

Abstract
Characterization of the conformational properties of unfolded proteins is essential for understanding the mechanisms of protein folding and misfolding. This information is also fundamental to determining the relationship between flexibility and function in the highly diverse families of intrinsically disordered proteins. Here we present a self-consistent model of conformational sampling of chemically denatured proteins in agreement with experimental data reporting on long-range distance distributions in unfolded proteins using small-angle x-ray scattering and nuclear magnetic resonance pulse-field gradient-based measurements. We find that standard statistical coil models, selected from folded protein databases with secondary structural elements removed, need to be refined to correct backbone dihedral angle sampling of denatured proteins, although they appear to be appropriate for intrinsically disordered proteins. For denatured proteins, pervasive increases in the sampling of more-extended regions of Ramachandran space {50°< ψ < 180°} throughout the peptide chain are found to be consistent with all experimental data. These observations are in agreement with previous conclusions derived from short-range nuclear magnetic resonance data from residual dipolar couplings, leading the way to a self-consistent description of denatured chains that is in agreement with short- and long-range data measured using both spectroscopic and scattering techniques.
Introduction
Despite the volume of structural information describing stable protein structures to high atomic resolution, the unfolded state occupies a conformational landscape that remains comparatively poorly understood. Development of an appropriate description of the conformational sampling of denatured proteins is essential for the study of protein folding, whereas the steadily increasing recognition of the biomedical relevance of natively unfolded proteins, for example in cancer-related signaling and neurodegenerative diseases (1–4), reinforce the need for relevant models of disordered molecular ensembles. Many intrinsically disordered proteins (IDPs) only adopt a stable conformation upon binding physiological partners (5–7), underlining the importance of understanding protein folding events occurring in the unfolded state.
Two models have been identified to explain the effects of chemical denaturants on folded proteins. The first invokes the different hydrogen-bonding properties of the denaturant relative to water in the vicinity of hydrophobic groups, such that it acts as a better solvent for hydrophobic groups that are normally buried in water-solvated proteins (8). An alternative model proposes that the denaturant binds directly to the protein backbone, presumably via hydrogen-bonding interaction with the amide groups, thereby destabilizing hydrogen-bonding networks that provide the origin of the stability of the native fold relative to the unfolded state (9). Both models stabilize the unfolded state in the presence of high concentrations of denaturant. The correct description of conformational sampling in the denatured state will surely provide crucial information on the molecular origins of chemical denaturation, thereby contributing to our understanding of protein folding and stability.
It has become clear over the last decade that rather than conforming to a simple random coil description, the conformational behavior of the unfolded protein can be extremely varied. The key question is not whether a protein is unfolded, rather how it is unfolded. To clarify these questions, conformational sampling in denatured proteins has been studied in detail using small-angle scattering (10,11), nuclear magnetic resonance (NMR) spectroscopy (12), and fluorescence correlation spectroscopy (13), as well as ensemble molecular simulation (14–19). Experimental and theoretical studies suggest that apparently diverse descriptions based on diverse experimental techniques probably derive from the sensitivity of the different approaches to local and long-range structure, underlining the strong complementarity of NMR and small-angle scattering techniques for the development of a self-consistent molecular description of the denatured state.
A popular approach used to analyze the conformational properties of disordered proteins is the use of explicit molecular descriptions of unfolded ensembles (17–22). To this end, explicit atomic-resolution ensemble models have been proposed on the basis of statistical coil sampling, extracted from backbone dihedral angles present in coil regions of protein databases. Amino-acid-specific behavior from these conformational distributions is then assumed to represent a Boltzmann distribution of dihedral angles in a statistical description of the unfolded polymer, such that an ensemble of conformers constructed on the basis of random sampling of these distributions can be considered to describe a representative ensemble. For these descriptions to yield useful information, they should be demonstrated to be accurate and relevant representations of the unfolded state. Ab initio and molecular dynamics simulations have indeed suggested that such approximations can be valid (23). In most cases, long-range interactions are reduced to a minimum, steric repulsion or excluded volume simplification, essentially to avoid chain overlap, with electrostatic and amino-acid-specific solvent effects ignored in the interest of efficiency. Nearest-neighbor effects can also be introduced to refine conformational behavior as a function of the nature of the neighboring amino acids.
One such approach, termed Flexible Meccano (FM; (24)), constructs conformational ensembles in this way. The algorithm uses a rudimentary spherical side-chain volume exclusion model, 20-amino-acid-specific conformational wells derived from coil regions of 500 high-resolution crystal structures, and additional conformational basins representing specific dihedral angle distributions such as pre-prolines, or prolines preceding prolines. The model has been validated against numerous types of experimental data: NMR residual dipolar couplings (RDCs) measured between covalently bound spins (24), reporting on orientational properties of internuclear vectors; NMR RDCs measured between protons (25), reporting on both distance and angular internuclear distribution functions; three-bond scalar couplings, reporting directly on backbone dihedral angles distributions (25); and small-angle scattering measurements, reporting on overall pairwise distribution functions and molecular dimensions (24). In addition, state-of-the-art molecular simulation technology, specifically the accelerated molecular dynamics approach (26,27) that allows for statistical sampling of the broad conformational subspace available to unfolded proteins, and critically, the reconstruction of Boltzmann-like sampling on an amino-acid-specific basis, reveals a very close reproduction of the conformational space described by the statistical coil database (28,29). These experimental and theoretical considerations allow us to propose that, although necessarily approximate, the FM approach provides a reasonable tool to allow the refinement of our understanding of the conformational properties of unfolded proteins in general and of denatured proteins in particular.
Partially aligned NMR spectroscopy has been shown to be a particularly powerful tool for studying molecular structure in both folded (30–32) and unfolded states (33–37). In recent years, we have used this parameter as a probe to derive an amino-acid-specific self-excluding model of the unfolded state. Explicit molecular ensemble analysis, using FM, successfully reproduced both spectroscopic and scattering data from protein X from Sendai virus, a protein that is natively partially unfolded (24), and RDCs from both apomyoglobin (34) and staphylococcal nuclease Δ131Δ (12) in high concentration of denaturant. This was subsequently used to identify conformational properties of several important IDPs involved in neurodegenerative diseases, the proteins τ (28) and α-synuclein (38), the tumor-suppressor p53 (29), and the unfolded C-terminal of the nucleoprotein of Sendai virus (39,40). Using the same conformational averaging approach, we have also recently developed further tools for refining the interpretation of small-angle x-ray scattering (SAXS) data from unfolded proteins in terms of average molecular dimensions (41) and the average surface accessibility of amino acids in unfolded proteins (42).
In a more recent study, up to seven RDCs per peptide unit could be determined for urea-unfolded ubiquitin, both between covalently bound and 1H-1H RDCs (25), allowing a detailed description of the angular and distance fluctuations of the unfolded state. The aim in this initial study was the calculation of complete conformational distributions of the unfolded or partially folded polypeptide chain that is in agreement with the available experimental data, resulting in the clear identification of enhanced sampling of two regions of Ramachandran space, in particular β-extended and polyproline II conformations. However, RDCs only report indirectly on the overall dimensions of the protein, thus a realistic description of the unfolded state should also reproduce experimental data reporting more directly on the overall dimensions of the protein. In this study, we demonstrate that this refined coil description provides a consistent description of the conformational behavior of chemically denatured proteins that is compatible with measurement of NMR RDCs, pulsed-field gradient (PFG) hydrodynamic measurements, and SAXS data.
Methods
The conformations used in this study to calculate the properties of the denatured state were built with Flexible Meccano (FM). To define the amino-acid-specific Ramachandran space, FM uses ϕ/ψ dihedral angles collected from 500 high-resolution (<1.8 Å) x-ray structures (43) where residues belonging to secondary structure elements were removed; this is the so-called standard (coil) library. Side chains for these conformations were added with the program SCCOMP (44).
SAXS properties of the different polypeptide chains were obtained from the averaging the individual scattering curves of 2000 atomic resolution conformations of each of the 23 proteins defined by their amino-acid sequence. The theoretical scattering profiles (s < 0.1 Å−1) for each conformation were calculated with the program CRYSOL (45), and averaged to yield a final SAXS curve for each protein. Random Gaussian noise of an amplitude of 2% was added to the curves to produce more experimental-like data. Rg values were derived by analyzing the initial part of the curve (sRg < 1.3) using Guinier's approach with the program PRIMUS (46).
Hydrodynamic properties of the denatured state were computed for eight proteins whose Rh values had been measured by PFG (47,48). For each of the eight proteins, an ensemble with 500 conformations was built as described above, and for each conformation the hydrodynamic radius was computed with a modified version of the program HYDROPRO (49) using an atomic element radius of 3.3 Å to define the volume of the conformation (50). The bead modeling strategy adopted by HYDROPRO has been proven to be efficient to calculate hydrodynamic properties for both rigid (51,52) and flexible (53) molecules. The averaged Rh was obtained as
| (1) |
where N is the number of conformations of the ensemble (500) and Rh,i is the hydrodynamic radius of the conformation i.
The same calculations of SAXS and hydrodynamic properties were repeated for six additional ensembles with an enhanced population of extended conformations {50° < ψ < 180°} in the Ramachandran space. Databases with X = 1.5, 2.0, 2.5, 3.0, 3.5, and 4.0 times' higher populations for this range of ψ were created. Explicitly the same conformations with {50° < ψ < 180°} were repeated X times in the database to increase their possibility of being selected. Final databases have 68.3%, 74.2%, 78.2%, 81.2%, 83.4%, and 85.2% of the conformations in the extended region, respectively.
Results and Discussion
The overall dimensions of chemically denatured proteins, expressed in terms of an effective ensemble radius of gyration Rg, has been predicted and then shown to display a simple power-law dependence on chain length N that is in agreement with random-coil polymer behavior (54,55),
| (2) |
where R0 is a constant that depends on the persistence length and ν is the exponential scaling factor. In a recent article, Kohn et al. measured and compiled the Rg obtained by SAXS for 28 proteins under different denaturing conditions, extracting a value of ν = 0.58 ± 0.11 (11). We have taken a set of 23 proteins from this list covering a broad range of chain lengths, from eight to 549 amino acids (see Table S1 in the Supporting Material). For each of the 23 amino-acid sequences, an ensemble of 2000 conformations was created using FM, and SAXS profiles were calculated from these ensembles (see Methods). The correlation between the experimental and simulated Rg values is shown in Fig. 1 a. A systematic deviation is observed, indicating that simulated ensembles underestimate the overall size of the denatured state.
Figure 1.
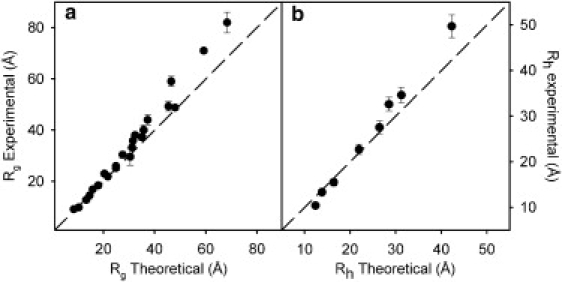
Correlation between experimental and theoretical values of (a) radius of gyration Rg, and (b) hydrodynamic radius Rh for the ensembles corresponding to the standard database. Dashed line represents slope equal to 1.0. Experimental values of Rg were obtained from Kohn et al. (11), and Rh values were obtained from Wilkins et al. (47) and Pan et al. (48).
Hydrodynamic measurements such as analytical ultracentrifugation, sedimentation coefficients, or translational diffusion coefficients have been used to obtain the hydrodynamic radius, Rh, for unstructured proteins (56,57). We have calculated the effective ensemble averaged hydrodynamic characteristics of eight proteins for which experimental data are available. The correlation between experimental and simulated averaged values is shown in Fig. 1 b. A systematic deviation on the correlation is observed when increasing the size of the polypeptide chain. This observation is coincident with the SAXS data and indicates an underestimation of the global structural parameters of the disordered chain under denaturing conditions when using the coil library to describe their conformational sampling.
The recent NMR study of urea-unfolded ubiquitin identified the need for an enhanced population of extended conformations, defined as a higher propensity to populate the region of Ramachandran space defined by {50° < ψ < 180°} (25). If true, this enhanced extendedness should be reflected in the overall dimensions of denatured proteins and therefore should be detectable by SAXS and hydrodynamic measurements. The overall population of extended conformations was systematically increased for each amino-acid-specific population in the database, by creating databases with X = 1.5, 2.0, 2.5, 3.0, 3.5, and 4.0 times higher populations for this range (see Methods). Taking into account that the coil database has 59.0% of conformations that are in the extended region, the new databases have 68.3%, 74.2%, 78.2%, 81.2%, 83.4%, and 85.2% of the conformations in the extended region, respectively. For each of the six new databases new ensembles of the same size for all proteins were computed and the Rg and Rh values were derived as explained for the standard (coil) database. The capacity of the ensembles to describe experimental Rg and Rh values was monitored via a reduced χ2 value computed as
| (3) |
where Rx,i is either Rg or Rh for protein i, NP is the total number of proteins for the SAXS (23) and for the PFG data (8). The resulting χ2 values for each database are shown in Fig. 2. Interestingly, for both experimentally measured parameters, Rg and Rh, the minimum value of the χ2 function is obtained for the 2.0 database. Rg shows more sensitivity to the degree of extendedness than Rh that has a very flat χ2 profile in the 68–78% range (1.5–2.5 databases). The presence of larger proteins in the dataset, and the different averaging processes governing the measured parameters, squared distance distribution functions compared to reciprocal, are possible reasons of this higher sensitivity found for Rg. The correlations between the experimental and simulated Rg and Rh values using the optimal database are displayed in Fig. 2, c and d, where a good agreement is observed. In this optimal database, the extended conformations represent 74.2% of the conformers—a significant enhancement when compared to the coil library, 59%. At this degree of extendedness, no systematic deviations are observed in the correlation plots, and both small peptides and large proteins are well described when using this database. Importantly, this optimal value of extendedness found by fitting overall parameters was also found when fitting a large dataset of residue-specific RDCs measured for urea-denatured ubiquitin (Fig. 3). It is also important to note that using this more extended conformational sampling gave very poor agreement for the RDCs (1DHN and 2DCHN) measured in the intrinsically disordered protein X (25). Conversely, the standard coil database provides excellent agreement of both RDCs measured for the same protein. These results strongly suggest the existence of distinct conformational sampling in intrinsically and chemically denatured proteins that probe more extended conformations.
Figure 2.
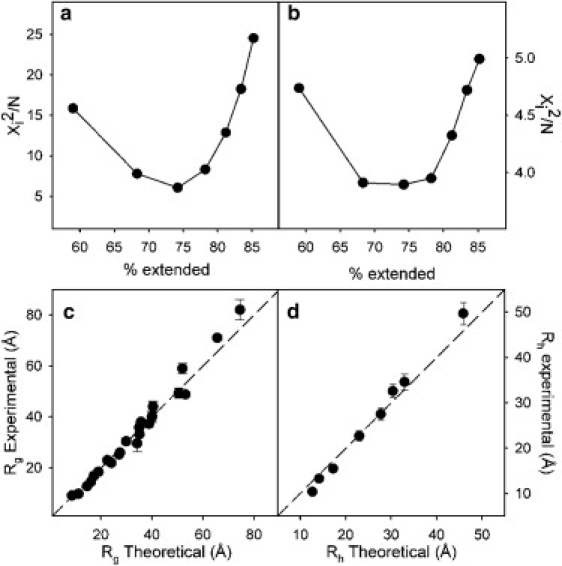
Optimal percentage of extended conformations. Agreement of the overall parameters (a) Rg and (b) Rh upon increasing the percentage of extended conformations in the database. Correlation between the experimental and theoretical (c) Rg and (d) Rh at the optimal database found, X = 2.0, which corresponds to a 74.2% of extended conformations. Dashed line again represents slope equal to 1.0.
Figure 3.
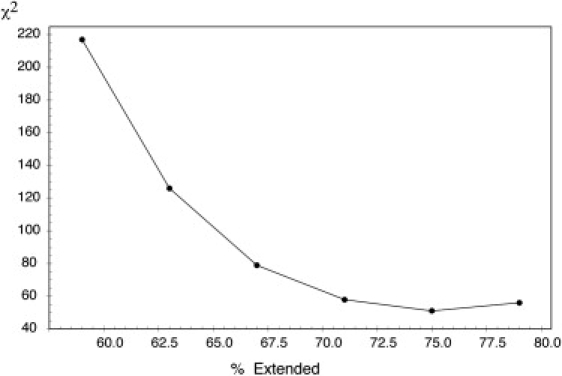
Reproduction of experimental residual dipolar couplings measured from urea unfolded ubiquitin at pH2 aligned in polyacrylamide gel using databases sampling different levels of extension. The χ2 is measured over seven types of coupling: 1DNH, 1DCαHα, 1DCαC′, DHNHα (i,i-1), DHNHN (i,i+1), DHNHN(i,i+2), and DHNHα. Data taken from Meier et al. (25).
It is interesting to compare the level of extension with similar databases extracted using comparable approaches. Removing sheet and helical regions from a compilation of high-resolution structures resulted in the development of a database with 55% of conformers in the extended region (58), while similar results were found by Griffiths-Jones et al. (54% in this region excluding glycine) (59), and from two similarly constructed earlier, smaller coil databases (60,61). Compared to 59% in our database, these numbers are therefore slightly less extended, indicating that the current findings—that more extended conformers are necessary for chemically denatured proteins—are generally relevant for coil database descriptions.
Our results clearly suggest that parameters describing Flory's relationship (Eq. 2) for chemically denatured proteins are different to these describing IDPs. A parameterization of Flory's equation for IDPs was obtained from the simulated Rg and Rh values using the coil database with the length of the biopolymer chains (see Fig. 4). From the fitting of the data to the logarithmic form of Eq. 2, ν-values of 0.522 ± 0.01 and 0.449 ± 0.01 for the Rg and Rh parameters were obtained, respectively, and the R0 values derived were 2.54 ± 0.01 and 3.53 ± 0.01 for the Rg and Rh, respectively. The ν-values obtained from the Rg and Rh data are notably smaller than the 0.6 and 0.588 values predicted for a random coil polymer with excluded volume (62), and experimentally calibrated values that are also close to these numbers (10,11,51).
Figure 4.
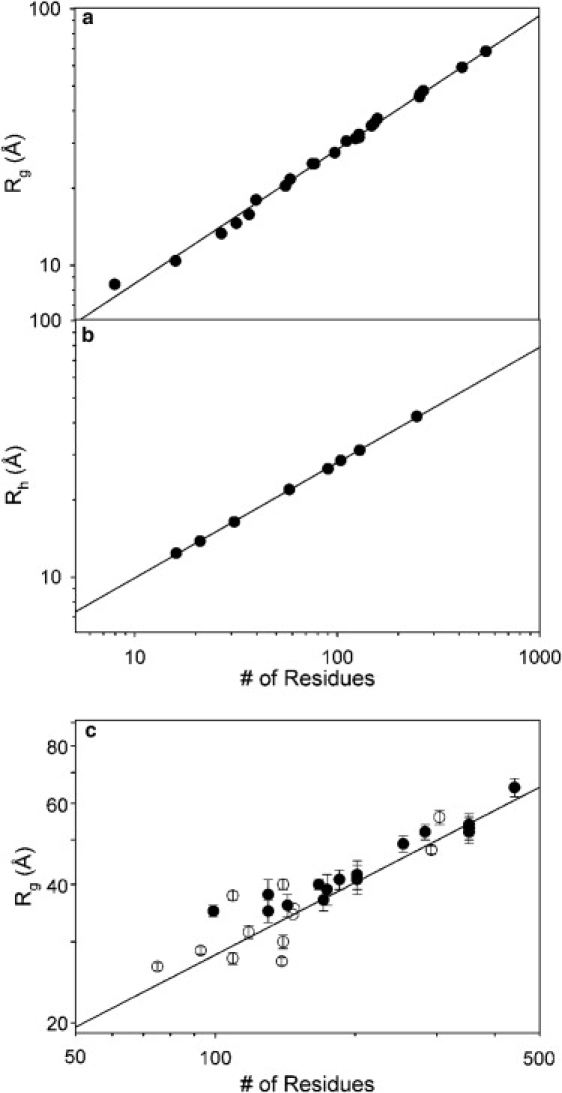
Relationship of the Rg (a) and Rh (b) values calculated using the standard database with the length of the protein chain N. From this correlation, the parameters of Flory's relationship (Eq. 1) were derived for both observables. From Rg, ν = 0.522 ± 0.01 and R0 = 2.54 ± 0.01; and from Rh, ν = 0.449 ± 0.01 and R0 = 3.53 ± 0.01. (c) Rg values compiled for protein τ (solid circles) and other IDPs (open circles), compared with the threshold values according to the calibration found in panel a (solid line). See the Supporting Material for the list of IDPs used in this plot.
The Rg values measured for different constructs of protein τ (63) and other IDPs have been collected from literature (see Table S2), and plotted as a function of the protein length in Fig. 4 c. The points are scattered around the line representing the previously described parameterization for IDPs indicating a broad agreement with the general law. Departures from the parameterized line are expected for IDPs, as they can often observe structuring phenomena that will modify the Rg values expected for a nonstructured chain of N residues. Rg values measured for different τ-protein constructs (63), solid circles in Fig. 4 c, appear above the line indicating that the conformational bias increases the overall stiffness of the protein. This general signature is more evident for K18 and K19 τ constructs that encompass the so-called repeat domain in which extended and turn regions have been identified in different NMR studies (28,64). Conversely, N-tail of measles virus nucleoprotein, a 139-residue-long IDP, appears as a more compact ensemble than the calibrated threshold (65). The SAXS data for this protein presents a Kratky plot with a clear bump at s = 0.08 Å−1 that indicates a certain degree of compactness in a concrete region of the chain. A bioinformatics analysis predicts a 16-residue-long fragment to have a strong tendency to form an α-helix that is probably the origin of this partial compactation (66).
Conclusions
In conclusion, by comparing explicit molecular simulation with extensive experimental data reporting on the overall dimensions of highly disordered chains, we are able to refine the coil database to a level that agrees simultaneously with different and highly complementary biophysical measurements of local and long-range structure. We find that Ramachandran sampling, described using popular definitions of the statistical coil model that simply remove secondary structural elements from high-resolution crystallographic structures, is insufficient to describe protein conformational sampling under strongly denaturing conditions. On the basis of small-angle scattering and NMR data, we define a self-consistent model that enhances access to β-extended and polyproline II regions of Ramachandran space within native amino-acid-specific conformational wells. It is interesting to speculate that this model agrees qualitatively with a mechanism of denaturation that implicates urea binding to the polypeptide backbone (9), causing steric hindrance that would favor more extended local conformations, although it certainly does not disprove the importance of the side-chain solvation model (8).
Supporting Material
One table is available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(09)01436-2.
Supporting Material
Acknowledgments
Authors acknowledge Prof. José García de la Torre (University of Murcia) for providing a modified version of HYDROPRO.
Financial support from the Spanish Ministry of Education-FEDER (grant No. BIO2007-63458) is gratefully acknowledged. P.B. holds a Ramón y Cajal contract that is partially financed by the Spanish Ministry of Education and the by funds provided to the Institute for Research in Biomedicine by the Generalitat de Catalunya. M.B. acknowledges support from grant No. ANR PCV 2008 PCV07-194985.
Contributor Information
Pau Bernadó, Email: pau.bernado@irbbarcelona.org.
Martin Blackledge, Email: martin.blackledge@ibs.fr.
References
- 1.Tompa P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002;27:527–533. doi: 10.1016/s0968-0004(02)02169-2. [DOI] [PubMed] [Google Scholar]
- 2.Uversky V.N. Natively unfolded proteins: a point where biology waits for physics. Protein Sci. 2002;11:739–756. doi: 10.1110/ps.4210102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fink A.L. Natively unfolded proteins. Curr. Opin. Struct. Biol. 2005;15:35–41. doi: 10.1016/j.sbi.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 4.Dyson H.J., Wright P.E. Intrinsically unstructured proteins and their functions. Chem. Rev. 2004;104:3607–3622. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 5.Fuxreiter M., Simon I., Friedrich P., Tompa P. Preformed structural elements feature in partner recognition by intrinsically unstructured proteins. J. Mol. Biol. 2004;338:1015–1026. doi: 10.1016/j.jmb.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 6.Vacic V., Oldfield C.J., Mohan A., Radivojac P., Cortese M.S. Characterization of molecular recognition features, MoRFs, and their binding partners. J. Protein Res. 2007;6:2351–2366. doi: 10.1021/pr0701411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wright P.E., Dyson H.J. Linking folding and binding for unstructured proteins. Curr. Opin. Struct. Biol. 2009;19:1–8. doi: 10.1016/j.sbi.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tanford C. Isothermal unfolding of globular proteins in aqueous urea solutions. J. Am. Chem. Soc. 1964;86:2050–2059. [Google Scholar]
- 9.Schellman J.A. Factors affecting the stability of hydrogen-bonded polypeptide structures in solution. J. Phys. Chem. 1958;62:1485–1494. [Google Scholar]
- 10.Millett I.S., Doniach S., Plaxco K.W. Toward a taxonomy of the denatured state: small angle scattering studies of unfolded proteins. Adv. Protein Chem. 2002;62:241–262. doi: 10.1016/s0065-3233(02)62009-1. [DOI] [PubMed] [Google Scholar]
- 11.Kohn J.E., Millett I.S., Jacob J., Zagrovic B., Dillon T.M. Random-coil behavior and the dimensions of chemically unfolded proteins. Proc. Natl. Acad. Sci. USA. 2004;101:12491–12496. doi: 10.1073/pnas.0403643101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shortle D., Ackerman M.S. Persistence of native-like topology in a denatured protein in 8 M urea. Science. 2001;293:487–489. doi: 10.1126/science.1060438. [DOI] [PubMed] [Google Scholar]
- 13.Lipman E.A., Schuler B., Bakajin O., Eaton W.A. Single-molecule measurement of protein folding kinetics. Science. 2003;301:1233–1235. doi: 10.1126/science.1085399. [DOI] [PubMed] [Google Scholar]
- 14.Smith L.J., Fiebig K.M., Schwalbe H., Dobson C.M. The concept of the random coil. Residual structure in peptides and denatured proteins. Fold. Des. 1996;1:R95–R106. doi: 10.1016/S1359-0278(96)00046-6. [DOI] [PubMed] [Google Scholar]
- 15.Pappu R.V., Srinivasan R., Rose G.D. The isolated-pair hypothesis is not valid for poly-peptide chains: implications for protein folding. Proc. Natl. Acad. Sci. USA. 2000;97:12565–12570. doi: 10.1073/pnas.97.23.12565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goldenberg D.P. Computational simulation of the statistical properties of unfolded proteins. J. Mol. Biol. 2003;326:1615–1633. doi: 10.1016/s0022-2836(03)00033-0. [DOI] [PubMed] [Google Scholar]
- 17.Fitzkee N.C., Rose G.D. Reassessing random-coil statistics in unfolded proteins. Proc. Natl. Acad. Sci. USA. 2004;101:12497–12502. doi: 10.1073/pnas.0404236101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ding F., Jha R.K., Dokholyan N.V. Scaling behavior and structure of denatured proteins. Structure. 2005;13:1047–1054. doi: 10.1016/j.str.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 19.Tran H.T., Wang X., Pappu R.V. Reconciling sequence-specific conformational preferences with generic behavior of denatured proteins. Biochemistry. 2005;44:11369–11380. doi: 10.1021/bi050196l. [DOI] [PubMed] [Google Scholar]
- 20.Zaman M.H., Shen M.-Y., Berry R.S., Freed K.F., Sosnick T.R. Investigations into sequence and conformational dependence of backbone entropy, inter-basin dynamics and the Flory isolated-pair hypothesis for peptides. J. Mol. Biol. 2003;331:693–771. doi: 10.1016/s0022-2836(03)00765-4. [DOI] [PubMed] [Google Scholar]
- 21.Avbelj F., Baldwin R.L. Role of backbone solvation and electrostatics in generating preferred peptide backbone conformations: distributions of ϕ. Proc. Natl. Acad. Sci. USA. 2003;100:5742–5747. doi: 10.1073/pnas.1031522100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fitzkee N.C., Rose G.D. Sterics and solvation winnow accessible conformational space for unfolded proteins. J. Mol. Biol. 2005;353:873–887. doi: 10.1016/j.jmb.2005.08.062. [DOI] [PubMed] [Google Scholar]
- 23.Mackerell A.D., Feig D., Brooks C.L. Improved treatment of the protein backbone in empirical force fields. J. Am. Chem. Soc. 2003;126:698–699. doi: 10.1021/ja036959e. [DOI] [PubMed] [Google Scholar]
- 24.Bernadó P., Blanchard L., Timmins P., Marion D., Ruigrok R.W.H. A structural model for unfolded proteins from residual dipolar couplings and small-angle x-ray scattering. Proc. Natl. Acad. Sci. USA. 2005;102:17002–17007. doi: 10.1073/pnas.0506202102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meier S., Grzesiek S., Blackledge M. Mapping the conformational landscape of urea-denatured ubiquitin using residual dipolar couplings. J. Am. Chem. Soc. 2007;129:9799–9807. doi: 10.1021/ja0724339. [DOI] [PubMed] [Google Scholar]
- 26.Hamelberg D., Mongan J., McCammon J.A. Accelerated molecular dynamics: a promising and efficient simulation method for biomolecules. J. Chem. Phys. 2004;120:11919–11929. doi: 10.1063/1.1755656. [DOI] [PubMed] [Google Scholar]
- 27.Markwick P.R.L., Bouvignies G., Blackledge M. Exploring multiple timescale motions in protein GB3 using accelerated molecular dynamics and NMR spectroscopy. J. Am. Chem. Soc. 2007;129:4724–4730. doi: 10.1021/ja0687668. [DOI] [PubMed] [Google Scholar]
- 28.Mukrasch M.D., Markwick P.R.L., Biernat J., von Bergen M., Bernadó P. Highly populated turn conformations in natively unfolded tau protein identified from residual dipolar couplings and molecular simulation. J. Am. Chem. Soc. 2007;129:5235–5243. doi: 10.1021/ja0690159. [DOI] [PubMed] [Google Scholar]
- 29.Wells M., Tidow H., Rutherford T.J., Markwick P., Jensen M.R. Structure of tumor suppressor p53 and its intrinsically disordered N-terminal transactivation domain. Proc. Natl. Acad. Sci. USA. 2008;105:5762–5767. doi: 10.1073/pnas.0801353105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tjandra N., Bax A. Direct measurement of distances and angles in biomolecules by NMR in a dilute liquid crystalline medium. Science. 1997;278:1111–1114. doi: 10.1126/science.278.5340.1111. [DOI] [PubMed] [Google Scholar]
- 31.Prestegard J.H., Bougault C.M., Kishore A.I. Residual dipolar couplings in structure determination of biomolecules. Chem. Rev. 2004;104:3519–3540. doi: 10.1021/cr030419i. [DOI] [PubMed] [Google Scholar]
- 32.Bouvignies G., Markwick P.R., Brüschweiler R., Blackledge M. Simultaneous determination of protein backbone structure and dynamics from residual dipolar couplings. J. Am. Chem. Soc. 2006;128:15100–15101. doi: 10.1021/ja066704b. [DOI] [PubMed] [Google Scholar]
- 33.Louhivouri M., Pääkkönen K., Fredriksson K., Permi P., Lounila J. On the origin of residual dipolar couplings from denatured proteins. J. Am. Chem. Soc. 2003;125:15647–15650. doi: 10.1021/ja035427v. [DOI] [PubMed] [Google Scholar]
- 34.Mohana-Borges R., Goto N.K., Kroon G.J., Dyson H.J., Wright P.E. Structural characterization of unfolded states of apomyoglobin using residual dipolar couplings. J. Mol. Biol. 2004;340:1131–1142. doi: 10.1016/j.jmb.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 35.Fieber W., Kristjansdottir S., Poulsen F.M. Short-range, long-range and transition state interactions in the denatured state of ACBP from residual dipolar couplings. J. Mol. Biol. 2004;339:1191–1199. doi: 10.1016/j.jmb.2004.04.037. [DOI] [PubMed] [Google Scholar]
- 36.Dames S.A., Aregger R., Vajpai N., Bernadó P., Blackledge M. Residual dipolar couplings in short peptides reveal systematic conformational preferences of individual amino acids. J. Am. Chem. Soc. 2006;128:13508–13514. doi: 10.1021/ja063606h. [DOI] [PubMed] [Google Scholar]
- 37.Meier S., Blackledge M., Grzesiek S. Conformational distributions of unfolded polypeptides from novel NMR techniques. J. Chem. Phys. 2008;128:052204. doi: 10.1063/1.2838167. [DOI] [PubMed] [Google Scholar]
- 38.Bernadó P., Bertoncini C.W., Griesinger C., Zweckstetter M., Blackledge M. Defining long-range order and short-range disorder in native α-synuclein using residual dipolar couplings. J. Am. Chem. Soc. 2005;127:17968–17969. doi: 10.1021/ja055538p. [DOI] [PubMed] [Google Scholar]
- 39.Jensen M.R., Houben K., Lescop E., Blanchard L., Ruigrok R.W.H. Quantitative conformational analysis of partially folded proteins from residual dipolar couplings: application to the molecular recognition element of Sendai virus nucleoprotein. J. Am. Chem. Soc. 2008;130:8055–8061. doi: 10.1021/ja801332d. [DOI] [PubMed] [Google Scholar]
- 40.Jensen M.R., Blackledge M. On the origin of NMR dipolar waves in transient helical elements of partially folded proteins. J. Am. Chem. Soc. 2008;130:11266–11267. doi: 10.1021/ja8039184. [DOI] [PubMed] [Google Scholar]
- 41.Bernadó P., Mylonas E., Petoukhov M.V., Blackledge M., Svergun D.I. Structural characterization of flexible proteins using small-angle x-ray scattering. J. Am. Chem. Soc. 2007;129:5656–5664. doi: 10.1021/ja069124n. [DOI] [PubMed] [Google Scholar]
- 42.Bernadó P., Blackledge M., Sancho J. Sequence-specific solvent accessibilities of protein residues in unfolded protein ensembles. Biophys. J. 2006;91:4536–4543. doi: 10.1529/biophysj.106.087528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lovell S.C., Davis I.W., Arendall W.B., de Bakker P.I.W., Word J.M. Structure validation by Cα geometry: ϕ,ψ and Cβ deviation. Proteins. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 44.Eyal E., Najmanovich R., McConkey B.J., Edelman M., Sobolev V. Importance of solvent accessibility and contact surfaces in modeling side-chain conformations in proteins. J. Comput. Chem. 2004;25:712–724. doi: 10.1002/jcc.10420. [DOI] [PubMed] [Google Scholar]
- 45.Svergun D.I., Barberato C., Koch M.H.J. CRYSOL—a program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Cryst. 1995;28:768–773. [Google Scholar]
- 46.Konarev P.V., Volkov V.V., Sokolova A.V., Koch M.H.J., Svergun D.I. PRIMUS—a Windows-PC based system for small-angle scattering data analysis. J. Appl. Cryst. 2003;36:1277–1278. [Google Scholar]
- 47.Wilkins D.K., Grimshaw S.B., Receveur V., Dobson C.M., Jones J.A. Hydrodynamic radii of native and denatured proteins measured by pulse field gradient NMR techniques. Biochemistry. 1999;38:16424–16431. doi: 10.1021/bi991765q. [DOI] [PubMed] [Google Scholar]
- 48.Pan H., Barany G., Woodward C. Reduced BPTI is collapsed. A pulsed field gradient NMR study of unfolded and partially folded bovine pancreatic trypsin inhibitor. Protein Sci. 1997;6:1985–1992. doi: 10.1002/pro.5560060919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.García de la Torre J., Huertas M.L., Carrasco B. Calculation of hydrodynamic properties of globular proteins from their atomic-level structure. Biophys. J. 2000;78:719–730. doi: 10.1016/S0006-3495(00)76630-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bernadó P., García de la Torre J., Pons M. Interpretation of 15N NMR relaxation data of globular proteins using hydrodynamic calculations with HYDRONMR. J. Biomol. NMR. 2002;23:139–150. doi: 10.1023/a:1016359412284. [DOI] [PubMed] [Google Scholar]
- 51.García de la Torre J., Bloomfield V.A. Hydrodynamic properties of complex, rigid, biological macromolecules. Theory and applications. Q. Rev. Biophys. 1981;14:81–139. doi: 10.1017/s0033583500002080. [DOI] [PubMed] [Google Scholar]
- 52.Carrasco B., García de la Torre J. Hydrodynamic properties of rigid particles. Comparison of different modeling and computational strategies. Biophys. J. 1999;76:3044–3057. doi: 10.1016/S0006-3495(99)77457-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.García de la Torre J., Pérez-Sánchez H.E., Ortega A., Hernández J.G., Fernández M.X. Calculation of the solution properties of flexible macromolecules: methods and applications. Eur. Biophys. J. 2003;32:477–486. doi: 10.1007/s00249-003-0292-0. [DOI] [PubMed] [Google Scholar]
- 54.Flory P.J. Cornell University Press; Ithaca, NY: 1953. Principles of Polymer Chemistry. [Google Scholar]
- 55.Tanford C., Kawahara K., Lapanje S., Hooker T.M., Zarlengo M.H. Proteins as random coils. 3. Optical rotatory dispersion in 6 M guanidine hydrochloride. J. Am. Chem. Soc. 1967;89:5023–5029. doi: 10.1021/ja00995a034. [DOI] [PubMed] [Google Scholar]
- 56.Jones J.A., Wilkins D.K., Smith L.J., Dobson C.M. Characterization of protein unfolding by NMR diffusion measurements. J. Biomol. NMR. 1997;10:199–203. [Google Scholar]
- 57.Choy W.-Y., Mulder F.A.A., Crowhurst K.A., Muhandiram D.R., Millett I.S. Distribution of molecular size within an unfolded state ensemble using small-angle x-ray scattering and pulse field gradient NMR techniques. J. Mol. Biol. 2002;316:101–112. doi: 10.1006/jmbi.2001.5328. [DOI] [PubMed] [Google Scholar]
- 58.Jha A.K., Colubri A., Zaman M.H., Koide S., Sosnick T.R. Helix, sheet, and polyproline II frequencies and strong nearest neighbor effects in a restricted coil library. Biochemistry. 2005;44:9691–9702. doi: 10.1021/bi0474822. [DOI] [PubMed] [Google Scholar]
- 59.Griffiths-Jones S.R., Sharman G.J., Maynard A.J., Searle M.S. Modulation of intrinsic ϕ,ψ propensities of amino acids by neighboring residues in the coil regions of protein structures: NMR analysis and dissection of a β-hairpin peptide. J. Mol. Biol. 1998;284:1597–1609. doi: 10.1006/jmbi.1998.2264. [DOI] [PubMed] [Google Scholar]
- 60.Swindells M.B., MacArthur M.W., Thornton J.M. Intrinsic ϕ,ψ propensities of amino-acids, derived from the coil regions of known structures. Nat. Struct. Biol. 1995;2:596–603. doi: 10.1038/nsb0795-596. [DOI] [PubMed] [Google Scholar]
- 61.Smith L.J., Bolin K.A., Schwalbe H., MacArthur M.W., Thornton J.M. Analysis of main-chain torsion angles in proteins-prediction of NMR coupling constants for native and random coil conformations. J. Mol. Biol. 1996;255:494–506. doi: 10.1006/jmbi.1996.0041. [DOI] [PubMed] [Google Scholar]
- 62.LeGuillou J.C., Zinn-Justin J. Critical exponents for the n-vector model in three dimensions from field theory. Phys. Rev. Lett. 1977;39:95–98. [Google Scholar]
- 63.Mylonas E., Hascher A., Bernadó P., Blackledge M., Mandelkow E. Domain conformation of τ-protein studied by solution small-angle x-ray scattering. Biochemistry. 2008;47:10345–10353. doi: 10.1021/bi800900d. [DOI] [PubMed] [Google Scholar]
- 64.Mukrasch M.D., Biernat J., von Bergen M., Griesinger C., Mandelkow E. Sites of τ important for aggregation populate β-structure and bind to microtubules and polyanions. J. Biol. Chem. 2006;280:24978–24986. doi: 10.1074/jbc.M501565200. [DOI] [PubMed] [Google Scholar]
- 65.Longhi S., Receveur-Bréchot V., Karlin D., Johansson K., Darbon H. The C-terminal domain of the measles virus nucleoprotein is intrinsically disordered and folds upon binding to the C-terminal moiety of the phosphoprotein. J. Biol. Chem. 2003;278:18638–18648. doi: 10.1074/jbc.M300518200. [DOI] [PubMed] [Google Scholar]
- 66.Bourhis J.-M., Receveur-Bréchot V., Oglesbee M., Zhang X., Buccellato M. The intrinsically disordered C-terminal domain of the measles virus nucleoprotein interacts with the C-terminal domain of the phosphoprotein via two distinct sites and remains predominantly unfolded. Protein Sci. 2005;14:1975–1992. doi: 10.1110/ps.051411805. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.