

Abstract
The critical importance of cytoskeletal function for correct neuronal migration during development of the cerebral cortex has been underscored by the identities of germline mutations underlying a number of human neurodevelopmental disorders. The proteins affected include TUBA1A, a major α-tubulin isoform, and microtubule-associated components such as doublecortin, and LIS1. Mutations in these genes are associated with the anatomical abnormality lissencephaly, which is believed to reflect failure of neuronal migration. An important recent observation has been the dependence of cortical neuronal migration upon acetylation of α-tubulin at lysine 40 by the histone acetyltransferase Elongator complex. Here, we describe a recognizable autosomal recessive syndrome, characterized by generalized polymicrogyria in association with optic nerve hypoplasia (PMGOH). By autozygosity mapping, we show that the molecular basis for this condition is mutation of the TUBA8 gene, encoding a variant α-tubulin of unknown function that is not susceptible to the lysine 40 acetylation that regulates microtubule function during cortical neuron migration. Together with the unique expression pattern of TUBA8 within the developing cerebral cortex, these observations suggest a role for this atypical microtubule component in regulating mammalian brain development.
Main Text
Polymicrogyria (PMG) is a malformation of the cerebral cortex in which the usual gyral pattern is replaced by numerous small infoldings, separated by sulci, which fuse inferiorly, and the normally six-layered cortex is replaced by a four-layered or unlayered cortex.1 PMG can be a focal lesion or a more generalized cortical abnormality and may be accompanied by other malformations, including agenesis of the corpus callosum, microcephaly, or cerebellar vermis hypoplasia. Localized PMG is most commonly perisylvian, but has been described in all areas of the brain, with its extent and location determining the neurological manifestations. Within a developmental classification of brain malformations, the PMG spectrum is believed to reflect abnormal late neuronal migration and cortical organization, unlike the lissencephalies, which are characterized by defective migration earlier in cortical development.1
Bilateral PMG has been recognized in various genetic disorders; mutations in RAB3GAP1 (MIM 602536),2 SRPX2 (MIM 300642),3 PAX6 (MIM 607108),4 TBR2 (MIM 604615),5 COL18A1 (MIM 120328),6 and KIAA1279 (MIM 609367)7 have been associated with PMG. However, these all appear to represent syndromes in which PMG is only an occasional feature. In contrast, it has been difficult to delineate clinical disorders in which PMG is a hallmark lesion, and genetic clues to the pathogenesis of PMG have consequently been lacking. One exception is the syndrome of bilateral frontoparietal polymicrogyria (BFPP [MIM 606854]), caused by mutations that interfere with the N-glycosylation of the GPR56 protein (GPR56 [MIM 604110]).8 Notably, though, the radiological appearance of BFPP resembles those of the so-called cobblestone malformations, the result of O-glycosylation defects in the developing brain.9 BFPP is in this respect rather distinct from other PMGs, which are thought to be the result of abnormal cortical organization.1
We assessed four children from two consanguineous families of Pakistani origin, all of whom presented with similar clinical features; the two families were not knowingly related to each other (Figure 1). Written informed consent for the studies was obtained from subjects or their parents, in accordance with a protocol approved by the Leeds (East) Research Ethics Committee (reference 07/H1306/113).
Figure 1.
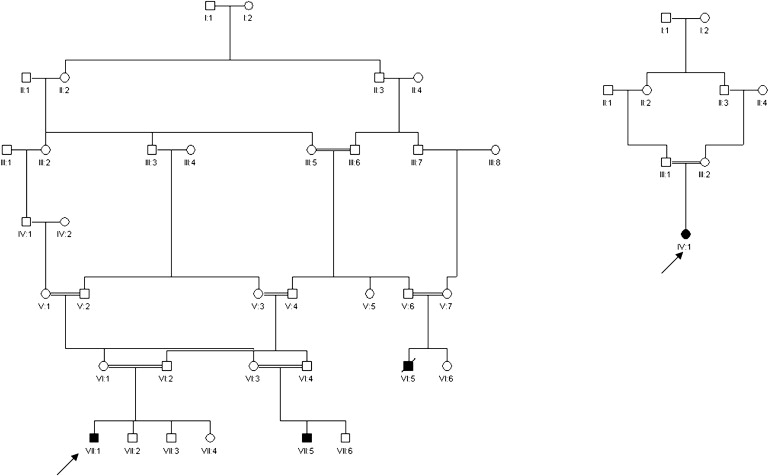
Pedigrees of Two Families with PMG and Optic Hypoplasia
In each case, the proband is indicated by an arrow.
The children had a constellation of severe developmental delay, hypotonia, and seizures. One child died at age 2 from an overwhelming respiratory infection, whereas the others have survived with profound neurological impairment. None of the children was dysmorphic; however, optic nerve hypoplasia was observed on ophthalmoscopy in all four subjects. Clinical details are found in Table 1.
Table 1.
Clinical Features in Affected Individuals
| Patient Information | ||||
|---|---|---|---|---|
| Patient | 1 | 2 | 3 | 4 |
| Family ID | Family 1 | Family 1 | Family 1 | Family 2 |
| Pedigree position | VII:1 | VII:5 | VI:5 | IV:1 |
| Age at last review | 30 months | 4.5 years | 12 years | 2.5 years |
| Clinical Feature | ||||
| Gestation | Term | Term | Term | Term |
| Delivery | Normal vaginal | Normal vaginal | Normal vaginal | Normal vaginal |
| Birthweight | 2975 g | 3200 g | N/A | 2675 g |
| Occipitofrontal circumference | 3° centile | 0.4° centile | 25°centile | 25° centile |
| Neonatal hypotonia | + | N/A | + | + |
| Obtunded reflexes | + | + | + | + |
| Seizure onset | 2 months | 5 days | 4 months | Six months |
| Seizure type | Infantile spasms | Tonic clonic | Tonic clonic | Tonic clonic |
| Speech | None | None | None | None |
| Motor development | Unable to sit | Unable to sit | Unable to sit | Unable to sit |
| Ophthalmoscopy | Optic nerve hypoplasia | Optic nerve hypoplasia | Optic nerve hypoplasia | Optic nerve hypoplasia |
| Neuroimaging | ||||
| Polymicrogyria | Extensive bilateral PMG; thickened gray matter | Extensive bilateral PMG, both anterior and posterior | Extensive bilateral PMG | Extensive bilateral PMG, both anterior and posterior |
| Corpus callosum | Thinning, marked posterior deficiency | Absent | Absent | Absent |
| Brainstem | Hypoplastic brainstem with loss of demarcation of pontomedullary junction | Loss of demarcation of the pontomedullary junction | Loss of demarcation of the pontomedullary junction | Loss of demarcation of the pontomedullary junction |
| Other features | Colpocephaly | Colpocephaly | Colpocephaly | |
+, feature present; N/A, information not available.
Neuroimaging revealed a pattern of extensive PMG, with a dysplastic or absent corpus callosum and hypoplastic optic nerves. In addition, there was a distinctive abnormality of the brainstem, in which the normal sharp demarcation between the pons and medulla was absent, with the pontine bulge extending too far caudally (Figure 2 and Figures S1–S6 available online).
Figure 2.
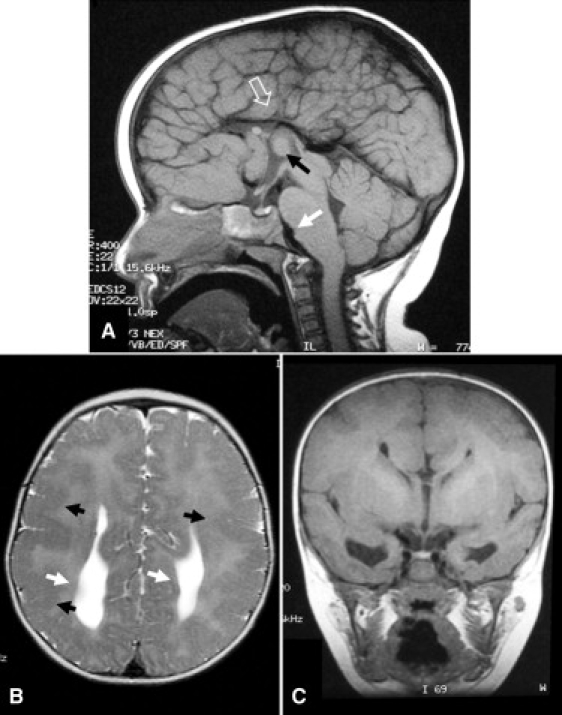
Magnetic Resonance Images of Affected Individual IV:1 from Family 2
(A) The corpus callosum is absent, as is the cingulate gyrus (hollow white arrow). Disorganized gyral folds radiate toward a high-riding third ventricle. A prominent massa intermedia is seen (black arrow). There is malformation of the brainstem, with loss of demarcation of the pontomedullary junction (solid white arrow).
(B) The cortex is thickened and the gyral pattern is abnormal. There is generalized PMG (black arrows), and agenesis of the corpus callosum has resulted in colpocephaly (white arrows).
(C) Coronal section at the level of the third ventricle, demonstrating absence of the corpus callosum with a “Viking helmet” configuration of the frontal horns and a high-riding third ventricle.
The structure of family 1 and the consanguinity in both families suggested an autosomal-recessive disorder. We therefore performed a SNP-microarray genome-wide homozygosity scan by using the Affymetrix Genome-Wide Human SNP Array 6.0. SNP genotypes were analyzed with AutoSNPa10 and IBDfinder software11 for identifying regions of concordant homozygosity in affected individuals from both families. Mutation analysis was performed by direct sequencing of purified genomic PCR products, using an Applied Biosystems 3130xl Genetic Analyzer (primer sequences and experimental conditions listed in Table S1. Anonymized control samples were screened by genoytping.
SNP genotyping revealed no call rates averaging 8.5%. Markers not called in all four cases were eliminated. A single 7.42 Mb region of concordant homozygosity on chromosome 22q11.2 in affected individuals, bounded by SNP markers rs4607032 (16.22 Mb) and rs139764 (23.64Mb), was identified. There were 929 SNPs within the shared minimal autozygous region. IBDfinder and AutoSNPa analysis revealed no other regions of shared concordant homozygosity between the four cases. The centromeric boundary of the shared region was defined by heterozygosity for rs4607032 in case 4 (individual IV:1 in family 2) and the telomeric boundary by heterozygous SNP rs139764 in case 1. Within this region, we also confirmed concordant homozygosity for microsatellite markers D22S420 (16.24 Mb), D22S427 (16.97 Mb), D22S264 (19.10 Mb), D22S446 (20.35 Mb), and D22S539 (20.59 Mb). The combined genotyping data are therefore strongly indicative of a common ancestral background for this autozygous region in both families.
The minimal autozygous interval contains more than 230 annotated genes (after exclusion of the immunoglobulin λ cluster). Of immediate note, however, was the α8-tubulin gene (TUBA8 [MIM 605742]).12 Dominant mutations in the α1-tubulin gene (TUBA1A [MIM 602529]) are known to cause lissencephaly associated with mild microcephaly and dysgenesis of the corpus callosum and brainstem hypoplasia.13–15 More recently, dominant mutations in TUBB2B (MIM 812650) resulting in an asymmetric frontal predominant PMG with corpus callosum dysgenesis have also been described.16 TUBA8 (known to be expressed in the brain12) was therefore an obvious candidate gene. Sequencing in the affected members of the two families revealed no coding region variants, but a homozygous 14 bp deletion in intron 1 of TUBA8 was identified (Figure 3A). This segregated with the disease in both families, and all obligate carriers were heterozygous for the mutation. The mutation was not present in 342 ethnically matched control chromosomes.
Figure 3.
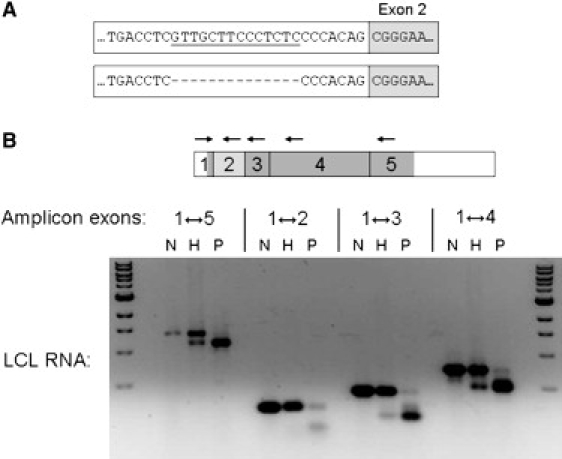
Splicing Mutation of TUBA8 in PMGOH-Affected Individuals
(A) Schematic to show site and extent of 14-bp deletion mutation.
(B) Reverse transcription PCR analysis of LCL RNA. The skipped exon 2 is shaded light gray, coding regions of other exons are shown in dark gray. The following abbreviations are used: N, normal control; H, heterozygote; and P, patient (homozygous).
The deletion lies 11 bp upstream of the exon 2 splice junction, eliminating a large portion of the acceptor site polypyrimidine tract. In silico analysis accordingly predicted that it would interfere with correct splicing of exon 2. To confirm this, we analyzed lymphoblastoid cell line (LCL) RNA from case 1 by using RT-PCR (Figure 3B). Total RNA from EBV-transformed lymphoblastoid cells was extracted with Trizol (Invitrogen, Renfrew, UK). Reverse transcription and PCR were performed with the Thermoscript RT-PCR system (Invitrogen Life Technologies), in accordance with the manufacturer's instructions. (Primer sequences and experimental conditions are listed in Table S1.)
The full-length TUBA8 transcript was found at greatly reduced level, the major species being a shortened transcript, confirmed by sequencing to lack exon 2. Similarly shortened RT-PCR products were obtained using various primer pairs flanking exon 2, in each case a low level of full-length transcript being detectable (Figure 3B). RT-PCR with one primer in exon 2 confirmed that a low level of correctly spliced exon 2-containing mRNA was detectable in the patient LCL. In his heterozygous father, RT-PCR product from the full-length transcript predominates over the shortened mutant product (even though the latter is expected to amplify more efficiently), implying greatly reduced level of the aberrantly spliced RNA, most likely because it is unstable.
We next investigated the expression pattern of Tuba8 in the developing mouse brain by using RNA in situ hybridization. In brief, digoxigenin-labeled Tuba8 riboprobes were generated by antisense (SP6) and sense (control, T7) in vitro transcription of a 443-bp region comprising the 3′ end of mouse Tuba8 cDNA (nt 990–1432 of NM_017379 inclusive), cloned into pGEM-T Easy (Promega UK, Southampton). Mouse embryos were collected under an approved animal research protocol. Tissue preparation and nonradioactive RNA in situ hybridization and visualization were performed as previously described.17
Tuba8 was found to be widely expressed in developing neural structures. At embryonic day (E) 13.5, Tuba8 is expressed in the cortical preplate (PP) and cingulate cortex (CiC).There is also evidence of weaker expression in the subventricular zone (SVZ)/mantle zone of the ganglionic eminence (GE) (Figures 4A and 4F); these cells will be undergoing final divisions and then making tangential migrations through the mantle zone to the cortex. By E15.5 (Figures 4B and 4G), the strongest expression is seen in the cortical plate (CP); there is fainter labeling in the SVZ and intermediate zone (IZ). There is now additional expression visible in the hypothalamus (Figure 4K). By E18.5, cortical expression is most intense in the upper layers and subplate (SP) (Figures 4C and 4H). There is strong expression in the areas CA1-3 of the hippocampus (Figure 4L). At P0, cortical expression is strongest in the dense cortical plate and subplate (Figures 4D and 4I). Hippocampal expression is more intense in areas CA1-3 than in the dentate gyrus (Figure 4M). At P8 (Figures 4E and 4J), lamination is almost complete and cortical expression is strongest in layers II–III and V and the subplate. There is also expression in the mediodorsal nuclei of the thalamus, the mitral cell layer of the olfactory bulb, and the external granular layer, molecular layer, and internal granular cell layer of the cerebellum (Figure 4O).
Figure 4.
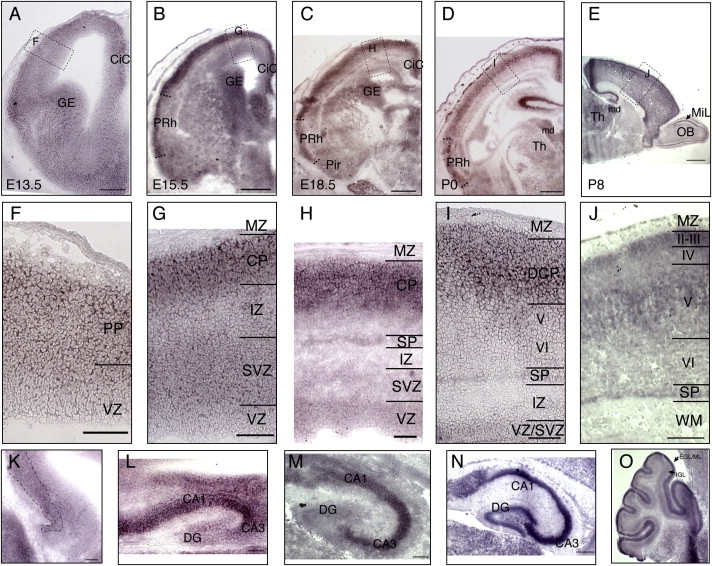
Tuba8 Expression Studied with In Situ Hybridization during Mouse Brain Development
(A–D) Coronal sections at E13.5, E15.5, E18, and P0.
(E) Sagittal section at P8. In (A)–(E), dashed boxes refer to areas shown enlarged in (F)–(J).
(K–N) Hippocampus at E15, E18, P0, and P8.
(O) Cerebellum at P8.
(A and F) At E13.5 there is generally stronger expression in the postmitotic layers of the telencephalon, such as dorsal cortex and cingulate cortex (CiC), ganglionic eminence (GE). There is also evidence of expression in migratory cells of the SVZ/mantle zone of the ganglionic eminence (GE).
(B and G) By E15.5 (B) the strongest expression is in the emerging cortical plate of the dorsal cortex, in perirhinal cortex (PRh), CiC and GE. High magnification (G) reveals most intense expression in cortical plate (CP), but there is also expression in the subventricular zone (SVZ), and intermediate zone (IZ).
(C and H) At E18.5 (C) the cortical expression becomes more intense in the upper layers and subplate, with relatively less expression in putative layer VI. The VZ has stronger expression at this age (H) than at E15.5 (G). The dorsal and perirhinal cortices have stronger expression than piriform cortex (Pir).
(D and I) At P0 the strongest cortical expression is in the layer of still migrating cortical neurons of the dense cortical plate (DCP) and subplate (SP). There is continued expression in the perirhinal cortex (PRh). The mediodorsal nuclei (md) of the thalamus (Th) have strong Tuba8 expression.
(E and J) By P8, the cortical lamination is close to completion. The strongest expression of Tuba8 is in layers II–III and V and the subplate. The expression in the dorsomedial thalamus has been maintained, and in the olfactory bulb (OB) expression can be seen in the mitral cell layer (MiL).
(K) The pyramidal layer of the hippocampus also starts to express Tuba8 from E15.
(L) At E 18 there is strong expression of Tuba8 in the CA1-3 region of the hippocampus, with lower expression in dentate gyrus (DG).
(M) At P0 there is strong Tuba8 expression in the hippocampus, more intense in regions CA1-3 than the dentate gyrus.
(N) At P8 in the hippocampus there is especially strong expression in area CA3, but there is expression in other regions, including the dentate gyrus (DG).
(O) Tuba8 expression is seen in the inner part of the external granular layer and possibly molecular layer (EGL/ML) and less intensely in the internal granular layer (IGL). Scale bars represent 150 μm in (A), 500 μm in (B)–(D), 1 mm in (E), 100 μm in (F)–(M), and 250 μm in (N) and (O). The following abbreviations are used: PP, preplate; MZ, marginal zone; and WM, white matter.
Very recently, de novo dominant mutations in a β-tubulin gene, TUBB2B, have been described in children who have an asymmetrical PMG, together with corpus callosum dysgenesis, abnormal basal ganglia, cerebellar dysplasia, and brainstem hypoplasia.16 There are similarities between the Tuba8 expression pattern we have observed with that reported for Tubb2b16 in the developing brain, but these are not maintained postnatally. At E15.5–E16.5, both Tuba8 and Tubb2b are expressed in the cortical plate and subplate, with little expression in the marginal zone and weaker expression in the ventricular and intermediate zones. In contrast, expression of the lissencephaly-associated Tuba1a protein is strong in the migratory cells of the intermediate zone at similar time points.14 Taken together, these data suggest that Tuba8, like Tubb2b, is strongly expressed in postmitotic cortical neurons.18–20 However, by P8, the strongest expression of Tuba8 is in cortical layers II–III and V and the subplate, whereas expression of Tubb2b decreases in the postnatal cortex.16 These data suggest that Tuba8 has a role in neuronal migration of deep-layer neurons but that its expression is further upregulated once the cells reach the cortical plate. This implies a role for Tuba8 in cortical organization not seen for either Tuba1a or Tubb2b.
The BFPP gene, Gpr56, which at E10–E16 is principally expressed in neuronal progenitors within the neurogenic compartments of the ventricular and subventricular zones, is absent from the cortical plate and intermediate zone.8 This is quite different from what we report for Tuba8. BFPP has a distinctive neuroradiological appearance that classifies it as one of the “cobblestone” defects of neuronal migration21.
The extensive expression of the tubulins during neurodevelopment is likely to reflect the essential structural and transport roles of microtubules in the cellular cytoskeleton during development, whereas the more restricted expression of GPR56 reflects its role in the maintenance of pial basement membrane integrity during the development of the cortex.22
The subcellular localization and function of α8-tubulin are unknown. Given that no antibodies specific for α8-tubulin exist, we created MDCKII cell lines stably expressing GFP-tagged TUBA8. A full-length human TUBA8 cDNA cassette was generated by reverse-transcription-PCR and fused in frame 3′ to the EGFP ORF of pEGFP-C1 (Clontech, Mountain View, CA). MDCKII cells (ECACC, Salisbury, UK) were cultured in MEM + 5% fetal bovine serum + 2 mM glutamine and transfected with Lipofectamine 2000 (Invitrogen Ltd., Renfrew, UK) in accordance with the manufacturers' instructions. Twenty-four hours after transfection, the medium was supplemented with 1 mg/ml G418 and refreshed every 2 days thereafter for 2 weeks. Surviving cells were then trypsinized and replated. Live imaging and processing for immunufluorescence after fixation in cold methanol were performed as described.23 Examination of these cells confirmed that the fusion protein incorporated into microtubules (Figure 5A), but at lower efficiency than GFP-tagged α1-tubulin in parallel transfection experiments (data not shown). The incorporation was therefore most evident in situations where microtubule bundles were prominent, such as during mitosis (Figures 5B and 5C). Immunostaining indicated that GFP-TUBA8 expression had no obvious gross impact on the general organization of the microtubule network in expressing cells (Figures 5D and 5E). These data therefore confirm that α8-tubulin participates in microtubule assembly in mammalian cells.
Figure 5.
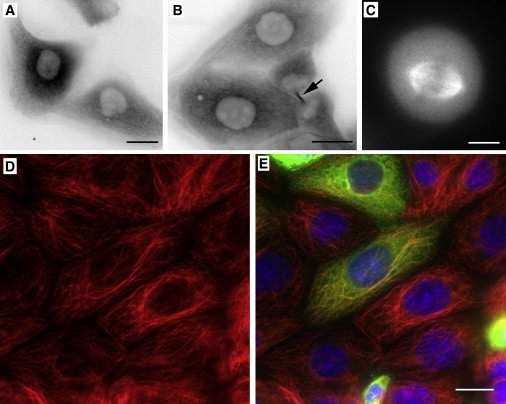
GFP-TUBA8 Incorporates into Microtubules within Cells
Individual frames from a time-lapse series of MDCKII cells stably transfected with GFP-TUBA8. The fusion protein incorporates weakly into microtubules (A). Microtubule incorporation was consequently most evident in cellular structures containing bundled microtubules, such as the midbody in telophase cells (B, arrow) or the mitotic spindle (metaphase cell shown in C). Images in (A) and (B) are shown in inverted grayscale for improving image contrast, whereas (C) is shown in grayscale. Transfected cells were also fixed and immunostained with antibodies to α-tubulin (D and E, red) and GFP (E, green). Nuclei were counterstained with DAPI (E, blue). (E) shows a merged image. No obvious abnormalities in microtubule organization were observed in cells expressing higher levels of GFP-TUBA8. Scale bars in (A), (B), and (E) represent 20 μm. The scale bar in (C) represents 10 μm.
TUBA8 is an evolutionary outlier within the mammalian α-tubulin family.12 Interestingly, its two most divergent regions of sequence are known in other family members to be subject to neurodevelopmentally important posttranslational modifications.24
The first of these modifications is specific excision and religation of a C-terminal tyrosine residue, whose status modulates the binding of CAP-Gly domain proteins to microtubule tips,25 thereby influencing morphological processes during neuronal differentiation.26 Tubulin tail modifications, including detyrosination, influence the activity of microtubule-severing factors that are involved in generating the noncentrosomal microtubule array in neurons.27 TUBA8 is unlikely to be subject to such modifications, given that its C-terminal residue is phenylalanine, as in S. cerevisiae α-tubulin (in which no evidence for an excision/religation cycle has been found).28
The second modification comprises acetylation of lysine 40, a residue conserved in all α-tubulins except TUBA8. Acetylation of α-tubulin lysine 40 by the Elongator complex is an important determinant of cortical neuronal migration in the intermediate zone.29 We have observed that expression of the nonacetylatable α8-tubulin is less prominent in these migratory neurons, in contrast to their high level of TUBA1A expression. TUBA8 expression is more marked in the postmigratory cortical plate neurons. Therefore, it may be that TUBA1A has a greater role in migration, whereas TUBA8 has a more prominent role in local adjustments in cell position after arrival in the cortical plate. In addition, though, optic nerve hypoplasia and callosal dysgenesis were consistent features in the patients described here. This may indicate that the TUBA8 mutations also impair axon outgrowth and/or establishment of connectivity. TUBA8 is now the third tubulin to be implicated in a specific aspect of cerebral cortical development. The distinctive phenotypes manifested by the patients harboring mutations in each of these three genes point to unique cellular roles for their respective gene products. The high degree of sequence similarity between members of the α-tubulin family presents a difficulty in dissecting out these unique features. For TUBA8, the important clue, that this isoform lacks the capability for important regulatory posttranslational modifications, offers a useful starting point in this regard.
Finally, the chromosomal localization of TUBA8 is noteworthy. It is the closest gene proximal to the low-copy repeat (LCR22-2, LCR-A)30 that mediates recurrent deletion of the DiGeorge/VCFS microdeletion region. Notably, a small proportion of children with the 22q11.2 microdeletion syndrome display PMG; indeed, this is possibly the single most common cause of PMG.31 TUBA8 was not observed to be deleted in a large DiGeorge/VCFS microdeletion series.32 It will therefore now be logical to assess the possibility that a positional effect on TUBA8 expression underlies the association of DiGeorge/VCFS and PMG.
All annotations and physical positions are recorded as in NCBI Genome Build 36.3.
Acknowledgments
This work was supported by funding from the UK Department of Health under the “New and Emerging Applications of Technology” (NEAT) scheme (H033; “New Technologies in Recessive Diseases”).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
AutoSNPa software, http://dna.leeds.ac.uk/autosnpa/
IBDFinder software, http://dna.leeds.ac.uk/ibdfinder/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim
References
- 1.Barkovich A.J., Kuzniecky R.I., Jackson G.D., Guerrini R., Dobyns W.B. A developmental and genetic classification for malformations of cortical development. Neurology. 2005;65:1873–1887. doi: 10.1212/01.wnl.0000183747.05269.2d. [DOI] [PubMed] [Google Scholar]
- 2.Aligianis I.A., Johnson C.A., Gissen P., Chen D., Hampshire D., Hoffmann K., Maina E.N., Morgan N.V., Tee L., Morton J. Mutations of the catalytic subunit of RAB3GAP cause Warburg Micro syndrome. Nat. Genet. 2005;37:221–223. doi: 10.1038/ng1517. [DOI] [PubMed] [Google Scholar]
- 3.Roll P., Rudolf G., Pereira S., Royer B., Scheffer I.E., Massacrier A., Valenti M.-P., Roeckel-Trevisiol N., Jamali S., Beclin C. SRPX2 mutations in disorders of language cortex and cognition. Hum. Mol. Genet. 2006;15:1195–1207. doi: 10.1093/hmg/ddl035. [DOI] [PubMed] [Google Scholar]
- 4.Glaser T., Ton C.C., Mueller R., Petzl-Erler M.L., Oliver C., Nevin N.C., Housman D.E., Maas R.L. Absence of PAX6 gene mutations in Gillespie syndrome (partial aniridia, cerebellar ataxia, and mental retardation) Genomics. 1994;19:145–148. doi: 10.1006/geno.1994.1024. [DOI] [PubMed] [Google Scholar]
- 5.Baala L., Briault S., Etchevers H.C., Laumonnier F., Natiq A., Amiel J., Boddaert N., Picard C., Sbiti A., Asermouh A. Homozygous silencing of T-box transcription factor EOMES leads to microcephaly with polymicrogyria and corpus callosum agenesis. Nat. Genet. 2007;39:454–456. doi: 10.1038/ng1993. [DOI] [PubMed] [Google Scholar]
- 6.Sertie A.L., Sossi V., Camargo A.A., Zatz M., Brahe C., Passos-Bueno M.R. Collagen XVIII, containing an endogenous inhibitor of angiogenesis and tumor growth, plays a critical role in the maintenance of retinal structure and in neural tube closure (Knobloch syndrome) Hum. Mol. Genet. 2000;9:2051–2058. doi: 10.1093/hmg/9.13.2051. [DOI] [PubMed] [Google Scholar]
- 7.Brooks A.S., Bertoli-Avella A.M., Burzynski G.M., Breedveld G.J., Osinga J., Boven L.G., Hurst J.A., Mancini G.M.S., Lequin M.H., de Coo R.F. Homozygous nonsense mutations in KIAA1279 are associated with malformations of the central and enteric nervous systems. Am. J. Hum. Genet. 2005;77:120–126. doi: 10.1086/431244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Piao X., Hill R.S., Bodell A., Chang B.S., Basel-Vanagaite L., Straussberg R., Dobyns W.B., Qasrawi B., Winter R.M., Innes A.M. G protein-coupled receptor-dependent development of human frontal cortex. Science. 2004;303:2033–2036. doi: 10.1126/science.1092780. [DOI] [PubMed] [Google Scholar]
- 9.Barresi R., Campbell K.P. Dystroglycan: From biosynthesis to pathogenesis of human disease. J. Cell Sci. 2006;119:199–207. doi: 10.1242/jcs.02814. [DOI] [PubMed] [Google Scholar]
- 10.Carr I.M., Flintoff K.J., Taylor G.R., Markham A.F., Bonthron D.T. Interactive visual analysis of SNP data for rapid autozygosity mapping in consanguineous families. Hum. Mutat. 2006;27:1041–1046. doi: 10.1002/humu.20383. [DOI] [PubMed] [Google Scholar]
- 11.Carr I.M., Sheridan E., Hayward B.E., Markham A.F., Bonthron D.T. IBDfinder and SNPsetter: Tools for pedigree-independent identification of autozygous regions in individuals with recessive inherited disease. Hum. Mutat. 2009;30:960–967. doi: 10.1002/humu.20974. [DOI] [PubMed] [Google Scholar]
- 12.Stanchi F., Corso V., Scannapieco P., Ievolella C., Negrisolo E., Tiso N., Lanfranchi G., Valle G. TUBA8: A new tissue-specific isoform of alpha-tubulin that is highly conserved in human and mouse. Biochem. Biophys. Res. Commun. 2000;270:1111–1118. doi: 10.1006/bbrc.2000.2571. [DOI] [PubMed] [Google Scholar]
- 13.Bahi-Buisson N., Poirier K., Boddaert N., Saillour Y., Castelnau L., Philip N., Buyse G., Villard L., Joriot S., Marret S. Refinement of cortical dysgeneses spectrum associated with TUBA1A mutations. J. Med. Genet. 2008;45:647–653. doi: 10.1136/jmg.2008.058073. [DOI] [PubMed] [Google Scholar]
- 14.Morris-Rosendahl D.J., Najm J., Lachmeijer A.M., Sztriha L., Martins M., Kuechler A., Haug V., Zeschnigk C., Martin P., Santos M. Refining the phenotype of alpha-1a Tubulin (TUBA1A) mutation in patients with classical lissencephaly. Clin. Genet. 2008;74:425–433. doi: 10.1111/j.1399-0004.2008.01093.x. [DOI] [PubMed] [Google Scholar]
- 15.Poirier K., Keays D.A., Francis F., Saillour Y., Bahi N., Manouvrier S., Fallet-Bianco C., Pasquier L., Toutain A., Tuy F.P.D. Large spectrum of lissencephaly and pachygyria phenotypes resulting from de novo missense mutations in tubulin alpha 1A (TUBA1A) Hum. Mutat. 2007;28:1055–1064. doi: 10.1002/humu.20572. [DOI] [PubMed] [Google Scholar]
- 16.Jaglin X.H., Poirier K., Saillour Y., Buhler E., Tian G., Bahi-Buisson N., Fallet-Bianco C., Phan-Dinh-Tuy F., Kong X.P., Bomont P. Mutations in the beta-tubulin gene TUBB2B result in asymmetrical polymicrogyria. Nat. Genet. 2009;41:746–752. doi: 10.1038/ng.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang W.-Z., Molnár Z. Dynamic pattern of mRNA expression of plasticity-related gene-3 (PRG-3) in the mouse cerebral cortex during development. Brain Res. Bull. 2005;66:454–460. doi: 10.1016/j.brainresbull.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 18.Kriegstein A.R., Noctor S.C. Patterns of neuronal migration in the embryonic cortex. Trends Neurosci. 2004;27:392–399. doi: 10.1016/j.tins.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 19.Marín O., Rubenstein J.L.R. Cell migration in the forebrain. Annu. Rev. Neurosci. 2003;26:441–483. doi: 10.1146/annurev.neuro.26.041002.131058. [DOI] [PubMed] [Google Scholar]
- 20.Métin C., Baudoin J.-P., Rakić S., Parnavelas J.G. Cell and molecular mechanisms involved in the migration of cortical interneurons. Eur. J. Neurosci. 2006;23:894–900. doi: 10.1111/j.1460-9568.2006.04630.x. [DOI] [PubMed] [Google Scholar]
- 21.Guerrini R., Dobyns W.B., Barkovich A.J. Abnormal development of the human cerebral cortex: Genetics, functional consequences and treatment options. Trends Neurosci. 2008;31:154–162. doi: 10.1016/j.tins.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 22.Jin Z., Tietjen I., Bu L., Liu-Yesucevitz L., Gaur S.K., Walsh C.A., Piao X. Disease-associated mutations affect GPR56 protein trafficking and cell surface expression. Hum. Mol. Genet. 2007;16:1972–1985. doi: 10.1093/hmg/ddm144. [DOI] [PubMed] [Google Scholar]
- 23.Langford K.J., Lee T., Askham J.M., Morrison E.E. Adenomatous polyposis coli localization is both cell type and cell context dependent. Cell Motil. Cytoskeleton. 2006;63:483–492. doi: 10.1002/cm.20139. [DOI] [PubMed] [Google Scholar]
- 24.Hammond J.W., Cai D., Verhey K.J. Tubulin modifications and their cellular functions. Curr. Opin. Cell Biol. 2008;20:71–76. doi: 10.1016/j.ceb.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peris L., Thery M., Fauré J., Saoudi Y., Lafanechère L., Chilton J.K., Gordon-Weeks P., Galjart N., Bornens M., Wordeman L. Tubulin tyrosination is a major factor affecting the recruitment of CAP-Gly proteins at microtubule plus ends. J. Cell Biol. 2006;174:839–849. doi: 10.1083/jcb.200512058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Erck C., Peris L., Andrieux A., Meissirel C., Gruber A.D., Vernet M., Schweitzer A., Saoudi Y., Pointu H., Bosc C. A vital role of tubulin-tyrosine-ligase for neuronal organization. Proc. Natl. Acad. Sci. USA. 2005;102:7853–7858. doi: 10.1073/pnas.0409626102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roll-Mecak A., Vale R.D. Structural basis of microtubule severing by the hereditary spastic paraplegia protein spastin. Nature. 2008;451:363–367. doi: 10.1038/nature06482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Badin-Larcon A.C., Boscheron C., Soleilhac J.M., Piel M., Mann C., Denarier E., Fourest-Lieuvin A., Lafanechere L., Bornens M., Job D. Suppression of nuclear oscillations in Saccharomyces cerevisiae expressing Glu tubulin. Proc. Natl. Acad. Sci. USA. 2004;101:5577–5582. doi: 10.1073/pnas.0307917101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Creppe C., Malinouskaya L., Volvert M.-L., Gillard M., Close P., Malaise O., Laguesse S., Cornez I., Rahmouni S., Ormenese S. Elongator controls the migration and differentiation of cortical neurons through acetylation of alpha-tubulin. Cell. 2009;136:551–564. doi: 10.1016/j.cell.2008.11.043. [DOI] [PubMed] [Google Scholar]
- 30.Shaikh T.H., Kurahashi H., Saitta S.C., O'Hare A.M., Hu P., Roe B.A., Driscoll D.A., McDonald-McGinn D.M., Zackai E.H., Budarf M.L. Chromosome 22-specific low copy repeats and the 22q11.2 deletion syndrome: Genomic organization and deletion endpoint analysis. Hum. Mol. Genet. 2000;9:489–501. doi: 10.1093/hmg/9.4.489. [DOI] [PubMed] [Google Scholar]
- 31.Robin N.H., Taylor C.J., McDonald-McGinn D.M., Zackai E.H., Bingham P., Collins K.J., Earl D., Gill D., Granata T., Guerrini R. Polymicrogyria and deletion 22q11.2 syndrome: window to the etiology of a common cortical malformation. Am. J. Med. Genet. A. 2006;140:2416–2425. doi: 10.1002/ajmg.a.31443. [DOI] [PubMed] [Google Scholar]
- 32.Jalali G.R., Vorstman J.A.S., Errami A., Vijzelaar R., Biegel J., Shaikh T., Emanuel B.S. Detailed analysis of 22q11.2 with a high density MLPA probe set. Hum. Mutat. 2008;29:433–440. doi: 10.1002/humu.20640. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.