for families 1–4 and Pakistani for family 5) was confirmed by single-strand conformation polymorphism (SSCP), performed as previously described,17 giving a power of 80%.21 The cDNA is numbered according to Ensembl transcript ID ENST00000378043, in which +1 is the A of the translation start codon.
Figure 1.
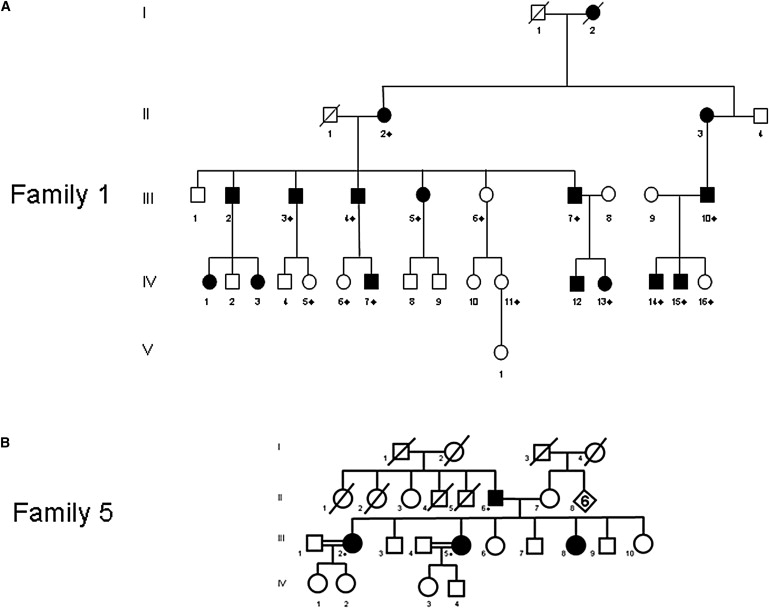
Pedigree of Families
(A) Family 1 had a history of adRP. The proband is IV-13.
(B) Family 5. The proband (III-5) was diagnosed with adRP, but sequencing revealed that all tested affected family members were homozygous for p.L140V. This suggests that the proband's mother is heterozygous for the same mutation. Neither the proband's mother nor any of her unaffected siblings have reported or been diagnosed with symptoms of RP, suggesting that in this family RP is recessively inherited. All ten coding exons and the flanking intronic sequences of BEST1 were sequenced in the probands from each family. Family members indicated with ⋄ were sequenced for the respective exon that was mutated in the family proband (exon 5 in family 1 and exon 6 in family 5).
Multiple alignment of human bestrophin-1 and nine orthologs was performed with ClustalW at BioEdit.
Cloning
Wild-type (WT) BEST1 in pAdlox was a kind gift from A Marmorstein (University of Arizona, USA). Additional mutant constructs for p.L140V, p.I205T, and p.D228N were generated by site-directed mutagenesis with the use of the QuickChange II Kit (Stratagene, Stockport, Cheshire, UK) in accordance with the manufacture's protocol. All generated constructs were confirmed by direct sequencing.
In Vitro Studies
Cell Culture
Human embryonic kidney 293 (HEK293) cells were cultured in minimum essential medium with Earle's salt and L-glutamate containing 9% fetal calf serum, 1% (v/v) nonessential amino acids, gentamicin (0.2 mg/ml), and penicillin G (100 U/ml) (all Invitrogen) at 37°C and 5% CO2. Madin-Darby canine kidney II (MDCK II) cells were cultured in Dulbecco's Modified Eagle Medium containing L-gultamine, D-glucose (4500 mg/l), and sodium pyruvate (110 mg/l) (Invitrogen) supplemented with 10% (v/v) fetal calf serum, 5% (v/v) penicillin/streptomycin, and 1% (v/v) nonessential amino acids (all from PAA Laboratories, Yeovil, Somerset, UK) at 37°C and 5% CO2.
Whole-Cell Patch Clamp
Whole-cell patch-clamping experiments on transiently transfected HEK293 cells were performed as previously described.17 Patch pipettes were manufactured from soda glass capillary tubes (VWR International, Lutterworth, Leicestershire, UK) on a PC10 two-stage vertical puller (Narishige, London). They had tip resistances from 2 to 4 MΩ when filled with the pipette solution. Conventional whole-cell recordings were made with the use of a Multiclamp 700B amplifier (MDS Analytical Technologies, Wokingham, Berkshire, UK) in the voltage-clamp mode. Voltage protocols were driven from a PC equipped with a Digidata interface and pClamp 9 software (MDS Analytical Technologies). The extracellular bath solution contained (mM) 140 NaCl, 2 CaCl2, 1 MgCl2, 10 glucose, 30 mannitol, 10 HEPES; pH 7.4 with NaOH. The pipette solution was a low Cl−, high Ca2+ solution containing (mM) 20 CsCl, 110 Cs aspartate, 2 MgCl2, 10 glucose, 10 HEPES, 10 EGTA, and 7.2 CaCl2; pH adjusted to 7.2 with CsOH. Free Ca2+ concentration was calculated to be approximately 0.5 μM with the use of the WEBMAXC program. Cs+ was included for the inhibition of K+ currents, and K+ was omitted from all solutions. Voltage was stepped from −120 to +80 mV in +20 mV increments and was applied for 450 ms from a holding potential of −50 mV. Results are presented as mean ± standard error of the mean (SEM). Differences between the data were analyzed by Kruskal-Wallis analysis of variance (ANOVA) followed by Dunn's multiple comparison.
Transient Transfection
MDCK II cells were seeded at a density of ∼1 × 106 cells per well on to 24 mm Transwell filters (Corning, Schiphol-Rijk, The Netherlands) 24 hr prior to transfection. Cells were transfected with 2 μg of plasmid DNA with the use of Lipofectamine 2000 Transfection Reagent (Invitrogen) in accordance with the manufacturer's instructions. HEK293 cells were transiently transfected as previously described.15 Cells were cotransfected with pAdlox vector containing WT or mutant BEST1 and the empty vector pEGFP-C1 in a 5:1 ratio with the use of Exgene500 in accordance with the manufacturer's instructions. Transfected cells were visualized by GFP fluorescence, and recordings were made only from these cells.
Immunofluorescence
Immunofluorescence experiments were performed at room temperature 48 hr after transfection. Primary antibodies were used at the following concentrations: mouse monoclonal anti-bestrophin-1 (NB300-164) at 1:2000 (Stratech Scientific, Newmarket, Suffolk, UK) and rabbit polyclonal anti-ZO-1 at 1:1000 (Invitrogen; 61-7300). Secondary antibodies were goat anti-mouse IgG1 Alexa Fluor 488 (Invitrogen; A21121), goat anti-rabbit IgG Alexa Fluor 594 (Invitrogen; A11012), and donkey anti-mouse IgG Texas Red (Stratech; NB120-7059). MDCK II cells grown on Transwell filters (Corning) were washed with phosphate-buffered saline (PBS), fixed with 4% paraformaldehyde (in PBS) for 20 min, washed with PBS (×3), quenched with 50 mM NH4Cl (in PBS) for 10 min, permeabilized with 0.1% (v/v) Trion X-100 for 10 min, and washed with PBS. Cells were then blocked for 30 min with 5% (v/v) fetal calf serum in PBS (FBS/PBS) and were then incubated with the appropriate primary antibody in FBS/PBS for 1 hr. Cells were then washed for 5 min with FBS/PBS (×3) before being incubated with the appropriate secondary antibody in FBS/PBS for 1 hr at room temperature and then washed with PBS at least three times for 5 min. The Transwell filters were then cut from the inserts and mounted onto microscope slides with AF1 anti-fadent solution (Citifluor, London, UK) with or without DAPI stain and sealed with nail varnish.
Images were collected with a Nikon C1 confocal on an upright 90i microscope with a 60 × 1.40 Plan Apo objective and 3× confocal zoom. The confocal settings were as follows: pinhole, 30 μm; scan speed, 400 Hz unidirectional; format, 1024 × 1024. Images for DAPI, Alexa Fluor 488, Alexa Fluor 594, and Texas Red were excited with the 405 nm, 488 nm, and 594 nm laser lines respectively. When 3D optical stacks were acquired, the confocal software (Nikon EZ-C1) was used for determining the optimal number of Z sections.
Results
Mapping of Family 1 to 11q13 and Sequencing of BEST1
A nonconsanguineous white family of European descent (family 1, Figure 1A) with a history of adRP underwent SNP array and linkage analysis for the identification of the causal gene. Parametric linkage analysis was carried out with seven affected and two unaffected family members. A maximum LOD score of 2.9 was obtained on chromosome 11, and no other chromosomes had a positive LOD score (Figures S1–S6, available online). Haplotype analysis showed a common inherited haplotype on chromosome 11, between markers rs536715 and rs17304039, giving a critical region of 64.9 Mb.
The critical region identified from linkage analysis in family 1 included BEST1 which is mutated in several other retinopathies. We therefore sequenced the coding region of BEST1 in the proband IV-13 and found the previously unreported heterozygous sequence variant c.614t>c (p.I205T) in exon 5. The mutation segregated with the adRP disease phenotype in nine other affected family members and was absent from five unaffected family members. No other genes in the critical region were screened.
Sequencing BEST1 in adRP and Concentric RP Panels
After the identification of a BEST1 mutation in this original adRP family, we sought to identify additional BEST1 mutations in a panel of 95 adRP patients in whom no mutations had been found in the following: RHO (MIM 180380), RDS (MIM 608133), ORP1 (MIM 603937) (c.2029c>t [p.R677X], c.2035c>t [p.R679X]), PAP1 (MIM 607331) (c.410a>t [p.H137L]), PRPF31 (MIM 600138) (c.527+3a>g; c.527+1g>t), IMPDH1 (MIM 180105) (exon 8), PRPF8 (MIM 600059) (exon 42), NRL (MIM 162080) (exon 1), NR2E3 (MIM 611131) (exons 2 and 3), and PRPF3 (MIM 601414) (exon 11). In addition, because of the phenotypic overlap between concentric RP22 and ADVIRC,15 we screened 12 individuals with concentric RP.
In the adRP panel, we identified mutations in two unrelated patients. The previously unreported BEST1 mutation, c.682g>a (p.D228N) in exon 6, was present as a heterozygous change in the proband and his mother (family 3). In family 5, a c.418c>g (p.L140V) homozygous change in exon 4 was found in the proband, an affected sister, and the father. The proband's parents were from the same village in Pakistan. We have previously found the p.L140V mutation in patients diagnosed with ARB (unpublished data), and it is likely that the phenotype seen in this family is actually ARB at an early time point (see Discussion). We also found two heterozygous mutations in apparently unrelated patients from the concentric RP panel. In family 2, the female proband and her brother had the same c.682g>a (p.D228N) change in exon 6 that we had seen in family 3. The female proband in family 4 was heterozygous for a c.680a>g (p.Y227C) mutation in exon 6 that has previously been reported as causal in two cases of VMD.23,24 All previously unreported sequence variations were absent from 210 ethnically matched control chromosomes, as determined by SSCP.
Clinical Details
Family 1: Original adRP Family, Heterozygous c.614t>c, p.I205T
Family 1 is a nonconsanguineous white family of European descent (family 1, Figure 1A) with a history of adRP. In all cases, affected individuals were noted to have reduced central vision beginning at age 5–10 years and were also noted to have night blindness. All patients were highly hyperopic. Visual symptoms were progressive, and blindness registration was generally around the end of the second decade of life. The proband (patient IV-13 in family 1, Figure 1A) fell down the stairs at age 13 mo, at which stage she was noted to have bilateral total serous retinal detachments associated with epiretinal membrane formation and severe retinal gliosis. The optic discs were pale, and there was no vessel attenuation. The remaining individuals have been examined in late childhood or adulthood; fundoscopy reveals a panretinal dystrophy associated with flecks in the midperiphery (autofluorescence imaging was not performed), retinal gliosis and severe vascular attenuation, but relatively little peripheral pigment deposition, which is not typical of classical RP but is consistent with a severe retinal dystrophy affecting both rods and cones (Figures 2A and 2B). All tested affected individuals had extinguished electroretinograms (ERGs) in the dark-adapted state with residual light-adapted responses. This was the case for all affected family members tested at all ages. As a result, EOG testing was not performed.
Figure 2.
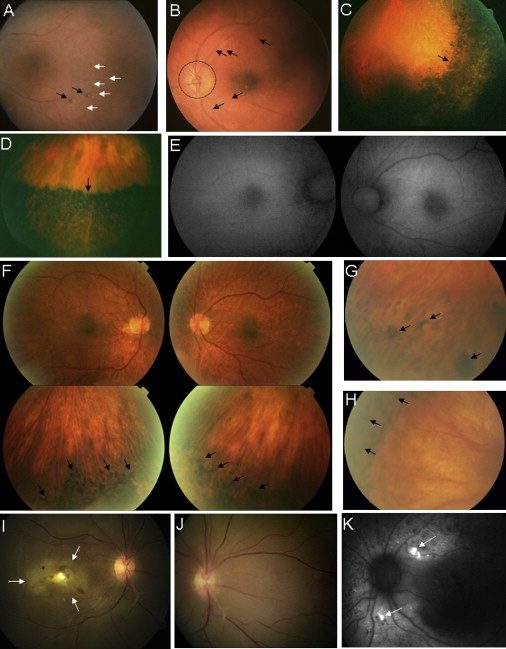
Retinal Images
(A and B) Family 1. Fundus images of left eyes of individual IV-15 (A), showing mid peripheral flecks (white arrows) and pigment clumping (black arrows), and of IV-1 (B), demonstrating severe vascular attenuation (arrows) and disc pallor (circled).
(C and D) Family 2. Fundus images of affected siblings, showing the dense pigmentary changes (black areas) that were present in all quadrants of the peripheral retina and the abrupt change to a more-normal-appearing central retina (arrows). Inferotemporal left retina of the female proband (P1) at age 30 years (C) and inferior left retina of the proband's brother (P2) at age 40 years (D).
(E and F) Family 3. Color fundus photos of the posterior pole (top) and inferior retina (bottom) of the right and left eyes of the proband in family 3 (F), with normal autofluorescence imaging of the posterior poles (E). Bone spiculation is seen in the peripheral retina only (arrows in F).
(G and H) Family 4. Fundus images of the left (G) and right (H) eyes of the proband. Note the abrupt demarcation between normal (light areas) and abnormal (dark areas) peripheral retina. RPE degeneration and bone-spicule-like pigment clumping (arrows) were present circumferentially in both eyes.
(I–K) Family 5. Proband (III-5), 23 years of age, showing total serous retinal detachment in the right eye (arrows), with central yellow deposit (I) and retinal detachment extending nasally (J). Autofluorescence imaging in individual III-2, showing increased signal in area of yellow deposits (arrows) (K).
Family 2: Concentric RP Panel, Heterozygous c.682g>a, p.D228N
Family 2 is a white family of European descent. Two siblings were diagnosed with RP at ∼25 years of age. At age 30 years, the female sibling (P1 in Milam et al.22) reported no night blindness or visual field loss. She had near-normal visual acuity, high myopia, some constriction of the visual field, and subnormal rod and cone ERGs. The male sibling (P2 in Milam et al.22) had longstanding complaints of peripheral visual loss but no night blindness at age 40 years. There was reduced visual acuity in the right eye as a result of a posterior subcapsular cataract, near-normal visual acuity in the left eye, constricted visual fields, and reduced rod and cone ERGs. Both patients had vessel attenuation, dense pigmentary change in all quadrants of the peripheral retina, and an abrupt change to a more-normal-appearing central retina (Figures 2C and 2D) (see also Figure 7 in Milam et al.22). There was no history of other affected family members, but given the possibility of an autosomal-dominant mode of inheritance in this family, it is of note that a fundus examination of the father revealed peripheral pigmentary changes in both eyes. The mother's clinical examination was normal. Samples for genotyping were not available from the parents.
Family 3: adRP Panel, Heterozygous c.682g>a, p.D228N
Family 3 is a white family of European descent. The proband reported the onset of night blindness in the fifth decade. He was noted to have a high degree of myopia, asteroid hyalosis, and reduced visual acuity and visual field at age 58 years. The maculae appeared normal on clinical examination, and this was supported by normal autofluorescence imaging (Figure 2E) and ocular coherence tomography. Intraretinal bone spicule pigmentation was present in the peripheral retina only but without a sharp demarcation between normal and abnormal retina (Figure 2F). The affected mother was also heterozygous for c.682g>a but was not seen in the clinic. Neither the proband nor his mother underwent EOG or ERG testing, because these could not be locally performed to ISCEV standards.
Family 4: Concentric RP Panel, Heterozygous c.680a>g, p.Y227C
Family 4 is a white family of European descent. The proband had minimal symptoms but was referred to the clinic at age 45 years after a retinal examination by her optometrist. Examination revealed an abrupt change from normal to abnormal retina in the periphery, suggestive of a bestrophinopathy (Figures 2G and 2H). After molecular testing and the identification of a heterozygous p.Y227C mutation adjacent to the other concentric RP p.D228N mutation reported here, the fundus images were found to be consistent with a diagnosis of concentric RP. The proband was unavailable for either ERG or EOG testing and has two affected daughters with a similar phenotype, who were unavailable for molecular testing.
Family 5: adRP Panel, Homozygous c.418c>g, p.L140V
Family 5 is a family of Pakistani origin. The proband (III-5 in family 5, Figure 1B) presented with night blindness and central vision loss in her right eye at the age of 11 years. Examination revealed a right total serous retinal detachment with bilateral central retinal yellow foveal deposits and macular edema (Figures 2I and 2J). Her visual acuities at 23 years of age were recorded as counting fingers (right eye) and 6/36 (left eye). Electrophysiology testing revealed no recordable pattern ERG, and extinguished or reduced rod “a” and “b” wave responses and maximal bright white flash ERGs in the right and left eyes, respectively. Cone responses were delayed with reduced amplitudes. Her father and two sisters were also affected. (family 5, Figure 1B). Her two affected sisters complained of nyctalopia and central visual loss as teenagers. Visual acuities were 6/12 (right) and 6/7.5 (left) for individual III-2 at age 30 years, and 6/12 (right) and 6/48 (left) for individual III-8 at age 18 years. The pattern ERGs were extinguished for subject III-8, and the rod and cone ERGs were also abnormal. The father, patient II-6, had visual acuities of 6/120 (right and left) at age 55 years. At 23 years of age, his visual acuities were severely reduced and electrophysiology testing revealed no recordable pattern ERG, extinguished or reduced rod “a” and “b” wave responses and maximal bright white flash ERGs, and delayed cone responses with reduced amplitudes.
All three affected siblings and their affected father have a similar phenotype, with bilateral macular edema and yellow deposits concentrated at the macula, scattered in the posterior pole, and characterized by an increased signal on autofluorescence (Figure 2K). Retinal degenerative changes were seen in the father, who presented at age 41 years with yellow deposits, intraretinal bone spicule pigmentation, and attenuated blood vessels.
Although the proband's parents were not of a consanguineous marriage, they were from the same village. All tested individuals (proband, father, and sister III-2) were homozygous for the mutation c.418c>g (p.L140V). No other family members were available for molecular testing.
Conservation of Mutated Residues
A multiple alignment of the region of bestrophin-1 containing the four RP mutations reported here with nine orthologs is shown in Figure 3. The tyrosine at position 227 and the aspartic acid at position 228 are both conserved down to Danio rerio. The leucine at position 140 is conserved down to Xenopus tropicalis but is a tyrosine in D. rerio. The isoleucine at position 205 is conserved in Pan troglodytes, Macaca mulatta, and Bos taurus but is a valine in the other six species.
Figure 3.

Multiple Alignment of Bestrophin-1 around the Residues Mutated in RP
The amino acid sequence of bestrophin-1 incorporating the four RP mutations (p.L140V, p.I205T, p.Y227C, p.D228N; shaded boxes) is shown for ten orthologs (Homo sapiens, Pan troglodytes, Macaca mulatta, Bos taurus, Canis familiaris, Mus musculus, Rattus norvegicus, Gallus gallus, Xenopus tropicalis, Danio rerio) The alignment was performed with ClustalW, and the sequence accession numbers are those quoted in Milenkovic et al.49
Whole-Cell Patch-Clamp Analysis of RP Mutant Isoforms
Cells expressing WT bestrophin-1 exhibited large, outwardly rectifying-currents (Figure 4B) in comparison to control cells transfected with eGFP (Figure 4A). Current magnitudes were very much smaller in cells transfected with the p.I205T mutant (Figure 4C). Current densities were only partially reduced in cells transfected with both p.I205T and WT bestrophin-1 (Figure 4D). The reversal potential was −19.5 ± 2.5 mV for the WT bestrophin-1 (Figure 4D). This value is toward the ECl (equilibrium potential for Cl− calculated with the Nernst equation) established by our experimental solutions (−34 mV) and is similar to values we have previously determined for bestrophin-1.17 The reversal potentials for the currents carried by p.I205T alone or by p.I205T plus WT were not significantly different from the WT value (p > 0.1) (Figure 4D).
Figure 4.
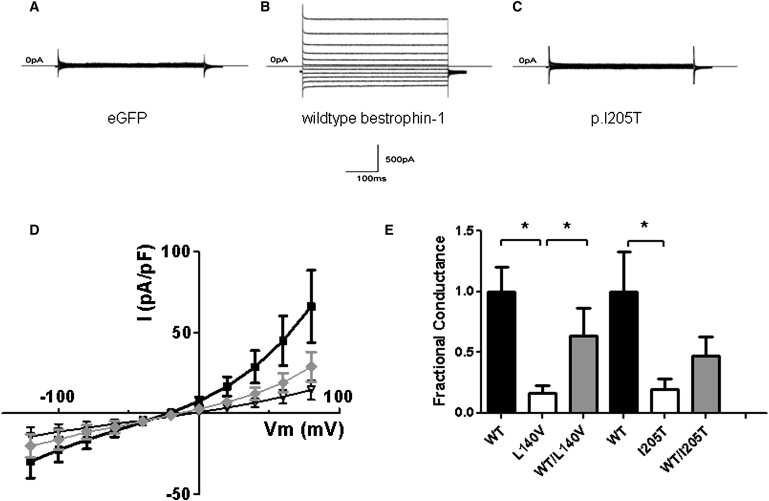
Cl− Channel Activity Associated with the Expression of p.I205T and p.L140V in HEK293 Cells
(A–C)Whole-cell current records in cells transfected with (A) eGFP, (B) WT bestrophin-1, and (C) p.I205T. Currents were evoked with 450 ms voltage steps (Vm) from −120 mV to 80 mV in increments of 20 mV. The holding Vm was −50 mV.
(D) Current density is plotted as a function of Vm for cells transfected with WT bestrophin-1 (■; n = 10), p.I205T (♦; n = 10), and WT plus p.I205T (∇; n = 7).
(E) Outward conductances measured for combinations of WT and mutant bestrophins. Conductances were measured between Vm = 60–80 mV, and the values were normalized to the mean WT conductance for each series of experiments. In the p.L140V series of experiments, the number of patched cells were as follows: WT, n = 14; p.I205T, n = 12; and WT plus p.I205T, n = 14. Results are presented as mean ± standard error of the mean. An asterisk denotes a significant difference (p < 0.05).
Whole-cell currents were greatly reduced or partially reduced in cells transfected with p.L140V alone or cotransfected with WT and p.L140V, respectively (Figure 4E). Conductances observed with the expression of p.L140V or p.I205T alone were significantly reduced in comparison to those observed in WT controls (p < 0.05). Cotransfection of either mutant with WT produced conductances that were not significantly reduced in comparison to controls (p > 0.1). However, the conductance observed with WT and p.L140V was significantly greater than that observed with p.L140V alone (p < 0.05) (Figure 4E).
Cellular Localization of RP Mutant Isoforms
The localization of WT and RP-associated bestrophin-1 mutant isoforms were determined in polarized epithelial MDCK II cells. WT bestrophin-1 localized to the basolateral membrane of MDCKII cells and was exclusively basal to the tight junction marker ZO-1 (Figure 5A). The p.I205T mutant isoform had a cell-membrane localization similar to that of the WT protein (Figure 5B). The localization of mutant isoforms p.L140V and p.D228N exhibited a significant intracellular localization (Figures 5C and 5D respectively), whereas the ARB mutation p.D312N has a cytoplasmic localization (Figure 5F). Images from the complete Z-stack were examined so that a lack of membrane localization at any lateral level was ensured.
Figure 5.
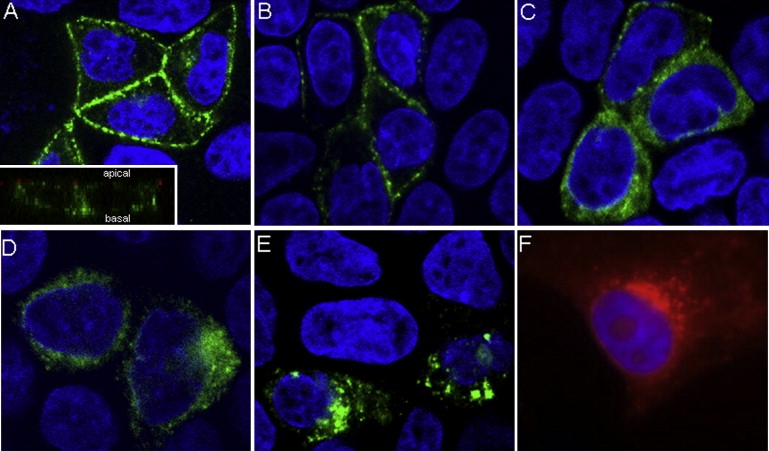
Cellular Localization of Bestrophin-1 Isoforms in MDCK II and HEK293 Cells
Transiently transfected MDCK II cells (A–D and F) were polarized on Transwell membranes before immunofluorescent labeling with bestrophin-1 antibody (green in A–D and F) and confocal microscopy. Nuclei were labeled with DAPI (blue). A representative lateral (Z-stack) image is shown. WT (A) and p.I205T (B) isoforms localized to the basolateral membrane. A vertical optical section shows that WT bestrophin-1 (green) localizes basal to the tight junction maker ZO-1 (red) (insert, A). The p.L140V (C) and p.D228N (D) isoforms exhibited a diffuse cytoplasmic localization. The p.D312N mutant isoform has a cytoplasmic localization (E). WT bestrophin-1 (red) expressed in HEK-293 cells does not traffic to the cell membrane (F).
Discussion
To our knowledge this is the first report of mutations in an RPE gene, BEST1, in patients diagnosed with adRP. Families 1, 3, and 4 carry three different mutations, two of them previously unreported, and have an autosomal-dominant mode of inheritance. Family 2 had two affected siblings with the same heterozygous mutation as that in family 3; the fact that their father was affected suggested an autosomal-dominant inheritance pattern. The fifth family (family 5) has a two-generation history of RP, which could be consistent with a dominant mode of inheritance (family 5, Figure 1B), but molecular analysis has shown the affected father and two affected daughters all to be homozygous for the missense mutation p.L140V. The mother, who is from the same village as the father, is presumed to be heterozygous for the mutation, and because neither the mother nor any of her other six children have reported or have been diagnosed with symptoms of RP, it must be concluded that in this family the disease is recessively inherited. Unfortunately, no other family members were available for molecular testing for confirmation of this.
The four RP mutations reported here all lie within the predicted cytoplasmic loop between transmembrane domains 2 and 3,25 and three of them are within 24 residues of each other, two being adjacent. This clustering within one domain of the bestrophin-1 protein suggests that a specific interaction or function is disrupted by these four mutations. To the best of our knowledge, this interaction domain has not been previously reported, because proteins previously identified as interacting with bestrophin-1, protein phosphatise 2A and the CaVβ subunit, interact with the cytoplasmic C-terminal tail, which is distal to the fourth transmembrane domain.26,27 The predicted cytoplasmic loop containing the four RP mutations also contains several other residues that are mutated in either VMD or ARB,28 suggesting that a particular function or interaction is disrupted when these residues are altered. With the exception of ADVIRC, the genotype-phenotype correlation with bestrophin-1 mutations is not straightforward. A very similar situation is seen in the peripherin/RDS gene, mutations in which cause macular dystrophies, cone and cone-rod dystrophy, and RP.29 Additionally, as with the bestrophinopathies, the phenotypes associated with peripherin/RDS mutations show a high degree of inter- and intrafamilial variation.29
Sequence analysis of ten bestrophin-1 orthologs revealed that three of the four residues (p.L140, p.Y227, and p.D228) that were altered in the RP patients were highly conserved, supporting the conclusion that the sequence alterations are pathogenic (Figure 3). The fourth residue, isoleucine at position 205, is conserved down to Bos taurus but is a valine in the other species analyzed (isoleucine and valine are both nonpolar, hydrophobic, and aliphatic residues). The change of this residue in family 1 from an isoleucine to a threonine alters the residue to a small polar one, which is likely to be functionally significant.
In this study, we examined the functional consequences of two of the bestrophin-1 mutations that cause RP on Cl− conductance and on protein localization: p.L140V (recessive) and p.I205T (dominant). Whole-cell patch-clamp data demonstrated that heterologous expression of the p.I205T and p.L140V mutations produced greatly reduced Cl− currents in comparison to the WT bestrophin-1. Cotransfection of the adRP p.I205T mutant isoform with WT bestrophin-1 resulted in a Cl− conductance that was less than 50% of the WT and was not significantly different from that measured in the presence of the mutant alone (p > 0.1). In contrast, cotransfection of the arRP p.L140V mutant isoform with WT bestrophin-1 resulted in a Cl− conductance that was about 60% of the WT and was significantly greater than the conductance measured with the mutant alone (p < 0.01; Figure 4). The behavior of the two mutant isoforms is therefore consistent with the dominant and recessive disorders with which they are associated.
We tested the effect of three of the RP mutations on the intracellular trafficking of bestrophin-1 by using polarized MDCK II cells. The choice of this model cell system is important. MDCK II cells are the most widely used mammalian cell system for studying cell trafficking and polarity because when they are grown on porous filters they model a physiological polarized system.30 Previous information about the trafficking and localization of bestrophin-1 is from experiments in nonpolarized HEK293 cells.31–34 These cells are easy to transfect and produce high levels of protein in comparison to more specialized cells, such as MDCK II cells. However, transient transfection in nonspecialized cells can lead to misinterpretation of protein localization,35 such that any conclusions drawn from such studies should be considered with due caution.36 WT bestrophin-1 does not traffic correctly in HEK293 cells (Figure 5E). The importance of studying the trafficking of proteins with a polarized distribution in a polarized cell model has been demonstrated for several integral membrane proteins, including CFTR. In HEK293 cells, some mutant CFTR isoforms traffic to the cell surface in quantities sufficient for generating specific currents via whole-cell patch-clamping experiments.37 However, when these mutant isoforms were studied in polarized MDCK II cells, they were not detectable at the cell surface.36,37 Similarly, bestrophin-1 p.D312N was detected at normal, or above normal, levels at the cell surface of HEK293 cells by a biotinylation assay,33 but we have found this isoform to be cytoplasmic in MDCK II cells (Figure 5F). Thus, for future bestrophin-1 trafficking studies, we suggest that the use of an appropriate cell system, such as the polarized MDCK II cells used here, should be a requirement. MDCK II cells do, however, express an endogenous Ca2+-activated Cl− conductance, so that HEK293 cells are still preferred for electrophysiological studies.38
Two of the three mutations analyzed, p.L140V (arRP) and p.D228N (adRP), caused bestrophin-1 to mislocalize from the basolateral membrane to the cytoplasm, and this is likely to contribute to their pathogenicity. Importantly, the p.I205T mutation that causes a more severe, earlier-onset, dominant retinal dystrophy had a similar localization to that of the WT protein (Figure 5), suggesting that membrane localization alone can explain neither the pathogenesis of this group of mutations nor the differences in Cl− conductance observed in this study.
The phenotype associated with each of the bestrophinopathies is distinct, and the lack of correlation between some mutations and their functional behavior implies that additional factors need to be considered. These could include a degree of redundancy provided by bestrophin-2.39 However, unpublished data cited by Marmorstein et al.40 show that bestrophin-2 is expressed not in the RPE but in the nonpigmented epithelial cells of both mouse and human retina and that a double knockout of Best1 and Best2 does not lead to either a retinal degeneration or an altered EOG. The null bestrophin-1 mouse and human have distinct phenotypes,9,17 so it is likely that other factors have a bearing on what phenotype is associated with a particular mutation. These include other genes, the environment, and how the cell machinery processes certain mutations. The importance of protein processing is illustrated by the example of claudin 16, which has a predicted topology similar to that of bestrophin-1. A study of 21 CLDN16 mutations that cause familial hypomagnesemia with hypercalciuria and nephrocalcinosis (MIM 248250) found that different mutations were processed in very different ways.41 Nine of the mutant isoforms were retained within the endoplasmic reticulum, and three accumulated in the Golgi complex. Two mutant isoforms were detected in lysosomes; of these one was retro-transported from the cell surface, and the second never made it to the cell surface at all. The remaining seven mutant isoforms were all correctly trafficked to tight junctions, although four were defective in paracellular Mg2+ transport.41
An interesting observation in the present study is the presence of retinal detachments in the probands of two families (1 and 5). In the case of family 1, the proband was found to have severe bilateral serous retinal detachments at the age of 13 months. Exudative detachment is a previously reported, but uncommon, feature of the pseudohypopyon stage of VMD.42–46 Basolateral membrane Cl− conductance regulates fluid transport across the RPE and, consequently, the volume of the subretinal space,47 and it has been suggested that disruption of fluid absorption across the RPE may be partly responsible for the etiology of diseases involving the RPE and photoreceptors. In the case of serous retinal detachments, this may occur by either a slowing or a reversal of fluid transport.47 Such a process has also been proposed as part of the mechanism underlying the accumulation of vitelliform pigment under the macula in the pseudohypopyon stage of VMD.6
The identification of the homozygous p.L140V mutation in a family diagnosed with arRP and ARB (our unpublished data) illustrates the increasingly complex clinical picture associated with bestrophin-1 mutations. For some mutations, such as p.L140V, it may be that the homozygous state can manifest as ARB, and given the phenotypic overlap between RP and ARB, it is also likely that clinicians may misdiagnose cases of ARB as RP if they are not fully aware of the ARB phenotype or see an ARB patient at later disease stages. The critical region identified from linkage analysis in family 1 overlaps with the locus for autosomal-dominant neovascular inflammatory vitreoretinopathy (ADNIV), which is characterized by retinal and iris neovascularization, abnormal retinal pigmentation, anterior chamber and vitreous inflammation, cystoid macular edema, vitreous hemorrhage, and traction retinal detachment.48 The ADNIV phenotype is rare but appears distinct from that seen in the family 1 patients who lack neovascularisation, vitreous hemorrhage, and traction retinal detachment. The ADNIV locus centers on marker D11S527, which is 14.6 Mb distal to BEST1. The phenotype and mapping data both suggest that it is unlikely that family 1 has ADNIV caused by mutations in an unknown gene.
This study reports four missense mutations, three previously unreported, that are the cause of autosomal-dominant, concentric, or autosomal-recessive RP and/or retinal dystrophies in five unrelated families. Phenotypic heterogeneity is not uncommon in ocular disease, as a review of the RetNet website clearly highlights. However, the situation with BEST1 is becoming increasingly complicated, because five separate ocular diseases are now ascribed to mutations in it (VMD, adult-onset VMD, ADVIRC, ARB, and RP) and the same mutation can be causal in multiple bestrophinopathies. Although the incidence of each of these bestrophinopathies is individually rare, the increasing number of phenotypes associated with BEST1 mutations suggests that it as an increasingly important cause of genetic ocular disease.
Supplemental Data
Supplemental Data include six figures and can be found with this article online at http://www.cell.com/AJHG.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
BioEdit (ClustalW), http://www.mbio.ncsu.edu/BioEdit/bioedit.html
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/omim/
WEBMAXC, http://www.stanford.edu/∼cpatton/webmaxc/webmaxcS.htm
Acknowledgments
A.E.D. is supported by the National Eye Research Centre (SCIAD 051). G.J.M. is supported by Fight for Sight. J.O'S. and A.J.L. are supported by the British Retinitis Pigmentosa Society. S.G.J. is funded by the Macula Vision Research Foundation. Funding was also provided by the Manchester Biomedical Research Centre, the Manchester Academic Health Sciences Centre (MAHSC), and the National Institute for Health Research (NIHR) Manchester Biomedical Research Centre. The confocal microscope used in this study was purchased with grants from the Biotechnology and Biological Sciences Research Council, The Wellcome Trust, and The University of Manchester Strategic Fund.
References
- 1.Sun H., Tsunenari T., Yau K.W., Nathans J. The vitelliform macular dystrophy protein defines a new family of chloride channels. Proc. Natl. Acad. Sci. USA. 2002;99:4008–4013. doi: 10.1073/pnas.052692999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Petrukhin K., Koisti M.J., Bakall B., Li W., Xie G.C., Marknell T., Sandgren O., Forsman K., Holmgren G., Andreasson S. Identification of the gene responsible for Best macular dystrophy. Nat. Genet. 1998;19:241–247. doi: 10.1038/915. [DOI] [PubMed] [Google Scholar]
- 3.Marmorstein A.D., Marmorstein L.Y., Rayborn M., Wang X.X., Hollyfield J.G., Petrukhin K. Bestrophin, the product of the Best vitelliform macular dystrophy gene (VMD2), localizes to the basolateral plasma membrane of the retinal pigment epithelium. Proc. Natl. Acad. Sci. USA. 2000;97:12758–12763. doi: 10.1073/pnas.220402097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bakall B., Marmorstein L.Y., Hoppe G., Peachey N.S., Wadelius C., Marmorstein A.D. Expression and localization of bestrophin during normal mouse development. Invest. Ophthalmol. Vis. Sci. 2003;44:3622–3628. doi: 10.1167/iovs.03-0030. [DOI] [PubMed] [Google Scholar]
- 5.Tsunenari T., Sun H., Williams J., Cahill H., Smallwood P., Yau K.W., Nathans J. Structure-function analysis of the bestrophin family of anion channels. J. Biol. Chem. 2003;278:41114–41125. doi: 10.1074/jbc.M306150200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hartzell H.C., Qu Z., Yu K., Xiao Q., Chien L.T. Molecular physiology of bestrophins: multifunctional membrane proteins linked to best disease and other retinopathies. Physiol. Rev. 2008;88:639–672. doi: 10.1152/physrev.00022.2007. [DOI] [PubMed] [Google Scholar]
- 7.Chien L.T., Zhang Z.R., Hartzell H.C. Single Cl- channels activated by Ca2+ in Drosophila S2 cells are mediated by bestrophins. J. Gen. Physiol. 2006;128:247–259. doi: 10.1085/jgp.200609581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosenthal R., Bakall B., Kinnick T., Peachey N., Wimmers S., Wadelius C., Marmorstein A., Strauss O. Expression of bestrophin-1, the product of the VMD2 gene, modulates voltage-dependent Ca2+ channels in retinal pigment epithelial cells. FASEB J. 2006;20:178–180. doi: 10.1096/fj.05-4495fje. [DOI] [PubMed] [Google Scholar]
- 9.Marmorstein L.Y., Wu J., McLaughlin P., Yocom J., Karl M.O., Neussert R., Wimmers S., Stanton J.B., Gregg R.G., Strauss O. The light peak of the electroretinogram is dependent on voltage-gated calcium channels and antagonized by bestrophin (Best-1) J. Gen. Physiol. 2006;127:577–589. doi: 10.1085/jgp.200509473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bear C.E., Li C., Kartner N., Bridges R.J., Jensen T.J., Ramjeesingh M., Riordan J.R. Purification and functional reconstitution of the cystic fibrosis transmembrane conductance regulator (CFTR) Cell. 1992;68:809–818. doi: 10.1016/0092-8674(92)90155-6. [DOI] [PubMed] [Google Scholar]
- 11.Choi J.Y., Muallem D., Kiselyov K., Lee M.G., Thomas P.J., Muallem S. Aberrant CFTR-dependent HCO-3 transport in mutations associated with cystic fibrosis. Nature. 2001;410:94–97. doi: 10.1038/35065099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reddy M.M., Light M.J., Quinton P.M. Activation of the epithelial Na+ channel (ENaC) requires CFTR Cl- channel function. Nature. 1999;402:301–304. doi: 10.1038/46297. [DOI] [PubMed] [Google Scholar]
- 13.Hartzell C., Qu Z., Putzier I., Artinian L., Chien L.T., Cui Y. Looking chloride channels straight in the eye: bestrophins, lipofuscinosis, and retinal degeneration. Physiology (Bethesda) 2005;20:292–302. doi: 10.1152/physiol.00021.2005. [DOI] [PubMed] [Google Scholar]
- 14.Gallemore R.P., Steinberg R.H. Effects of DIDS on the chick retinal pigment epithelium. II. Mechanism of the light peak and other responses originating at the basal membrane. J. Neurosci. 1989;9:1977–1984. doi: 10.1523/JNEUROSCI.09-06-01977.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yardley J., Leroy B.P., Hart-Holden N., Lafaut B.A., Loeys B., Messiaen L.M., Perveen R., Reddy M.A., Bhattacharya S.S., Traboulsi E. Mutations of VMD2 splicing regulators cause nanophthalmos and autosomal dominant vitreoretinochoroidopathy (ADVIRC) Invest. Ophthalmol. Vis. Sci. 2004;45:3683–3689. doi: 10.1167/iovs.04-0550. [DOI] [PubMed] [Google Scholar]
- 16.Kaufman S.J., Goldberg M.F., Orth D.H., Fishman G.A., Tessler H., Mizuno K. Autosomal dominat vitreochoroidopathy. Arch. Ophthalmol. 1982;100:272–278. doi: 10.1001/archopht.1982.01030030274008. [DOI] [PubMed] [Google Scholar]
- 17.Burgess R., Millar I.D., Leroy B.P., Urquhart J.E., Fearon I.M., De Baere E., Brown P.D., Robson A.G., Wright G.A., Kestelyn P. Biallelic mutation of BEST1 causes a distinct retinopathy in humans. Am. J. Hum. Genet. 2008;82:19–31. doi: 10.1016/j.ajhg.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guziewicz K.E., Zangerl B., Lindauer S.J., Mullins R.F., Sandmeyer L.S., Grahn B.H., Stone E.M., Acland G.M., Aguirre G.D. Bestrophin gene mutations cause canine multifocal retinopathy: a novel animal model for best disease. Invest. Ophthalmol. Vis. Sci. 2007;48:1959–1967. doi: 10.1167/iovs.06-1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin–rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 20.Barrett J.C., Fry B., Maller J., Daly M.J. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 21.Collins J.S., Schwartz C.E. Detecting Polymorphisms and Mutations in Candidate Genes. Am. J. Hum. Genet. 2002;71:1251–1252. doi: 10.1086/344344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Milam A.H., De Castro E.B., Smith J.E., Tang W.X., John S.K., Gorin M.B., Stone E.M., Aguirre G.D., Jacobson S.G. Concentric retinitis pigmentosa: clinicopathologic correlations. Exp. Eye Res. 2001;73:493–508. doi: 10.1006/exer.2001.1059. [DOI] [PubMed] [Google Scholar]
- 23.Marquardt A., Stohr H., Passmore L.A., Kramer F., Rivera A., Weber B.H. Mutations in a novel gene, VMD2, encoding a protein of unknown properties cause juvenile-onset vitelliform macular dystrophy (Best's disease) Hum. Mol. Genet. 1998;7:1517–1525. doi: 10.1093/hmg/7.9.1517. [DOI] [PubMed] [Google Scholar]
- 24.Lotery A.J., Munier F.L., Fishman G.A., Weleber R.G., Jacobson S.G., Affatigato L.M., Nichols B.E., Schorderet D.F., Sheffield V.C., Stone E.M. Allelic variation in the VMD2 gene in best disease and age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 2000;41:1291–1296. [PubMed] [Google Scholar]
- 25.Milenkovic V.M., Rivera A., Horling F., Weber B.H.F. Insertion and topology of normal and mutant bestrophin-1 in the endoplasmic reticulum membrane. J. Biol. Chem. 2007;282:1313–1321. doi: 10.1074/jbc.M607383200. [DOI] [PubMed] [Google Scholar]
- 26.Marmorstein A.D., McLaughlin P.J., Stanton J., Yan L., Crabb J.W., Marmorstein L.Y. Bestrophin interacts with the b-catalytic subunit of protein phosphatase 2A. Invest. Ophthalmol. Vis. Sci. 2002;43:U818. [Google Scholar]
- 27.Yu K., Xiao Q., Cui G., Lee A., Hartzell H.C. The best disease-linked Cl- channel hBest1 regulates Ca V 1 (L-type) Ca2+ channels via src-homology-binding domains. J. Neurosci. 2008;28:5660–5670. doi: 10.1523/JNEUROSCI.0065-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boon C.J., Klevering B.J., Leroy B.P., Hoyng C.B., Keunen J.E., den Hollander A.I. The spectrum of ocular phenotypes caused by mutations in the BEST1 gene. Prog. Retin. Eye Res. 2009;28:187–205. doi: 10.1016/j.preteyeres.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 29.Boon C.J., den Hollander A.I., Hoyng C.B., Cremers F.P., Klevering B.J., Keunen J.E. The spectrum of retinal dystrophies caused by mutations in the peripherin/RDS gene. Prog. Retin. Eye Res. 2008;27:213–235. doi: 10.1016/j.preteyeres.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 30.Mostov K., Su T., ter Beest M. Polarized epithelial membrane traffic: conservation and plasticity. Nat. Cell Biol. 2003;5:287–293. doi: 10.1038/ncb0403-287. [DOI] [PubMed] [Google Scholar]
- 31.Xiao Q., Prussia A., Yu K., Cui Y.Y., Hartzell H.C. Regulation of bestrophin Cl channels by calcium: role of the C terminus. J. Gen. Physiol. 2008;132:681–692. doi: 10.1085/jgp.200810056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu K., Cui Y., Hartzell H.C. The Bestrophin Mutation A243V, Linked to Adult-Onset Vitelliform Macular Dystrophy, Impairs Its Chloride Channel Function. Invest. Ophthalmol. Vis. Sci. 2006;47:4956–4961. doi: 10.1167/iovs.06-0524. [DOI] [PubMed] [Google Scholar]
- 33.Yu K., Qu Z., Cui Y., Hartzell H.C. Chloride channel activity of bestrophin mutants associated with mild or late-onset macular degeneration. Invest. Ophthalmol. Vis. Sci. 2007;48:4694–4705. doi: 10.1167/iovs.07-0301. [DOI] [PubMed] [Google Scholar]
- 34.Qu Z., Cheng W., Cui Y., Cui Y., Zheng J. Human disease-causing mutations disrupt an N-C-terminal interaction and channel function of bestrophin 1. J. Biol. Chem. 2009;284:16473–16481. doi: 10.1074/jbc.M109.002246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cordat E. Unraveling trafficking of the kidney anion exchanger 1 in polarized MDCK epithelial cells. Biochem. Cell Biol. 2006;84:949–959. doi: 10.1139/o06-200. [DOI] [PubMed] [Google Scholar]
- 36.Krasnov K.V., Tzetis M., Cheng J., Guggino W.B., Cutting G.R. Localization studies of rare missense mutations in cystic fibrosis transmembrane conductance regulator (CFTR) facilitate interpretation of genotype-phenotype relationships. Hum. Mutat. 2008;29:1364–1372. doi: 10.1002/humu.20866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mickle J.E., Milewski M., Macek M., Cutting G.R. Effects of Cystic Fibrosis and Congenital Bilateral Absence of the Vas Deferens-Associated Mutations on Cystic Fibrosis Transmembrane Conductance Regulator-Mediated Regulation of Separate Channels. Am. J. Hum. Genet. 2000;66:1485–1495. doi: 10.1086/302893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mendes F., Wakefield J., Bachhuber T., Barroso M., Bebok Z., Penque D., Kunzelmann K., Amaral M.D. Establishment and characterization of a novel polarized MDCK epithelial cellular model for CFTR studies. Cell. Physiol. Biochem. 2005;16:281–290. doi: 10.1159/000089857. [DOI] [PubMed] [Google Scholar]
- 39.Barro-Soria R., Schreiber R., Kunzelmann K. Bestrophin 1 and 2 are components of the Ca(2+) activated Cl(-) conductance in mouse airways. Biochim. Biophys. Acta. 2008;1783:1993–2000. doi: 10.1016/j.bbamcr.2008.06.016. [DOI] [PubMed] [Google Scholar]
- 40.Marmorstein A.D., Cross H.E., Peachey N.S. Functional roles of bestrophins in ocular epithelia. Prog. Retin. Eye Res. 2009;28:206–226. doi: 10.1016/j.preteyeres.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kausalya P.J., Amasheh S., Gunzel D., Wurps H., Muller D., Fromm M., Hunziker W. Disease-associated mutations affect intracellular traffic and paracellular Mg2+ transport function of Claudin-16. J. Clin. Invest. 2006;116:878–891. doi: 10.1172/JCI26323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pianta M.J., Aleman T.S., Cideciyan A.V., Sunness J.S., Li Y., Campochiaro B.A., Campochiaro P.A., Zack D.J., Stone E.M., Jacobson S.G. In vivo micropathology of Best macular dystrophy with optical coherence tomography. Exp. Eye Res. 2003;76:203–211. doi: 10.1016/s0014-4835(02)00280-4. [DOI] [PubMed] [Google Scholar]
- 43.Men G., Batioglu F., Ozkan S.S., Atilla H., Ozdamar Y., Aslan O. Best's vitelliform macular dystrophy with pseudohypopyon: an optical coherence tomography study. Am. J. Ophthalmol. 2004;137:963–965. doi: 10.1016/j.ajo.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 44.Vedantham V., Ramasamy K. Optical coherence tomography in Best's disease: an observational case report. Am. J. Ophthalmol. 2005;139:351–353. doi: 10.1016/j.ajo.2004.07.039. [DOI] [PubMed] [Google Scholar]
- 45.Pierro L., Tremolada G., Introini U., Calori G., Brancato R. Optical coherence tomography findings in adult-onset foveomacular vitelliform dystrophy. Am. J. Ophthalmol. 2002;134:675–680. doi: 10.1016/s0002-9394(02)01685-9. [DOI] [PubMed] [Google Scholar]
- 46.Jaffe G.J., Schatz H. Histopathologic features of adult-onset foveomacular pigment epithelial dystrophy. Arch. Ophthalmol. 1988;106:958–960. doi: 10.1001/archopht.1988.01060140104034. [DOI] [PubMed] [Google Scholar]
- 47.Gallemore R.P., Hughes B.A., Miller S.S. Retinal pigment epithelial transport mechanisms and their contributions to the electroretinogram. Prog. Retin. Eye Res. 1997;16:509–566. [Google Scholar]
- 48.Stone E.M., Kimura A.E., Folk J.C., Bennett S.R., Nichols B.E., Streb L.M., Sheffield V.C. Genetic linkage of autosomal dominant neovascular inflammatory vitreoretinopathy to chromosome 11q13. Hum. Mol. Genet. 1992;1:685–689. doi: 10.1093/hmg/1.9.685. [DOI] [PubMed] [Google Scholar]
- 49.Milenkovic V.M., Langmann T., Schreiber R., Kunzelmann K., Weber B.H. Molecular evolution and functional divergence of the bestrophin protein family. BMC Evol. Biol. 2008;8:72. doi: 10.1186/1471-2148-8-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.