

Abstract
Despite a primary tumor-suppressor role, there is compelling evidence suggesting that TGF-β can promote tumor growth, invasion and metastasis in advanced stages of colorectal cancer. Blocking these tumor-promoting effects of TGF-β provides a potentially important therapeutic strategy for the treatment of colorectal cancer. However, little is known about how the inhibitors of TGF-β receptor kinases affect colorectal cancinogenesis in vivo. Here, we have observed that a novel dual kinase inhibitor of TGF-β type I and type II receptors, LY2109761, inhibits TGF-β-mediated activation of Smad and non-Smad pathways in CT26 colon adenocarcinoma cells having K-Ras mutation. The inhibitor attenuates the oncogenic effects of TGF-β on cell migration, invasion and tumorigenicity of CT26 cells. Furthermore, LY2109761 decreases liver metastases and prolongs survival in an experimental metastasis model. These findings suggest that the dual kinase inhibitor LY2109761 has potential therapeutic value for metastatic colorectal cancer.
Keywords: TGF-β, colon cancer, liver metastasis, LY2109761, bioluminescence
1. Introduction
Metastasis of colorectal cancer to the liver presents a significant challenge to physicians due to the limited efficacy of conventional cancer therapies. In order to reduce morbidity and to improve the survival rate of colon cancer, it is essential to develop therapeutics that specifically targets the signaling pathways including TGF-β pathway that are inappropriately activated in colorectal cancer cells. TGF-β family of polypeptides regulates a wide variety of biological functions including cell proliferation, differentiation, migration, cell survival, angiogenesis and immunosurveillance. The multifunctional effects of TGF-β are elicited through an oligomeric complex between the type I (TβRI) and type II (TβRII) serine-threonine kinase receptor. In response to TGF-β, TβRII phosphorylates TβRI. TβRI then activates Smad2 and Smad3, which associates with Smad4 and translocates to the nucleus, where it modulates transcription of TGF-β target genes. Additionally, TGF-β can induce non-Smad pathways including p38MAPK, ERK, PI3K, JNK, and Rho, which are important for pro-oncogenic activities [1, 2].
TGF-β signaling can function both as a tumor suppressor and a tumor promoter in a wide array of cancers. Prior to initiation and during the early stage of cancer progression, TGF-β acts as a tumor suppressor by inhibiting cellular proliferation and inducing apoptosis through Smad-dependent signaling [3]. Loss of responsiveness to the growth-inhibitory effects of TGF-β occurs in many cancer cell types such as pancreatic, breast and colorectal carcinoma cells. In colon cancer, loss of TGF-β sensitivity is frequently due to loss or mutation of known components of the TGF-β signaling pathway, notably TβRII and Smad4 [1, 3]. As such, many dedifferentiated, late-stage tumors are resistant to growth inhibition by TGF-β, and secreted TGF-β promotes oncogenic functions through activation of non-Smad signaling pathways.
Several recent studies suggest that small molecule inhibitors of TGF-β may be a useful therapeutic tool for the treatment of cancers. Previously, we demonstrated that the TβRI inhibitor, SB-431542, reduces the tumor-promoting effects of TGF-β including cell motility, migration, invasion and tumorigenicity [4]. The inhibitory effects of TGF-β small molecule inhibitors have also been reported to reduce metastasis of breast and pancreatic cancer cells [5, 6]. Recent reports have shown that LY2109761, a TGF-β dual receptor kinase inhibitor, can reduce migration and invasion of hepatocellular carcinoma cells and suppress pancreatic cancer metastasis [7, 8]. However, nothing is presently known about the effects of LY2109761 on colorectal cancer metastasis to the liver. In the present study, we demonstrate that LY2109761 reduces TGF-β-mediated cell migration, invasion, tumorigenicity and metastasis of the mouse colon carcinoma cell line, CT26.
2. Materials and methods
2.1. Cell line, reagents and antibodies
CT26 cells were derived from an undifferentiated murine adenocarcinoma that was induced by rectal injection of N-nitroso-N-methylurethane in Balb/c mice (American Type Culture Collection, Rockville, MD). CT26 Cells were maintained in RPMI medium containing 10% fetal bovine serum (FBS) and penicillin-streptomycin. CT26-Luc cells, which stably express firefly luciferase, were maintained in the above medium containing G418 (200 μg/ml). To generate the luciferase expression construct, the SV40 promoter element was excised from a pGL3-luc promoter vector by HindIII and BamH1 digestion. The SV40 promoter was then subcloned into the neomycin resistant pGL4-Luc vector (Promega). The resultant plasmid, pGL4-Neo-Luc was transfected into CT26 cells and positive selection was carried out by treating the cultures with media containing G418 (200 μg/ml). Several clones were isolated from the polyclonal population and the expression of luciferase was verified by bioluminescence and reporter assays. Four stable CT26-Luc clones expressed high levels of luciferase. The subsequent experiments were conducted using one of these clones. The mouse colon adenocarcinoma cell line, MC38 (kindly provided by Dr. Lee Gorden at the Vanderbilt University Ingram-Cancer Center) [10] were maintained in DMEM mediun containing 10% fetal bovine serum (FBS) and penicillin-streptomycin. TGF-β1 was purchased from R&D Systems (Minneapolis, MN). The TβRI and TβRII kinase inhibitor, LY2109761, was kindly provided by Dr. Jonathan M. Yingling at Eli Lilly Pharmaceuticals. Anti Smad2, Smad3 (Zymed Laboratories Inc.), Smad4, ERK, c-Jun (Santa Cruz Biotechnology Inc.), phospho-Smad2, phospho-ERK, phospho-c-Jun (Cell Signaling) and β-actin antibodies (Sigma Biochemicals) were used for western blot and immunoprecipitation analyses.
2.2. Extraction of genomic DNA
CT26 cells were lysed in 1 ml of lysis buffer (10 mM Tris-HCl, 10 mM EDTA, 0.5% SDS) containing 200 μg/ml Proteinase K and incubated at 55° C for 3 hours. NaCl (0.2M final) and RNAse A (25 μg/ml) were added and incubated overnight at 37° C. DNA was extracted twice with phenol:chloroform, precipitated with 100% ethanol, and resuspended in TE buffer.
2.3. PCR and sequence analyses
PCR analysis for exon-1 of mouse K-Ras was performed in a 25 μl reaction mixture containing 300 ng genomic DNA, 0.5 μl of 10 mM dNTP mixture, 2 μl of 25 mM MgCl2, 25 pmol of each forward (5′-GGCTGTTTAGATCAACAAGCTAAAT–3′) and reverse (5′-AAGCGCACGCAGACTGTAG-3′) primers and 0.5 μl (2.5 units) of Taq DNA polymerase (Invitrogen, CA). Thermal cycling was initiated with a 3 min denaturation at 95°C followed by 30 cycles with 95°C for 45 sec, 52°C for 45 sec and 72°C for 1 min 30 sec followed by extension for 10 min at 72°C. Specific PCR product (∼400 bp) was sequenced and mutations in codons 12, 13 and 61 of the K-Ras gene were examined
2.4. Immunoprecipitation and western blot analyses
CT26 cells were treated with 5 ng/ml of TGF-β1 and solubilized in lysis buffer as described previously [4]. For immunoprecipitation assays, lysates prepared from TGF-β1 (10 ng/ml) treated CT26 cells were incubated with anti Smad2 and Smad3 antibodies for 2 hours at 4°C. Immune complexes were precipitated, washed, and analyzed by western blotting with mouse anti-Smad4 antibodies.
2.5. [3H]Thymidine incorporation assay
CT26 cells were seeded into 24 well-plates at a density of 3×104 cells/well and treated with either TGF-β1 (5 ng/ml), LY2109761 (10 μM) or both TGF-β1 and LY2109761 in RPMI medium containing 10% FBS. After 25 hours, cells were labeled with [3H]thymidine (NEN, Boston, MA) at a dose of 4 μCi/well for 2 hours. Cells were then fixed in 10% cold trichloroacetic acid (TCA), washed, and lysed in 0.2 N NaOH. Radioactivity incorporated into TCA-insoluble [3H]thymidine was analyzed in a scintillation counter.
2.6. Cell counting assay
CT26 cells were seeded at a density of 8×103 in 12-well plates. Cells were treated with either TGF-β1 (5 ng/ml), LY2109761 (10 μM), or a combination of TGF-β1 and LY2109761 in RPMI medium. TGF-β1 and/or LY2109761 containing media was changed every 48 hours for a total of 5 days. Cells were counted every day and the average cell numbers from triplicate wells were presented.
2.7 ELISA for TGF-β1
1×105 cells from each pool of CT26 and MC38 were seeded into each well of 12-well plates and incubated for 24 hours in serum free medium. One hundred microliters of each supernatant media was activated and used for a TGF-β1 assay according to the manufacturer's instructions (R&D Systems).
2.8. Cell migration assay
CT26 cells (4×104) were seeded in the upper chamber of 8-μM pore transwells coated with collagen. Cells were allowed to migrate towards 7% serum containing medium with 5 ng/ml TGF-β1, LY2109761 (10 μM) or both, for 24 hours. The cells that passed through the membrane were fixed with 4% paraformaldehyde and stained with 1% crystal violet. Six random fields of view in each well were selected for counting. Each data point represents the average number from three wells.
2.9. Cell invasion assay
For invasion assays, the upper surface of 8-μM pore transwell chambers were overlaid with 100 μg of collagen dissolved in 100 μl of PBS or 50 μl (∼100 μg) of diluted matrigel (BD Biosciences, San Diego, CA) solution. Thereafter, 40,000 (collagen invasion) or 20,000 CT26 cells (matrigel invasion) in 100 μl RPMI media containing 0.2% BSA were seeded into the upper chambers of transwell inserts. 7% FBS containing media with 5 ng/ml TGF-β1 in the presence or absence of LY2109761 (10 μM) was used as a chemoattractant in the lower chamber, and then incubated at 37°C for 21 hours (collagen invasion) or 28 hours (matrigel invasion). Cells that invaded through the collagen or matrigel barrier were processed and analyzed as described in the migration assay above.
2.10. Wound healing assay
Confluent CT26 cells were pretreated with Mitomycin C (1 μg/ml) for 3 hours. A straight wound line of cell-free area was made in the monolayer of cells. Wounded cells were treated with 5 ng/ml TGF-β1, LY2109761 (10 μM), or both, and the wound line was monitored for 36 hours. Pictures were taken at a magnification of 200X.
2.11. Soft agarose assay
The soft agarose assay was performed as described previously [4]. Briefly, CT26 cells (1.5×104) were suspended in 1 ml of 0.4% sea plaque agarose containing 10% FBS and then plated on top of 1 ml of semisolid 0.8% agarose in 35-mm plates. Cells were treated with TGF-β1 (5 ng/ml), LY2109761 (10 μM) or both every 48 hours for two weeks. Colonies grown on soft agarose were counted and pictures of colonies were shown.
2.12. Tumorigenicity assay
CT26 cells (5×105) were injected subcutaneously behind the anterior forelimb of Balb/c nude mice as described previously [9]. Mice were divided into control (n=5) and LY2109761-treated (n=5) groups. LY2109761 (50 mg/kg) was orally administered twice daily. Tumor volumes were calculated and the growth curves for tumors were plotted as the mean ± SD of tumors from five mice.
2.13. In vivo experimental model of liver metastasis of colorectal cancer
Balb/c mice were divided into two groups: control (n=5) and LY2109761-treated (n=5). Mice were anesthetized using 2% isoflurane and an incision was made in the middle of the abdomen. 1×105 CT26 cells stably expressing firefly luciferase (CT26-Luc) were injected into mice spleens as described previously [10]. LY2109761 was dissolved in the SX-1292 oral vehicle (1% sodium carboxymethylcellulose, 0.5% sodium lauryl sulfate, and 0.05% antifoam; Eli Lilly) and administered orally (50 mg/kg) to the treatment group twice a day for 23 days. Liver metastases were monitored using bioluminescence imaging. Briefly, anesthetized mice were intraperitoneally injected with 200 μl of 15 mg/ml Beetle Luciferin (Promega) dissolved in water. Bioluminescent images were acquired using the IVIS Imaging System (Xenogen) 5 min after intraperitonial injection of Beetle Luciferin. Analyses were performed with LIVINGIMAGE software (Xenogen) by measuring photon flux. Imaging was performed every five days interval. At the end, mice were sacrificed and tumor tissue was removed for further analyses.
2.14. Survival assay
Mouse survival was determined by injecting CT26 cells into the spleens of Balb/c mice as described above. LY2109761 (50 mg/kg) was administered orally twice daily. Mice exhibiting signs of serious illness were sacrificed. The mean survival time for each group of mice was calculated and survival was evaluated by log-rank test.
3. Results
3.1. CT26 cells contain an oncogenic K-Ras mutation and LY2109761 inhibits TGF-β-mediated Smad and non-Smad signaling
Oncogenic K-Ras mutation (40-50%) is thought to be critical for colorectal carcinogenesis due to its ability to activate growth pathways and inhibit the tumor suppressor function of TGF-β. The most frequent type of mutation involves G to A transition and G to T transversion at the second position of codon 12 in the K-Ras gene [11]. Mutations of codon 13 in exon-1 and codon 61 in exon-2 have also been reported. To identify whether K-Ras mutations are present in mouse colon carcinoma CT26 cells, we performed PCR analyses of K-Ras exon-1 and exon-2 using genomic DNA from CT26 cells. Sequencing analyses revealed a G to A transition mutation of the K-Ras gene at codon 12 (GGT) (Fig. 1A), whereas no mutations were found at codon 13 and 61 (data not shown). These results suggest that CT26 cells contain an activating mutation of K-Ras at codon 12.
Fig. 1.
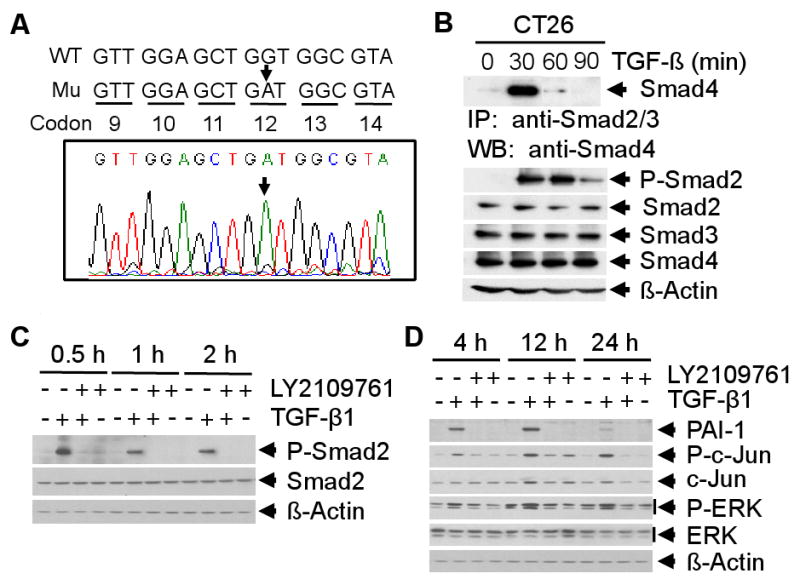
Sequence analysis of the K-Ras gene in CT 26 cells and the effects of LY2109761 on TGF-β-induced Smad-dependent and -independent pathways. A, Sequence analyses of PCR product from exon-1 of K-Ras using mouse genomic DNA as template shows a G to A substitution in codon 12 (GGT to GAT) (indicated by arrow). B, Lysates from TGF-β treated CT26 cells were immunoprecipitated with anti-Smad2/Smad3 polyclonal antibodies followed by western blotting with anti-Smad4 antibody (top panel). The expression of Smad proteins was analyzed (bottom section). C, CT26 cells were treated with TGF-β1 (5 ng/ml), LY2109761 (10 μM), or both for 0.5, 1 and 2 hours. Cell lysates were analyzed by western blotting with anti-phospho-Smad2 and anti-Smad2 antibodies. D, CT26 cells were treated for 4, 12 and 24 hours as indicated, and cell lysates were analyzed by western blotting with antibodies against PAI-1, phospho-c-Jun, c-Jun, phospho-ERK and ERK. Each experiment was repeated 3 times.
To determine whether the Smad-dependent TGF-β pathway is intact in the CT26 cell line and whether the TGF-β receptor kinase inhibitor (TRKI), LY2109761, inhibits this pathway, we first tested the ability of functional complex formation between Smad2, Smad3 and Smad4 using lysates from TGF-β-treated CT26 cells. Equal amount of each lysate was analyzed for Smad complex formation by immunoprecipitation using anti-Smad2 and anti-Smad3 antibodies followed by western blotting with anti-Smad4 antibody. TGF-β induced Smad complex formation within 30 minutes after treatment (Fig. 1B, top panel). We also observed that TGF-β significantly induced Smad2 phosphorylation within 30 minutes (Fig. 1B, bottom panel). To determine the role of LY2109761 on TGF-β-induced phosphorylation of Smad2, CT26 cells were treated with TGF-β, LY2109761, or both, and cell lysates were analyzed by western blotting. While total Smad2 was unchanged irrespective of treatments, we observed that LY2109761 completely blocked TGF-β-induced phosphorylation of Smad2 (Fig. 1C). In addition, we observed that LY2109761 inhibited expression of the TGF-β-inducible protein PAI-1 (Fig. 1D). These results suggest that LY2109761 inhibits TGF-β/Smad-dependent signaling by inhibiting phosphorylation of Smad2.
In addition to Smad signaling, TGF-β can also induce other non-Smad signaling pathways such as p38MAPK, ERK, PI3K, JNK. To determine whether LY2109761 can inhibit these effects of TGF-β on non-Smad signaling pathways, we tested the phosphorylation of ERK, JNK, AKT and p38-MAPK by western blot analyses using lysates from CT26 cells that were treated with TGF-β, LY2109761 or both. TGF-β induced phosphorylation of c-Jun and ERK in a time-dependent manner (Fig. 1D) and LY2109761 treatment inhibited TGF-β mediated activation of c-jun and ERK. No detectable changes in the levels of phospho-p38 and phospho-AKT were observed with TGF-β treatment (data not shown). These results suggest that LY2109761 can inhibit activation of oncogenic Smad-independent TGF-β signaling pathways.
3.2. LY2109761 inhibits TGF-β-induced cell migration and invasion
TGF-β stimulates chemotaxis and migration of tumor cells. To determine whether LY2109761 has any effect on TGF-β-induced motility of CT26 cells, we performed in vitro transwell migration, invasion and wound closure assays. We observed that TGF-β enhanced migration of CT26 cells by four-fold through a polycarbonate membrane, whereas LY2109761 treatment completely abrogated TGF-β-induced migration of CT26 cells (Fig. 2A). TGF-β increased invasion of CT26 cells by 2-fold through collagen layer and 1.8-fold through matrigel barrier (Fig. 2B and C). LY2109761 inhibited TGF-β-induced invasion of CT26 cells. We next examined the effect of LY2109761 on motility of CT26 cells in a wound healing assay. TGF-β accelerated wound closure within 36 hours, whereas treatment with LY2109761 inhibited TGF-β-induced cell motility (Fig. 2D). These results suggest that LY2109761 efficiently inhibits TGF-β induced migration and invasion of CT26 cells.
Fig. 2.
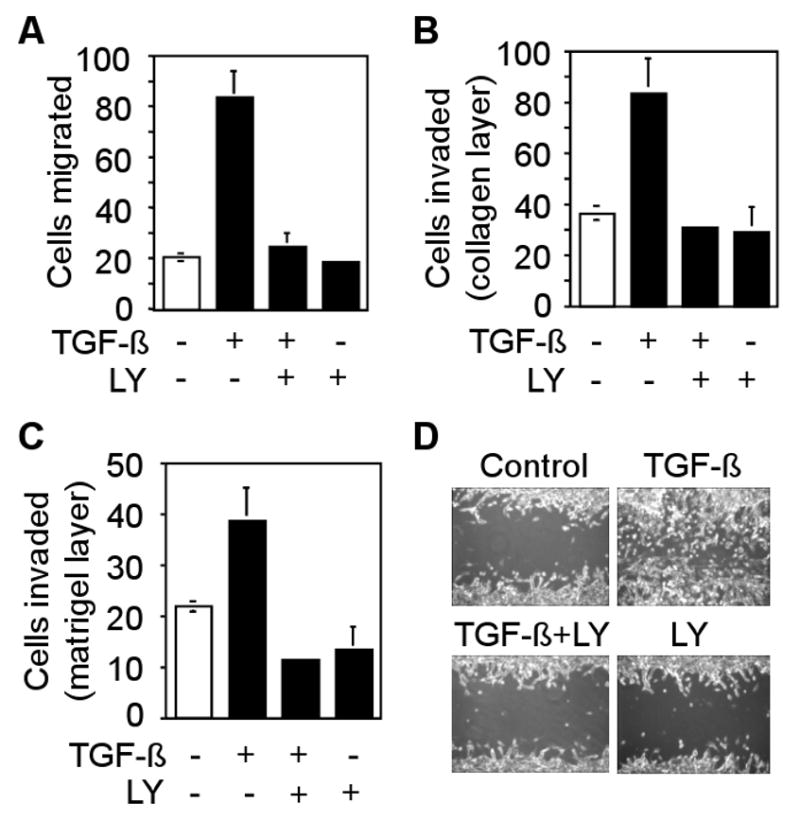
Effect of LY2109761 on TGF-β-induced migration, invasion and wound healing. A, CT26 cells were allowed to migrate through 8-μM pores in transwell chambers containing TGF-β (5 ng/ml), LY2109761 (10 μM) or both TGF-β and LY2109761. Cells that migrated through the pores were fixed, stained and counted. Individual data points represent the mean ± SD of three independent wells. B and C, CT26 cells were allowed to pass through a collagen-layer (B) or a matrigel-barrier (C) in transwell chambers. Cells that invaded through the filter were fixed, stained and counted. Individual data point represents the mean ± SD of three independent wells. D, CT26 cells were pretreated with Mitomycin C (1μg/ml) for 3 hours before wounding. Wounded cells were treated with 5 ng/ml TGF-β1 for 36 hours in presence or absence of LY2109761. Phase contrast images are shown.
3.3. TGF-β has no significant effect on the growth of CT26 cells
One of the most important biological effects of TGF-β is its ability to inhibit proliferation of epithelial cells. However, under transforming conditions, the growth of tumor cells is occasionally stimulated by TGF-β. To test whether CT26 cells are growth inhibited by TGF-β, we first performed a [3H]thymidine incorporation assay. We observed that TGF-β marginally inhibits thymidine incorporation in CT26 cells. Although the effects of exogenous TGF-β on CT26 cells were not statistically significant, this minor effect of TGF-β was blocked by LY2109761 (Fig. 3A). The effects of TGF-β and LY2109761 on growth of CT26 cells were also evaluated by cell counting. Similarly, we observed that TGF-β marginally inhibits growth of CT26 cells, whereas LY2109761 alone has no effect on the growth of these cells (Fig. 3B). To examine the possibility that the lack of growth inhibition is due to saturation of the TGF-β receptors with secreted TGF-β, we performed ELISA assays using culture medium from CT26 and control MC38 cells. We observed that both CT26 and MC38 cells produced a significant amount of TGF-β (Fig. 3C). These results suggest that TGF-β has no significant effect on the growth of CT26 cells.
Fig. 3.

Effects of TGF-β and LY2109761 on the growth of CT26 cells. A, [3H]thymidine incorporation assay. CT26 cells were treated with TGF-β (5 ng/ml) in presence or absence of LY2109761 (10 μM) for 25 hours and then treated for an additional 2 hours with [3H]thymidine. Cells were fixed in trichloroacetic acid (TCA), washed, and lysed in 0.2 N NaOH. Radioactivity incorporated into TCA-insoluble [3H]thymidine was measured by scintillation counting. Individual data points are the mean ± SD of triplicate determinations. *P>0.05, compared with untreated control, Student's t-test. B, Cell counting assay. CT26 cells were seeded into each well of 12-well plates and then treated with TGF-β (5 ng/ml) in presence or absence of LY2109761 (10 μM) for five days. Cells were counted every day and the individual data points are presented as the mean ± S.D. of triplicate determinations. *P>0.05, compared with untreated control, Student's t-test. C, ELISA for TGF-β1. CT26 and MC38 cells were serum-starved for 24 hours. Supernatant media was activated and used for a TGF-β1 ELISA. Individual data points are representative of the mean ± SD of three individual measurements. Each experiment was repeated at least three times.
3.4. LY2109761 inhibits tumorigenicity of CT26 cells in vitro and in vivo
A common characteristic of cancer cells is its ability to grow in an anchorage-independent manner. To determine the effect of the inhibitor on TGF-β-induced anchorage-independent growth of CT26 cells, we performed an in vitro soft agarose assay. We observed that TGF-β enhanced colony formation both in size and number in soft agarose, whereas LY2109761 reduced TGF-β-induced colony formation (Fig. 4A and B). To further examine the effect of LY2109761 on tumorigenicity in vivo, we performed tumor allograft studies by inoculating CT26 cells into the anterior forelimb of Balb/c nude mice. Mice were divided into control (untreated) and LY2109761-treated groups. LY2109761 was orally administered twice a day to the experimental group and the effect of LY2109761 on tumor growth is shown in Fig. 4C. LY2109761 reduced tumor growth in treated mice relative to the control mice (Fig. 4C). The efficacy of LY2109761 in inhibiting TGF-β signaling in vivo was examined by western blot analyses using tumor lysates from control and LY2109761-treated mice. Specifically, we observed decreased levels of Smad2 phosphorylation in tumor lysates of LY2109761-treated mice when compared with control mice (Fig. 4D). These results suggest that partial inhibition of TGF-β signaling can decrease tumorigenicity of CT26 cells.
Fig. 4.
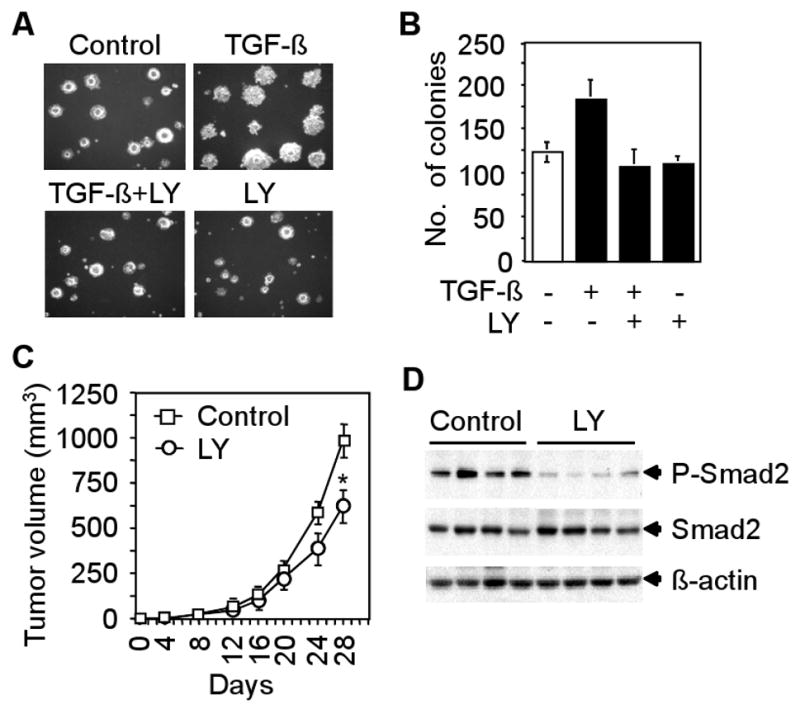
Effect of LY2109761 on tumorigenicity. A and B, CT26 cells were plated in soft agarose and treated with 5 ng/ml TGF-β in presence or absence of LY2109761 (10 μM) every 48 hours for 2 weeks. Pictures of colonies are shown. Colonies were counted and each data point represents the mean ± SD of triplicate plates. C, 5×105 cells were injected subcutaneously into the right flank of Balb/c nude mice. These mice were divided into control (n=5) and LY2109761-treated (n=5) groups. LY2109761 (50 mg/kg) was administered orally twice a day. Growth curves for tumors were plotted from the mean volume ± SD of tumors. Above experiments were repeated three times. *P<0.05, compared with untreated control, Student's t-test. D, Western blot analyses using lysates from four tumors of control and LY2109761-treated mice showing the levels of anti-phospho-Smad2 and total Smad2.
3.5. LY2109761 treatment reduces colon cancer liver metastasis and prolongs the survival of metastatic tumor-bearing mice
To test the therapeutic potential of LY2109761 under physiologically relevant conditions, we employed an experimental model for liver metastasis by splenic injection of CT26 cells in Balb/c mice. For this experiment, we generated stable CT26 clones that constitutively expressed firefly luciferase. The stable clone that expressed the highest level of luciferase (CT26-Luc) was used in the splenic injection metastasis model. Mice treated with LY2109761 showed significantly reduced liver metastases as monitored by bioluminescence imaging (Fig. 5A). The regression in liver metastasis by LY2109761 (*P <0.01) when compared with vehicle control is shown in Fig. 5B. Using the above metastasis model system, we determined the effect of LY2109761 on survival of mice following injection of CT26-Luc cells into spleens of Balb/c mice. The mean survival of LY2109761-treated mice were prolonged to 35.2 days compared with 24.5 days in control mice (Fig. 5C; *P <0.001, log rank test). The overall survival at 30 days was 85.71% in LY2109761-treated mice and 0% in control mice. These data indicate that attenuating TGF-β signaling by LY2109761 reduces colon cancer liver metastasis, and prolongs survival of mice bearing tumors. Taken together, our results demonstrate that the TβRI and TβRII dual receptor kinase inhibitor, LY2109761 may have potential for therapeutic application in reducing TGF-β-mediated tumor progression and metastasis in vivo.
Fig. 5.
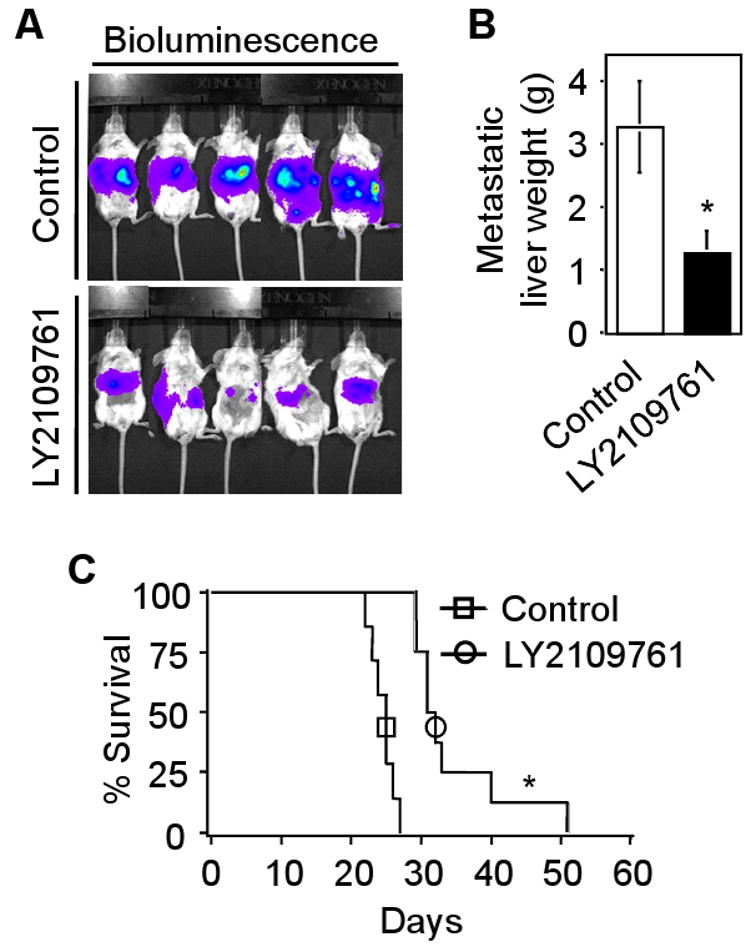
LY2109761 reduces liver metastasis and prolongs survival of Balb/c mice. A, 1×105 CT26-Luc cells were injected into spleens of Balb/c mice and mice assigned to the treatment group were given LY2109761 (50 mg/kg) orally twice daily. Bioluminescence images, taken after 23 days of splenic injection, are presented. B, Data represents average liver weight from control and LY2109761-treated mice (*P<0.01 compared to control, Student's t-test). C, To determine the survival of mice treated with LY2109761, mice were randomly divided into control group (n=7) and LY2109761-treated group (n=8). Mice were sacrificed when they were moribund and the day of sacrifice was considered the day of death for survival evaluation. Data represents the survival curve of control and LY2109761-treated mice (*P<0.001 compared to control, log-rank test). Each experiment was repeated three times with similar results.
4. Discussion
TGF-β plays an important role in cancer progression and increased production of TGF-β in human tumors is correlated with poor prognosis. In most human cancers, cells first become resistant to TGF-β-induced growth inhibition and apoptosis. In the later stages of cancer progression, high levels of TGF-β can promote tumorigenicity in an autocrine and/or paracrine manner by promoting invasion and metastasis [3, 12]. Therefore, the rational design of inhibitors that specifically counteract the tumor-promoting effects of TGF-β may be useful as a therapeutic strategy for the treatment of human cancers. Thus far, the small molecule inhibitors of TGF-β receptor kinases have demonstrated superior efficacy due to increased tissue penetration and cellular permeability [13]. Although several small molecule inhibitors are in pre-clinical studies, the utility of these inhibitors in the prevention of colon cancer metastasis has not been fully determined. Herein, we report that the TRKI, LY2109761 reduces liver metastasis of mouse colon cancer.
In this study, we have used murine colon carcinoma CT26 cells that are aggressive in carcinogenesis, and metastasize to the liver in an experimental splenic injection model. While our studies suggest that CT26 cells are responsive to TGF-β, we did not observe significant growth inhibition in the presence of exogenous TGF-β (Fig. 3A and B). Mutation in the Ras proto-oncogene can be found in about 50% of colorectal cancers, and circumstantial evidence suggest that the cross talk between TGF-β and oncogenic Ras may be required for its pro-oncogenic effects [14]. Interestingly, we found an activating mutation in codon 12 of K-Ras in CT26 cells (Fig.1A) that may play an important role in carcinogenesis of CT26 cells. It is possible that activation of Ras is associated with the resistance of these cells to TGF-β mediated growth inhibition, although the Smad signaling is intact in these cells as assessed by Smad2 phosphorylation as well as functional complex formation between Smad2, Smad3 and Smad4 (Fig. 1B and C).
Growing evidence indicates that activation of non-Smad signaling pathways like p38MAPK, ERK, PI3K, JNK, or Rho are involved in the pro-oncogenic functions of TGF-β [2]. We have observed that TGF-β mediated activation of ERK and JNK pathways in CT26 cells is inhibited by LY2109761 (Fig. 1D). Furthermore, LY2109761 treatment reduces the effects of TGF-β on migration, invasion (Fig. 2) and tumorigenicity of these cells in vitro (Fig. 4A and B). In vivo, we have determined that LY2109761 reduces subcutaneous growth of CT26 cells in Balb/c nude mice (Fig. 4C). Inhibition of endogenous TGF-β signaling in tumor tissues from the treated mice was confirmed by western blot analysis of phosphorylated Smad2 (Fig. 4D). We have observed that CT26 cells produce a significant amount of TGF-β (Fig. 3C). This secreted TGF-β may stimulate signaling pathways in vivo which leads to aggressive tumor growth in mice. Our data also demonstrates that treatment with LY2109761 decreases the size of liver metastases (Fig. 5A and B) and prolongs survival of treated mice relative to the control mice (Fig. 5C). It is possible that the aggressiveness in metastasis of these cells is due to the concerted effects of oncogenic Ras and the pro-oncogenic effects of TGF-β. These findings demonstrate that LY2109761 can reduce metastasis of colon cancer cells to the liver by attenuating TGF-β-induced oncogenic signaling pathways.
Acknowledgments
This work was supported by R01 CA95195 and CA113519, NCI SPORE grant in lung cancer (5P50CA90949, project# 4) and a Clinical Innovator Award from Flight Attendant Medical Research Institute (to P.K.D). We are grateful to Jennifer E. Reiner and Nilesh D. Kashikar for critical reading of the manuscript.
Footnotes
Conflicts of Interest Statement: None Declared
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signaling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 2.Moustakas A, Heldin CH. Non-Smad TGF-beta signals. J Cell Sci. 2005;118:3573–3584. doi: 10.1242/jcs.02554. [DOI] [PubMed] [Google Scholar]
- 3.Bierie B, Moses HL. Tumour microenvironment: TGF-beta: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer. 2006;6:506–520. doi: 10.1038/nrc1926. [DOI] [PubMed] [Google Scholar]
- 4.Halder SK, Beauchamp RD, Datta PK. A specific inhibitor of TGF-beta receptor kinase, SB-431542, as a potent antitumor agent for human cancers. Neoplasia. 2005;7:509–521. doi: 10.1593/neo.04640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gaspar NJ, Li L, Kapoun AM, Medicherla S, Reddy M, Li G, O'Young G, Quon D, Henson M, Damm DL, Muiru GT, Murphy A, Higgins LS, Chakravarty S, Wong DH. Inhibition of transforming growth factor beta signaling reduces pancreatic adenocarcinoma growth and invasiveness. Mol Pharmacol. 2007;72:152–161. doi: 10.1124/mol.106.029025. [DOI] [PubMed] [Google Scholar]
- 6.Ge R, Rajeev V, Ray P, Lattime E, Rittling S, Medicherla S, Protter A, Murphy A, Chakravarty J, Dugar S, Schreiner G, Barnard N, Reiss M. Inhibition of growth and metastasis of mouse mammary carcinoma by selective inhibitor of transforming growth factor-beta type I receptor kinase in vivo. Clin Cancer Res. 2006;12:4315–4330. doi: 10.1158/1078-0432.CCR-06-0162. [DOI] [PubMed] [Google Scholar]
- 7.Fransvea E, Angelotti U, Antonaci S, Giannelli G. Blocking transforming growth factor-beta up-regulates E-cadherin and reduces migration and invasion of hepatocellular carcinoma cells. Hepatology. 2008;47:1557–1566. doi: 10.1002/hep.22201. [DOI] [PubMed] [Google Scholar]
- 8.Melisi D, Ishiyama S, Sclabas GM, Fleming JB, Xia Q, Tortora G, Abbruzzese JL, Chiao PJ. LY2109761, a novel transforming growth factor beta receptor type I and type II dual inhibitor, as a therapeutic approach to suppressing pancreatic cancer metastasis. Mol Cancer Ther. 2008;7:829–840. doi: 10.1158/1535-7163.MCT-07-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Halder SK, Beauchamp RD, Datta PK. Smad7 induces tumorigenicity by blocking TGF-β-induced growth inhibition and apoptosis. Exp Cell Res. 2005;307:231–246. doi: 10.1016/j.yexcr.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 10.Gorden DL, Fingleton B, Crawford HC, Jansen DE, Lepage M, Matrisian LM. Resident stromal cell-derived MMP-9 promotes the growth of colorectal metastases in the liver microenvironment. Int J Cancer. 2007;121:495–500. doi: 10.1002/ijc.22594. [DOI] [PubMed] [Google Scholar]
- 11.Akagi K, Uchibori R, Yamaguchi K, Kurosawa K, Tanaka Y, Kozu T. Characterization of a novel oncogenic K-ras mutation in colon cancer. Biochem Biophys Res Commun. 2007;352:728–732. doi: 10.1016/j.bbrc.2006.11.091. [DOI] [PubMed] [Google Scholar]
- 12.Tobin SW, Douville K, Benbow U, Brinckerhoff CE, Memoli VA, Arrick BA. Consequences of altered TGF-beta expression and responsiveness in breast cancer: evidence for autocrine and paracrine effects. Oncogene. 2002;21:108–118. doi: 10.1038/sj.onc.1205026. [DOI] [PubMed] [Google Scholar]
- 13.Yingling JM, Blanchard KL, Sawyer JS. Development of TGF-beta signalling inhibitors for cancer therapy. Nat Rev Drug Discov. 2004;3:1011–1022. doi: 10.1038/nrd1580. [DOI] [PubMed] [Google Scholar]
- 14.Datta PK, Mann JR. Transforming growth factor-β (TGF-β) signaling inhibitors in cancer therapy. In: Jakowlew SB, editor. Transforming growth factor-β in cancer therapy, volume II, Cancer treatment and therapy. Human press; 2008. pp. 573–587. [Google Scholar]