

Abstract
A modified α-N-acetylgalactosaminidase (NAGA) with α-galactosidase A (GLA)-like substrate specificity was designed on the basis of structural studies and was produced in Chinese hamster ovary cells. The enzyme acquired the ability to catalyze the degradation of 4-methylumbelliferyl-α-D-galactopyranoside. It retained the original NAGA's stability in plasma and N-glycans containing many mannose 6-phosphate (M6P) residues, which are advantageous for uptake by cells via M6P receptors. There was no immunological cross-reactivity between the modified NAGA and GLA, and the modified NAGA did not react to serum from a patient with Fabry disease recurrently treated with a recombinant GLA. The enzyme cleaved globotriaosylceramide (Gb3) accumulated in cultured fibroblasts from a patient with Fabry disease. Furthermore, like recombinant GLA proteins presently used for enzyme replacement therapy (ERT) for Fabry disease, the enzyme intravenously injected into Fabry model mice prevented Gb3 storage in the liver, kidneys, and heart and improved the pathological changes in these organs. Because this modified NAGA is hardly expected to cause an allergic reaction in Fabry disease patients, it is highly promising as a new and safe enzyme for ERT for Fabry disease.
Introduction
Fabry disease (MIM 301500) is an X-linked genetic disease caused when a deficiency of α-galactosidase A (GLA, MIM 300644, EC3.2.1.22) activity results in lysosomal accumulation of globotriaosylceramide (Gb3; also called ceramide trihexoside, CTH).1 The disease exhibits a wide range of clinical phenotypes, from the early-onset severe “classic form” to the late-onset moderate one called the “variant form.” Having no GLA activity, patients with the classic form develop pain in the peripheral extremities, hypohidrosis, angiokeratomas, and corneal opacities, and there is also renal and cardiac involvement. Patients with the variant form have residual GLA activity and develop milder clinical manifestations, sometimes limited to heart disorders.1 Recently, a high incidence (1 in ∼3,000) of this disease was revealed in newborn screening,2 and the clinical importance of this disease is being increasingly recognized.
So far, two different recombinant GLAs have been developed for enzyme replacement therapy (ERT) for Fabry disease: agalsidase beta (Fabrazyme; Genzyme Therapeutics, Cambridge, MA), generated in Chinese hamster ovary (CHO) cells,3,4 and agalsidase alfa (Replagal; Shire HGT, Cambridge, MA), produced in cultured human fibroblasts.5 They are intravenously injected into patients with Fabry disease every 2 weeks and are expected to be incorporated into cells via mannose 6-phosphate (M6P) receptors on the plasma membrane and then transported to lysosomes.6 Although many patients have been successfully treated with these recombinant GLA proteins, many problems remain to be resolved, i.e., (1) these drugs are unstable in blood7, (2) they are not well incorporated into target organs, including the kidneys and heart7, and (3) because most male patients with Fabry disease do not have any GLA proteins, repeated injection of recombinant enzymes frequently results in an allergic reaction.4,5 Furthermore, development of antibodies against the recombinant GLAs sometimes decreases the effects of the enzymes.8–10
In this study, we altered the substrate specificity of α-N-acetylgalactosaminidase (NAGA, MIM 104170, EC3.2.1.49), which had been previously called α-galactosidase B11 because the modified enzyme is similar in protein structure to NAGA but displays catalytic activity toward Gb3, by making structure-based amino acid substitutions in the active site to develop a new recombinant enzyme for ERT for Fabry disease.
Material and Methods
The study involving the human serum and cultured fibroblasts was approved by the ethical committee of our institute. The study involving mice was performed according to the rules drawn up by the animal care committee of our institute.
Molecular Design of a Modified NAGA
Structural modeling of human NAGA was performed with molecular modeling software Jackal.12 The crystal structure of chicken NAGA (PDB code: 1KTB)13 was used as a template, and energy minimization was performed. The root-mean-square gradient value was set at 0.05 kcal/mol/Å. The new structure was then compared with the human GLA structure (PDB code: 1R47)14, and the difference between the structures of their respective active sites was examined. Then, target amino acid residues for altering the substrate specificity were determined, and a structural model of the modified NAGA was built with TINKER.15–18 Then, cDNA for the modified NAGA (DDBJ: AB522713) was designed.
GLA and NAGA Assays and Protein Determination
For fluorometric measurement of GLA-degrading activity, 4-methylumbelliferyl (MU)-α-D-galactopyranoside (Calbiochem, LaJolla, CA) was used as a substrate, and N-acetyl-D-galactosamine (Sigma-Aldrich, St. Louis, MO) was used as an inhibitor of NAGA.19 NAGA-degrading activity was fluorometrically measured with MU-α-D-N-acetylgalactosamine (MU-2-acetamide-2-deoxy-α-D-galactopyranoside; Toronto Research Chemicals, North York, ON, Canada) as a substrate.20 Both the GLA and the NAGA assay were conducted with a Wallac 1420 ARVO MX multilabel counter (Perkin Elmer, Waltham, MA) at excitation and emission wavelengths of 355 nm and 460 nm, respectively. Protein concentrations were determined with a Micro BCA Protein Assay Reagent Kit (PIERCE, Rockfold, IL), and bovine serum albumin was used as a standard.
Preparation of a Retrovirus Vector for Expression of the Modified NAGA
Human NAGA cDNA (Open Biosystems, Huntsville, AL; GenBank accession: BC000095, I.M.A.G.E.: 3504221) was used as a template. The coding region for human NAGA was amplified by PCR with the following primers and then introduced into a retrovirus vector, pCX4neo21: NAGA sense, 5′-GATGCTGCTGAAGACATGGCTCTT-3′; and NAGA antisense, 5′-TCACTGCTGGGACTACTCCAGGTT-3′. To generate a modified NAGA with altered substrate specificity, we performed site-directed mutagenesis with a Gene Tailor site-directed mutagenesis kit (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. pCX4neo-NAGA was used as a template with the following primers: S188E sense, 5′-CCCATCGCCTTCTCCTGCGAGTGGCCAGCCTATGA-3′; S188E antisense, 5′-GCAGGAGAAGGCGATGGGGCGGCCTGT-3′. Then pCXneo-NAGA S188E was used as a template with the following primers: A191L sense, 5′-TTCTCCTGCGAGTGGCCACTCTATGAAGGCGGCCT-3′; and A191L antisense, 5′-TGGCCACTCGCAGGAGAAGGCGATGGGG-3′. The italicized nucleotides are the mutated sites. The site-directed mutagenesis was confirmed via sequencing in both directions.
Retrovirus-Mediated Gene Transfer to Human Fabry Fibroblasts
Retrovirus vector plasmids, pCX4neo-NAGA and pCX4neo-modified NAGA, were used for transfection of Phoenix Ampho, an amphotropic human leukemia virus-packaging cell line,22 by means of Dofect-GT1 (Dojindo, Kumamoto, Japan) according to the manufacturer's directions. Two days after the transfection, culture supernatants were collected, filtered, supplemented with 2 μg/ml polybrene (Sigma-Aldrich), and then used for infection. Infection was carried out through the addition of a retrovirus solution of pCX4neo-NAGA or pCX4neo-modified NAGA to the culture medium of Fabry fibroblasts. Two days after the infection, infected cells were selected in 250 μg/ml of G418 disulfate (GIBCO, Grand Island, NY) for 14 days or more. The G418-resistant populations of human Fabry fibroblasts were used for determining the MU-α-D-galactopyranoside- and MU-α-D-N-acetylgalactosamine-degrading activities.
Preparation of a Modified NAGA Construct via a Glutamine Synthetase Gene-Expression System
pCX4neo-modified NAGA was used as a template. The coding region for the modified NAGA was amplified by PCR with the following primers: HindIII-NAGA sense, 5′-CCCAAGCTTGATGCTGCTGAAGACAGTGCTCTT-3′; and EcoRI-NAGA antisense, 5′-GGAATTCTCACTGCTGGGACATCTCCAGGTT-3′. The italicized nucleotides are the HindIII and EcoRI sites, respectively. PCR fragments of the modified NAGA cDNA were introduced into the pEE14.4 vector (Lonza Biologics, Allendale, NJ). Furthermore, so that the secretion would be enhanced, a modified NAGA was generated by replacement of the amino terminus (amino acids 1–17) with the signal peptide of human GLA (amino acids 1–31). Overlap extension was performed by PCR. First, to generate a PCR product of the GLA signal, we amplified the GLA signal sequence by performing PCR with the following primers: Hind III-GLA sense, 5′-CCCAAGCTTATGCAGCTGAGGAACCCAGAA-3′; and NAGA-GLA antisense, 5′-GTCCAGTGCTCTAGCCCCAG-3′. pCXN2-Gal, which cloned the full-length human GLA cDNA23, was used as a template. Second, to generate a PCR product of the modified NAGA minus its signal sequence, we amplified it by conducting PCR with the following primers: GLA-NAGA sense, 5′-TCACTGCTGGGACATCTCCAGGTT-3′; and EcoRI-NAGA antisense, 5′-GGAATTCTCACTGCTGGGACATCTCCAGGTT-3′. pCX4neo-modified NAGA was used as a template. The italicized nucleotides and the underlined nucleotides are the restriction sites and overlapping regions, respectively. Third, to generate a modified NAGA with the GLA signal sequence, we performed overlap extension by conducting PCR with Hind III-GLA sense and EcoRI-NAGA antisense primers and using the first and second PCR products as templates. The PCR fragment of GLA signal-modified NAGA cDNA was introduced into the pEE14.4 vector.
Generation of Stable Cell Lines Expressing the Modified NAGA
CHO cells stably expressing the modified NAGA were generated with a glutamine synthetase gene-expression system (Lonza Biologics) and cloned according to the manufacturer's protocol.
Purification of the Modified NAGA
CHO cells stably expressing the modified NAGA and secreting it into the medium were at first cultured in Dulbecco's modified Eagle's medium without glutamine (GIBCO) but containing 10% dialyzed fetal bovine serum (FBS), glutamine synthetase supplement (SAFC Bioscience, Leneva, KS), and 1 μM L-methionine sulfoximine (MSX; Sigma-Aldrich) at 37°C in an incubator containing 5% CO2. Then the medium was changed to CD Opti-CHO medium (GIBCO) containing 1% dialyzed FBS, glutamine synthetase supplement, and 1 μM MSX, and the conditioned culture medium was harvested every week. The collected medium was clarified, concentrated via an ultrafiltration membrane (Millipore Corporation, Bedford, MA), and then precipitated with ammonium sulfate. The precipitate was dissolved, then subjected to column chromatography on HiLoad 26/10 phenyl-Sepharose, HiLoad 26/10 SP-Sepharose, and HiLoad 26/10 Q-Sepharose HP columns (GE Healthcare Bio-Sciences, Piscataway, NJ), and the fractions showing MU-α-D-galactopyranoside-degrading activity were collected.
Biochemical Analyses of the Modified NAGA
The purity and molecular mass of the modified NAGA were determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) followed by staining with Coomassie brilliant blue R. Deglycosylation of the enzyme was performed with an enzymatic deglycosylation kit (Prozyme, San Lendodro, CA) including glycopeptidase F (PNGase F). As controls, placenta NAGA (a gift from Dr. A. Tsuji, University of Tokushima), agalsidase beta, and agalsidase alfa were used. The enzyme proteins were electrophoresed on a Tris-glycine polyacrylamide gel (Cosmo-bio, Tokyo, Japan) and then stained with Coomassie brilliant blue R. Immunoblotting analyses with anti-NAGA polyclonal antibodies20 and anti-GLA polyclonal antibodies24 were performed. The monosaccharide compositions of N-glycans of the modified NAGA were determined by means of capillary electrophoresis with a P/ACE MDQ equipped with a laser-induced fluorescence detector (Beckman Coulter, Fullerton, CA) after digestion with PNGase F (TAKARA BIO, Shiga, Japan). GLA-degrading activity was measured with MU-α-D-galactopyranoside as a substrate, and NAGA-degrading activity was measured with MU-α-D-N-acetylgalactosamine as a substrate as described above. We performed a kinetic experiment with the modified NAGA and various concentrations of the substrate to determine the Vmax and Km values; agalsidase beta and agalsidase alfa served as controls, as described previously.25
Examination of Immune Reaction of the Modified NAGA to Fabry Serum
To determine whether the modified NAGA reacts with anti-GLA serum, we performed a solid-phase enzyme-linked immunosorbent assay (ELISA). A serum sample was obtained from a Fabry patient who had been repeatedly injected with agalsidase beta (titer of the antibodies against the recombinant GLAs, 12,800×). In brief, a 96-well flat-bottom microplate for ELISA (Maxisorp; Nunk, Rockilde, Denmark) was coated with 100 μl of modified NAGA (1, 3, and 10 μg/ml), agalsidase beta (1 and 3 μg/ml), and agalsidase alfa (1 and 3 μg/ml) at 4°C overnight. After the wells were washed with phosphate-buffered saline (PBS), 250 μl of Block Ace (diluted 1:4; DS Pharma Biomedical, Osaka, Japan) was added to each well as a blocking solution, and incubation followed for 1 hr at room temperature. After removal of the blocking solution, each well was washed with 0.1% Tween 20 in PBS (PBS-T). Then 150 μl of the patient's serum (diluted 1:1,000 or 1:10,000) was added to each well, and incubation followed for 1 hr at room temperature. The wells were then washed, incubated in 200 μl of peroxidase-conjugated anti-human IgG (diluted 1:50,000; Jackson Immuno Research, West Grove, PA) for 1 hr at room temperature, washed again, and then incubated in 100 μl peroxidase substrate (SAT Blue; Dojindo) for 5 min at room temperature. Then, 50 μl of a stopping solution (2 M H2SO4) was added, and the absorbance of each well was measured by means of a microplate reader (Benchmark; BioRad, Hercules, CA).
Stability of the Modified NAGA
To assess the stability of the modified NAGA, agalsidase beta, and agalsidase alfa at various pHs, we added each of these to 10 mM sodium citrate buffer (pH 4.0), 10 mM sodium acetate buffer (pH 4.5 and 5.0), 10 mM citric acid/sodium phosphate buffer (pH 5.5, 6.0 and 6.5), or 10 mM phosphate buffer (pH 7.0, 7.5 and 8.0) so that the result was 2 μmol/h/ml of MU-α-D-galactopyranoside-degrading activity. Incubation followed at 37°C for 16 hr. The enzyme activity at time zero was taken to be 100% under each pH condition in a buffer, and the stabilities of the modified NAGA, agalsidase beta, and agalsidase alfa were calculated as the ratio (percent) of the enzyme activity after a 16 hr incubation to the level at time zero. Furthermore, we examined the stabilities of the enzymes under pH 4.5 and 7.0 conditions in a buffer and in plasma at 37°C by following the time course. The stabilities of the enzymes were expressed as the ratio (percent) of the enzyme activity at a particular incubation time point to the value at time zero.
Examination of the Effect of the Modified NAGA on Cultured Fabry Fibroblasts
The effect of the modified NAGA on cleavage of the Gb3 that had accumulated in cultured fibroblasts from a patient with Fabry disease was examined. Previously, Ries et al. reported that the GLA activity in plasma reached 1–10 μmol/h/ml within 30 min after the administration of agalsidase alfa to Fabry patients.26 Therefore, we added the modified NAGA and each of the recombinant GLAs to the culture medium of cells to obtain a concentration of 5 μmol/h/ml. For examination of the inhibitory effect of M6P on the cellular uptake of the modified NAGA, Fabry fibroblasts were cultured in medium containing 5 mM M6P and the modified NAGA. The cells were cultured for 72 hr, and then immunostaining of the accumulated Gb3 with an anti-Gb3 mouse monoclonal antibody (mAb)27 was performed, as described previously.7,24
Examination of the Effect of the Modified NAGA on Fabry Mice
Fabry mice (GLA knock-out mice donated by A.B. Kulkarni and T. Ohshima, et al.)28,29 and wild-type C57BL/6 mice were used in this experiment. We examined the biodistribution of the enzyme. In a previously reported animal experiment30, a single dose of recombinant GLA at 0.3–10 mg/kg body weight was used for a biodistribution study. Here, a single dose (2 mmol/h/kg body weight) of the modified NAGA was injected into a tail vein of each Fabry mouse. As controls, agalsidase beta and agalsidase alfa were injected into litter-matched Fabry mice; the activity of the injected enzyme was the same as for the modified NAGA.7,24 The mice were sacrificed 1 hr after administration of the enzymes, and then the MU-α-D-galactopyranoside-degrading activity in the liver, kidneys, and heart was measured.
To examine cleavage of the Gb3 accumulated in organs, we repeatedly injected Fabry mice with the modified NAGA (2 mmol/h/kg body weight) every day for seven days and sacrificed mice 24 hr after the last injection. The liver, kidneys, and heart were then removed as samples for immunostaining, biochemical Gb3-determination, and electron microscopy. For immunohistochemical analysis, frozen sections of 8 μm thickness were fixed with 4% PFA in PBS for 30 min and then blocked with 10% M.O.M. mouse Ig Blocking Reagent (Vector Laboratories, Burlingame, CA) in PBS for 2 hr. The specimens were then incubated with a hybridoma supernatant containing anti-Gb3 (IgG isotype) mouse mAb27 at 4°C overnight. After being washed in PBS-T, the specimens were further incubated with an Alexa Fluor 488-conjugated goat anti-mouse IgG F(ab′)2 (diluted 1:1,000; Molecular Probes, Eugene, OR) for 1 hr. The specimens were embedded in PermaFlour (Thermo Electron Corporation, MA). The stained specimens were observed under a microscope (Axiovert 135; Carl Zeiss, Oberkochen, Germany) equipped with an Axio Vision 3.1 system. For determination of their Gb3 levels, the liver, kidneys, and heart were analyzed by means of thin-layer chromatography (TLC) followed by densitometry, as described previously.31 For morphological analysis, electron microscopy was performed as described previously.7,24
Statistical Analyses
Data are expressed as means ± standard deviation (SD). We performed statistical analyses with a Student's t test. Values were considered statistically significant at p < 0.05.
Results
Structure-Based Design of a Modified NAGA
A structural model of human NAGA was built on the basis of the crystallographic structure data for chicken NAGA, which exhibits 75% amino acid identity (Figure 1B). The resulting model was compared with the crystal structure of human GLA (Figure 1A), especially regarding the active site. It was revealed that Glu203 and Leu206 play important roles in the recognition of a galactose residue in GLA (Figure 1D) and that Ser188 and Ala191 play important roles in the recognition of an N-acetylgalactosamine residue in NAGA (Figure 1E). Therefore, we replaced Ser188 and Ala191 of NAGA with Glu and Leu, respectively, to alter the substrate specificity and thereby designed a modified NAGA (Figures 1C and 1F).
Figure 1.
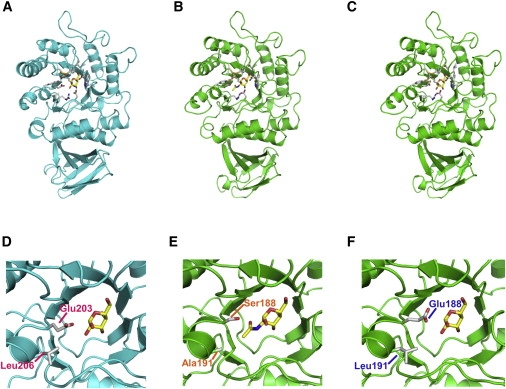
Structures of GLA, NAGA, and the Modified NAGA
The whole structures and those of the active sites of GLA (A and D), NAGA (B and E), and the modified NAGA (C and F) are shown as ribbon models. Each is presented as a complex with its substrate.
Expression of the Modified NAGA cDNA in Cultured Fabry Fibroblasts
On the basis of this structure-based design, we at first constructed a cDNA encoding the modified NAGA and having its own signal sequence, introduced it into an expression vector, and then expressed it in cultured fibroblasts from a patient with Fabry disease. An expression study showed that the two amino acid substitutions in the active site of NAGA resulted in alteration of its substrate preference (Table 1).
Table 1.
Expression Study on the NAGA and Modified NAGA cDNAs
| Cells | Introduced cDNA | MU-α-D-Galactopyranoside-Degrading Activity (nmol/h/mg protein) | MU-α-D-N-Acetylgalactosamine-Degrading Activity (nmol/h/mg protein) |
|---|---|---|---|
| Wild type | - | 30 | 2.5 × 102 |
| Kanzaki | - | 40 | <1.0 |
| Fabry | - | 2.0 | 2.3 × 102 |
| NAGA | 13 | 1.3 × 104 | |
| modified NAGA | 5.5 × 102 | 1.0 × 102 |
Wild type, human cultured fibroblasts from an apparently normal subject; Kanzaki, cultured fibroblasts from a patient with Kanzaki disease (inherited NAGA deficiency); and Fabry, cultured fibroblasts from a patient with Fabry disease.
Production and Purification of the Modified NAGA
To produce a sufficient amount of the modified NAGA protein in CHO cells, the cDNA encoding the modified NAGA and having its own signal sequence was introduced into an expression vector, pEE14.4. Furthermore, to enhance the secretion of the enzyme, we replaced the signal sequence region of NAGA with that of GLA, which should be cleaved at an early stage of biosynthesis. Then, the vector was transfected into CHO cells. CHO cells stably expressing the modified enzyme were obtained via a glutamine synthetase gene expression system. The productivity was estimated to be approximately 10 mg/liter.
Then, the enzyme was purified from conditioned culture medium containing the modified NAGA protein by means of column chromatography. The purification degree was 560-fold, and the recovery of the enzyme protein was 38% (Table 2).
Table 2.
Purification of the Modified NAGA from Conditioned Culture Medium
| Total Protein (g) | Total Activity (mmol/h) | Specific Activity (μmol/h/mg) | Relative Purity | Yield (Percent) | |
|---|---|---|---|---|---|
| Medium concentration | 61 | 55 | 0.9 | 1.0 | 100 |
| Dialyzed 50% | 28 | 53 | 1.9 | 2.1 | 96 |
| (NH4)2SO4 fraction | |||||
| Phenyl-Sepharose | 3.3 | 34 | 10 | 12 | 63 |
| SP-Sepharose | 0.13 | 22 | 1.7 × 102 | 1.9 × 102 | 40 |
| Flow-through | |||||
| Q-Sepharose | 0.042 | 21 | 5.0 × 102 | 5.6 × 102 | 38 |
Enzymological Characterization of the Modified NAGA
The purified modified NAGA was enzymologically characterized. The degrading activity toward MU-α-D-galactopyranoside, an artificial substrate for GLA, of the modified NAGA was 0.5 mmol/h/mg protein; this value was about a quarter of those of agalsidase beta and agalsidase alfa. The Km value of the modified NAGA was 4.8 mM, which was slightly higher than those of agalsidase beta and agalsidase alfa (4.0 mM). The modified NAGA hardly exhibited any hydrolyzing activity toward MU-α-N-acetylgalactosamine, an artificial substrate for NAGA (Table 3).
Table 3.
Enzymological Parameters of the Modified NAGA
|
MU-α-D-Galactopyranoside-Degrading Activity |
MU-α-D-N-Acetylgalactosamine-Degrading Activity |
|||
|---|---|---|---|---|
| Specific Activity (mmol/h/mg) | Vmax (mmol/h/mg) | Km (mM) | Specific Activity (μmol/h/mg) | |
| Modified NAGA | 0.5 | 1.0 | 4.8 | 3.0 |
| Placenta NAGAa | <0.01 | - | - | 24 |
| Agalsidase beta | 2.3 | 3.8 | 4.0 | <1.0 |
| Agalsidase alfa | 2.1 | 3.8 | 4.0 | <1.0 |
As controls, NAGA from placenta, agalsidase beta, and agalsidase alfa were used.
Stored at −20°C for 30 yr.
The modified NAGA was detected as two or more broad bands on SDS-PAGE, but it only gave one band after deglycosylation, suggesting that it has heterogeneous sugar chains (Figure 2). The results of immunoblotting revealed that there was no immunological cross-reactivity between the modified NAGA and a recombinant GLA protein (Figure 3). The content of M6P, which is very important for incorporation of the enzyme into cells, was 5.1 mol/mol protein for the modified NAGA, and this value was higher than those for both agalsidase beta (3.6 mol/mol protein) and agalsidase alfa (2.1 mol/mol protein) (p < 0.05, t test, Table 4).
Figure 2.
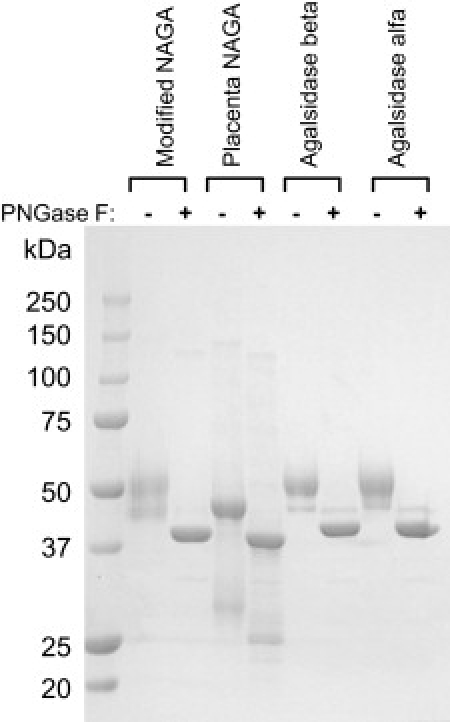
Coomassie Brilliant Blue R Staining of the Modified NAGA on a Tris-Glycine Polyacrylamide Gel
Before the electrophoresis, the enzyme was treated (PNGase F+) or not treated (PNGase F−) with glycopeptidase F. As controls, NAGA purified from human placenta (Placenta NAGA), agalsidase beta (Agalsidase beta), and agalsidase alfa (Agalsidase alfa) were used.
Figure 3.
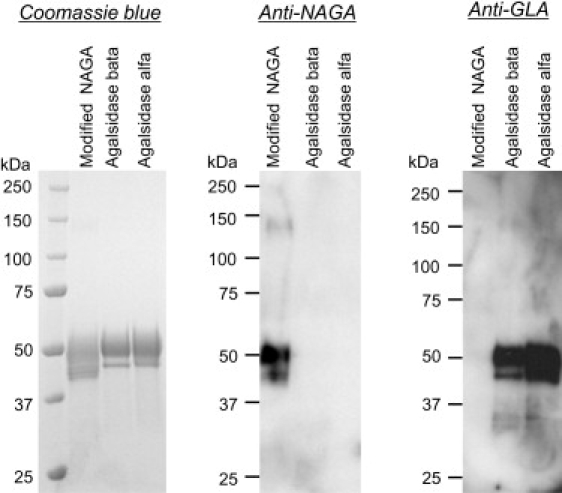
Immunoblotting of the Modified NAGA, Agalsidase Beta, and Agalsidase Alfa
The modified NAGA, agalsidase beta, and agalsidase alfa were separated on a Tris-glycine polyacrylamide gel and stained with Coomassie brilliant blue R, and then immunoblotting analyses with anti-NAGA and anti-GLA polyclonal antibodies were performed. The results showed that there was no immunological cross-reactivity between the modified NAGA and the recombinant GLAs, i.e., agalsidase beta and agalsidase alfa. Figure labels are as follows: Coomassie blue, staining of the gel with Coomassie brilliant blue R; Anti-NAGA, immunoblotting with anti-NAGA polyclonal antibodies; and Anti-GLA, immunoblotting with anti-GLA polyclonal antibodies.
Table 4.
Monosaccharide Analysis of the Modified NAGA
| Modified NAGA | Agalsidase Beta | Agalsidase Alfa | |
|---|---|---|---|
| N-Acetylglucosamine | 34 ± 2.0 | 19 ± 8.0 | 18 ± 4.0 |
| Mannose | 44 ± 14 | 24 ± 14 | 24 ± 3.0 |
| Fucose | 6.5 ± 0.6 | 3.5 ± 1.6 | 4.1 ± 0.6 |
| Galactose | 13 ± 0.2 | 11 ± 6.4 | 12 ± 1.8 |
| Mannose 6-phosphate | 5.1 ± 1.2a | 3.6 ± 2.2 | 2.1 ± 0 |
| Sialic acid | 1.1 ± 0.4 | 6.1 ± 0.2 | 4.8 ± 0.8 |
All data are expressed as means ± SD (n = 4). As controls, agalsidase beta and agalsidase alfa were used.
The mannose 6-phosphate content of the modified NAGA is higher than that of agalsidase beta or agalsidase alfa (p < 0.05, t test).
The stability of the modified NAGA in buffer and human plasma was examined in vitro. The results revealed that the modified NAGA was more stable under both acidic and neutral pH conditions in a buffer (p < 0.05, t test, Figures 4A–4C) and also in human plasma (p < 0.05, t test, Figure 4D) than agalsidase beta and agalsidase alfa.
Figure 4.
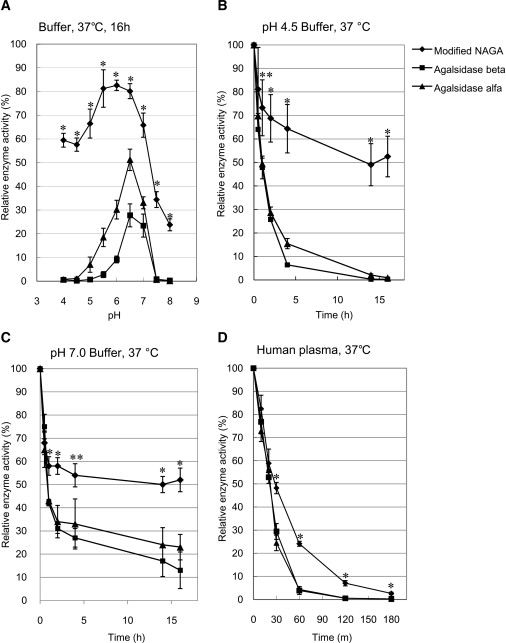
Stability of the Modified NAGA
The modified NAGA was added to buffers of the indicated pH values and incubated at 37°C for 16 hr, and then MU-α-D-galactopyranoside-degrading activity was determined (A). We examined the stability of the modified NAGA under pH 4.5 (B) and pH 7.0 (C) conditions in a buffer and in plasma (D) at 37°C by following the time course after the addition of the enzyme to the buffers and plasma. As controls, agalsidase beta and agalsidase alfa were used. Error bars represent means ± SD (n = 3). The modified NAGA is more stable than the recombinant GLAs. ∗p < 0.01 in comparisons to both agalsidase beta and agalsidase alfa; ∗∗p < 0.05 in comparisons to both agalsidase beta and agalsidase alfa, t test.
Immunoreactivity of the Modified NAGA to the Serum from a Fabry Patient
The immunoreactivity of the modified NAGA to the serum from a Fabry patient who had been repeatedly injected with agalsidase beta was examined by means of ELISA. The serum strongly reacted to the recombinant GLAs, agalsidase beta and agalsidase alfa, but not to the modified NAGA (Figure 5).
Figure 5.
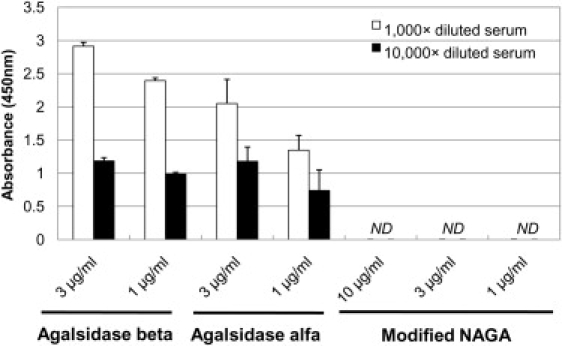
Immune Reaction of the Modified NAGA to Serum from a Fabry Patient
We performed ELISA to determine whether the modified NAGA immunologically reacted to serum from a Fabry patient who had been repeatedly injected with agalsidase beta. Although the serum (titer of the antibodies against GLA, 12,800×) strongly reacted to agalsidase beta and agalsidase alfa, it did not react to the modified NAGA. Open columns: 1,000× diluted patient's serum. Closed columns: 10,000× diluted patient's serum. ND: Not detected. Error bars represent means ± SD (n = 3).
Effect of the Modified NAGA on Cultured Fabry Fibroblasts
The effect of the modified NAGA on cleavage of the Gb3 that had accumulated in Fabry fibroblasts was examined by means of immunostaining for Gb3 and was compared with the effects of agalsidase beta and agalsidase alfa. The results are shown in Figure 6. The modified NAGA and the two recombinant GLAs were each added to the culture medium at the concentration giving an activity level of 5 μmol/h/ml. The cells were cultured at 37°C for 72 hr and then stained with anti-Gb3 mouse mAb. The immunofluorescence of Gb3 apparently decreased with the addition of the modified NAGA, and there were no differences in Gb3-degrading activity between the modified NAGA and the two recombinant GLAs. The cleavage of the accumulated Gb3 by the modified NAGA was almost completely inhibited by the addition of 5 mM M6P, suggesting that incorporation of the modified NAGA depends on M6P receptors.
Figure 6.
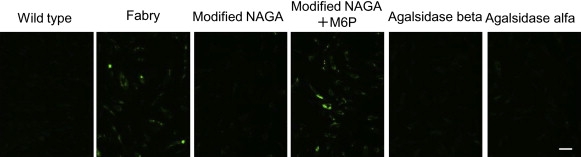
Immunostaining for the Gb3 Accumulated in Fabry Fibroblasts
Cultured Fabry fibroblasts were stained with anti-Gb3 mAb after addition of the modified NAGA (Modified NAGA), the modified NAGA and 5 mM M6P (Modified NAGA+M6P), agalsidase beta (Agalsidase beta), or agalsidase alfa (Agalsidase alfa). As controls, wild-type cells (Wild-type) and untreated Fabry cells (Fabry) were examined. Each of the enzymes was added to the culture medium to give a concentration of 5 μmol/h/ml, and the cells were cultured for 72 hr. The Gb3 that had accumulated in Fabry fibroblasts was cleaved on administration of the modified NAGA as well as the recombinant GLAs, but not on coadministration of the modified NAGA and M6P. The scale bar represents 50 μm.
Effect of the Modified NAGA on Fabry Mice
We injected the modified NAGA into the tail veins of Fabry mice to compare its therapeutic effect with those of agalsidase beta and agalsidase alfa. Because the specific enzyme activity differed among the enzymes, we injected amounts of the enzymes exhibiting almost the same activity (2 mmol/h/kg body weight) into litter-matched Fabry mice for comparison, according to the method previously reported.7,24
At 1 hr after administration of a single dose of each enzyme, the mice were sacrificed, and MU-α-D-galactopyranoside-degrading activity in the liver, kidneys, and heart was determined. An apparent increase in the enzyme activity was observed in these organs of Fabry mice after the modified NAGA administration. The enzyme activity increased up to 6, 4, and 5 times that in the liver, kidneys, and heart, respectively, of the wild-type mice. The increases in the enzyme activity were greater in the kidneys (p = 0.005 and 0.003 for a comparison with agalsidase beta and agalsidase alfa, respectively, t test) and heart (p = 0.001 and 0.003 for a comparison with agalsidase beta and agalsidase alfa, respectively, t test), and similar in the liver (Figure 7).
Figure 7.
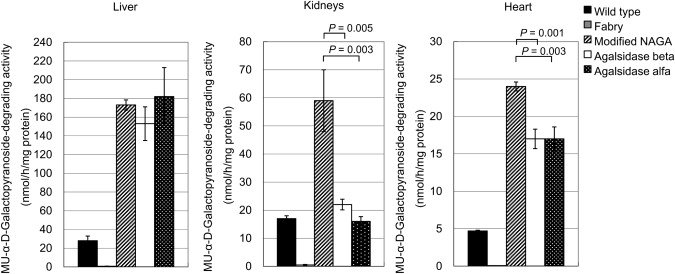
MU-α-D-Galactopyranoside-Degrading Activity (nmol/h/mg protein) in Organs from Fabry Mice Treated with a Single Dose of the Modified NAGA, Agalsidase Beta, or Agalsidase Alfa
Fabry mice were injected with a single dose (2 mmol/h/kg body weight) of each of the enzymes and then sacrificed 1 hr after the administration. Wild-type, wild-type mice; Fabry, untreated Fabry mice; Modified NAGA, Fabry mice treated with the modified NAGA; Agalsidase beta, Fabry mice treated with agalsidase beta; and Agalsidase alfa, Fabry mice treated with agalsidase alfa. Error bars represent means ± SD (n = 3). The increases in the enzyme activity upon administration of the modified NAGA are greater in the kidneys (p = 0.005 and 0.003 in comparison to agalsidase beta and agalsidase alfa, respectively, t test) and heart (p = 0.001 and 0.003 in comparison to agalsidase beta and agalsidase alfa, respectively, t test) tissues than in the cases of the recombinant GLAs.
The effect of administration of the modified NAGA on degradation of the Gb3 that had accumulated in tissues was examined after repeated injection of the enzyme at 2 mmol/h/kg body weight every day for 7 days, followed by killing of the mice 24 hr after the last injection. Immunohistochemical analysis revealed that the Gb3 that had accumulated in the liver was almost completely hydrolyzed. In both the kidney and heart tissues, the Gb3 immunofluorescence was apparently decreased. These findings were essentially the same as those in the cases of administration of agalsidase beta and agalsidase alfa (Figure 8). The results of TLC analysis also revealed that the modified NAGA cleaved the Gb3 accumulated in the liver, kidneys, and heart (Table 5).
Figure 8.
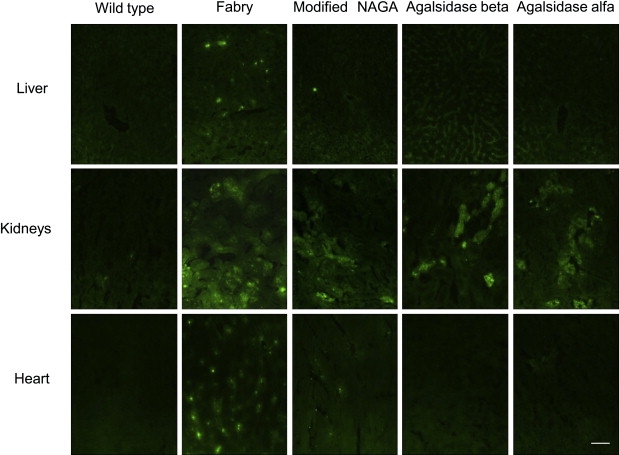
Immunohistochemical Analyses of the Accumulated Gb3 in Organs of Fabry Mice and Its Degradation by the Modified NAGA, Agalsidase Beta, and Agalsidase Alfa
Fabry mice were repeatedly injected with the modified NAGA, agalsidase beta, or agalsidase alfa, and then immunostaining for Gb3 was performed. Wild-type, a wild-type mouse; Fabry, an untreated Fabry mouse; Modified NAGA, a Fabry mouse treated with the modified NAGA; Agalsidase beta, a Fabry mouse treated with agalsidase beta; and Agalsidase alfa, a Fabry mouse treated with agalsidase alfa. The scale bar represents 50 μm.
Table 5.
Gb3 Levels in Organs from a Fabry Mouse Treated with the Modified NAGA
| Liver | Kidneys | Heart | |
|---|---|---|---|
| Wild-Type | |||
| 0.15 | 0.48 | 0.06 | |
| Fabry | |||
| Untreated | 0.82 | 2.0 | 0.69 |
| Treated | 0.22 | 1.4 | 0.40 |
Values are given in μg/mg wet weight.
Morphological analysis revealed that many lamellar inclusion bodies, including deposits of Gb3, were present in Kupffer cells of the liver, renal tubular cells of the kidneys, and pericytes of the heart and that they were markedly decreased after repeated administration of the modified NAGA, as in the cases of agalsidase beta and agalsidase alfa (Figure 9).
Figure 9.
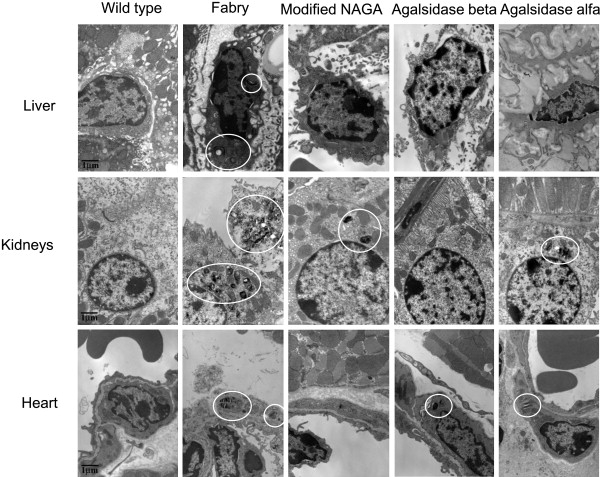
Morphological Effects of Repeated Administration of the Modified NAGA, Agalsidase Beta, and Agalsidase Alfa
The enzymes were repeatedly injected into Fabry mice, and then liver, kidney, and heart tissues were examined by electron microscopy. The circles indicate pathological changes resulting from Fabry disease (inclusion bodies). The scale bar represents 1μm.
Discussion
Repeated administration of recombinant GLAs to male patients with Fabry disease frequently causes infusion reactions, mainly allergic ones,4,5 because most male Fabry patients do not have any GLA proteins. IgG antibodies to recombinant GLAs develop in the majority of male Fabry patients treated with these recombinant GLAs32,although female Fabry patients having some GLA proteins rarely develop antibodies to the enzymes. It has been reported that even the development of IgE antibodies can occur.33 These antibodies probably decrease the effect of ERT for Fabry disease.8–10
We therefore paid attention to NAGA. NAGA catalyzes the hydrolysis of the terminal α-N-acetylgalactosamine of glycoconjugates, including polysaccharides, glycolipids, and glycoproteins.11,34 Human NAGA and GLA genes exhibit remarkable homology of their predicted amino acid sequences, and a deficiency of NAGA leads to very rare genetic diseases, i.e., Schindler disease (MIM 609241) and Kanzaki disease (MIM 609242), neurological and skin disorders, respectively.34 Their substrate specificities differ only in the presence of an N-acetyl or hydroxyl moiety, respectively, at position 2 of the galactose ring. GLA and NAGA resemble each other in structure, but their immunogenicities are different, and there is no immunological cross-reactivity between them.35,36 Therefore, we compared a homology model of human NAGA with the crystallographic structure of human GLA and predicted amino acid residues that could alter the substrate specificity of NAGA to that of GLA. On the basis of the results of structural analysis, we designed a modified NAGA with two amino acid substitutions, Ser188Glu and Ala191Leu, and an expression study revealed that the modified NAGA strongly degraded MU-α-D-galactopyranoside, one of the preferred substrates of GLA (Table 1).
Because the whole structure of the modified NAGA is not altered, although the substrate-recognition region inside the molecule is moderately changed, the administration of the modified NAGA to Fabry patients, who usually have endogenous NAGA protein, should hardly cause an allergic reaction to administered modified NAGA. It is known that NAGA is heat stable, i.e., that GLA loses approximately 80% of its activity but NAGA retains more than 50% of its activity when heated to 55°C in pH 6–7 buffer for approximately 30 min.11 Furthermore, the number of sugar chains attached to NAGA (10 mol/mol protein)37 is larger than the number attached to GLA (6 mol/mol protein).1 So, the modified NAGA is expected to have clinical advantages for ERT for Fabry disease.
First, we generated stable CHO cell lines expressing the modified NAGA. To enhance the secretion of the modified NAGA into the culture medium, we replaced its signal sequence with that of GLA because the modified NAGA with its own signal sequence was retained in cells (data not shown). The productivity of the modified NAGA is 10 mg/liter under the culture conditions on a laboratory scale, and it could be further improved by the development of an industrial production system in the future.
A purification study followed by biochemical characterization revealed that the modified NAGA was an N-glycoprotein having heterogeneous sugar chains (Figure 2) and that its M6P residue content was higher than that of agalsidase beta or agalsidase alfa (p < 0.05, t test, Table 4). Because M6P residues are essential for the incorporation of a modified NAGA into cells via M6P receptors on the cell membrane (Figure 6), as in the cases of recombinant GLAs,38,39 a modified NAGA having many M6P residues would be beneficial for ERT. Furthermore, the modified NAGA was more stable than agalsidase beta and agalsidase alfa both in buffers (p < 0.05, t test, Figures 4A–4C) and in human plasma (p < 0.05, t test, Figure 4D), as expected. This would also be beneficial for ERT. Immunological examinations revealed that the modified NAGA did not cross-react with recombinant GLAs (Figure 3) and that it did not react to the serum from a Fabry patient who had been repeatedly injected with agalsidase beta (Figure 5). Regarding the specific activity of the modified NAGA, it was lower than those of the recombinant GLAs. This might be due to the small differences in the structures of the active-site pocket and the surface of the molecule between the modified NAGA and the GLAs because the structures of these regions should be associated with substrate recognition. This would be disadvantageous for a modified NAGA. The lysosomal degradation of Gb3 requires saposin B, a nonenzymic activator that presents Gb3 to the catalytic site of GLA.40 Because saposin B is essentially required for enzymic Gb3 degradation in cells, it may also play a role in the degradation of Gb3 by a modified NAGA. Coadministration of saposin B with the modified NAGA might accelerate its degradation of the Gb3 accumulated in Fabry cells.
After confirmation of the effect of the modified NAGA on cultured Fabry fibroblasts (Figure 6), we injected the modified NAGA into Fabry mice and examined both the incorporation of the enzyme into organs and its Gb3-degrading activity. The modified NAGA was successfully incorporated into the liver, kidneys, and heart, the Gb3 deposited in these organs was apparently cleaved or decreased (Figure 8 and Table 5), and the specific pathological changes were improved (Figure 9), as in the cases of the administration of the recombinant GLAs.
In conclusion, we produced a modified NAGA exhibiting catalytic activity toward Gb3 and MU-α-D-galactopyranoside by means of a structure-based design. The enzyme has many advantages because of its high stability and high M6P content and the low possibility of the occurrence of an allergic reaction, although these characteristics should be confirmed in future clinical studies. The modified NAGA is highly promising as a new enzyme for ERT for Fabry disease, and such structure-based design of modified lysosomal enzymes should be useful for the development of ERT for lysosomal storage diseases.
Web Resources
The URLs for the data presented herein are as follows:
DNA Data Bank of JAPAN (DDBJ), http://www.ddbj.nig.ac.jp
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim
Protein Data Bank (PDB), http://www.rcsb.org/pdb/home/home.do
Acknowledgments
We would like to thank S. Aikawa and F. Matsuzawa (Altif Laboratories) for discussion; A. Tsuji (The University of Tokushima) for the NAGA purified from placenta and anti-NAGA polyclonal antibodies; A.B. Kulkarni and T. Oshima (National Institutes of Health) for the Fabry mice; B. Honig (Columbia University) for the Jackal software; and J. Ponder (Washington University) for the TINKER program. This work was supported by the Japan Science and Technology Agency (ID: 1805, H.S.); the Japan Society for the Promotion of Science (ID: 18390303, H.S.); the Ministry of Health and Welfare of Japan (H.S.); the High-Tech Research Center Project of the Ministry of Education, Science, Sports and Culture of Japan (H.S.); and the Program for Promotion of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation (ID: 09-15, H.S.).
References
- 1.Desnick R.J., Ioannou Y.A., Eng C.M. alpha-Galactosidase A deficiency: Fabry disease. In: Scriver C.R., Beaudet A.L., Sly W.S., Valle D., editors. The Metabolic and Molecular Bases of Inherited Disease. Eighth Edition. McGraw-Hill; New York, USA: 2001. pp. 3733–3774. [Google Scholar]
- 2.Spada M., Paqgliardini S., Yasuda M., Tukel T., Thiagarajan G., Sakuraba H., Ponzone A., Desnick R.J. High incidence of later-onset Fabry disease revealed by newborn screening. Am. J. Hum. Genet. 2006;79:31–40. doi: 10.1086/504601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eng C.M., Banikazemi M., Gordon R.E., Goldman M., Phelps R., Kim L., Gass A., Winston J., Dikman S., Fallon J.T. A phase 1/2 clinical trial of enzyme replacement in Fabry disease: Pharmacokinetic, substrate clearance, and safety studies. Am. J. Hum. Genet. 2001;68:711–722. doi: 10.1086/318809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eng C.M., Guffon N., Wilcox W.R., Germain D.P., Lee P., Waldek S., Caplan L., Linthorst G.E., Desnick R.J. Safety and efficacy of recombinant human alpha-galactosidase A replacement therapy in Fabry's disease. N. Engl. J. Med. 2001;345:9–16. doi: 10.1056/NEJM200107053450102. [DOI] [PubMed] [Google Scholar]
- 5.Schiffmann R., Murray G.J., Treco D., Daniel P., Sellos-Moura M., Myers M., Quirk J.M., Zirzow G.C., Borowski M., Loveday K. Infusion of alpha-galactosidase A reduces tissue globotriaosylceramide storage in patients with Fabry disease. Proc. Natl. Acad. Sci. USA. 2000;97:365–370. doi: 10.1073/pnas.97.1.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kornfeld S., Sly W.S. I-cell disease and pseudo-Hurler polydystrophy: Disorders of lysosomal enzyme phosphorylation and localization. In: Scriver C.R., Beaudet A.L., Sly W.S., Valle D., editors. The Metabolic and Molecular Bases of Inherited Disease. Eighth Edition. McGraw-Hill; New York: 2001. pp. 3469–3482. [Google Scholar]
- 7.Sakuraba H., Murata-Ohsawa M., Kawashima I., Tajima Y., Kotani M., Ohshima T., Chiba Y., Takashiba M., Jigami Y., Fukushige T. Comparison of the effects of agalsidase alfa and agalsidase beta on cultured human Fabry fibroblasts and Fabry mice. J. Hum. Genet. 2006;51:180–188. doi: 10.1007/s10038-005-0342-9. [DOI] [PubMed] [Google Scholar]
- 8.Ohashi T., Sakuma M., Kitagawa T., Suzuki K., Ishige N., Eto Y. Influence of antibody formation on reduction of globotriaosylceramide (GL-3) in urine from Fabry patients during agalsidase beta therapy. Mol. Genet. Metab. 2007;92:271–273. doi: 10.1016/j.ymgme.2007.06.013. [DOI] [PubMed] [Google Scholar]
- 9.Ohashi T., Iizuka S., Ida H., Eto Y. Reduced alpha-Gal A enzyme activity in Fabry fibroblast cells and Fabry mice tissues induced by serum from antibody positive patients with Fabry disease. Mol. Genet. Metab. 2008;94:313–318. doi: 10.1016/j.ymgme.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 10.Vedder A.C., Breunig F., Donker-Koopman W.E., Mills K., Young E., Winchester B., Ten Berge I.J., Groener J.E., Aerts J.M., Wanner C. Treatment of Fabry disease with different dosing regimens of agalsidase: Effects on antibody formation and GL-3. Mol. Genet. Metab. 2008;94:319–325. doi: 10.1016/j.ymgme.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 11.Dean K.J., Sweeley C.C. Fabry disease: α-galactosidase A deficiency. In: Glew R.H., Peters S.P., editors. Practical Enzymology of the Sphingolipidoses. Alan R. Liss; New York: 1977. pp. 173–216. [Google Scholar]
- 12.Petrey D., Xiang Z., Tang C.L., Xie L., Gimpelev M., Mitros T., Soto C.S., Goldsmith-Fischman S., Kernytsky A., Schlessinger A. Using multiple structure alignments, fast model building, and energetic analysis in fold recognition and homology modeling. Proteins. 2003;53:430–435. doi: 10.1002/prot.10550. [DOI] [PubMed] [Google Scholar]
- 13.Garman S.C., Hannick L., Zhu A., Garboczi D.N. The 1.9 Å structure of α-N-acetylgalactosaminidase: Molecular basis of glycosidase deficiency diseases. Structure. 2002;10:425–434. doi: 10.1016/s0969-2126(02)00726-8. [DOI] [PubMed] [Google Scholar]
- 14.Garman S.C., Garboczi D.N. The molecular defect leading to Fabry disease: Structure of human alpha-galactosidase. J. Mol. Biol. 2004;337:319–335. doi: 10.1016/j.jmb.2004.01.035. [DOI] [PubMed] [Google Scholar]
- 15.Dudek M.J., Ponder J.W. Accurate modeling of the intramolecular electrostatic energy of proteins. J. Comput. Chem. 1995;16:791–816. [Google Scholar]
- 16.Kong M.J., Ponder J.W. Calculation of the reaction field due to off-center point multipoles. J. Chem. Phys. 1997;107:481–492. [Google Scholar]
- 17.Pappu R.V., Hart R.K., Ponder J.W. Analysis and application of potential energy smoothing for global optimization. J. Phys. Chem. B. 1998;102:9725–9742. [Google Scholar]
- 18.Ren P., Ponder J.W. Polarizable atomic multipole water model for molecular mechanics simulation. J. Phys. Chem. B. 2003;107:5933–5947. [Google Scholar]
- 19.Mayes J.S., Scheerer J.B., Sifers R.N., Donaldson M.L. Differential assay for lysosomal α-galactosidases in human tissues and its application to Fabry's disease. Clin. Chim. Acta. 1981;112:247–251. doi: 10.1016/0009-8981(81)90384-3. [DOI] [PubMed] [Google Scholar]
- 20.Hu P., Reuser A.J., Janse H.C., Kleijer W.J., Schindler D., Sakuraba H., Tsuji A., Suzuki Y., van Diggelen O.P. Biosynthesis of human α-N-acetylgalactosaminidase: Defective phosphorylation and maturation in infantile α-NAGA deficiency. Biochem. Biophys. Res. Commun. 1991;175:1097–1103. doi: 10.1016/0006-291x(91)91678-6. [DOI] [PubMed] [Google Scholar]
- 21.Akagi T., Sasai K., Hanafusa H. Refractory nature of normal human diploid fibroblasts with respect to oncogene-mediated transformation. Proc. Natl. Acad. Sci. USA. 2003;100:13567–13572. doi: 10.1073/pnas.1834876100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Swift S., Lorens J., Achacoso P., Nolan G.P. Rapid production of retroviruses for efficient gene delivery to mammalian cells using 293T cell-based systems. In: Coligan J.E., editor. Current Protocols in Immunology. John Wiley and Sons; New York: 2001. pp. 10.17.14–10.17.29. [DOI] [PubMed] [Google Scholar]
- 23.Okumiya T., Ishii S., Takenaka T., Kase R., Kamei S., Sakuraba H., Suzuki Y. Galactose stabilizes various missense mutants of α-galactosidase in Fabry disease. Biochem. Biophys. Res. Commun. 1995;214:1219–1224. doi: 10.1006/bbrc.1995.2416. [DOI] [PubMed] [Google Scholar]
- 24.Sakuraba H., Chiba Y., Kotani M., Kawashima I., Ohsawa M., Tajima Y., Takaoka Y., Jigami Y., Takahashi H., Hirai Y. Corrective effect on Fabry mice of yeast recombinant human α-galactosidase with N-linked sugar chains suitable for lysosomal delivery. J. Hum. Genet. 2006;51:341–352. doi: 10.1007/s10038-006-0369-6. [DOI] [PubMed] [Google Scholar]
- 25.Sugawara K., Tajima Y., Kawashima I., Tsukimura T., Saito S., Ohno K., Iwamoto K., Kobayashi T., Itoh K., Sakuraba H. Molecular interaction of imino sugars with human alpha-galactosidase: Insight into the mechanism of complex formation and pharmacological chaperone action in Fabry disease. Mol. Genet. Metab. 2009;96:233–238. doi: 10.1016/j.ymgme.2008.12.017. [DOI] [PubMed] [Google Scholar]
- 26.Ries M., Clarke J.T., Whybra C., Mehta A., Loveday K.S., Brady R.O., Beck M., Schiffmann R. Enzyme replacement in Fabry disease: Pharmacokinetics and pharmacodynamics of agalsidase alfa in children and adolescents. J. Clin. Pharmacol. 2007;47:1222–1230. doi: 10.1177/0091270007305299. [DOI] [PubMed] [Google Scholar]
- 27.Kotani M., Kawashima I., Ozawa H., Ogura K., Ariga T., Tai T. Generation of one set of murine monoclonal antibodies specific for globo-series glycolipids: Evidence for differential distribution of the glycolipids in rat small intestine. Arch. Biochem. Biophys. 1994;310:89–96. doi: 10.1006/abbi.1994.1144. [DOI] [PubMed] [Google Scholar]
- 28.Ohshima T., Murray G.J., Swaim W.D., Longenecker G., Quirk J.M., Cardarelli C.O., Sugimoto Y., Pastan I., Gottesman M.M., Brady R.O. alpha-Galactosidase A deficient mice: A model of Fabry disease. Proc. Natl. Acad. Sci. USA. 1997;94:2540–2544. doi: 10.1073/pnas.94.6.2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ohshima T., Schiffmann R., Murray G.J., Kopp J., Quirk J.M., Stahl S., Chan C.-C., Zerfas P., Tao-Cheng J.-H., Ward J.M. Aging accentuates and bone marrow transplantation ameliorates metabolic defects in Fabry disease mice. Proc. Natl. Acad. Sci. USA. 1999;96:6423–6427. doi: 10.1073/pnas.96.11.6423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ioannou Y.A., Zeidner K.M., Gordon R.E., Desnick R.J. Fabry disease: Preclinical studies demonstrate the effectiveness of α-galactosidase A replacement in enzyme-deficient mice. Am. J. Hum. Genet. 2001;68:14–25. doi: 10.1086/316953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takahashi H., Hirai Y., Migita M., Seino Y., Fukuda Y., Sakuraba H., Kase R., Kobayashi T., Hashimoto Y., Shimada T. Long-term systemic therapy of Fabry disease in a knockout mouse by adeno-associated virus-mediated muscle-directed gene transfer. Proc. Natl. Acad. Sci. USA. 2002;99:13777–13782. doi: 10.1073/pnas.222221899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Linthorst G.E., Hollak C.E., Donker-Koopman W.E., Strijland A., Aerts J.M. Enzyme therapy for Fabry disease: Neutralizing antibodies toward agalsidase alpha and beta. Kidney Int. 2004;66:1589–1595. doi: 10.1111/j.1523-1755.2004.00924.x. [DOI] [PubMed] [Google Scholar]
- 33.Bodensteiner D., Scott C.R., Sims K.B., Shepherd G.M., Cintron R.D., Germain D.P. Successful reinstitution of agalsidase beta therapy in Fabry disease patients with previous IgE-antibody or skin-test reactivity to the recombinant enzyme. Genet. Med. 2008;10:353–358. doi: 10.1097/GIM.0b013e318170f868. [DOI] [PubMed] [Google Scholar]
- 34.Desnick R.J., Schindler D. alpha-N-Acetylgalactosaminidase deficiency: Schindler disease. In: Scriver C.R., Beaudet A.L., Sly W.S., Valle D., editors. The Metabolic and Molecular Bases of Inherited Disease. Eighth Edition. McGraw-Hill; New York: 2001. pp. 3483–3505. [Google Scholar]
- 35.Beutler E., Kuhl W. Purification and properties of human alpha-galactosidases. J. Biol. Chem. 1972;247:7195–7200. [PubMed] [Google Scholar]
- 36.Schram A.W., Hamers M.N., Tager J.M. The identity of alpha-galactosidase B from human liver. Biochim. Biophys. Acta. 1977;482:138–144. doi: 10.1016/0005-2744(77)90361-8. [DOI] [PubMed] [Google Scholar]
- 37.Ohta M., Ohnishi T., Ioannou Y.A., Hodgson M.E., Matsuura F., Desnick R.J. Human α-N-acetylgalactosaminidase: Site occupancy and structure of N-linked oligosaccharide. Glycobiology. 2000;10:251–261. doi: 10.1093/glycob/10.3.251. [DOI] [PubMed] [Google Scholar]
- 38.Chiba Y., Sakuraba H., Kotani M., Kase R., Kobayashi K., Takeuchi M., Ogasawara S., Maruyama Y., Nakajima T., Takaoka Y. Production in yeast of α-galactosidase A, a lysosomal enzyme applicable to enzyme replacement therapy for Fabry disease. Glycobiology. 2002;12:821–828. doi: 10.1093/glycob/cwf096. [DOI] [PubMed] [Google Scholar]
- 39.Kawashima I., Watabe K., Tajima Y., Fukushige T., Kanzaki T., Kanekura T., Sugawara K., Ohyanagi N., Suzuki T., Togawa T. Establishment of immortalized Schwann cells from Fabry mice and their low uptake of recombinant α-galactosidase. J. Hum. Genet. 2007;52:1018–1025. doi: 10.1007/s10038-007-0210-x. [DOI] [PubMed] [Google Scholar]
- 40.Sandhoff K., Kolter T., Harzer K. Sphingolipid activator proteins. In: Scriver C.R., Beauded A.L., Sly W.S., Valle D., editors. The Metabolic and Molecular Bases of Inherited Disease. Eighth Edition. McGraw-Hill; New York: 2001. pp. 3371–3388. [Google Scholar]