

Abstract
Rhodobacter capsulatus regulates many metabolic processes in response to the level of environmental oxygen and the energy state of the cell. One of the key global redox regulators of the cell’s metabolic physiology is the sensor kinase RegB that controls the synthesis of numerous energy generation and utilization processes. In this study, we have succeeded in purifying full-length RegB containing six transmembrane-spanning elements. Exogenous addition of excess oxidized coenzyme Q1 is capable of inhibiting RegB autophosphorylation ~6-fold. However, the addition of reduced coenzyme Q1 exhibits no inhibitory effect on kinase activity. A ubiquinone-binding site, as defined by azidoquinone photo affinity cross-linking, was determined to lie within a periplasmic loop between transmembrane helices 3 and 4 that contains a fully conserved heptapeptide sequence of GGXXNPF. Mutation of the phenylalanine in this heptapeptide renders RegB constitutively active in vivo, indicating that this domain is responsible for sensing the redox state of the ubiquinone pool and subsequently controlling RegB autophosphorylation.
Rhodobacter capsulatus is a purple non-sulfur photosynthetic bacterium that exhibits remarkable metabolic diversity. Aerobically, R. capsulatus is capable of using an electron transport chain with oxygen as a terminal electron acceptor that drives the generation of a proton membrane potential (1). If oxygen is not present, R. capsulatus can also perform anaerobic respiration by reducing other compounds such as dimethyl sulfoxide (1). In addition to respiratory functions, R. capsulatus is capable of converting light energy to chemical energy via synthesis of a bacterial photosystem (1). Synthesis of the photosystem is under redox control, such that it is only synthesized under oxygen limiting conditions. R. capsulatus also demonstrates redox control of additional components needed for cellular processes such as carbon fixation, nitrogen fixation, and hydrogen utilization (2, 3). Remarkably, the redox-responding two-component signal transduction cascade, RegB/ RegA, is responsible for the transcriptional control of all of the above components (4). Disruptive mutations in either the sensor kinase, RegB, or in the cognate response regulator, RegA, results in cells that are altered in their ability to respire, photosynthesize, and fix carbon and nitrogen (4).
Nearly all of the metabolic events that are part of the RegB/RegA regulon affect cellular redox chemistry with most of these processes obtaining their reducing power from the ubiquinol pool. Carbon and nitrogen fixation are also very effective electron sinks that utilize large amounts of reducing power to drive their metabolic processes. Given that redox chemistry is a common thread to the majority of RegB/RegA-regulated processes, it has been proposed that RegB may interact with, and monitor, the redox state of a component of the electron transport chain. Specifically, ubiquinones and cytochrome cbb3 oxidase have been suggested components that may interact with RegB and modulate its kinase activity (4, 5). There is also evidence that RegB contains a redox active cysteine residue that when oxidized inhibits kinase activity (6). Neither of these modes of regulation are mutually exclusive. For example, the widely studied global sensor kinase ArcB from Escherichia coli controls a number of cellular processes in response to changes in oxygen tension. It has been proposed that activity of ArcB is regulated by two redox active cysteines that form intermolecular disulfide bonds when oxidized by ubiquinone (7, 8). Because the oxidation-reduction state of the ubiquinone pool fluctuates greatly based on oxygen tension, it has been proposed that an interaction of ArcB with the ubiquinone pool ultimately regulates the activity of this kinase (7, 8).
Given that RegB is a membrane-bound sensor kinase, one can presume that it is in close contact with the ubiquinone pool and thus could potentially monitor the oxidation/reduction state of this membrane soluble electron carrier. In purple bacteria, the oxidation state of the ubiquinone pool is known to dramatically fluctuate, becoming predominantly oxidized under aerobic respiratory growth and predominantly reduced under anaerobic photosynthetic growth conditions (9). Given that purple bacteria have a large ubiquinone pool (10) that undergoes significant changes in redox potential (9), it is apparent that this molecule would be an excellent indicator of changes in environmental oxygen tension.
In this study, we describe the isolation and biochemical characterization of a full-length membrane-spanning version of RegB. We provide in vitro evidence that oxidized ubiquinone is a potent inhibitor of RegB. A ubiquinone-binding site was identified by performing photo affinity cross-linking with a ubiquinone analog that binds to a universally conserved heptapeptide sequence GGXXNPF in the membrane-spanning input domain of RegB. Mutational analysis of this heptapeptide sequence results in dramatic derepression of RegB autophosphorylation in vivo, resulting in elevated expression of photosynthesis genes even in the presence of oxygen.
EXPERIMENTAL PROCEDURES
Strain, Media and Growth Conditions
R. capsulatus strain, SB1003 was used as the parent strain for all of the mutants generated for this study. All of the R. capsulatus strains were grown at 34 °C in either RCV or PY, with spectinomycin and gentamycin concentrations of 10 and 1.5 μg/ml, respectively. E. coli strain DH5α was grown at 37 °C in Luria broth for all cloning procedures, and strain BL21 (λde3) was grown in Terrific broth for protein overexpression, with kanamycin, spectinomycin, and ampicillin concentrations of 50, 50, and 150 μg/ml, respectively.
Plasmid Construction
An expression plasmid for full-length RegB was constructed by PCR of the regB gene using primers; RegBFullNcoI (5′-TACCATGGTGAGGGCTGTCGACC) and RegBFullXhoI (5′-ATCTCGAGGGCGGTGATCGGAACATTC) with genomic template DNA obtained from wild type cells. The PCR product was cloned into the NcoI and XhoI sites of pET28.
A chromosomal in-frame deletion of regB was constructed by first PCR amplifying the entire regB gene plus 500 bp upstream and downstream of the regB open reading frame using primers RegBdbSacF (5′-TAGAGCTCCATCAGATCGGTGAATTC) and RegBdbXbaR (5′-TATCTAGACCGCATCAGATCCAGTTC). The amplified fragment was cloned into pCRScript SK+ (Stratagene) forming plasmid pCRScriptRegB+ and sequenced to ensure correct amplification. Plasmid pCRScriptRegB+ was then used as a PCR template for construction of the in-frame deletion of regB and as a template to generate regB point mutations.
The in-frame regB deletion was constructed by PCR amplifying 500-bp segments of DNA flanking the regB gene using primers RegBdbSacF (5′-TAGAGCTCCATCAGATCGGTGAATTC) and RegBdelUp (5′-ATGATATCCATATCTGCCATCGTCG) for the upstream region and RegBdbXbaR (5′-TATCTAGACCGCATCAGATCCAGTTC) and RegBdelDown (5′-ATGATATCGCCTGATGATCACAATCG) for the downstream region with plasmid pCRScriptRegB+ as a template. EcoRV sites were engineered into the RegBdelUP and RegBdelDown primers, such that when the two PCR products were ligated together they generated an in-frame deletion of regB that extended from the first to last codon of RegB. The deletion product was then cloned into the corresponding sites of the suicide vector pZJD29a.2 Single recombinants were first selected by gentamycin resistance followed by plating on PY plates containing 5% sucrose to select for double recombinant cells that had lost the suicide vector.
Point mutations in the ubiquinone-binding domain of RegB were generated using a PCR technique as described previously (11). The N110A mutation was generating using primer N-AForsew (5′-GCTGAACGCCCCCTTCGCGC) and a reverse complement of this primer, N-ARevsew. The F112A mutation was generated using primer F-AForsew (5′-AACCCCGCCGCGCTTTTGATC) and a reverse complement of this primer, F-ARevsew. The mutant PCR products were then cloned into the suicide vector, pZJD1, using primer encoded XbaI, SacI restriction sites.2 The clones were transformed into SM10 λpir and sequenced to ensure the presence of the mutation and absence of secondary mutations in regB. These mutant plasmids were then conjugated into the regB deletion strain with single recombinants selected by spectinomycin resistance. A control strain that recombined a wild type copy of regB into the regB deletion strain was also generated using the same technique.
Purification of Full-length RegB
The cell cultures were grown at 37 °C in Terrific broth with moderate shaking until they reached an A600 of 0.4–0.6, at which time isopropyl β-d-thiogalactopyranoside was added to a final concentration of 75 μm. Protein overexpression proceeded for 4 h at 37 °C before the cells were harvested by centrifugation and resuspended in 100 ml of 10 mm Tris, pH 8, 100 mm NaCl until no visible debris remained. The cells were lysed by two passages through the M-110L Micro Fluidizer Processor at 2000 p.s.i. (Microfluidics) with the cell lysate clarified by centrifugation at 11,000 × g for 30 min at 4 °C. The supernatant was then centrifuged at 150,000 × g for 1 h at 4 °C to pellet inner membranes that contained full-length RegB as based on Western blot analysis (data not shown). The inner membrane pellet was solubilized by addition of 2 ml of solubilization buffer composed of 20 mm Tris, pH 8.0, 20 mm imidazole, 300 mm NaCl, 20% glycerol, and 1% n-dodecyl β-d-maltoside/liter of lysed E. coli cells. The membrane solubilization solution was shaken in a 50-ml conical tube attached to an orbital shaker rotating at 100 rpm for 1 h at 24 °C. The solubilized membranes were then centrifuged at 150,000 × g for 1 h at 4 °C to remove insoluble debris. The supernatant containing full-length RegB was then diluted 20-fold in wash buffer comprised of 20 mm Tris, pH 8.0, 20 mm imidazole, 150 mm NaCl, and 10% glycerol and allowed to incubate with 1 ml of settled charged nickel resin (Novagen) for 1 h at room temperature shaking at 40 rpm on an orbital shaker. The nickel resin was then pelleted by centrifugation at 1,000 × g for 5 min at 4 °C. The supernatant was discarded, and the resin was applied to a disposable column and washed with 50 column volumes of wash buffer comprised of 20 mm Tris, pH 8.0, 20 mm imidazole, 150 mm NaCl, and 10% glycerol at 24 °C. When purifying full-length RegB for use with azido-Q3 binding experiments, the wash buffer step was modified by the addition of a second wash step of 50 column volumes of wash buffer containing 1% sodium cholate to remove and replace protein-bound n-dodecyl β-d-maltoside that interferes with azido-Q binding. Full-length RegB was then eluted with 10 ml of elution buffer comprised of 20 mm Tris, pH 8.0, 200 mm imidazole, 150 mm NaCl, and 10% glycerol at 24 °C with 1-ml fractions collected and analyzed for protein content using the Bio-Rad assay. Fractions containing protein were separated by SDS-PAGE to confirm the presence of RegB. Appropriate fractions were pooled and dialyzed against 10 mm Tris, pH 8.0, 150 mm NaCl, 50% glycerol at 4 °C and then stored at −20 °C until the kinase assays were completed.
Kinase Assays
The kinase assays were completed as described by Bird et al. (12). For ubiquinone addition experiments, benzoquinone (Sigma catalog number D9150) or coenzyme Q1 (Sigma catalog number C7956) was solubilized in 95% ethanol at a concentration of 167 μm and added to replicate kinase with one assay containing only the solvent (<1% ethanol) and the other reaction containing solvent plus either benzoquinone or coenzyme Q1. In cases where ubiquinone was reduced to ubiquinol, 10 mm dithiothreitol was added to the ubiquinone prior to addition to the kinase assays.
Affinity Binding of [3H]Azidoquinone
Tritium-labeled azidoquinone ([3H]azido-Q) was synthesized as described previously (13). Affinity cross-linking of [3H]azido-Q to sodium cholate-purified full-length RegB was performed by mixing 42 mm of [3H]azido-Q that was solubilized in 95% ethanol with 42 μm of full-length RegB in a buffer comprised of 20 mm Tris, pH 8.0, 150 mm NaCl, and 30% glycerol. The final concentration of ethanol in the mixture was kept lower than 5% to prevent denaturation of the protein. After incubation for 60 min at 4 °C in the dark, the protein and [3H]azido-Q was placed in a 2-mm light path quartz cuvette and exposed to long wavelength UV light (Spectroline EN-14, 365-nm-long wavelength, 23 watts) at 0 °C for 20 min at a distance of 4 cm from the light source. Unbound [3H]azido-Q was then separated from UV cross-linked [3H]azido-Q-RegB by paper chromatography using chloroform:methanol (2:1, v/v) as a solvent system. [3H]azido-Q-RegB remained at the paper origin, whereas unbound [3H]azido-Q migrated at the solvent front. Chromatographically purified [3H]azido-Q-RegB was then solubilized from the paper matrix by the addition of elution buffer comprised of 30 mm Tris-Cl, pH 7.4, 0.5% SDS, 1 m urea and then subjected to protease K digestion at 37 °C for 6 h using a protease K:RegB ratio of 1:50 (w/w). The protease K-digested sample was then subjected to HPLC separation on a Supelcosil™ LC-308 (C8, 5-μm particles, 300-Å pores, 4.6-mm inner diameter, 25-cm length) using a gradient formed from 0.1% trifluoroacetic acid and 90% acetonitrile containing 0.1% trifluoroacetic acid with a flow rate of 0.8 ml/min with 0.4-ml fractions collected. The fractions recovered from the HPLC chromatograms were analyzed for peptide patterns by UV absorbance recorded with a Waters 996 diode array detector and the presence of [3H] by scintillation counting in a Packard Tri-Carb 1900CA scintillation analyzer. Fractions from several independent HPLC chromatograms that contained 3H protein profiles were then subjected to amino-terminal sequence analysis at the Molecular Biology Resource Facility of the Saint Francis Hospital of Tulsa Medical Research Institute, University of Oklahoma Health Science Center, under the supervision of Dr. Ken Jackson.
Spectroscopy and Northern Blot Analysis
Spectral analysis of R. capsulatus cell membranes were performed as previously published (6). For Northern blot analysis, cellular RNA was isolated using a RNeasy Mini kit (Qiagen). The RNA content was assayed by absorbance at 280 nm and then normalized for ribosomal RNA content after electrophoretic separation and visualization of 16 S rRNA on a formamide gel. The Northern blot analysis was performed as described previously (14). The RNA blot was probed with a digoxigenin-labeled (Roche Applied Science catalog number 1-636-090) puf or puc probe for 16 h at 65 °C. The digoxigenin-labeled puf and puc probes were generated by PCR from genomic template using primers PufQup (5′-TGGGTAGCGTCTACCTGCTG), PufAdown (5′-CCCGCAACGCTCAGTAACGC), PucUP (5′-CTCCCATAGTGCGTCTCACG), and PucDown (5′-TTACTGAGCCGGCGCAACAGC), respectively. Detection of the digoxigenin-labeled probe was carried out as recommenced by the manufacturer of the Dig luminescent detection kit (Roche Applied Science catalog number 1-636-514) with hybridization levels visualized using radiographic film and quantitated using a Fluorchem 5500 imaging system from Alpha Innotech. The 16 S ribosomal RNA was also quantitated and used to normalize Northern blot hybridization levels.
RESULTS
Purification of Full-length Membrane-spanning RegB
A specific involvement of the membrane-spanning domain of RegB in redox sensing has not been extensively explored beyond the construction of a few mutations in the membrane-spanning region, several of which affect phosphorylation (15). To biochemically explore whether the membrane-spanning domain controls RegB activity, we expressed His6-tagged full-length RegB in E. coli and isolated full-length protein from crude membrane fractions after solubilization with the detergent n-dodecyl β-d-maltoside (see “Experimental Procedures”). Detergent solubilized full-length RegB was then purified by nickel ion chromatography and visualized by SDS-PAGE (Fig. 1A). The most prominent band on the gel corresponds to a RegB monomer at 47 kDa and a RegB dimer at 98 kDa, which is close to the calculated monomer and dimer size for RegB at 50.1 and 100.2 kDa. Western blot analysis using an anti-His tag antibody (Santa Cruz Biotechnology catalog number SC803) verified the identity of these bands as His-tagged RegB (data not shown).
FIGURE 1. SDS-PAGE analysis and autophosphorylation assays of RegB.
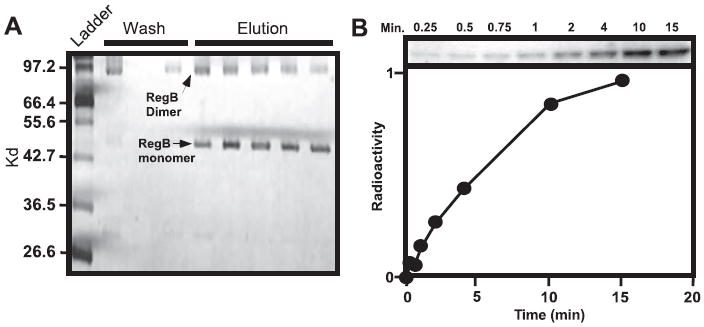
A, solubilized and purified RegB separated by SDS-PAGE. Lane 1 contains the molecular weight mass followed by the wash fraction and the elution fraction after the addition of imidazole. B, autophosphorylation of purified monomer RegB using γ-32P-labeled ATP as a tracer. The aliquots were removed at 0.25, 0.5, 0.75, 1, 2, 4, 10, and 15 min after the addition of ATP, and the reaction was quenched with SDS-PAGE loading buffer before being separated by SDS-PAGE. The graph below the autoradiograph represents the arbitrary units of 32P incorporation derived from phosphorimaging data analysis.
Autophosphorylation assays were performed on full-length RegB in the presence of [γ-32P]ATP with aliquots being removed at intervals ranging from 0.25 to 15 min (Fig. 1B). The autoradiograph in Fig. 1B demonstrates that purified full-length RegB exhibits excellent autophosphorylation with activity detected as early as 0.25 min, linear within the first 2 min, and reaching a plateau in ~10 min. Similar phosphorylation was observed with a soluble truncated form of RegB (RegBs), which does not contain the membrane-spanning domain (6) as well as the full-length RegB homolog from Rhodobacter sphaeroides (16). This indicates that the mild detergent, n-dodecyl β-d-maltoside does not compromise the structural integrity of RegB nor significantly impact the level of activity of the protein.
RegB Autophosphorylation Is Regulated By The Redox State of Ubiquinone
To address the role of ubiquinone in controlling kinase activity, we added a 50-fold molar excess of coenzyme Q1, which is a close analog of ubiquinone containing only one isoprenoid side chain unit. When an oxidized form of this soluble quinone analog was allowed to incubate with RegB for 20 min prior to the initiation of autophosphorylation, there was a 5.4-fold decrease in autophosphorylation levels after the reactions reached plateau (Fig. 2A). Half-maximal kinase inhibition was achieved at a 20-fold molar excess of ubiquinone to RegB (data not shown). Note that there is a vast excess of quinones over that of the molar amount of RegB/cell (10), so it is not surprising that a 20-fold excess of coenzyme Q1 is needed to observe half-maximal inhibition. A similar inhibitory affect on RegB activity was observed with the addition of oxidized benzoquinone, which contains only the polar head group of coenzyme Q1, suggesting that the redox active head group of ubiquinone possesses the inhibitory signal (data not shown). Finally, kinase assays in the presence of a 50-fold excess of reduced coenzyme Q1 (ubiquinol) does not affect the autophosphorylation activity of full-length RegB (Fig. 2B). Thus, only the presence of oxidized quinone has an inhibitory affect on RegB kinase activity.
FIGURE 2. RegB autophosphorylation assays in the presence of ubiquinone.
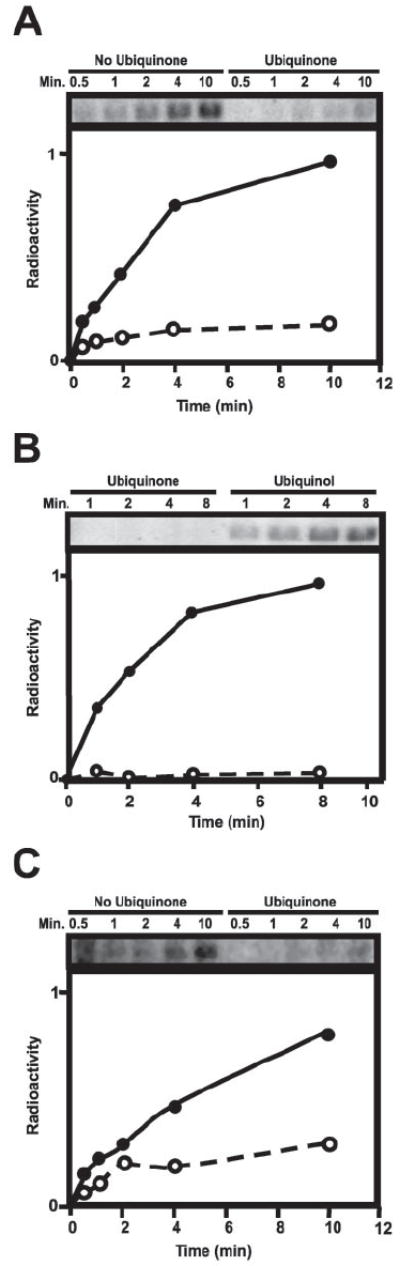
A, autophosphorylation assays of RegB in the presence and absence of ubiquinone (Q1) at a 50-fold molar excess to RegB. Ethanol was added to the RegB sample lacking ubiquinone to account for the ethanol concentration of the ubiquinone treated sample. The two reactions were incubated for 20 min at 37 °C before the addition of [γ-32P]ATP. Aliquots were removed at 0.5, 1, 2, 4, and 10 min and quenched with SDS-PAGE loading buffer before being separated by SDS-PAGE. B, autophosphorylation assays of RegB in the presence of either oxidized ubiquinone or reduced ubiquinol (Q1). RegB was incubated in the presence of a 50-fold molar excess of ubiquinone or ubiquinol at 37 °C for 20 min prior to the addition of [γ-32P]ATP. Aliquots were removed at 1, 2, 4, and 8 min and quenched with SDS-PAGE loading buffer before being separated by SDS-PAGE. C, autophosphorylation assays of C265SA mutant RegB in the presence and absence of ubiquinone (Q1) at a 50-fold molar excess to RegB. Ethanol was added to the RegB sample lacking ubiquinone to account for the ethanol concentration of the ubiquinone treated sample. The two reactions were incubated for 20 min at 37 °C before the addition of [γ-32P]ATP. Aliquots were removed at 0.5, 1, 2, 4, and 10 min and quenched with SDS-PAGE loading buffer before being separated by SDS-PAGE. All of the autophosphorylation assays were visualized and quantitated using the Typhoon phosphorimaging system (Amersham Biosciences).
Involvement of Cys265
We previously demonstrated that RegB contains a fully conserved redox active cysteine (Cys265), located in the cytoplasmic domain between the H-box site of phosphorylation and the ATP-binding cassette (6). In vitro evidence indicated that oxidation of Cys265 leads to significant inhibition of kinase activity of the truncated cytosolic form of RegB (RegBs) (6). However, mutational analysis of Cys265 in full-length RegB only partially abrogated redox control in vivo, suggesting that additional factors beyond that of Cys265 were involved in controlling RegB kinase activity (6). Thus, redox regulation of full-length RegB autophosphorylation could be a result of altering the oxidation state of a bound ubiquinone, the redox state of Cys265, or both.
To differentiate these possibilities, we isolated a full-length version of RegB that contained a cysteine to alanine mutation at amino acid 265 (C265A) and then assayed this mutant form of full-length RegB for redox-mediated changes in autophosphorylation in vitro. The results of these assays demonstrate that the addition of oxidized coenzyme Q1 to full-length C265A RegB still results in inhibition of autophosphorylation (Fig. 2C). Thus, oxidized coenzyme Q1 is not simply affecting the redox state of Cys265. This indicates that redox control of wild type full-length RegB most likely involves both an alteration of the redox state of Cys265 as well as direct inhibition of activity by oxidized ubiquinone.
Identification of the Ubiquinone-binding Site
A tritium-labeled photo affinity analog of ubiquinone, 3-azido-2-methyl-5-methoxy-[3H]6-decyl-1,4-benzoquinone (azido-Q) was used to identify a ubiquinone-binding site in RegB. This azidoubiquinone derivative and related analogs have been widely used to identify Q-binding domains present in NADH-Q oxidoreductase (17), disulfide bond protein B (18), succinate-Q oxidoreductases (19-21), cytochrome bc1 complexes (13, 22-26), and plastoquinone-binding sites in the cytochrome b6f complex from spinach chloroplasts (27). The binding of this quinone analog was performed by equilibrating purified full-length RegB with 20-fold molar excess of azido-Q for 1 h at 0 °C in the dark. The RegB-quinone analog complex was then illuminated with long wavelength UV light (365 nm) for 20 min to initiate photoaffinity cross-linking. Electrophoretically and chromatographically separated azido-Q-treated RegB demonstrates good incorporation of tritium, indicating that azido-Q undergoes efficient cross-linking to RegB (data not shown).
Kinase assays indicate that the activity of RegB is decreased >95% after photoaffinity cross-linking in the presence of azido-Q (Fig. 3A). Inactivation is not due to the inhibition of RegB by photolyzed products of azido-Q, because no inhibition of activity is observed when azido-Q is first photolyzed in the absence of RegB and then mixed with the enzyme. Inactivation is also not due to protein damage by UV radiation because illumination of the enzyme alone does not affect activity. To further establish that inactivation of RegB activity is a direct result of covalent linkage of azido-Q, we performed kinase assays on RegB-azido-Q complexes that had undergone different periods of illumination. As shown in Fig. 3A, when RegB is treated with 20-fold molar excess of azido-Q and then illuminated for different time periods, activity decreases as the time of illumination increases. Moreover, the amount of azido-Q incorporated into RegB parallels the extent of inactivation, until maximum incorporation and maximum inactivation is reached at 20 min of illumination. Although illumination for longer than 20 min causes no further decrease in activity, azido-Q uptake continues with a slower rate, indicating that further incorporation is due to nonspecific binding of azido-Q to the protein. Note that a control sample containing the same composition of RegB, buffer, ethanol, solvent etc, without azido-Q exhibits negligible (<5%) loss of kinase activity when illuminated under identical time periods. Thus, we can conclude that inactivation of RegB activity in the presence of azido-Q is a direct result of specific covalent binding of the quinone analog to full-length RegB.
FIGURE 3. Azido-Q uptake of RegB and the HPLC profile of digested RegB.
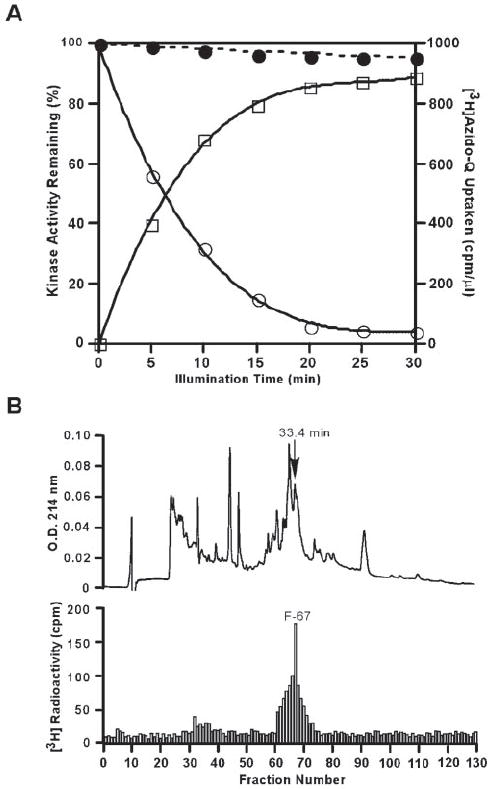
A, effect of illumination time on azido-Q uptake and inactivation of the sensor kinase RegB. Purified RegB in 20 mm Tris-Cl, pH 8.0, containing 150 mm NaCl and 30% glycerol was incubated with azido-Q in ethanol (open circles and open squares) or ethanol only (solid circles) for 1 h at 4 °C in the dark. The samples were then illuminated with long wavelength UV light for the indicated times at 0 °C. The determination of activity (open circles and solid circles) and radioactivity (open squares) were performed as described under “Experimental Procedures.” B, 3H radioactivity distribution in an HPLC chromatogram of a protease K-digest of [3H]azido-Q-labeled RegB. The labeled protein was digested, fractioned, and assayed for radioactivity as described under “Experimental Procedures.”
Peptide fragments of RegB were isolated after photoaffinity labeling with [3H]azido-Q to ascertain whether the quinone analog is binding to a specific region of RegB. For this analysis, we photo affinity-labeled RegB with [3H]azido-Q, separated unbound [3H]azido-Q from RegB-[3H]azido-Q, proteolytically digested RegB-[3H]azido-Q with protease K, and then separated the proteolytic fragments by HPLC. The HPLC elution profile in Fig. 3B shows that a major distribution of 3H radioactivity occurs at fraction 67 with a retention time of 33.4 min. This labeling result was replicated several times with independent RegB isolates. To ensure purity, the peptide was subjected to a second round of HPLC purification using the same eluting program and buffer. A single peak with the same retention time of 33.4 min was subsequently recovered from which a partial NH2-terminal amino acid sequence was found to be NH2-SLTGGLNNPF, which corresponds to amino acid residues 103–112 of RegB. Mass spectral analysis determined that the size of the Q-binding peptide was 2366.1 daltons, indicating that the Q-labeled peptide is composed of 20 amino acid residues from Ser103 to Ile122.
The location of the putative Q-binding domain in RegB (Fig. 4A) is at the junction of the second periplasmic loop formed by transmembrane helices 3 and 4 toward the middle of transmembrane helix 4. Inspection of the sequence of 20 different RegB homologs from a diverse number of α, β, and γ-proteobacterial species present in genome databases indicates that this fragment contains a universally (100%) conserved heptapeptide sequence, GGXXNPF that presumably forms a ubiquinone-binding pocket (Fig. 4B).
FIGURE 4. Topological representation and homolog pile up of the transmembrane-spanning domains of RegB.
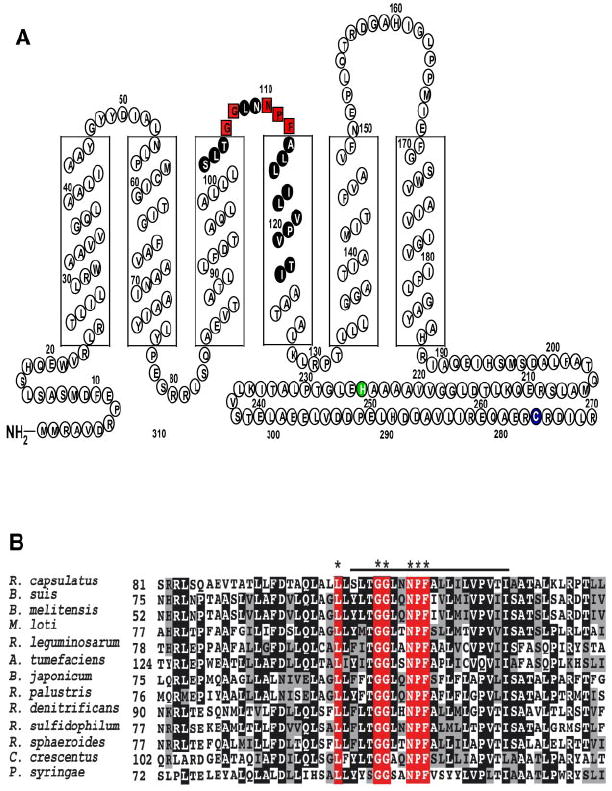
A, RegB has six transmembrane-spanning domains that anchor it to the bacterial membrane. The boxed red letters represent the 100% conserved pentapeptide ubiquinone-binding motif. The amino acids of the peptide fragment identified by HPLC and azido-Q cross-linking experiments are colored black. The site of phosphorylation is colored green, and the redox active Cys265 is colored blue. Amino acid residues past 270 were omitted for simplicity. B, RegB homologs were selected from Brucella suis, Brucella melitensis, Mesorhizobium loti, Rhizobium leguminosarum, Agrobacterium tumefaciens, Bradyrhizobium japonicum, Rhodopseudomonas palustris, Roseobacter denitrificans, Rhodovulum sulfidophilum, R. sphaeroides, R. capsulatus, Caulobacter crescentus, and Pseudomonas syringae. The asterisks denote the 100% conserved residues in the newly identified ubiquinone-binding site. The alignment was created using ClustalW and the ALN file was imported into BoxShade to color code the conserved residues in black, similar residues in gray, and nonconserved residues in white. The line above the alignment denotes the identified ubiquinone-binding peptide.
In Vivo Characterization of Ubiquinone-binding Site Mutants
We addressed the physiological significance of the azido-Q binding heptapeptide sequence GGXXNPF by constructing alanine substitution mutations of the Asn and Phe residues that convert the GGXXNPF heptapeptide sequence to GGXXAPF (mutation N110A) and GGXXNPA (mutation F112A). These mutations were chosen on the supposition that the heptapeptide forms a ubiquinone-binding pocket with Asn110, providing hydrogen bond interactions, and Phe112, providing π-π interactions with the ubiquinone polar head group. regB constructs containing these mutations were cloned into a suicide vector and then recombined into the chromosome of an R. capsulatus strain that contains a chromosomal deletion of regB. As a control, we also recombined a wild type copy of the regB gene into the same regB deletion strain. Because the RegB-RegA regulon controls expression of a large number of aerobic and anaerobic processes (4), we attempted to grow wild type and mutant recombinants under a variety of conditions. In comparison with the wild type control that grew in all tested conditions, the N110A alanine substitution mutants were only viable when grown anaerobically (photosynthetically) on agar-solidified RCV minimal medium. Under this condition, colonies of the N110A strain were significantly darker in appearance than wild type recombinants (supplemental Fig. S1), which is significant given that RegA is a potent anaerobic activator of photosynthesis gene expression when phosphorylated by RegB. Because the N110A mutation would not grow on rich medium or aerobically on minimal medium, this strain was not further characterized.
In contrast to poor viability of the N110A mutation, the F112A recombinants were viable under several growth conditions. When grown on RCV minimal medium, the F112A mutant grew well photosynthetically, producing large colonies after 3 days of growth. Under aerobic growth conditions the F112A mutant strain exhibited slightly smaller colonies. In addition, the F112A mutant exhibited elevated photosystem synthesis as evidenced by more colony pigmentation under both aerobic and photosynthetic (supplemental Fig. S1) growth conditions. When grown on nutrient-rich PY medium, the F112A mutant has reduced growth rate as well as more pigmentation than the wild type recombinant (data not shown). To accurately assess photosystem production, we performed spectral scans of membrane fractions from wild type and F112A mutant recombinant strains that were grown aerobically, semi-aerobically, and photosynthetically (anaerobic) on RCV minimal medium. The noticeably darker colony pigmentation of the F112A mutant was corroborated by spectral scans, which demonstrate that this mutation results in 94, 61, and 14% increases in photosystem production under aerobic, semi-aerobic, and photosynthetic growth conditions relative to the wild type recombinant strain, respectively (Fig. 5, A–C). The dramatic elevation in aerobic photosystem production of the F112A mutation is significant given that wild type RegB kinase activity is normally low under this growth condition. Thus, the F112A mutation appears to disrupt the ability of oxidized ubiquinone to repress RegB autophosphorylation in vivo.
FIGURE 5. Spectral scans of wild type and RegB F112A R. capsulatus cells.
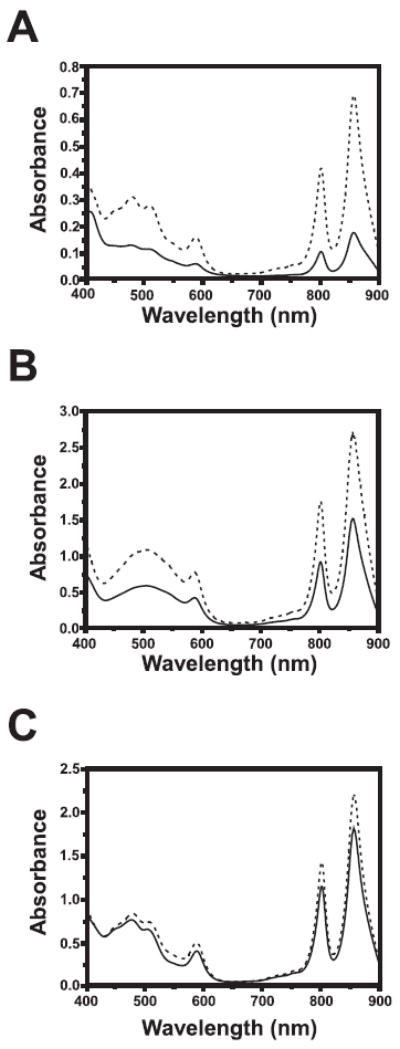
R. capsulatus strains SB1003 and RegB F-A were grown to the same cell density under aerobic, semi-aerobic, and photosynthetic conditions. The cells were then lysed, clarified and spectrally scanned from 400 to 900 nm. A, aerobically grown cells. B, semi-aerobically gown cells. C, photosynthetically grown cells. The solid lines denote SB1003, whereas the dashed lines denote the RegB F112A mutant strain.
The RegB-RegA regulon is known to directly control expression of the puf operon that encodes for the light harvesting I and reaction center proteins, as well as the puc operon that encodes the light harvesting II polypeptides. To show a direct effect on gene expression, we performed Northern blot analysis of puf and puc mRNA extracted from both the F112A mutant and the wild type control recombinant strains grown under both aerobic and anaerobic growth conditions. In agreement with the spectral data, mRNA levels of puf and puc operons were highly elevated under aerobic growth conditions in the F112A mutant, with puf expression elevated 2.5-fold and puc expression elevated 6.5-fold (Fig. 6). Interestingly, puf and puc expression were also elevated 1.6-and 1.9-fold under photosynthetic growth conditions, respectively, indicating that wild type RegB is not fully derepressed under photosynthetic growth conditions (Fig. 6). This suggests that the ubiquinone pool is not fully reduced even under the tested anaerobic growth conditions.
FIGURE 6. Northern blot analysis of puf and puc operon expression.
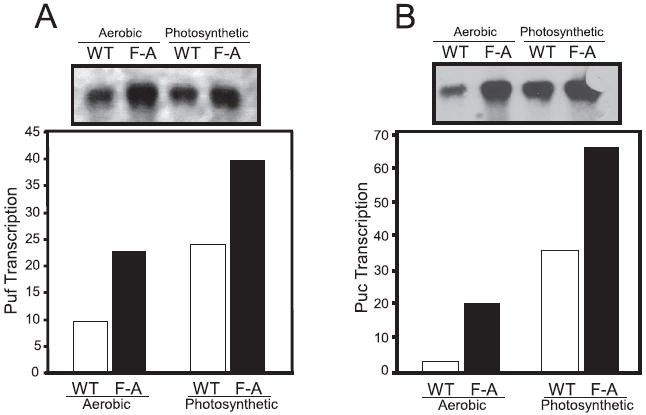
The puf (A) and puc (B) Northern blot autoradiograph of RNA isolated from aerobic and anaerobic grown wild type (WT) and the F112A mutant (FA). The RNA was probed using a digoxigenin-labeled probe and visualized using x-ray film. The data were then plotted as a bar graph. RNA transcript for puf and puc were normalized using 16 S ribosomal RNA levels as a control. Northern blots were carried out three times independent times with three different RNA preparations. The data were consistent between all three replicas.
DISCUSSION
The RegB/RegA signal transduction system regulates the expression of multiple cellular processes such as respiration, photosynthesis, hydrogen utilization, carbon fixation, and nitrogen fixation (4). Many of these processes use redox chemistry to catalyze specific reactions and thus affect the energy state of the cell. The common link for all RegB controlled processes is the membrane-bound electron/proton carrier, ubiquinone. The ubiquinone derivative found in Rhodobacter species is ubiquinone-10, which is produced at extremely high levels with the pool localized to the cytoplasmic/intracytoplasmic membrane, which is also the site of RegB localization (9). Furthermore, the redox balance of the ubiquinone pool is known to dramatically shift in photosynthetic bacteria from a predominantly oxidized state under aerobic conditions to being predominantly reduced under photosynthetic (anaerobic) conditions (9). The abundance and dramatic changes in the oxidation/reduction state of the ubiquinone pool makes it an extremely attractive sensory molecule for RegB to monitor the energy status of the cell and subsequently regulate synthesis of the photosystem, components of the respiratory chain and energy utilizing fixation pathways.
This study reveals that the conserved motif GGXXNPF, located between transmembrane 3 and 4 of RegB (Fig. 4), constitutes a ubiquinone-binding site. A previously reported Pho fusion study on RegB places the ubiquinone-binding domain at the junction of the second periplasmic loop and the beginning of the membrane interface of TM4 (28). This placement is in close agreement with TopPred, which predicts that the ubiquinone-binding domain is housed within the bacterial membrane at the beginning of TM4. The membrane location of the predicted RegB ubiquinone-binding site is also consistent with that of other ubiquinone-binding domains that are typically located within or very near the membrane. Our in vitro photoaffinity cross-linking results with [3H]azido-Q, and in vivo mutational analysis of this region are also supported by Oh et al. (15), who used multiple (five consecutive) Ala substitution analyses to identify a region extending from TM3 to TM4 as being potentially involved in the redox responsive behavior of RegB in R. sphaeroides (15). Our study confirms that this region is involved in redox sensing and has further assigned this region as a highly conserved ubiquinone-binding site.
Models of Quinone Inactivation of RegB
One can envision several ways that ubiquinone may affect the kinase activity of RegB. Binding of this cofactor could be promoting an allosteric modification of the tertiary structure of RegB ultimately inhibiting RegB autophosphorylation. Alternatively, ubiquinone could be promoting physical oxidation of one or more residues in RegB. Neither mechanism is mutually exclusive, leaving open the possibility that ubiquinone could regulate kinase activity via both allosteric and redox mechanisms. In regards to an allosteric mechanism, one can envision that the aromatic side group of Phe112 could stack with the para-hydroxybenzoate ring of ubiquinone via stabilizing π-π interactions between these rings. Asn110 could also act as a hydrogen bond donor to the oxidized ubiquinone, stabilizing ubiquinone-protein interactions. Indeed, a hydrogen bond interaction of Asn110 to ubiquinone could be a switch that leads to allosteric modification of RegB structure. Under anaerobic conditions, when the ubiquinone pool is shifted toward the doubly protonated ubiquinol state, hydrogen bond interactions between Asn110 and the ubiquinone could be lost. This could lead to rearrangement of hydrogen bonds in RegB, resulting in a change in structure that favors autophosphorylation.
The results of our previous study on a truncated version of RegB indicated that alterations of the redox state of Cys265 also affected autophosphorylation in vitro (6). Interestingly, mutational analysis of Cys265, showed attenuated, but not absent, redox control of RegB activity in vivo, suggesting that there are additional redox inputs controlling RegB activity (6). The results of this study indicate that the redox state of the ubiquinone pool constitutes an additional mechanism of controlling activity of RegB in response to changes in cellular redox.
The predicted membrane topology of RegB places the GGXXNPF quinone-binding domain in a short periplasmic loop near TM4, which is contrasted by Cys265 that is located in the cytosolic domain between the site of phosphorylation and the kinase domain (6). This would indicate that ubiquinone may not be directly affecting the redox state of Cys265 and that Cys265 may instead be a separate redox input to control the activity of RegB perhaps by direct oxidation of Cys265 via oxygen.
Even though continued studies will be needed to reveal the underlying mechanism by which ubiquinone and Cys265 together inhibit RegB autophosphorylation, the relation between ubiquinone and redox active cysteine residues is not unprecedented. In fact, two such examples are well described in the scientific literature. The disulfide bond-generating protein, DsbB has the ability to interact with ubiquinone and use the oxidative power of that ubiquinone to induce a disulfide bond in adjacent cysteine residues (29). Recently, the sensor kinase, ArcB from E. coli was also shown to use the oxidation power of ubiquinone to generate an intermolecular disulfide bond that diminished kinase activity (7). These two studies, in concert with the data presented in this manuscript, provide a compelling argument that the interaction between cysteine residues and ubiquinone may be a common theme for redox-sensing and -responding mechanisms in biology.
In addition to ArcB, the sensor kinases BvgS and EvgS from Bordetella pertussis and E. coli, respectively also exhibit decreased autophosphorylation in the presence of ubiquinone in vitro (30). These two kinases have a domain structure that is very similar to that of ArcB with two transmembrane-spanning helices followed by a cytoplasmic Per Arnt Sim (PAS) domain and then a kinase domain. In ArcB, the PAS domain contains two cysteine residues that form intermolecular disulfide bonds in response to the addition of ubiquinone (7). It has been inferred that the PAS domain is responsible for ubiquinone binding; however, direct evidence for binding of ubiquinone to the ArcB PAS domain has not been provided. In addition, mutations in putative ubiquinone-binding residues of the BvgS PAS domain had little effect on ubiquinone inhibition of kinase activity (30). Interestingly, all of the published in vitro analysis on ArcB, BvgS, and EvgS has been performed using truncated forms of these protein that lack their transmembrane-spanning domains (7, 8, 30). Key to the success of this study was the purification of full-length RegB that contains six transmembrane-spanning helices. The utilization of ubiquinone as a cofactor to regulate RegB activity is not surprising given that an interaction of RegB with the ubiquinone pool would clearly provide a mechanism by which this sensor kinase could control expression of downstream genes that ultimately affect the redox state of the cell. As is the case with other sensor kinases, it remains to be determined how binding of this cofactor ultimately leads to inhibition of kinase activity.
Supplementary Material
Acknowledgments
We thank Danielle Swem for technical support and for helpful discussions.
Footnotes
This work was supported by National Institutes of Health Grants GM30721 (to C.-A. Y.) and GM53940 (to C. E. B.).
The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1.
Z. Jiang and C. Bauer, unpublished data.
The abbreviations used are: azido-Q, azidoquinone; HPLC, high pressure liquid chromatography; TM, transmembrane domain; PAS, Per Arnt Sim.
Supplemental Material can be found at: http://www.jbc.org/cgi/content/full/M50968/DC1
References
- 1.Madigan MT, Gest H. Arch Microbiol. 1978;117:119–122. doi: 10.1007/BF00402298. [DOI] [PubMed] [Google Scholar]
- 2.Dubbs JM, Bird TH, Bauer CE, Tabita FR. J Biol Chem. 2000;275:19224–19230. doi: 10.1074/jbc.M002125200. [DOI] [PubMed] [Google Scholar]
- 3.Elsen S, Dischert W, Colbeau A, Bauer CE. J Bacteriol. 2000;182:2831–2837. doi: 10.1128/jb.182.10.2831-2837.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Elsen S, Swem LR, Swem DL, Bauer CE. Microbiol Mol Biol Rev. 2004;68:263–279. doi: 10.1128/MMBR.68.2.263-279.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oh JI, Ko IJ, Kaplan S. Biochemistry. 2004;43:7915–7923. doi: 10.1021/bi0496440. [DOI] [PubMed] [Google Scholar]
- 6.Swem LR, Kraft BJ, Swem DL, Setterdahl AT, Masuda S, Knaff DB, Zaleski JM, Bauer CE. EMBO J. 2003;22:4699–4708. doi: 10.1093/emboj/cdg461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Malpica R, Franco B, Rodriguez C, Kwon O, Georgellis D. Proc Natl Acad Sci U S A. 2004;101:13318–13323. doi: 10.1073/pnas.0403064101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Georgellis D, Kwon O, Lin EC. Science. 2001;292:2314–2316. doi: 10.1126/science.1059361. [DOI] [PubMed] [Google Scholar]
- 9.Parson W. In: The Photosynthetic Bacteria. Clayton RK, Sistrom WR, editors. Plenum Press; New York: 1978. pp. 455–469. [Google Scholar]
- 10.Bolton JR. In: The Photosynthetic Bacteria. Clayton RK, Sistrom WR, editors. Plenum Press; New York: 1978. pp. 419–429. [Google Scholar]
- 11.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Gene (Amst) 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 12.Bird TH, Du S, Bauer CE. J Biol Chem. 1999;274:16343–16348. doi: 10.1074/jbc.274.23.16343. [DOI] [PubMed] [Google Scholar]
- 13.Yu L, Yang FD, Yu CA. J Biol Chem. 1985;260:963–973. [PubMed] [Google Scholar]
- 14.Buggy JJ, Sganga MW, Bauer CE. J Bacteriol. 1994;176:6936–6943. doi: 10.1128/jb.176.22.6936-6943.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oh JI, Ko IJ, Kaplan S. J Bacteriol. 2001;183:6807–6814. doi: 10.1128/JB.183.23.6807-6814.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Potter CA, Ward A, Laguri C, Williamson MP, Henderson PJ, Phillips-Jones MK. J Mol Biol. 2002;320:201–213. doi: 10.1016/S0022-2836(02)00424-2. [DOI] [PubMed] [Google Scholar]
- 17.Gong X, Xie T, Yu L, Hesterberg M, Scheide D, Friedrich T, Yu CA. J Biol Chem. 2003;278:25731–25737. doi: 10.1074/jbc.M302361200. [DOI] [PubMed] [Google Scholar]
- 18.Xie T, Yu L, Bader MW, Bardwell JC, Yu CA. J Biol Chem. 2002;277:1649–1652. doi: 10.1074/jbc.M108697200. [DOI] [PubMed] [Google Scholar]
- 19.Lee GY, He DY, Yu L, Yu CA. J Biol Chem. 1995;270:6193–6198. doi: 10.1074/jbc.270.11.6193. [DOI] [PubMed] [Google Scholar]
- 20.Shenoy SK, Yu L, Yu CA. J Biol Chem. 1997;272:17867–17872. doi: 10.1074/jbc.272.28.17867. [DOI] [PubMed] [Google Scholar]
- 21.Yang X, Yu L, He D, Yu CA. J Biol Chem. 1998;273:31916–31923. doi: 10.1074/jbc.273.48.31916. [DOI] [PubMed] [Google Scholar]
- 22.Yu L, Yang FD, Yu CA, Tsai AL, Palmer G. Biochim Biophys Acta. 1986;848:305–311. doi: 10.1016/0005-2728(86)90204-5. [DOI] [PubMed] [Google Scholar]
- 23.Yu L, Xu JX, Haley PE, Yu CA. J Biol Chem. 1987;262:1137–1143. [PubMed] [Google Scholar]
- 24.Yang FD, Yu L, Yu CA, Lorence RM, Gennis RB. J Biol Chem. 1986;261:14987–14990. [PubMed] [Google Scholar]
- 25.Welter R, Gu LQ, Yu L, Yu CA, Rumbley J, Gennis RB. J Biol Chem. 1994;269:28834–28838. [PubMed] [Google Scholar]
- 26.He DY, Yu L, Yu CA. J Biol Chem. 1994;269:2292–2298. [PubMed] [Google Scholar]
- 27.Doyle MP, Li LB, Yu L, Yu CA. J Biol Chem. 1989;264:1387–1392. [PubMed] [Google Scholar]
- 28.Ouchane S, Kaplan S. J Biol Chem. 1999;274:17290–17296. doi: 10.1074/jbc.274.24.17290. [DOI] [PubMed] [Google Scholar]
- 29.Bader MW, Xie T, Yu CA, Bardwell JC. J Biol Chem. 2000;275:26082–26088. doi: 10.1074/jbc.M003850200. [DOI] [PubMed] [Google Scholar]
- 30.Bock A, Gross R. Eur J Biochem. 2002;269:3479–3484. doi: 10.1046/j.1432-1033.2002.03029.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.