

Abstract
Neurodegenerative diseases are characterized by selective and progressive loss of specific populations of neurons, which determines the clinical presentation. The same neuronal populations can be affected in a number of different disorders. Given that the clinical presentation reflects the particular population of neurons that are targets of the disease process, it is clear that for any given clinical syndrome, more than one neurodegenerative disease can account for the clinical syndrome. Because of this clinical ambiguity, for the purpose of this brief review neurodegenerative disorders are classified according to the underlying molecular pathology rather than their clinical presentation. The major neurodegenerative diseases can be classified into amyloidoses, tauopathies, α-synucleinopathies and TDP-43 proteinopathies.
Keywords: Amyloidosis, tauopathy, synucleinopathy, TDP-43 proteinopathy
Introduction
By definition a neurodegenerative disease is one in which there is selective and progressive loss of specific populations of neurons for reasons that in most cases remain unknown. The goals of research on neurodegenerative disorders are to determine the molecular basis of selective vulnerability and common final pathways of progressive neuronal loss. In the most common neurodegenerative disorders there are biochemical changes in a specific protein that often produces characteristic inclusion bodies within neurons or glia, or both. The particular population of neurons that are vulnerable in each disorder determines the clinical presentation, and each specific disorder is defined by a combination of clinical, pathologic and biochemical features. There are genetic underpinnings in most of the common neurodegenerative disorders, but only a small fraction of cases are due to causative mutations in defined genes.
The same neuronal populations can be affected in a number of different disorders. For example, neurons in the hippocampus and brainstem monoaminergic nuclei are vulnerable in a wide range of distinct clinicopathologic entities. Given that the clinical presentation reflects the particular population of neurons that are targets of the disease process, it is clear that for any given clinical syndrome, there will usually be more than one neurodegenerative disease that can account for the clinical syndrome. Because of this ambiguity, for the purpose of this brief review neurodegenerative disorders are classified according to the underlying molecular pathology rather than their clinical presentation.
Molecular classification of neurodegenerative disorders
Table 1 is an abbreviated list of neuro-degenerative disorders classified by the major molecular abnormality, with a greatly simplified listing of anatomical and clinical features.
Table 1.
Classification of major neurodegenerative disorders
| Disorder | Anatomy | Major clinical feature | |
|---|---|---|---|
| Amyloidoses | |||
| Aβ | Alzheimer disease | Corticolimbic | Dementia |
| ABri | Familial British dementia | Corticolimbic & cerebellar | Dementia & ataxia |
| PrP | Creutzfeldt-Jakob disease | Cortical & basal ganglia | Dementia & movement disorder |
| Gerstmann-Straussler-Scheinker | Cortical & Cerebellar | Ataxia & dementia | |
| Tauopathies | |||
| 4R | Progressive supranuclear palsy | Basal ganglia & brainstem | Parkinsonism |
| Corticobasal degeneration | Cortical & basal ganglia | Focal cortical syndrome & parkinsonism | |
| Argyrophilic grain disease | Limbic | Amnestic cognitive impairment | |
| 3R | Pick's disease | Corticolimbic | Dementia & focal cortical syndromes |
| 3R+4R | Tangle predominant dementia | Limbic | Dementia |
| Guam Parkinson dementia complex | Cortex & brainstem | Dementia & parkinsonism | |
| Synucleinopathies | |||
| Parkinson disease | Brainstem | Parkinsonism | |
| Dementia with Lewy bodies | Corticolimbic & brainstem | Dementia & parkinsonism | |
| Multiple system atrophy | Basal ganglia, brainstem & cerebellum | Parkinsonism & ataxia | |
| TDP-43 proteinopathies | |||
| Amyotrophic lateral sclerosis | Motor neurons | Spasticity & weakness | |
| Frontotemporal lobar degeneration with ubiquitinated inclusions | Cortex & basal ganglia | Dementia & focal cortical syndromes |
Amyloidoses
The presence of abnormal proteins with specific properties defines the amyloidoses, of which Alzheimer disease (AD) is the most common. Amyloid is a generic name for proteins with common physicochemical properties (e.g., Congo red birefringence) due to abnormal conformation, with cross beta-pleated sheets, which gives the protein a propensity to form fibrils and to aggregate, most often as extracellular deposits. The amyloidoses are sometimes referred to as beta-fibrilloses to reflect this molecular property [1, 2]. The amyloid protein in AD, Aβ, is derived from a precursor protein, amyloid precursor protein (APP) by regulated intramembranous proteolysis [3]. By most accounts this process is considered to be fundamental to the pathogenesis of AD [4]. Deposits of Aβ are not found only in AD, but also in elderly nondemented individuals [5], sometimes in great numbers in a process we call pathological aging [6]. Recently, it has become possible to detect Aβ deposits in the brains of living subjects with positron emission tomography, and these studies have also shown that a significant number of clinically normal elderly individuals have Aβ deposits [7, 8]. Whether this represents preclinical AD remains to be determined. Deposits of Aβ are also found in other neurodegenerative disorders, as a function of age and apolipoprotein E ε4 carrier state [9], which is the major genetic risk factor for AD [10]. Amyloid plaques are often abundant in dementia with Lewy bodies (DLB) [11].
Amyloid of a different molecular nature accumulates in a rare form of dementia originally described in British families [12]. The mutation in familial British dementia (FBD) introduces a stop codon in the normal protein, ABri, creating a truncated protein with amyloidogenic properties. Amyloid deposits in FBD are in blood vessels and adjacent tissue and are numerous in cortex and cerebellum (Figure 1). Like AD, FBD is associated with neurofibrillary tangles composed of tau protein (see below), but unlike AD tangles are relatively restricted to the medial temporal lobe, while they are widespread in the cortex in AD [13].
Figure 1.
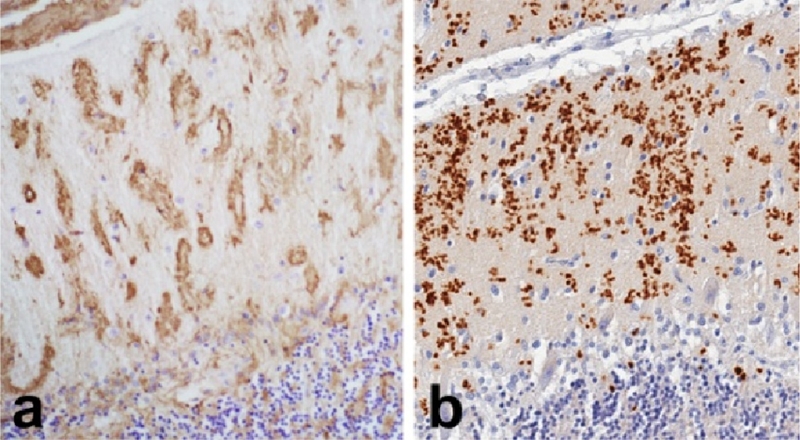
Amyloidoses: Familial British dementia (a) and familial Creutzfeldt-Jakob disease (b) have cerebellar amyloid deposits composed on non-Aß amyloid, ABri and PrP.
Another rare form of non-Alzheimer degenerative dementia associated with non-Aβ amyloid deposits is Creutzfeldt-Jacob disease (CJD). The protein deposited in tissue is an abnormal conformer of a normal cellular protein, PrP, referred to as PrPres [14], reflecting the fact that the abnormal protein is resistant to proteolytic degradation. Amyloid deposits composed of PrPres are particularly common in familial variants of CJD with insertion mutations in the prion gene (PRNP) [15] and in Gerstmann-Straussler-Scheinker syndrome (GSS) [16], but are also found as dense kuru-like plaques in sporadic CJD. The latter term reflects the fact that similar amyloid plaques are detected in kuru, a form of transmissible prion disease associated with ritualistic cannibalism in the Fore population of Papua New Guinea [17]. Sporadic CJD is classified based upon the nature of the electrophoretic mobility of PrPres and the genotype at codon 129 of PRNP [18]. Kuru-type plaques are most common in cases with heterozygosity at codon 129 (M/V) and type 2 PrPres [18]. Amyloid plaques, in particular multicentric plaques with peri-plaque vacuolation (“florid plaques”) are a characteristic feature of variant CJD, which is linked to bovine spongiform encephalopathy [19, 20].
Like FBD, some cases of GSS are associated with neurofibrillary tangles similar to those in AD [21, 22]. Given the widespread distribution of amyloid plaques and neurofibrillary tangles, such cases can be mistaken for an unusual variant of AD [23]. The presence of neurofibrillary pathology in amyloidoses due to distinct molecular forms of amyloid (Aβ, ABri and PrPres) is a strong argument that in these disorders, neurofibrillary pathology is being driven by factors directly related to the amyloid formation and can be considered “secondary tauopathies.”
Tauopathies
The major structural protein of neurofibrillary tangles is the microtubule associated protein, tau [24]. Tau is a heat-resistant phospho-protein that promotes microtubule polymerization and stabilization. Once considered to be relatively restricted to neurons [25], it is now known that tau accumulates not only in neurons in neurofibrillary tangles, but also in glia in a wide range of neurodegenerative disorders and in the aging brain. That tau pathology is fundamentally important has been proven unequivocally since mutations in the gene for tau (MAPT) cause neurodegeneration in humans [26] and in transgenic animal models over expressing mutant tau [27].
Disorders in which tau pathology is considered to be the major contributing factor to neuro-degeneration are referred to as “primary tauopathies.” Tau protein in the brain is heterogeneous due to alternative splice forms, as well as post-translational modifications, including phosphorylation. In neurodegenerative diseases tau protein has an abnormal conformation and abnormal solubility properties that favor its aggregation and fibril formation, similar to amyloid, except that the fibrils are not in the extracellular space, but within the cytoplasm of the affected cells. Exon 10 of MAPT is alternatively spliced to generate tau species with either three or four conserved ∼30 amino acid repeats in the microtubule binding domain of tau protein [28], referred to as 3R and 4R tau. There is preferential accumulation of 3R or 4R tau in various tauopathies, providing an additional subclassification of the tauopathies. In AD neurofibrillary pathology is composed of an equimolar ratio of 3R and 4R tau [29].
Argyrophilic grain disease (AGD)
The most common of the primary tauopathies are 4R tauopathies, and all disorders of this type are associated with both neuronal and glial tau inclusions. The most common of these is argyrophilic grain disease (AGD), which increases in frequency with age and is detected in about 5% of autopsies of individuals with late onset dementia [30, 31]. It is common in mild cognitive impairment [32], and it may co-exist with other degenerative disorders, particularly the 4R tauopathies, such as corticobasal degeneration (CBD) and progressive supranuclear palsy (PSP) [33].
The characteristic lesion in AGD is the comma shaped or grain-like structure in the neuropil of the medial temporal lobe [34]. Grains are aggregates of tau in dendritic processes of neurons [35]. Neuronal tau is accompanied by tau-positive oligodendroglia (“coiled bodies” [36]) and ramified astrocytes [37]. Ballooned neurons are also common [38]. Many cases of AGD have varying degrees of Alzheimer type neurofibrillary degeneration, and it can be difficult to detect AGD in cases with severe Alzheimer type pathology [34]. Use of immunohistochemistry with an antibody specific for 4R tau [30] permits detection of AGD even in advanced AD, since grains and glial lesions are selectively labeled [39] (Figure 2a and b). Using this technique demonstrates AGD in more than 25% of AD cases, with increasing frequency with increased age.
Figure 2.
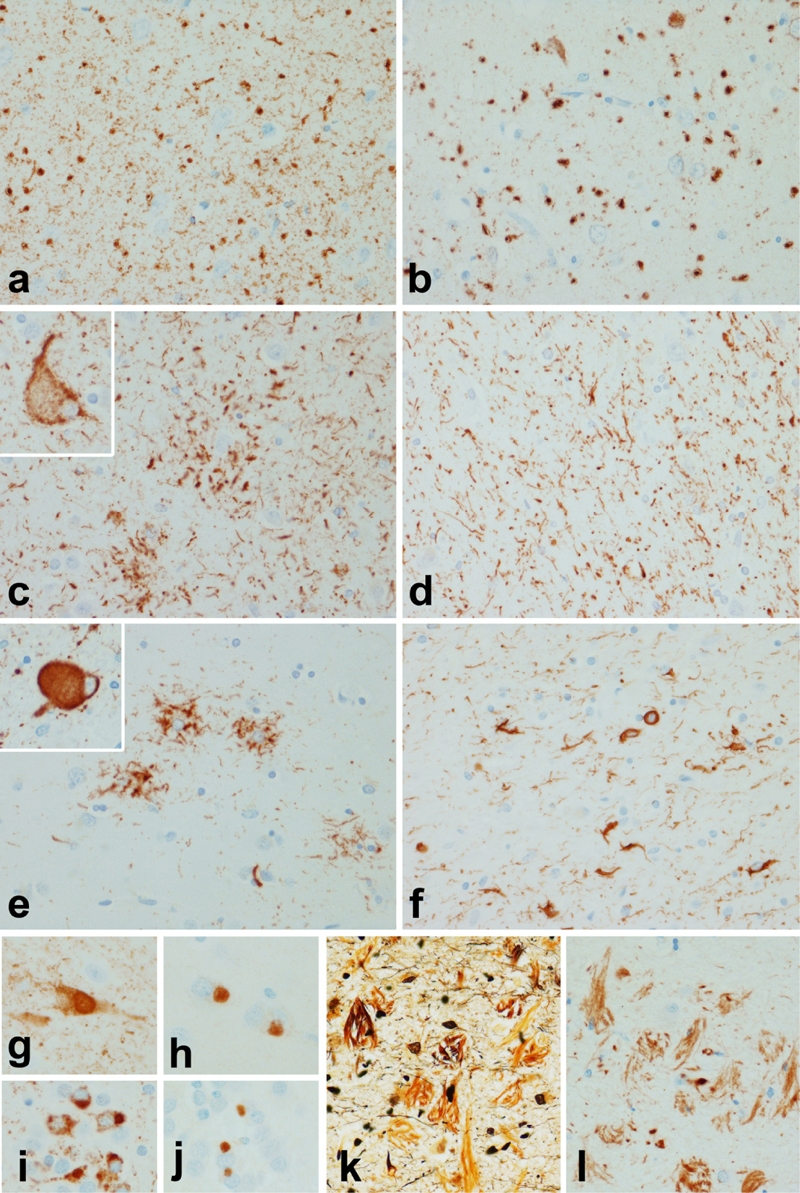
Tauopathies: a & b Argyrophilic grain disease (AGD); c & d Corticobasal degeneration (CBD); e & f Progressive supranuclear palsy (PSP); g-j Pick's disease (PiD); k & l Guam Parkinson dementia complex (PDC). The neuropil of AGD has small round inclusions in neuronal processes (a) that are composed of 4R tau as shown with a monoclonal antibody specific to 4R tau (b). The histologic hallmarks of CBD are astrocytic plaques, which appears as a cluster of short tau positive processes around a central astrocyte (c), ballooned neurons (inset) and thread-like processes in both gray and white matter (d). In PSP the characteristic astrocytic lesion appears as a tuft of abnormal fibers (3), globose neurofibrillary tangles (inset) are the typical neuronal lesion and oligodendroglia in white matter have inclusions referred to as “coiled bodies (f). The defining histologic lesion in PiD is the Pick bodies shown with phospho-tau immunohistochemistry in pyramidal neurons of the hippocampus (g) and granular neurons of the dentate gyrus (i). The inclusions are composed to 3R tau as demonstrated with a monoclonal antibody to 3R tau (h and j). In Guam PDC there are numerous neurofibrillary tangles in cortex and hippocampus (k & l) with many of the tangles released into the extracellular compartment after neuronal death (Bielschowsky stain in k). The tangles are positive for 3R and 4R tau, but extracellular tangles show preferential immunoreactivity for 3R tau (l).
Clinicopathologic correlations in AGD are challenging due in part to the fact that it is rarely a pure pathologic process, and since it is most common in the very old where other pathologic processes increase in frequency with advancing age [40]. The predilection of this pathology to the medial temporal lobe would suggest that an amnestic clinical syndrome should be common in AGD, and in patients with amnestic mild cognitive impairment who come to autopsy, AGD is sometimes found [41]. Saito and co-workers have suggested a staging scheme for AGD, with extension to other limbic related structures in advanced stages of the disease; rarely AGD spreads beyond limbic lobe structures, so-called “diffuse AGD,” in patients with dementia [42].
Corticobasal degeneration (CBD)
CBD is a 4R tauopathy that has a range of clinical presentations due to the fact that it is associated with focal cortical degeneration, the distribution of which varies from person to person for reasons that still remain to be explained. In the classical presentation, which is referred to as the “corticobasal syndrome (CBS),” there is asymmetrical cortical degeneration of the superior frontal gyrus and superior parietal lobule and the patient has asymmetrical rigidity and apraxia, often with dystonia and alien limb sign [43]. More often there is focal atrophy of the frontal lobes producing frontal lobe dementia or of language areas producing progressive nonfluent aphasia [44]. The characteristic pathology is phospho-tau accumulation in cell processes of neurons and astrocytes in the cortex, basal ganglia, thalamus and brainstem [45]. The most specific lesion in the neuropathologic diagnosis of CBD is the astrocytic plaque [46] (Figure 2c), which is not seen in other disorders [47]. Ballooned neurons, also known as swollen achromatic neurons [48] (Figure 2c, inset), are usually numerous in affected cortical areas. On the other hand, research criteria for CBD emphasize presence of abnormal tau-positive, thread-like processes in both gray and white matter of cortical and subcortical regions (Figure 2d) , a feature that has been validated as diagnostically useful [45].
Progressive supranuclear palsy (PSP)
Progressive supranuclear palsy in most cases presents as an atypical parkinsonism with axial rigidity, postural instability and unexplained falls, with most patients also developing progressive vertical gaze palsy (for which the disorder is named), dysarthria and dysphagia [49]. Other clinical presentations are also recognized, including dementia [50], speech apraxia [51], corticobasal syndrome [52] and pure akinesia with gait failure [53, 54]. In a subset of patients the clinical features initially are similar to those in Parkinson disease, so-called “PSP-P” [55]. The distribution of tau pathology determines the particular clinical presentation; some cases have severe brainstem involvement (e.g., pure akinesia) and others have severe cortical involvement (e.g., dementia, corticobasal syndrome [56] and speech apraxia).
The core neuroanatomical regions affected in all cases of PSP include the basal ganglia, subthalamic nucleus and the substantia nigra [57]. Pathology of the cerebellar dentate nucleus and the outflow pathway (dentato-rubro-thalamic pathway) is usually severe and associated with profound atrophy of the superior cerebellar peduncle [58], which can be used as a biologic marker of disease progression with structural imaging [59].
The hallmark neuronal lesion is the globose neurofibrillary tangle (Figure 2e, inset), while tuft-shaped astrocytes or tufted astrocytes (Figure 2e) are the most characteristic glial lesion in PSP [60]. Tufted astrocytes are most abundant in the motor cortex and the corpus striatum. Neuronal loss and gliosis is most marked in the substantia nigra and subthalamic nucleus, where many thread-like processes and oligodendroglial coiled bodies are often found (Figure 2f). In PSP threads and coiled bodies are found together, while in CBD many threads are detected in the near complete absence of coiled bodies (compare Figure 2d and 2f).
Pick's disease (PiD)
PiD is a rare cause of frontal lobe dementia, accounting for less than 5% in autopsy series of dementia [61]. It is associated with circumscribed “lobar” atrophy; like CBD the distribution of focal cortical degeneration determines the presentation. Behavioral and personality deterioration deficits are typical in cases with frontotemporal atrophy, while frontoparietal atrophy presents with apraxia or aphasia similar to CBD. When the amnestic symptoms prevail the clinical diagnosis is often initially AD. It is a disorder that affects men and women equally and is usually a “presenile dementia” with age of onset younger than 65 years. Mutations in the tau gene (MAPT) account for most cases of pathologically confirmed cases of familial PiD [62, 63].
The cardinal neuropathologic features are circumscribed cortical atrophy associated with neuronal loss, gliosis and argyrophilic, round intraneuronal inclusions (Pick bodies). Pyramidal neurons in the hippocampus and granular neurons in the dentate fascia are particularly vulnerable (Figure 2g and i). Pick bodies are composed of tau protein enriched in 3R tau, which can be shown with biochemical studies [64], or more recently with antibodies specific to tau isoforms [65] (Figure 2h and j). Less specific features include leukoencephalopathy and ballooned cortical neurons (Pick cells), which are similar to those found in CBD. Glial reaction is often pronounced in affected cerebral gray and white matter. Tau-immunoreactive glial inclusions are sometimes present in PiD [63]. Interestingly, the glial lesions contain predominantly 4R tau, which may contribute to the variability in the ratio between 3R and 4R tau observed in some cases of Pick's disease [66]. Involvement of the deep gray matter and the brainstem is typical, with a predilection for the monoaminergic nuclei [67]. Neuro-chemical studies demonstrate deficits in intrinsic cortical neurotransmitter systems (e.g., somatostatin), but usually less involvement of transmitters in systems projecting to the cortex, such as the cholinergic neurons of the basal nucleus of Meynert [68].
Tangle predominant dementia
Tangle predominant dementia is a disorder of the very old (80 years and greater), where it may account for more than 5% of dementia cases [69–73]. It is associated with predominantly an amnestic clinical syndrome and can sometimes be differentiated from AD because of this [70], although it still remains an entity that is better known to neuro-pathologists than to clinicians. Unlike AD, it is not associated with increased frequency of apolipoprotein E ε4 allele [71–73].
Pathologically, it is characterized by diffuse cerebral atrophy with the most severe atrophy in the medial temporal lobe, which corresponds to the distribution of neuro-fibrillary tangles. Tangles are most dense in the hippocampus, amygdala and medial temporal cortex, with fewer in convexity cortices. There are usually no or very few neocortical neuritic plaques, but there may be diffuse amyloid deposits [74], which are diagnostically nonspecific, since they can be found in large numbers in the brains of neurologically normal elderly individuals (i.e. pathological aging) as mentioned previously [6]. The tangles are histologically and biochemically similar to those in AD, with an admixture of 3R and 4R tau [69] (Figure 2k & l). Many of the tangles are extracellular “ghost” tangles. For reasons that remain unclear extracellular tangles preferentially are immunoreactive for 3R tau (Figure 2l), while intracellular tangles have a mixture of 3R and 4R tau [69].
Guam Parkinson dementia complex (PDC)
Guam PDC is an endemic disease affecting the Chamorro people of Guam characterized by progressive dementia and parkinsonism first described by Hirano and colleagues [75, 76]. A similar disorder occurs in the Kii peninsula in Japan [77]. In both populations, the disorder clusters in families and there is also increased frequency of motor neuron disease. The etiology is unknown and despite more than three decades of research, a genetic cause is unknown [78].
Pathologically, Guam PDC is characterized by diffuse cerebral atrophy with degeneration of brainstem monoaminergic nuclei. In areas of cortical and subcortical degeneration, neurons have tangles similar to those in AD [79], with 3R and 4R tau. Some cases have Lewy bodies [80], but they are usually restricted to the amygdala, as is common in AD [81]. Recently, TAR DNA binding protein of 43 kDa (TDP-43) has been shown to be present in most cases of Guam PDC [82, 83]. In cases with motor neuron disease, the TDP-43 pathology resembles than seen in sporadic amyotrophic lateral sclerosis (see below), but in PDC cases lacking motor neuron pathology TDP-43 pathology is present in cortical and subcortical areas in the form neuronal cytoplasmic inclusions, dystrophic neurites and oligodendroglia inclusions [83].
Synucleinopathies
Alpha-synuclein is a member of a family of proteins that also contains β-synuclein and γ-synuclein that are pleiotropic in terms of function [84]. In the central nervous system α-synuclein has been implicated in several disorders. It was originally discovered as a non-amyloid component of senile plaques that was enriched in presynaptic termini [85, 86], but little attention was paid to it until mutations were discovered in the gene for α-synuclein (SNCA) in familial Parkinson disease (PD) [87] and it was found to be the major structural protein in Lewy bodies, the hallmark histopathologic lesion in PD and dementia with Lewy bodies (DLB) [88]. Availability of antibodies to α-synuclein proved essential to the greater recognition of the importance of Lewy body disorders and the presence of α-synuclein in other neurodegenerative disorders, the synucleinopathies, which includes multiple system atrophy and neuroaxonal dystrophies.
Lewy body disorders
As noted above, Lewy bodies are the histologic hallmark of PD, but they are found in other disorders, as well, including as many as 10% of neurologically normal elderly over age 60 years, where they are considered coincidental [89]. They are the sine qua non of DLB and have been noted in a subset of other neurodegenerative disorders, such as AGD, PSP, CBD and PiD [90]. In these disorders they are considered to be coincidental [91]. Lewy bodies are also common in AD, particularly in the amygdala [81, 92], where up to 50% of AD cases are positive [93]. Lewy bodies and α-synuclein immunoreactive axonal spheroids have been described in some of the neuroaxonal dystrophies, particularly neurodegeneration with brain iron, formerly known as Hallervorden-Spatz disease [94, 95].
PD is a disorder characterized by bradykinesia, tremor and rigidity with gait and balance disorders. Motor deficits in PD are associated with loss of substantia nigra dopaminergic neurons [96]. Much current interest in PD is focused on non-motor aspects of the illness, such as hyposmia, autonomic dysfunction and sleep disorders that may precede motor problems by decades [97–99], as well as on cognitive deficits that occur late in the disease course in about 40% of cases [100]. Braak and co-workers have proposed a staging scheme for PD in which early pathology is in peripheral autonomic nervous system, with later involvement of the olfactory bulb and the lower brainstem autonomic and sleep related nuclei, spreading in a caudal-to-rostral manner, ending with widespread cortical involvement [101]. This scheme fits with premotor aspects of PD and late developing dementia in PD [101–103]. The scheme posits a direct cell-to-cell transmission of a causative agent to account for the interrelation of vulnerable neuronal populations. Recently, in vitro studies have provided support for the notion that abnormal conformers of α-synuclein can be transmitted from cell-to-cell [104], which may explain the intriguing observation that Lewy bodies are found in engrafted tissue many years after fetal tissue transplants for treatment of PD [105, 106]. The results suggest that α-synuclein may have properties similar to other transmissible amyloid proteins, such as prion protein [107].
The Lewy body is a concentric hyaline perikaryal inclusion (Figure 3a) that is immunereactive for α-synuclein, but similar inclusions are also present in neuronal cell processes as so-called intraneuritic Lewy bodies. Less well defined inclusions, so-called cortical Lewy bodies are found in the cortex in PD with dementia (PDD) and in DLB (Figure 3b). DLB is a disorder characterized by dementia with visual hallucinations, fluctua-tions and parkinsonism [108, 109]. It is distinguished from PDD by the temporal sequence of cognitive impairment with respect to parkinsonism, being early in DLB and late in PDD [110]. Pathologically, most cases of rigorously diagnosed DLB have diffuse cortical Lewy bodies with mild Alzheimer type pathology (Braak stage IV or less with mostly diffuse type amyloid plaques) [111].
Figure 3.
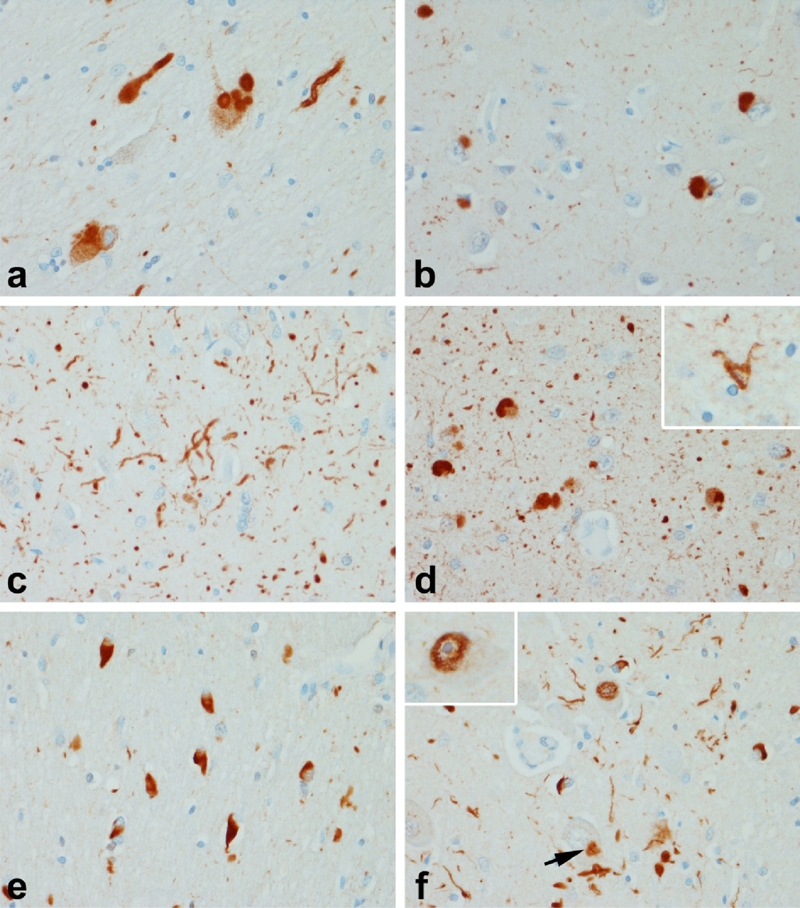
Synucleinopathies: a-d Lewy body disease (LBD); e & f Multiple system atrophy (MSA). Vulnerable neurons in LBD, such as the basal nucleus of Meynert (a) have dense round inclusions (Lewy bodies) as well as Lewy bodies with axons (a). In cases with dementia, there are also neuronal inclusions in cortical neurons (b), as well as many neurites in the hippocampus (c) and amygdala (d). The morphology of the neurites in the amygdala are often grain-like and have been referred to as Lewy dots to distinguish them from argyrophilic grains. In some cases there are sparse oligodendroglial inclusions (inset in d), which are clearly different from the glial cytoplasmic inclusions (GCI) that are the hallmark of MSA (e). GCI are abundant in the basal ganglia, pons (e), medulla and cerebellum. In addition to GCI neuronal inclusions (arrow in f) are present in many cases and are usually most numerous in the pontine base (f) where they are accompanied by dystrophic neurites and synuclein positive fibrillar inclusions within some neuronal nuclei (inset).
The largest burden of abnormal α-synuclein in DLB, as well as in PD and PDD, is not in Lewy bodies, but rather dystrophic neurites, so-called Lewy neurites. Lewy neurites are curvilinear or dot-like processes [112] that are found in regions with the highest density of Lewy bodies, such as limbic cortex and amygdala (Figure 3d). They are also found in most cases of PDD and DLB in the CA2/3 sector of the hippocampus [113, 114] (Figure 3c). The density of cortical Lewy bodies and neurites correlates with cognitive impairment in some studies [115–118]. On the other hand, some studies fail to find a clear correlation [119–121]. In many cases of PD, α-synuclein is also present in small glial cells consistent with oligodendroglia (Figure 3d, inset); these can be particularly numerous in early onset PD due to mutations in SNCA [122]. The glial lesions in PD and DLB are never as numerous as in multiple system atrophy (MSA).
Multiple system atrophy (MSA)
MSA is a sporadic synucleinopathy characterized by autonomic dysfunction, parkin-sonism and cerebellar ataxia, associated with neurodegeneration of the substantia nigra, basal ganglia, pontine nuclei, inferior olivary nucleus and the cerebellum [123]. Depending upon the prevailing clinical features, it is sub-classified as MSA-P (for parkinsonism) and MSA-C (for cerebellar ataxia). While it is usually sporadic, there are recent reports suggesting that variants in SNCA may be associated with increased risk for MSA [124].
Neurodegeneration in MSA is associated with extensive α-synuclein pathology in oligo-dendrocytes, so-called glial cytoplasmic inclusions (GCI) [125] (Figure 3e). How glial pathology is linked to neuronal loss remains to be determined. Neuronal inclusions and dystrophic neurites (Figure 3d) are detected in most cases, but they are highly variable in density, not clearly associated with neuronal loss and largely confined to the putamen, pontine nuclei and inferior olive. In some cases there are α-synuclein immunoreactive intranuclear inclusions [126] (Figure 3d, inset).
TDP-43 proteinopathies
Transactive response DNA binding protein of 43 kDa (TDP-43) has structural properties similar to heterogenous nuclear ribonucleo-proteins, including RNA binding domains, which appear necessary in its role in transcriptional regulation [127]. More recently, TDP-43 has been shown to be a component of RNA granules [128], which play a critical role in cellular response to cell stress by arresting translation [129]. Interest in TDP-43 grew when it was shown to be a component of the neuronal inclusions of amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration with ubiquitinated inclusions (FTLD-U) [130]. While initially considered specific to ALS and FTLD-U, it has become clear that TDP-43 immunoreactive neuronal inclusions are found in other disorders, such as AD and hippocampal sclerosis [131], Guam PDC [82] and LBD [132].
Amyotrophic lateral sclerosis (ALS)
ALS is a neurodegenerative disease process known for its selective involvement of upper and lower motor neurons, but increasingly it is has been shown to be a multisystem degeneration with pathology in extra-motor locations [133]. As is true for most neurodegenerative diseases, a small subset of ALS is due to genetic mutations. Autosomal dominant forms of ALS are due to mutations in superoxide dismutase 1 (SOD1) [134], angiogenic (ANG) [135] and TARDBP, the gene for TDP-43 on chromosome 1 [136]. Recently, mutations in TARDBP have been reported in a family with frontotemporal dementia [137], but most mutations in TARDBP are associated with ALS.
In addition to neuronal loss, affected neurons in ALS have characteristic inclusions bodies, including Bunina bodies, Lewy-like hyaline inclusions and skein-like inclusions [133]. Bunina bodies are eosinophilic granular cytoplasmic inclusions that are found in degenerating motor neurons and are immune-reactive for cystatin C [138]. The hyaline neuronal and glial inclusions in SOD1-linked familial ALS are immunoreactive for SOD1, but not TDP-43 [139]. In contrast, Lewy-like hyaline inclusions and skein-like inclusions in ALS are TDP-43-positive (Figure 4).
Figure 4.
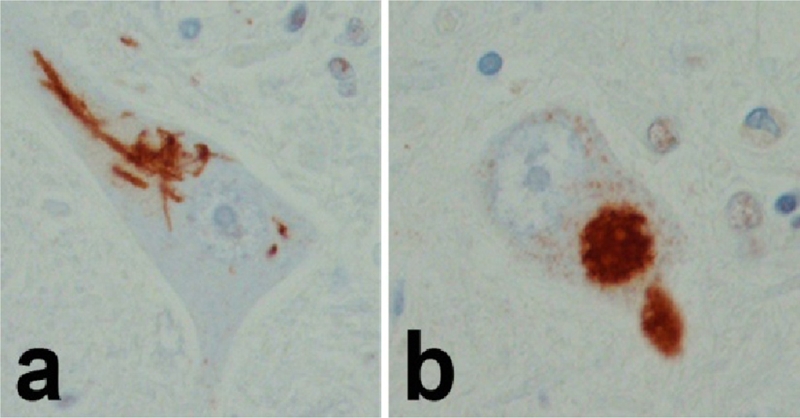
TDP-43 in ALS: Both skein-like (a) and Lewy-like hyaline inclusions (b) are positive for TDP-43.
Frontotemporal lobar degeneration with ubiquitin inclusions (FTLD-U)
Frontotemporal lobar degeneration (FTLD) is a generic term for the group of non-Alzheimer degenerative dementias with focal cortical pathology in frontal and temporal lobes [140]. It encompasses a range of different clinical syndromes – behavioral variant fronto-temporal dementia (FTDbv), progressive nonfluent aphasia (PNFA), semantic dementia (SD) and corticobasal syndrome (CBS) – and a range of different pathologies. The most common is FTLD-U, with tauopathies considered slightly less common and including PSP, CBD and PiD [141]. Mutations in MAPT account for a subset of the familial FTLD, all of which are associated with tauopathies (FTLD-TAU) [26]. Mutations in the gene for progranulin (GRN) account for most of the cases of familial FTLD-U [142]. A rare cause of familial FTLD-U is mutation in valosin containing protein (VCP), which produces dementia with Paget's disease of bone and inclusion body myositis (IBM) [143]. In this disorder there are also TDP-43 positive neuronal inclusions, with many neuronal intranuclear inclusions the most characteristic feature [144].
Rare causes of FTLD include disorders associated with neuronal inclusions composed of neuronal intermediate filament proteins, including alpha-internexin [145]. There are also several case reports of atypical MSA presenting with frontal dementia and mimicking PiD except that the inclusion bodies contain α-synuclein rather than tau [146, 147].
A recently recognized class of FTLD has neuronal inclusions composed of the protein FUS (“fused in sarcoma”) [148], which is also a rare cause of familial ALS [149]. Like TDP-43, FUS is an RNA binding protein that is normally located in the nucleus, with relocation to cytoplasmic inclusions in disease.
Rare cases of FTLD-U have ubiquitinated inclusions for which the protein remains to be determined. One of these is associated with mutations in endosomal ESCRTIII-complex subunit CHMP2B [150]. Sparse TDP-43 ubiquitin positive inclusions are detected in this disorder, but largely confined to the hippocampal dentate fascia [151].
Given the heterogeneity of the pathology of FTLD a proposed nomenclature for this group of disorders is shown in Table 2 [152]. We have taken the liberty of adding FTLD-AS (for FTLD associated with α-synuclein), since the scheme was meant to be updated as the protein in the ubiquitin immunoreactive neuronal inclusions was discovered, as is the case for the recently added FTLD-FUS [148].
Table 2.
Classification of frontotemporal lobar degenerations (adapted from [152])
| FTLD-Molecular abnormality | Genetic loci |
|---|---|
| FTLD-TDP | |
| Frontotemporal lobar degeneration with TDP-43 inclusions | |
| Subtype 1 (associated with SD) | None known |
| Subtype 2 (associated with MND) | Chromosome 9 |
| Subtype 3 (associated with FTDbv and PNFA) | GRN |
| Subtype 4 (associated with Paget's and IBM) | VCP |
| FTLD-TAU | |
| Frontotemporal lobar degeneration with tauopathy | MAPT |
| Pick's disease (3R tauopathy) | |
| Corticobasal degeneration (4R tauopathy) | |
| Progressive supranuclear palsy | |
| Multisystem tauopathy (4R tauopathy) | |
| FTLD-IF | |
| Frontotemporal lobar degeneration with intermediate filament inclusions | None known |
| FTLD-FUS | |
| Frontotemporal lobar degeneration with FUS inclusions | FUS |
| FTLD-UPS | |
| Frontotemporal lobar degeneration with inclusions composed of ubiquitin and other components of ubiquitin-proteasome system (e.g., P62-sequestosome) | CHMP2b |
| FTLD-AS | |
| Frontotemporal lobar degeneration with inclusions composed of α-synuclein (atypical Pick's disease) | SNCA? |
| FTLD-NI | |
| Frontotemporal lobar degeneration with no inclusions | None known |
FTLD-TDP Subtype 1 = Mackenzie Type 2; FTLD-TDP Subtype 2 = Mackenzie Type 3; FTLD-TDP Subtype 3 = Mackenzie Type 1; FTLD-TDP Subtype 4 = no Mackenzie type assigned.
Subclassification of FTLD-TDP
The FTLD-TDP group of disorders has been sub-classified based upon characteristic appearance and distribution of the TDP-43 inclusions [153, 154]. We recently expanded the analyses to include multiple subcortical regions of analysis, as was also done by Alafuzoff and co-workers [155]. Like that study, we also found that the classification scheme originally proposed by Mackenzie and co-workers had the best clinicopathologic correlations, and it also was associated with distinctive subcortical pathology [156]. The Mackenzie classification scheme originally took into account only two regions – cortex and hippocampus [154]. We have added amygdala, basal ganglia, thalamus, midbrain and medulla. Mackenzie type 1 (similar to Cairns type 3 [153]) is characterized by superficial cortical spongiosis with pleo-morphic TDP-43 neuronal inclusions and short curvilinear neurites in the same superificial layers (Figure 5b). The dentate fascia neurons are affected and have crescent shaped or round inclusions (Figure 5a). A distinctive histologic feature is the presence of delicate thin neurites in the pyramidal layer of the hippocampus, first reported by Hatanpaa [157] (Figure 5c). This form of FTLD-TDP is associated with the most widespread pathology, and there are often inclusions in the amygdala, basal ganglia, thalamus and brainstem. The clinical presentation is FTDbv, PNFA or occasionally CBS. Mutations in GRN are associated with this pathology and all such cases have neuronal intranuclear inclusions [158, 159].
Figure 5.
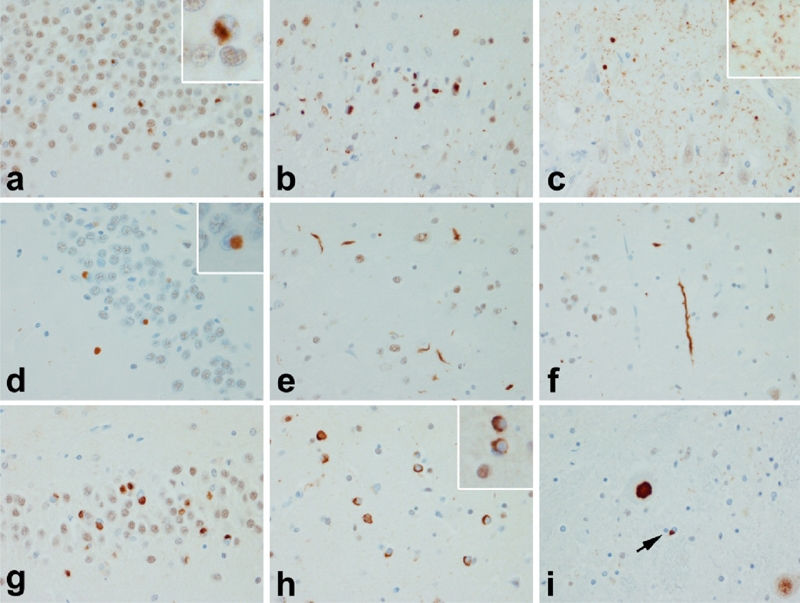
TDP-43 proteinopathies: a, b & c FTLD-TDP Type 1; d, e & f FTLD-TDP Type 2; g, h & I FTLD-TDP Type 3. Distinct patterns of TDP-43 pathology define subtypes of TDP-43 proteinopathies. In Type 1 there is widespread pathology in forebrain and hindbrain structures, with neurites and neuronal cytoplasmic inclusions (NCI) in hippocampal dentate fascia (a) and neocortex (b). A characteristic feature of many Type 1 cases is the presence of many small fine neurites in the pyramidal layer of the hippocampus (c). In Type 2 there are round dense NCI in the hippocampal dentate fascia (d) (as well as in the amygdala and basal ganglia), but predominantly long thick neurites in the cortex (e & f). The pathology is minimal in the hindbrain in Type 2. In Type 3 cases the predominant pathology is NCI with a paucity of dystrophic neurites. In addition to the hippocampus (g) and cortex (h), NCI are found in motor neurons of the brainstem and spinal cord (i). Inclusions in Type 3 are similar to those in ALS, with more widespread forebrain involvement in cases with dementia than in those with only motor neuron signs.
Mackenzie type 2 (Cairns type 1) FTLD-TDP is associated with temporal atrophy, especially affecting the dominant hemisphere and associated with SD. The hallmark histo-pathologic lesions are long, thick neurites (Figure 5f) with no cortical laminar predilection (Figure 5e) often involving the lower cortical layers. There are also Pick-body like inclusions in the dentate fascia (Figure 5d), amygdala and basal ganglia. This form of FTLD-TDP has minimal pathology in diencephalon or brainstem.
Mackenzie type 3 (Cairns type 1) is associated with FTLD with motor neuron disease and ALS. There are TDP-43 inclusions in neuronal cell bodies (Figure 5g), but few or no neurites. Some of the inclusions have diffuse granular cytoplasmic staining typical of “pre-inclusions’ (Figure 5h). The dentate fascia is variably affected. This form of FTLD-TDP has a predilection for the frontal cortex and has highly variable involvement in the lower neuroaxis, the exception being the regular involvement of the hypoglossal nucleus (Figure 5i), which is affected in cases with motor neuron. Glial inclusions (Figure 5i, arrow) are common in this type of FTLD-TDP.
TDP-43 pathology in AD
TDP-43 immunoreactive neurons are sometimes detected in the setting of other disorders, particularly AD [131, 160–162], where it may be seen in neurons with neurofibrillary tangles [131]. In many of these cases the TDP-43 pathology is relatively restricted to the limbic lobe and not associated with many dystrophic neurites. It is thus, similar to Mackenzie type 3, but given the co-localization in neurons with tau tangles and the absence of TDP-43 pathology in motor neurons, it is actually a distinct type of TDP-43 proteinopathy. At present, this tangle-associated TDP-43 has not been incorporated into classification schemes for FTLD-TDP and it could be argued that it is a distinct process from FTLD-TDP, analogous to the presence of α-synuclein in limbic lobe neurons, sometimes in neurons with tangles, in AD [81]. The proportion of AD cases with TDP-43 pathology ranges from 20 to 50% depending upon whether tangle-associated TDP-43 is included [131, 160, 163]. Except for our study, where we did not include tangle-associated TDP-43, none of the other reports has made this distinction. It is of interest that TDP-43 associated with Lewy body disorders is also so some extent a function of concurrent AD pathology [132, 160, 161]. Tangle associated TDP-43 appears to be a phenomenon that is relatively unique to 3R+4R tauopathies, such as AD, Guam PDC and in tangle predominant dementia. In a large series of PSP cases (over 250 cases), we have not seen TDP-43 pathology, except in cases with concurrent AD or hippocampal sclerosis (unpublished observations).
Hippocampal sclerosis (HpScl)
Hippocampal sclerosis (HpScl) is a pathologic finding characterized by neuronal loss in the subiculum and CA1 of the hippocampus. It is a common finding in elderly subjects with dementia, either alone or more often accompanied by other pathologic processes [164–168]. It is even more frequent in FTLD-U, where over 70% of cases have HpScl [169]. Conversely, in studies of HpScl, up to 12% of cases have ubiquitin-immunoreactive inclu-sions similar to FTLD-U [170]. Due to this strong association, when HpScl is detected, it is important to rule out FTLD-U. In our series of HpScl we detected TDP-43 immunoreactivity, most often similar to Mackenzie type 1, in over 70% of cases [131]. In contrast to FTLD-TDP, TDP-43 pathology in HpScl may be limited to limbic lobe structures, particularly the amygdala and entorhinal cortex, rather than being more widespread [131]. This suggests that TDP-43 in the setting of HpScl may be a forme fruste of FTLD-TDP. It is important to emphasize that HpScl that occurs in the setting of temporal lobe epilepsy or after cardiac arrest and anoxic brain injury is negative for TDP-43 [131, 171, 172].
When TDP-43 occurs in the setting of other well recognized neurodegenerative disorders, such as AD and HpScl, the significance of this finding is debated. Does it represent concurrent FTLD-TDP or co-deposition of fibrillogenic proteins in a vulnerable set of neurons? Experimental observations by Zhang, and co-workers suggests that TDP-43 becomes more fibrillogenic when it undergoes cleavage and that this cleavage can be promoted by apoptosis [173]. That TDP-43 is vulnerable to proteolytic cleavage comes from global mapping of proteolytic events in apoptosis [174]; TDP-43 is one of the many proteins that is cleaved during apoptosis. In addition, carboxyl terminal fragments of TDP-43 have been shown to be more toxic than full length protein and to have greater propensity to form inclusions [175, 176]. Thus, it could be hypothesized that under certain conditions of cell stress that lead to activation of proteolysis associated with programmed cell death, TDP-43 is cleaved. The cleaved TDP-43 fragments subsequently assemble into filaments aggregates [177]. Thus, TDP-43 inclusions could have more specificity with respect to mechanism of neurodegeneration than to disease type [178].
Concluding remarks
This brief overview of select aspects of non-AD neurodegenerative diseases highlights some common features of these clinically and pathologically diverse disorders. Among these key principles is the importance of abnormal protein conformers, particularly conformers with amyloid-like beta-sheet secondary structure that have a propensity to aggregate either in the extracellular domain or within the cytoplasm of neurons or glia, or both. Another important point is that for most, but not all, of these disorders mutations are found in the gene that encodes the abnormal protein that is found in these aggregates and genetic variants in these genes contribute to increased risk for the disease in sporadic cases. Several examples can be cited: mutations in MAPT give rise to FTLD-TAU [179], while genetic variants in MAPT predispose to the tauopathies PSP and CBD [180]; mutations in SNCA give rise to familial PD [87], while genetic variants in SNCA predispose to the α-synucleinopathies PD [181] and MSA [124]; while progranulin does not accumulate in FTLD-TDP, mutations in GRN give rise to FTLD-TDP [142] and genetic variants in GRN predispose to sporadic FTLD-TDP [182]. Further studies are needed to determine what determine non-genetic risk factors and if there are common mechanisms for selective neuronal vulnerability that is the defining feature of these disorders.
Acknowledgments
The authors thank Virginia Philips, Linda Rousseau and Monica Castanedes-Casey for their expert technical assistance and Wen-Lang Lin for ultrastructural studies. The author acknowledges the assistance of Dr. Tamas Revesz, University College London, for providing tissue samples of FBD and John Steele for providing tissue samples from Guam PDC. This study was supported by NIH grants P50-NS40256, P50-AG25711, P50-AG16574 and P01-AG17216, as well as the State of Florida Alzheimer Disease Initiative, Society for Progressive Supranuclear Palsy and Mayo Foundation for Education and Research.
References
- 1.Glenner GG. Amyloid deposits and amyloidosis: the beta-fibrilloses (second of two parts) N Engl J Med. 1980;302:1333–1343. doi: 10.1056/NEJM198006123022403. [DOI] [PubMed] [Google Scholar]
- 2.Glenner GG. Amyloid deposits and amyloidosis. The beta-fibrilloses (first of two parts) N Engl J Med. 1980;302:1283–1292. doi: 10.1056/NEJM198006053022305. [DOI] [PubMed] [Google Scholar]
- 3.Gu Y, Sanjo N, Chen F, Hasegawa H, Petit A, Ruan X, Li W, Shier C, Kawarai T, Schmitt-Ulms G, Westaway D, St George-Hyslop P, Fraser PE. The presenilin proteins are components of multiple membrane-bound complexes that have different biological activities. J Biol Chem. 2004;279:31329–31336. doi: 10.1074/jbc.M401548200. [DOI] [PubMed] [Google Scholar]
- 4.Hardy J. Has the amyloid cascade hypothesis for Alzheimer's disease been proved? Curr Alzheimer Res. 2006;3:71–73. doi: 10.2174/156720506775697098. [DOI] [PubMed] [Google Scholar]
- 5.Braak H, Braak E. Frequency of stages of Alzheimer-related lesions in different age cat-egories. Neurobiol Aging. 1997;18:351–357. doi: 10.1016/s0197-4580(97)00056-0. [DOI] [PubMed] [Google Scholar]
- 6.Dickson DW, Crystal HA, Mattiace LA, Masur DM, Blau AD, Davies P, Yen SH, Aronson MK. Identification of normal and pathological aging in prospectively studied nondemented elderly humans. Neurobiol Aging. 1992;13:179–189. doi: 10.1016/0197-4580(92)90027-u. [DOI] [PubMed] [Google Scholar]
- 7.Aizenstein HJ, Nebes RD, Saxton JA, Price JC, Mathis CA, Tsopelas ND, Ziolko SK, James JA, Snitz BE, Houck PR, Bi W, Cohen AD, Lopresti BJ, De Kosky ST, Halligan EM, Klunk WE. Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch Neurol. 2008;65:1509–1517. doi: 10.1001/archneur.65.11.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reiman EM, Chen K, Liu X, Bandy D, Yu M, Lee W, Ayutyanont N, Keppler J, Reeder SA, Langbaum JB, Alexander GE, Klunk WE, Mathis CA, Price JC, Aizenstein HJ, De Kosky ST, Caselli RJ. Fibrillar amyloid-beta burden in cognitively normal people at 3 levels of genetic risk for Alzheimer's disease. Proc Natl Acad Sci U S A. 2009;106:6820–6825. doi: 10.1073/pnas.0900345106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Josephs KA, Tsuboi Y, Cookson N, Watt H, Dickson DW. Apolipoprotein E epsilon 4 is a determinant for Alzheimer-type pathologic features in tauopathies, synucleinopathies, and frontotemporal degeneration. Arch Neurol. 2004;61:1579–1584. doi: 10.1001/archneur.61.10.1579. [DOI] [PubMed] [Google Scholar]
- 10.Strittmatter WJ, Roses AD. Apolipoprotein E and Alzheimer disease. Proc Natl Acad Sci USA. 1995;92:4725–4727. doi: 10.1073/pnas.92.11.4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dickson DW, Crystal H, Mattiace LA, Kress Y, Schwagerl A, Ksiezak-Reding H, Davies P, Yen SH. Diffuse Lewy body disease: light and electron microscopic immunocytochemistry of senile plaques. Acta Neuropathol. 1989;78:572–584. doi: 10.1007/BF00691284. [DOI] [PubMed] [Google Scholar]
- 12.Vidal R, Frangione B, Rostagno A, Mead S, Revesz T, Plant G, Ghiso J. A stop-codon mutation in the BRI gene associated with familial British dementia. Nature. 1999;399:776–781. doi: 10.1038/21637. [DOI] [PubMed] [Google Scholar]
- 13.Holton JL, Ghiso J, Lashley T, Rostagno A, Guerin CJ, Gibb G, Houlden H, Ayling H, Martinian L, Anderton BH, Wood NW, Vidal R, Plant G, Frangione B, Revesz T. Regional distribution of amyloid-Bri deposition and its association with neurofibrillary degeneration in familial British dementia. Am J Pathol. 2001;158:515–526. doi: 10.1016/S0002-9440(10)63993-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ironside JW, Head MW, Bell JE, McCardle L, Will RG. Laboratory diagnosis of variant Creutzfeldt-Jakob disease. Histopathology. 2000;37:1–9. doi: 10.1046/j.1365-2559.2000.00946.x. [DOI] [PubMed] [Google Scholar]
- 15.Capellari S, Vital C, Parchi P, Petersen RB, Ferrer X, Jarnier D, Pegoraro E, Gambetti P, Julien J. Familial prion disease with a novel 144-bp insertion in the prion protein gene in a Basque family. Neurology. 1997;49:133–141. doi: 10.1212/wnl.49.1.133. [DOI] [PubMed] [Google Scholar]
- 16.Bugiani O, Giaccone G, Piccardo P, Morbin M, Tagliavini F, Ghetti B. Neuropathology of Gerstmann-Straussler-Scheinker disease. Microsc Res Tech. 2000;50:10–15. doi: 10.1002/1097-0029(20000701)50:1<10::AID-JEMT3>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 17.Gajdusek DC, Zigas V. Kuru; clinical, pathological and epidemiological study of an acute progressive degenerative disease of the central nervous system among natives of the Eastern Highlands of New Guinea. Am J Med. 1959;26:442–469. doi: 10.1016/0002-9343(59)90251-7. [DOI] [PubMed] [Google Scholar]
- 18.Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O, Zerr I, Budka H, Kopp N, Piccardo P, Poser S, Rojiani A, Streichemberger N, Julien J, Vital C, Ghetti B, Gambetti P, Kretzschmar H. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol. 1999;46:224–233. [PubMed] [Google Scholar]
- 19.Bruce ME, Will RG, Ironside JW, McConnell I, Drummond D, Suttie A, McCardle L, Chree A, Hope J, Birkett C, Cousens S, Fraser H, Bostock CJ. Transmissions to mice indicate that ‘new variant’ CJD is caused by the BSE agent. Nature. 1997;389:498–501. doi: 10.1038/39057. [DOI] [PubMed] [Google Scholar]
- 20.Collinge J, Sidle KC, Meads J, Ironside J, Hill AF. Molecular analysis of prion strain vari-ation and the aetiology of ‘new variant’ CJD. Nature. 1996;383:685–690. doi: 10.1038/383685a0. [DOI] [PubMed] [Google Scholar]
- 21.Ghetti B, Dlouhy SR, Giaccone G, Bugiani O, Frangione B, Farlow MR, Tagliavini F. Gerstmann-Straussler-Scheinker disease and the Indiana kindred. Brain Pathol. 1995;5:61–75. doi: 10.1111/j.1750-3639.1995.tb00578.x. [DOI] [PubMed] [Google Scholar]
- 22.Giaccone G, Tagliavini F, Verga L, Frangione B, Farlow MR, Bugiani O, Ghetti B. Neuro-fibrillary tangles of the Indiana kindred of Gerstmann-Straussler-Scheinker disease share antigenic determinants with those of Alzheimer disease. Brain Res. 1990;530:325–329. doi: 10.1016/0006-8993(90)91304-y. [DOI] [PubMed] [Google Scholar]
- 23.Azzarelli B, Muller J, Ghetti B, Dyken M, Conneally PM. Cerebellar plaques in familial Alzheimer's disease (Gerstmann-Straussler-Scheinker variant?) Acta Neuropathol. 1985;65:235–246. doi: 10.1007/BF00687003. [DOI] [PubMed] [Google Scholar]
- 24.Grundke-Iqbal I, Iqbal K, Quinlan M, Tung YC, Zaidi MS, Wisniewski HM. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J Biol Chem. 1986;261:6084–6089. [PubMed] [Google Scholar]
- 25.Binder LI, Frankfurter A, Rebhun LI. The distribution of tau in the mammalian central nervous system. J Cell Biol. 1985;101:1371–1378. doi: 10.1083/jcb.101.4.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, Hillebrand M, Joosse M, Kwon JM, Nowotny P, Che LK, Norton J, Morris JC, Reed LA, Trojanowski J, Basun H, Lannfelt L, Neystat M, Fahn S, Dark F, Tannenberg T, Dodd PR, Hayward N, Kwok JB, Schofield PR, Andreadis A, Snowden J, Craufurd D, Neary D, Owen F, Oostra BA, Hardy J, Goate A, van Swieten J, Mann D, Lynch T, Heutink P. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 27.Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn-Hardy K, Paul Murphy M, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. 2000;25:402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- 28.Andreadis A, Brown WM, Kosik KS. Structure and novel exons of the human tau gene. Biochemistry. 1992;31:10626–10633. doi: 10.1021/bi00158a027. [DOI] [PubMed] [Google Scholar]
- 29.Goedert M, Spillantini MG, Potier MC, Ulrich J, Crowther RA. Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain. Embo J. 1989;8:393–399. doi: 10.1002/j.1460-2075.1989.tb03390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Togo T, Sahara N, Yen SH, Cookson N, Ishizawa T, Hutton M, de Silva R, Lees A, Dickson DW. Argyrophilic grain disease is a sporadic 4-repeat tauopathy. J Neuropathol Exp Neurol. 2002;61:547–556. doi: 10.1093/jnen/61.6.547. [DOI] [PubMed] [Google Scholar]
- 31.Tolnay M, Clavaguera F. Argyrophilic grain disease: a late-onset dementia with distinctive features among tauopathies. Neuropathology. 2004;24:269–283. doi: 10.1111/j.1440-1789.2004.00591.x. [DOI] [PubMed] [Google Scholar]
- 32.Jicha GA, Petersen RC, Knopman DS, Boeve BF, Smith GE, Geda YE, Johnson KA, Cha R, Delucia MW, Braak H, Dickson DW, Parisi JE. Argyrophilic grain disease in demented subjects presenting initially with amnestic mild cognitive impairment. J Neuropathol Exp Neurol. 2006;65:602–609. doi: 10.1097/01.jnen.0000225312.11858.57. [DOI] [PubMed] [Google Scholar]
- 33.Togo T, Cookson N, Dickson DW. Argyrophilic grain disease: neuropathology, frequency in a dementia brain bank and lack of relationship with apolipoprotein. E Brain Pathol. 2002;12:45–52. doi: 10.1111/j.1750-3639.2002.tb00421.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Braak H, Braak E. Argyrophilic grain disease: frequency of occurrence in different age categories and neuropathological diagnostic criteria. J Neural Transm. 1998;105:801–819. doi: 10.1007/s007020050096. [DOI] [PubMed] [Google Scholar]
- 35.Tolnay M, Mistl C, Ipsen S, Probst A. Argyrophilic grains of Braak: occurrence in dendrites of neurons containing hyperphosphorylated tau protein. Neuropathol Appl Neurobiol. 1998;24:53–59. doi: 10.1046/j.1365-2990.1998.00090.x. [DOI] [PubMed] [Google Scholar]
- 36.Braak H, Braak E. Argyrophilic grains: characteristic pathology of cerebral cortex in cases of adult onset dementia without Alzheimer changes. Neurosci Lett. 1987;76:124–127. doi: 10.1016/0304-3940(87)90204-7. [DOI] [PubMed] [Google Scholar]
- 37.Botez G, Probst A, Ipsen S, Tolnay M. Astrocytes expressing hyperphosphorylated tau protein without glial fibrillary tangles in argyrophilic grain disease. Acta Neuropathol. 1999;98:251–256. doi: 10.1007/s004010051077. [DOI] [PubMed] [Google Scholar]
- 38.Tolnay M, Probst A. Ballooned neurons expressing alphaB-crystallin as a constant feature of the amygdala in argyrophilic grain disease. Neurosci Lett. 1998;246:165–168. doi: 10.1016/s0304-3940(98)00250-x. [DOI] [PubMed] [Google Scholar]
- 39.Fujino Y, Wang DS, Thomas N, Espinoza M, Davies P, Dickson DW. Increased frequency of argyrophilic grain disease in Alzheimer disease with 4R tau-specific immunohistochemistry. J Neuropathol Exp Neurol. 2005;64:209–214. doi: 10.1093/jnen/64.3.209. [DOI] [PubMed] [Google Scholar]
- 40.Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology. 2007;69:2197–2204. doi: 10.1212/01.wnl.0000271090.28148.24. [DOI] [PubMed] [Google Scholar]
- 41.Petersen RC, Parisi JE, Dickson DW, Johnson KA, Knopman DS, Boeve BF, Jicha GA, Ivnik RJ, Smith GE, Tangalos EG, Braak H, Kokmen E. Neuropathologic features of amnestic mild cognitive impairment. Arch Neurol. 2006;63:665–672. doi: 10.1001/archneur.63.5.665. [DOI] [PubMed] [Google Scholar]
- 42.Maurage CA, Sergeant N, Schraen-Maschke S, Lebert F, Ruchoux MM, Sablonniere B, Pasquier F, Delacourte A. Diffuse form of argyrophilic grain disease: a new variant of four-repeat tauopathy different from limbic argyrophilic grain disease. Acta Neuropathol. 2003;106:575–583. doi: 10.1007/s00401-003-0762-6. [DOI] [PubMed] [Google Scholar]
- 43.Litvan I, Grimes DA, Lang AE. Phenotypes and prognosis: clinicopathologic studies of corticobasal degeneration. Adv Neurol. 2000;82:183–196. [PubMed] [Google Scholar]
- 44.Kertesz A, Martinez-Lage P, Davidson W, Munoz DG. The corticobasal degeneration syndrome overlaps progressive aphasia and frontotemporal dementia. Neurology. 2000;55:1368–1375. doi: 10.1212/wnl.55.9.1368. [DOI] [PubMed] [Google Scholar]
- 45.Dickson DW, Bergeron C, Chin SS, Duyckaerts C, Horoupian D, Ikeda K, Jellinger K, Lantos PL, Lippa CF, Mirra SS, Tabaton M, Vonsattel JP, Wakabayashi K, Litvan I. Office of Rare Diseases neuropathologic criteria for cortico-basal degeneration. J Neuropathol Exp Neurol. 2002;61:935–946. doi: 10.1093/jnen/61.11.935. [DOI] [PubMed] [Google Scholar]
- 46.Feany MB, Dickson DW. Widespread cytoskeletal pathology characterizes cortico-basal degeneration. Am J Pathol. 1995;146:1388–1396. [PMC free article] [PubMed] [Google Scholar]
- 47.Komori T, Arai N, Oda M, Nakayama H, Mori H, Yagishita S, Takahashi T, Amano N, Murayama S, Murakami S, Shibata N, Kobayashi M, Sasaki S, Iwata M. Astrocytic plaques and tufts of abnormal fibers do not coexist in corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol. 1998;96:401–408. doi: 10.1007/s004010050911. [DOI] [PubMed] [Google Scholar]
- 48.Rebeiz JJ, Kolodny EH, Richardson EP., Jr Corticodentatonigral degeneration with neuronal achromasia: a progressive disorder of late adult life. Trans Am Neurol Assoc. 1967;92:23–26. [PubMed] [Google Scholar]
- 49.Steele JC, Richardson JC, Olszewski J. Progressive Supranuclear Palsy. a Heterogeneous Degeneration Involving the Brain Stem, Basal Ganglia and Cerebellum with Vertical Gaze and Pseudobulbar Palsy, Nuchal Dystonia and Dementia. Arch Neurol. 1964;10:333–359. doi: 10.1001/archneur.1964.00460160003001. [DOI] [PubMed] [Google Scholar]
- 50.Bigio EH, Brown DF, White CL., 3rd Progressive supranuclear palsy with dementia: cortical pathology. J Neuropathol Exp Neurol. 1999;58:359–364. doi: 10.1097/00005072-199904000-00006. [DOI] [PubMed] [Google Scholar]
- 51.Josephs KA, Boeve BF, Duffy JR, Smith GE, Knopman DS, Parisi JE, Petersen RC, Dickson DW. Atypical progressive supranuclear palsy underlying progressive apraxia of speech and nonfluent aphasia. Neurocase. 2005;11:283–296. doi: 10.1080/13554790590963004. [DOI] [PubMed] [Google Scholar]
- 52.Josephs KA, Katsuse O, Beccano-Kelly DA, Lin WL, Uitti RJ, Fujino Y, Boeve BF, Hutton ML, Baker MC, Dickson DW. Atypical progressive supranuclear palsy with cortico-spinal tract degeneration. J Neuropathol Exp Neurol. 2006;65:396–405. doi: 10.1097/01.jnen.0000218446.38158.61. [DOI] [PubMed] [Google Scholar]
- 53.Ahmed Z, Josephs KA, Gonzalez J, DelleDonne A, Dickson DW. Clinical and neuropathologic features of progressive supra-nuclear palsy with severe pallido-nigroluysial degeneration and axonal dystrophy. Brain. 2008;131:460–472. doi: 10.1093/brain/awm301. [DOI] [PubMed] [Google Scholar]
- 54.Williams DR, Holton JL, Strand K, Revesz T, Lees AJ. Pure akinesia with gait freezing: a third clinical phenotype of progressive supranuclear palsy. Mov Disord. 2007;22:2235–2241. doi: 10.1002/mds.21698. [DOI] [PubMed] [Google Scholar]
- 55.Williams DR, de Silva R, Paviour DC, Pittman A, Watt HC, Kilford L, Holton JL, Revesz T, Lees AJ. Characteristics of two distinct clinical phenotypes in pathologically proven progressive supranuclear palsy: Richardson's syndrome and PSP-parkinsonism. Brain. 2005;128:1247–1258. doi: 10.1093/brain/awh488. [DOI] [PubMed] [Google Scholar]
- 56.Tsuboi Y, Josephs KA, Boeve BF, Litvan I, Caselli RJ, Caviness JN, Uitti RJ, Bott AD, Dickson DW. Increased tau burden in the cortices of progressive supranuclear palsy presenting with corticobasal syndrome. Mov Disord. 2005;20:982–988. doi: 10.1002/mds.20478. [DOI] [PubMed] [Google Scholar]
- 57.Hauw JJ, Daniel SE, Dickson D, Horoupian DS, Jellinger K, Lantos PL, McKee A, Tabaton M, Litvan I. Preliminary NINDS neuro-pathologic criteria for Steele-Richardson-Olszewski syndrome (progressive supranuclear palsy) Neurology. 1994;44:2015–2019. doi: 10.1212/wnl.44.11.2015. [DOI] [PubMed] [Google Scholar]
- 58.Tsuboi Y, Slowinski J, Josephs KA, Honer WG, Wszolek ZK, Dickson DW. Atrophy of superior cerebellar peduncle in progressive supranuclear palsy. Neurology. 2003;60:1766–1769. doi: 10.1212/01.wnl.0000068011.21396.f4. [DOI] [PubMed] [Google Scholar]
- 59.Nilsson C, Markenroth Bloch K, Brockstedt S, Latt J, Widner H, Larsson EM. Tracking the neurodegeneration of parkinsonian disorders – a pilot study. Neuroradiology. 2007;49:111–119. doi: 10.1007/s00234-006-0165-1. [DOI] [PubMed] [Google Scholar]
- 60.Yamada T, McGeer PL, McGeer EG. Appearance of paired nucleated, Tau-positive glia in patients with progressive supranuclear palsy brain tissue. Neurosci Lett. 1992;135:99–102. doi: 10.1016/0304-3940(92)90145-w. [DOI] [PubMed] [Google Scholar]
- 61.Barker WW, Luis CA, Kashuba A, Luis M, Harwood DG, Loewenstein D, Waters C, Jimison P, Shepherd E, Sevush S, Graff-Radford N, Newland D, Todd M, Miller B, Gold M, Heilman K, Doty L, Goodman I, Robinson B, Pearl G, Dickson D, Duara R. Relative frequencies of Alzheimer disease, Lewy body, vascular and frontotemporal dementia, and hippocampal sclerosis in the State of Florida Brain Bank. Alzheimer Dis Assoc Disord. 2002;16:203–212. doi: 10.1097/00002093-200210000-00001. [DOI] [PubMed] [Google Scholar]
- 62.Bronner IF, ter Meulen BC, Azmani A, Severijnen LA, Willemsen R, Kamphorst W, Ravid R, Heutink P, van Swieten JC. Hereditary Pick's disease with the G272V tau mutation shows predominant three-repeat tau pathology. Brain. 2005;128:2645–2653. doi: 10.1093/brain/awh591. [DOI] [PubMed] [Google Scholar]
- 63.Hogg M, Grujic ZM, Baker M, Demirci S, Guillozet AL, Sweet AP, Herzog LL, Weintraub S, Mesulam MM, LaPointe NE, Gamblin TC, Berry RW, Binder LI, de Silva R, Lees A, Espinoza M, Davies P, Grover A, Sahara N, Ishizawa T, Dickson D, Yen SH, Hutton M, Bigio EH. The L266V tau mutation is associated with frontotemporal dementia and Pick-like 3R and 4R tauopathy. Acta Neuropathol. 2003;106:323–336. doi: 10.1007/s00401-003-0734-x. [DOI] [PubMed] [Google Scholar]
- 64.Buee L, Delacourte A. Comparative bio-chemistry of tau in progressive supranuclear palsy, corticobasal degeneration, FTDP-17 and Pick's disease. Brain Pathol. 1999;9:681–693. doi: 10.1111/j.1750-3639.1999.tb00550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.de Silva R, Lashley T, Strand C, Shiarli AM, Shi J, Tian J, Bailey KL, Davies P, Bigio EH, Arima K, Iseki E, Murayama S, Kretzschmar H, Neumann M, Lippa C, Halliday G, MacKen-zie J, Ravid R, Dickson D, Wszolek Z, Iwatsubo T, Pickering-Brown SM, Holton J, Lees A, Revesz T, Mann DM. An immunohistochemical study of cases of sporadic and inherited frontotemporal lobar degeneration using 3R- and 4R-specific tau monoclonal antibodies. Acta Neuropathol. 2006;111:329–340. doi: 10.1007/s00401-006-0048-x. [DOI] [PubMed] [Google Scholar]
- 66.Zhukareva V, Mann D, Pickering-Brown S, Uryu K, Shuck T, Shah K, Grossman M, Miller BL, Hulette CM, Feinstein SC, Trojanowski JQ, Lee VM. Sporadic Pick's disease: a tauopathy characterized by a spectrum of pathological tau isoforms in gray and white matter. Ann Neurol. 2002;51:730–739. doi: 10.1002/ana.10222. [DOI] [PubMed] [Google Scholar]
- 67.Yoshimura N. Topography of Pick body distribution in Pick's disease: a contribution to understanding the relationship between Pick's and Alzheimer's diseases. Clin Neuropathol. 1989;8:1–6. [PubMed] [Google Scholar]
- 68.Hansen LA, Deteresa R, Tobias H, Alford M, Terry RD. Neocortical morphometry and cholinergic neurochemistry in Pick's disease. Am J Pathol. 1988;131:507–518. [PMC free article] [PubMed] [Google Scholar]
- 69.Iseki E, Yamamoto R, Murayama N, Minegishi M, Togo T, Katsuse O, Kosaka K, Akiyama H, Tsuchiya K, de Silva R, Andrew L, Arai H. Immunohistochemical investigation of neuro-fibrillary tangles and their tau isoforms in brains of limbic neurofibrillary tangle dementia. Neurosci Lett. 2006;405:29–33. doi: 10.1016/j.neulet.2006.06.036. [DOI] [PubMed] [Google Scholar]
- 70.Ikeda K, Akiyama H, Arai T, Oda T, Kato M, Iseki E, Kosaka K, Wakabayashi K, Takahashi H. Clinical aspects of ‘senile dementia of the tangle type’– a subset of dementia in the senium separable from late-onset Alzheimer's disease. Dement Geriatr Cogn Disord. 1999;10:6–11. doi: 10.1159/000017091. [DOI] [PubMed] [Google Scholar]
- 71.Ikeda K, Akiyama H, Arai T, Sahara N, Mori H, Usami M, Sakata M, Mizutani T, Wakabayashi K, Takahashi H. A subset of senile dementia with high incidence of the apolipo-protein E epsilon2 allele. Ann Neurol. 1997;41:693–695. doi: 10.1002/ana.410410522. [DOI] [PubMed] [Google Scholar]
- 72.Jellinger KA, Attems J. Neurofibrillary tangle-predominant dementia: comparison with classical Alzheimer disease. Acta Neuropathol. 2007;113:107–117. doi: 10.1007/s00401-006-0156-7. [DOI] [PubMed] [Google Scholar]
- 73.Jellinger KA, Bancher C. Senile dementia with tangles (tangle predominant form of senile dementia) Brain Pathol. 1998;8:367–376. doi: 10.1111/j.1750-3639.1998.tb00160.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Braak H, Braak E. Neurofibrillary changes confined to the entorhinal region and an abundance of cortical amyloid in cases of presenile and senile dementia. Acta Neuropathol. 1990;80:479–486. doi: 10.1007/BF00294607. [DOI] [PubMed] [Google Scholar]
- 75.Hirano A, Kurland LT, Krooth RS, Lessell S. Parkinsonism-dementia complex, an endemic disease on the island of Guam. I. Clinical features. Brain. 1961;84:642–661. doi: 10.1093/brain/84.4.642. [DOI] [PubMed] [Google Scholar]
- 76.Hirano A, Malamud N, Kurland LT. Parkinsonism-dementia complex, an endemic disease on the island of Guam. II. Pathological features. Brain. 1961;84:662–679. doi: 10.1093/brain/84.4.662. [DOI] [PubMed] [Google Scholar]
- 77.Kuzuhara S, Kokubo Y. Atypical parkinsonism of Japan: amyotrophic lateral sclerosis-parkinsonism-dementia complex of the Kii peninsula of Japan (Muro disease): an update. Mov Disord. 2005;20(Suppl 12):S108–113. doi: 10.1002/mds.20548. [DOI] [PubMed] [Google Scholar]
- 78.Morris HR, Steele JC, Crook R, Wavrant-De Vrieze F, Onstead-Cardinale L, Gwinn-Hardy K, Wood NW, Farrer M, Lees AJ, McGeer PL, Siddique T, Hardy J, Perez-Tur J. Genome-wide analysis of the parkinsonism-dementia complex of Guam. Arch Neurol. 2004;61:1889–1897. doi: 10.1001/archneur.61.12.1889. [DOI] [PubMed] [Google Scholar]
- 79.Buee-Scherrer V, Buee L, Hof PR, Leveugle B, Gilles C, Loerzel AJ, Perl DP, Delacourte A. Neurofibrillary degeneration in amyotrophic lateral sclerosis/parkinsonism-dementia complex of Guam. Immunochemical characterization of tau proteins. Am J Pathol. 1995;146:924–932. [PMC free article] [PubMed] [Google Scholar]
- 80.Forman MS, Schmidt ML, Kasturi S, Perl DP, Lee VM, Trojanowski JQ. Tau and alpha-synuclein pathology in amygdala of Parkinsonism-dementia complex patients of Guam. Am J Pathol. 2002;160:1725–1731. doi: 10.1016/s0002-9440(10)61119-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Uchikado H, Lin WL, DeLucia MW, Dickson DW. Alzheimer disease with amygdala Lewy bodies: a distinct form of alpha-synucleinopathy. J Neuropathol Exp Neurol. 2006;65:685–697. doi: 10.1097/01.jnen.0000225908.90052.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hasegawa M, Arai T, Akiyama H, Nonaka T, Mori H, Hashimoto T, Yamazaki M, Oyanagi K. TDP-43 is deposited in the Guam parkinsonism-dementia complex brains. Brain. 2007;130:1386–1394. doi: 10.1093/brain/awm065. [DOI] [PubMed] [Google Scholar]
- 83.Geser F, Winton MJ, Kwong LK, Xu Y, Xie SX, Igaz LM, Garruto RM, Perl DP, Galasko D, Lee VM, Trojanowski JQ. Pathological TDP-43 in parkinsonism-dementia complex and amyotrophic lateral sclerosis of Guam. Acta Neuropathol. 2008;115:133–145. doi: 10.1007/s00401-007-0257-y. [DOI] [PubMed] [Google Scholar]
- 84.Clayton DF, George JM. The synucleins: a family of proteins involved in synaptic function, plasticity, neurodegeneration and disease. Trends Neurosci. 1998;21:249–254. doi: 10.1016/s0166-2236(97)01213-7. [DOI] [PubMed] [Google Scholar]
- 85.Iwai A, Masliah E, Sundsmo MP, De Teresa R, Mallory M, Salmon DP, Saitoh T. The synaptic protein NACP is abnormally expressed during the progression of Alzheimer's disease. Brain Res. 1996;720:230–234. doi: 10.1016/0006-8993(96)00014-5. [DOI] [PubMed] [Google Scholar]
- 86.Masliah E, Iwai A, Mallory M, Ueda K, Saitoh T. Altered presynaptic protein NACP is associated with plaque formation and neuro-degeneration in Alzheimer's disease. Am J Pathol. 1996;148:201–210. [PMC free article] [PubMed] [Google Scholar]
- 87.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 88.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 89.Gibb WR. Idiopathic Parkinson's disease and the Lewy body disorders. Neuropathol Appl Neurobiol. 1986;12:223–234. doi: 10.1111/j.1365-2990.1986.tb00136.x. [DOI] [PubMed] [Google Scholar]
- 90.Popescu A, Lippa CF, Lee VM, Trojanowski JQ. Lewy bodies in the amygdala: increase of alpha-synuclein aggregates in neurodegenerative diseases with tau-based inclusions. Arch Neurol. 2004;61:1915–1919. doi: 10.1001/archneur.61.12.1915. [DOI] [PubMed] [Google Scholar]
- 91.Uchikado H, Dickson DW. Presence of Lewy bodies in progressive supranuclear palsy represents an independent disease process. J Neuropathol Exp Neurol. 2005;64:450–450. doi: 10.1097/01.jnen.0000218449.17073.43. [DOI] [PubMed] [Google Scholar]
- 92.Lippa CF, Schmidt ML, Lee VM, Trojanowski JQ. Antibodies to alpha-synuclein detect Lewy bodies in many Down's syndrome brains with Alzheimer's disease. Ann Neurol. 1999;45:353–357. doi: 10.1002/1531-8249(199903)45:3<353::aid-ana11>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 93.Hamilton RL. Lewy bodies in Alzheimer's disease: a neuropathological review of 145 cases using alpha-synuclein immunohistochemistry. Brain Pathol. 2000;10:378–384. doi: 10.1111/j.1750-3639.2000.tb00269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Newell KL, Boyer P, Gomez-Tortosa E, Hobbs W, Hedley-Whyte ET, Vonsattel JP, Hyman BT. Alpha-synuclein immunoreactivity is present in axonal swellings in neuroaxonal dystrophy and acute traumatic brain injury. J Neuropathol Exp Neurol. 1999;58:1263–1268. doi: 10.1097/00005072-199912000-00007. [DOI] [PubMed] [Google Scholar]
- 95.Arawaka S, Saito Y, Murayama S, Mori H. Lewy body in neurodegeneration with brain iron accumulation type 1 is immunoreactive for alpha-synuclein. Neurology. 1998;51:887–889. doi: 10.1212/wnl.51.3.887. [DOI] [PubMed] [Google Scholar]
- 96.Greffard S, Verny M, Bonnet AM, Beinis JY, Gallinari C, Meaume S, Piette F, Hauw JJ, Duyckaerts C. Motor score of the Unified Parkinson Disease Rating Scale as a good predictor of Lewy body-associated neuronal loss in the substantia nigra. Arch Neurol. 2006;63:584–588. doi: 10.1001/archneur.63.4.584. [DOI] [PubMed] [Google Scholar]
- 97.Boeve BF, Silber MH, Ferman TJ, Lucas JA, Parisi JE. Association of REM sleep behavior disorder and neurodegenerative disease may reflect an underlying synucleinopathy. Mov Disord. 2001;16:622–630. doi: 10.1002/mds.1120. [DOI] [PubMed] [Google Scholar]
- 98.Miyamoto T, Miyamoto M, Inoue Y, Usui Y, Suzuki K, Hirata K. Reduced cardiac 123I-MIBG scintigraphy in idiopathic REM sleep behavior disorder. Neurology. 2006;67:2236–2238. doi: 10.1212/01.wnl.0000249313.25627.2e. [DOI] [PubMed] [Google Scholar]
- 99.Stiasny-Kolster K, Doerr Y, Moller JC, Hoffken H, Behr TM, Oertel WH, Mayer G. Combination of ‘idiopathic’ REM sleep behaviour disorder and olfactory dysfunction as possible indicator for alpha-synucleinopathy demonstrated by dopamine transporter FPCIT-SPECT. Brain. 2005;128:126–137. doi: 10.1093/brain/awh322. [DOI] [PubMed] [Google Scholar]
- 100.Aarsland D, Perry R, Brown A, Larsen JP, Ballard C. Neuropathology of dementia in Parkinson's disease: a prospective, community-based study. Ann Neurol. 2005;58:773–776. doi: 10.1002/ana.20635. [DOI] [PubMed] [Google Scholar]
- 101.Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 102.Braak H, Bohl JR, Muller CM, Rub U, de Vos RA, Del Tredici K. Stanley Fahn Lecture 2005: The staging procedure for the inclusion body pathology associated with sporadic Parkinson's disease reconsidered. Mov Disord. 2006;21:2042–2051. doi: 10.1002/mds.21065. [DOI] [PubMed] [Google Scholar]
- 103.Braak H, Del Tredici K. Neuroanatomy and pathology of sporadic Parkinson's disease. Adv Anat Embryol Cell Biol. 2009;201:1–119. [PubMed] [Google Scholar]
- 104.Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci USA. 2009;106:13010–13015. doi: 10.1073/pnas.0903691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, Lashley T, Quinn NP, Rehncrona S, Bjorklund A, Widner H, Revesz T, Lindvall O, Brundin P. Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host-to-graft disease propagation. Nat Med. 2008;14:501–503. doi: 10.1038/nm1746. [DOI] [PubMed] [Google Scholar]
- 106.Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson's disease. Nat Med. 2008;14:504–506. doi: 10.1038/nm1747. [DOI] [PubMed] [Google Scholar]
- 107.Frost B, Diamond MI. The expanding realm of prion phenomena in neurodegenerative disease. Prion. 2009;3:74–77. doi: 10.4161/pri.3.2.8754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.McKeith I, Mintzer J, Aarsland D, Burn D, Chiu H, Cohen-Mansfield J, Dickson D, Dubois B, Duda JE, Feldman H, Gauthier S, Halliday G, Lawlor B, Lippa C, Lopez OL, Carlos Machado J, O'Brien J, Playfer J, Reid W. Dementia with Lewy bodies. Lancet Neurol. 2004;3:19–28. doi: 10.1016/s1474-4422(03)00619-7. [DOI] [PubMed] [Google Scholar]
- 109.McKeith IG, Dickson DW, Lowe J, Emre M, O'Brien JT, Feldman H, Cummings J, Duda JE, Lippa C, Perry EK, Aarsland D, Arai H, Ballard CG, Boeve B, Burn DJ, Costa D, Del Ser T, Dubois B, Galasko D, Gauthier S, Goetz CG, Gomez-Tortosa E, Halliday G, Hansen LA, Hardy J, Iwatsubo T, Kalaria RN, Kaufer D, Kenny RA, Korczyn A, Kosaka K, Lee VM, Lees A, Litvan I, Londos E, Lopez OL, Minoshima S, Mizuno Y, Molina JA, Mukaetova-Ladinska EB, Pasquier F, Perry RH, Schulz JB, Trojanowski JQ, Yamada M. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65:1863–1872. doi: 10.1212/01.wnl.0000187889.17253.b1. [DOI] [PubMed] [Google Scholar]
- 110.Lippa CF, Duda JE, Grossman M, Hurtig HI, Aarsland D, Boeve BF, Brooks DJ, Dickson DW, Dubois B, Emre M, Fahn S, Farmer JM, Galasko D, Galvin JE, Goetz CG, Growdon JH, Gwinn-Hardy KA, Hardy J, Heutink P, Iwatsubo T, Kosaka K, Lee VM, Leverenz JB, Masliah E, McKeith IG, Nussbaum RL, Olanow CW, Ravina BM, Singleton AB, Tanner CM, Trojanowski JQ, Wszolek ZK. DLB and PDD boundary issues: diagnosis, treatment, molecular pathology, and biomarkers. Neurology. 2007;68:812–819. doi: 10.1212/01.wnl.0000256715.13907.d3. [DOI] [PubMed] [Google Scholar]
- 111.Fujishiro H, Ferman TJ, Boeve BF, Smith GE, Graff-Radford NR, Uitti RJ, Wszolek ZK, Knopman DS, Petersen RC, Parisi JE, Dickson DW. Validation of the neuropathologic criteria of the third consortium for dementia with Lewy bodies for prospectively diagnosed cases. J Neuropathol Exp Neurol. 2008;67:649–656. doi: 10.1097/NEN.0b013e31817d7a1d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Saito Y, Kawashima A, Ruberu NN, Fujiwara H, Koyama S, Sawabe M, Arai T, Nagura H, Yamanouchi H, Hasegawa M, Iwatsubo T, Murayama S. Accumulation of phosphorylated alpha-synuclein in aging human brain. J Neuropathol Exp Neurol. 2003;62:644–654. doi: 10.1093/jnen/62.6.644. [DOI] [PubMed] [Google Scholar]
- 113.Dickson DW, Ruan D, Crystal H, Mark MH, Davies P, Kress Y, Yen SH. Hippocampal degeneration differentiates diffuse Lewy body disease (DLBD) from Alzheimer's disease: light and electron microscopic immunocyto-chemistry of CA2–3 neurites specific to DLBD. Neurology. 1991;41:1402–1409. doi: 10.1212/wnl.41.9.1402. [DOI] [PubMed] [Google Scholar]
- 114.Dickson DW, Schmidt ML, Lee VM, Zhao ML, Yen SH, Trojanowski JQ. Immunoreactivity profile of hippocampal CA2/3 neurites in diffuse Lewy body disease. Acta Neuropathol. 1994;87:269–276. doi: 10.1007/BF00296742. [DOI] [PubMed] [Google Scholar]
- 115.Bertrand E, Lechowicz W, Szpak GM, Lewandowska E, Dymecki J, Wierzba-Bobrowicz T. Limbic neuropathology in idiopathic Parkinson's disease with concomitant dementia. Folia Neuropathol. 2004;42:141–150. [PubMed] [Google Scholar]
- 116.Apaydin H, Ahlskog JE, Parisi JE, Boeve BF, Dickson DW. Parkinson disease neuropathology: later-developing dementia and loss of the levodopa response. Arch Neurol. 2002;59:102–112. doi: 10.1001/archneur.59.1.102. [DOI] [PubMed] [Google Scholar]
- 117.Churchyard A, Lees AJ. The relationship be-tween dementia and direct involvement of the hippocampus and amygdala in Parkinson's disease. Neurology. 1997;49:1570–1576. doi: 10.1212/wnl.49.6.1570. [DOI] [PubMed] [Google Scholar]
- 118.Hurtig HI, Trojanowski JQ, Galvin J, Ewbank D, Schmidt ML, Lee VM, Clark CM, Glosser G, Stern MB, Gollomp SM, Arnold SE. Alpha-synuclein cortical Lewy bodies correlate with dementia in Parkinson's disease. Neurology. 2000;54:1916–1921. doi: 10.1212/wnl.54.10.1916. [DOI] [PubMed] [Google Scholar]
- 119.Colosimo C, Hughes AJ, Kilford L, Lees AJ. Lewy body cortical involvement may not always predict dementia in Parkinson's disease. J Neurol Neurosurg Psychiatry. 2003;74:852–856. doi: 10.1136/jnnp.74.7.852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Parkkinen L, Kauppinen T, Pirttila T, Autere JM, Alafuzoff I. Alpha-synuclein pathology does not predict extrapyramidal symptoms or dementia. Ann Neurol. 2005;57:82–91. doi: 10.1002/ana.20321. [DOI] [PubMed] [Google Scholar]
- 121.Zaccai J, Brayne C, McKeith I, Matthews F, Ince PG. Patterns and stages of alpha-synucleinopathy: Relevance in a population-based cohort. Neurology. 2008;70:1042–1048. doi: 10.1212/01.wnl.0000306697.48738.b6. [DOI] [PubMed] [Google Scholar]
- 122.Gwinn-Hardy K, Mehta ND, Farrer M, Maraganore D, Muenter M, Yen SH, Hardy J, Dickson DW. Distinctive neuropathology revealed by alpha-synuclein antibodies in hereditary parkinsonism and dementia linked tochromosome 4p. Acta Neuropathol (Berl) 2000;99:663–672. doi: 10.1007/s004010051177. [DOI] [PubMed] [Google Scholar]
- 123.Gilman S, Low PA, Quinn N, Albanese A, Ben-Shlomo Y, Fowler CJ, Kaufmann H, Klockgether T, Lang AE, Lantos PL, Litvan I, Mathias CJ, Oliver E, Robertson D, Schatz I, Wenning GK. Consensus statement on the diagnosis of multiple system atrophy. J Neurol Sci. 1999;163:94–98. doi: 10.1016/s0022-510x(98)00304-9. [DOI] [PubMed] [Google Scholar]
- 124.Scholz SW, Houlden H, Schulte C, Sharma M, Li A, Berg D, Melchers A, Paudel R, Gibbs JR, Simon-Sanchez J, Paisan-Ruiz C, Bras J, Ding J, Chen H, Traynor BJ, Arepalli S, Zonozi RR, Revesz T, Holton J, Wood N, Lees A, Oertel W, Wullner U, Goldwurm S, Pellecchia MT, Illig T, Riess O, Fernandez HH, Rodriguez RL, Okun MS, Poewe W, Wenning GK, Hardy JA, Singleton AB, Gasser T. SNCA variants are associated with increased risk for multiple system atrophy. Ann Neurol. 2009;65:610–614. doi: 10.1002/ana.21685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lantos PL. The definition of multiple system atrophy: a review of recent developments. J Neuropathol Exp Neurol. 1998;57:1099–1111. doi: 10.1097/00005072-199812000-00001. [DOI] [PubMed] [Google Scholar]
- 126.Lin WL, De Lucia MW, Dickson DW. Alpha-synuclein immunoreactivity in neuronal nuclear inclusions and neurites in multiple system atrophy. Neurosci Lett. 2004;354:99–102. doi: 10.1016/j.neulet.2003.09.075. [DOI] [PubMed] [Google Scholar]
- 127.Wang IF, Wu LS, Shen CK. TDP-43: an emerging new player in neurodegenerative diseases. Trends Mol Med. 2008;14:479–485. doi: 10.1016/j.molmed.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 128.Wang IF, Wu LS, Chang HY, Shen CK. TDP-43, the signature protein of FTLD-U, is a neu-ronal activity-responsive factor. J Neurochem. 2008;105:797–806. doi: 10.1111/j.1471-4159.2007.05190.x. [DOI] [PubMed] [Google Scholar]
- 129.Yamasaki S, Anderson P. Reprogramming mRNA translation during stress. Curr Opin Cell Biol. 2008;20:222–226. doi: 10.1016/j.ceb.2008.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Ubiquitinated TDP-43 in fronto-temporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 131.Amador-Ortiz C, Lin WL, Ahmed Z, Personett D, Davies P, Duara R, Graff-Radford NR, Hutton ML, Dickson DW. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer's disease. Ann Neurol. 2007;61:435–445. doi: 10.1002/ana.21154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Nakashima-Yasuda H, Uryu K, Robinson J, Xie SX, Hurtig H, Duda JE, Arnold SE, Siderowf A, Grossman M, Leverenz JB, Woltjer R, Lopez OL, Hamilton R, Tsuang DW, Galasko D, Masliah E, Kaye J, Clark CM, Montine TJ, Lee VM, Trojanowski JQ. Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases. Acta Neuropathol. 2007;114:221–229. doi: 10.1007/s00401-007-0261-2. [DOI] [PubMed] [Google Scholar]
- 133.Kato S, Shaw P, Wood-Allum C, Leigh PN, Shaw C. Amyotrophic lateral sclerosis. In: Dickson DW, editor. Neurodegeneration: The molecular pathology of dementia and movement disorders. Basel: ISN Neuropath Press; 2003. pp. 350–368. [Google Scholar]
- 134.Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 135.Greenway MJ, Andersen PM, Russ C, Ennis S, Cashman S, Donaghy C, Patterson V, Swingler R, Kieran D, Prehn J, Morrison KE, Green A, Acharya KR, Brown RH, Jr, Hardiman O. ANG mutations segregate with familial and ‘sporadic’ amyotrophic lateral sclerosis. Nat Genet. 2006;38:411–413. doi: 10.1038/ng1742. [DOI] [PubMed] [Google Scholar]
- 136.Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, Baralle F, de Belleroche J, Mitchell JD, Leigh PN, Al-Chalabi A, Miller CC, Nicholson G, Shaw CE. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kovacs GG, Murrell JR, Horvath S, Haraszti L, Majtenyi K, Molnar MJ, Budka H, Ghetti B, Spina S. TARDBP variation associated with frontotemporal dementia, supranuclear gaze palsy, and chorea. Mov Disord. 2009 doi: 10.1002/mds.22697. [DOI] [PubMed] [Google Scholar]
- 138.Okamoto K, Hirai S, Amari M, Watanabe M, Sakurai A. Bunina bodies in amyotrophic lateral sclerosis immunostained with rabbit anti-cystatin C serum. Neurosci Lett. 1993;162:125–128. doi: 10.1016/0304-3940(93)90576-7. [DOI] [PubMed] [Google Scholar]
- 139.Mackenzie IR, Bigio EH, Ince PG, Geser F, Neumann M, Cairns NJ, Kwong LK, Forman MS, Ravits J, Stewart H, Eisen A, McClusky L, Kretzschmar HA, Monoranu CM, Highley JR, Kirby J, Siddique T, Shaw PJ, Lee VM, Trojanowski JQ. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol. 2007;61:427–434. doi: 10.1002/ana.21147. [DOI] [PubMed] [Google Scholar]
- 140.McKhann GM, Albert MS, Grossman M, Miller B, Dickson D, Trojanowski JQ. Clinical and pathological diagnosis of frontotemporal dementia: report of the Work Group on Fronto-temporal Dementia and Pick's Disease. Arch Neurol. 2001;58:1803–1809. doi: 10.1001/archneur.58.11.1803. [DOI] [PubMed] [Google Scholar]
- 141.Wider C, Wszolek ZK. Etiology and pathophysiology of frontotemporal dementia, Parkinson disease and Alzheimer disease: lessons from genetic studies. Neurodegener Dis. 2008;5:122–125. doi: 10.1159/000113680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D, Berger Z, Eriksen J, Robinson T, Zehr C, Dickey CA, Crook R, McGowan E, Mann D, Boeve B, Feldman H, Hutton M. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–919. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- 143.Watts GD, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D, Pestronk A, Whyte MP, Kimonis VE. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet. 2004;36:377–381. doi: 10.1038/ng1332. [DOI] [PubMed] [Google Scholar]
- 144.Forman MS, Mackenzie IR, Cairns NJ, Swanson E, Boyer PJ, Drachman DA, Jhaveri BS, Karlawish JH, Pestronk A, Smith TW, Tu PH, Watts GD, Markesbery WR, Smith CD, Kimonis VE. Novel ubiquitin neuropathology in frontotemporal dementia with valosin-containing protein gene mutations. J Neuropathol Exp Neurol. 2006;65:571–581. doi: 10.1097/00005072-200606000-00005. [DOI] [PubMed] [Google Scholar]
- 145.Cairns NJ, Grossman M, Arnold SE, Burn DJ, Jaros E, Perry RH, Duyckaerts C, Stankoff B, Pillon B, Skullerud K, Cruz-Sanchez FF, Bigio EH, Mackenzie IR, Gearing M, Juncos JL, Glass JD, Yokoo H, Nakazato Y, Mosaheb S, Thorpe JR, Uryu K, Lee VM, Trojanowski JQ. Clinical and neuropathologic variation in neuronal intermediate filament inclusion disease. Neurology. 2004;63:1376–1384. doi: 10.1212/01.wnl.0000139809.16817.dd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Horoupian DS, Dickson DW. Striatonigral degeneration, olivopontocerebellar atrophy and “atypical” Pick disease. Acta Neuropathol (Berl) 1991;81:287–295. doi: 10.1007/BF00305870. [DOI] [PubMed] [Google Scholar]
- 147.Konagaya M, Sakai M, Matsuoka Y, Konagaya Y, Hashizume Y. Multiple system atrophy with remarkable frontal lobe atrophy. Acta Neuropathol. 1999;97:423–428. doi: 10.1007/s004010051008. [DOI] [PubMed] [Google Scholar]
- 148.Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, Mackenzie IR. Frontotemporal lobar degeneration with FUS pathology. Brain. 2009 doi: 10.1093/brain/awp214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kwiatkowski TJ, Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, Hosler BA, Cortelli P, de Jong PJ, Yoshinaga Y, Haines JL, Pericak-Vance MA, Yan J, Ticozzi N, Siddique T, McKenna-Yasek D, Sapp PC, Horvitz HR, Landers JE, Brown RH., Jr Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- 150.Skibinski G, Parkinson NJ, Brown JM, Chakrabarti L, Lloyd SL, Hummerich H, Nielsen JE, Hodges JR, Spillantini MG, Thusgaard T, Brandner S, Brun A, Rossor MN, Gade A, Johannsen P, Sorensen SA, Gydesen S, Fisher EM, Collinge J. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in fronto-temporal dementia. Nat Genet. 2005;37:806–808. doi: 10.1038/ng1609. [DOI] [PubMed] [Google Scholar]
- 151.Holm IE, Englund E, Mackenzie IR, Johannsen P, Isaacs AM. A reassessment of the neuropathology of frontotemporal dementia linked to chromosome 3. J Neuropathol Exp Neurol. 2007;66:884–891. doi: 10.1097/nen.0b013e3181567f02. [DOI] [PubMed] [Google Scholar]
- 152.Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J, Kovacs GG, Ghetti B, Halliday G, Holm IE, Ince PG, Kamphorst W, Revesz T, Rozemuller AJ, Kumar-Singh S, Akiyama H, Baborie A, Spina S, Dickson DW, Trojanowski JQ, Mann DM. Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: consensus recommendations. Acta Neuropathol. 2009;117:15–18. doi: 10.1007/s00401-008-0460-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Cairns NJ, Bigio EH, Mackenzie IR, Neumann M, Lee VM, Hatanpaa KJ, White CL, 3rd, Schneider JA, Grinberg LT, Halliday G, Duyckaerts C, Lowe JS, Holm IE, Tolnay M, Okamoto K, Yokoo H, Murayama S, Woulfe J, Munoz DG, Dickson DW, Ince PG, Trojanowski JQ, Mann DM. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 2007;114:5–22. doi: 10.1007/s00401-007-0237-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Mackenzie IR, Baborie A, Pickering-Brown S, Du Plessis D, Jaros E, Perry RH, Neary D, Snowden JS, Mann DM. Heterogeneity of ubiquitin pathology in frontotemporal lobar degeneration: classification and relation to clinical phenotype. Acta Neuropathol. 2006;112:539–549. doi: 10.1007/s00401-006-0138-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Pikkarainen M, Hartikainen P, Alafuzoff I. Neuropathologic features of frontotemporal lobar degeneration with ubiquitin-positive inclusions visualized with ubiquitin-binding protein p62 immunohistochemistry. J Neuropathol Exp Neurol. 2008;67:280–298. doi: 10.1097/NEN.0b013e31816a1da2. [DOI] [PubMed] [Google Scholar]
- 156.Josephs KA, Stroh A, Dugger B, Dickson DW. Evaluation of subcortical pathology and clinical correlations in FTLD-U subtypes. Acta Neuropathol. 2009;118:349–358. doi: 10.1007/s00401-009-0547-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Hatanpaa KJ, Bigio EH, Cairns NJ, Womack KB, Weintraub S, Morris JC, Foong C, Xiao G, Hladik C, Mantanona TY, White CL., 3rd TAR DNA-binding protein 43 immunohistochemistry reveals extensive neuritic pathology in FTLD-U: a midwest-southwest consortium for FTLD study. J Neuropathol Exp Neurol. 2008;67:271–279. doi: 10.1097/NEN.0b013e31816a12a6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Mackenzie IR, Baker M, Pickering-Brown S, Hsiung GY, Lindholm C, Dwosh E, Gass J, Cannon A, Rademakers R, Hutton M, Feldman HH. The neuropathology of fronto-temporal lobar degeneration caused by mutations in the progranulin gene. Brain. 2006;129:3081–3090. doi: 10.1093/brain/awl271. [DOI] [PubMed] [Google Scholar]
- 159.Josephs KA, Ahmed Z, Katsuse O, Parisi JF, Boeve BF, Knopman DS, Petersen RC, Davies P, Duara R, Graff-Radford NR, Uitti RJ, Rademakers R, Adamson J, Baker M, Hutton ML, Dickson DW. Neuropathologic features of frontotemporal lobar degeneration with ubiquitin-positive inclusions with progranulin gene (PGRN) mutations. J Neuropathol Exp Neurol. 2007;66:142–151. doi: 10.1097/nen.0b013e31803020cf. [DOI] [PubMed] [Google Scholar]
- 160.Arai T, Mackenzie IR, Hasegawa M, Nonoka T, Niizato K, Tsuchiya K, Iritani S, Onaya M, Akiyama H. Phosphorylated TDP-43 in Alzheimer's disease and dementia with Lewy bodies. Acta Neuropathol. 2009;117:125–136. doi: 10.1007/s00401-008-0480-1. [DOI] [PubMed] [Google Scholar]
- 161.Higashi S, Iseki E, Yamamoto R, Minegishi M, Hino H, Fujisawa K, Togo T, Katsuse O, Uchikado H, Furukawa Y, Kosaka K, Arai H. Concurrence of TDP-43, tau and alpha-synuclein pathology in brains of Alzheimer's disease and dementia with Lewy bodies. Brain Res. 2007;1184C:284–294. doi: 10.1016/j.brainres.2007.09.048. [DOI] [PubMed] [Google Scholar]
- 162.Josephs KA, Whitwell JL, Knopman DS, Hu WT, Stroh DA, Baker M, Rademakers R, Boeve BF, Parisi JE, Smith GE, Ivnik RJ, Petersen RC, Jack CR, Jr, Dickson DW. Abnormal TDP-43 immunoreactivity in AD modifies clinic-pathologic and radiologic phenotype. Neurology. 2008;70:1850–1857. doi: 10.1212/01.wnl.0000304041.09418.b1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Hu WT, Josephs KA, Knopman DS, Boeve BF, Dickson DW, Petersen RC, Parisi JE. Temporal lobar predominance of TDP-43 neuronal cytoplasmic inclusions in Alzheimer disease. Acta Neuropathol. 2008;116:215–220. doi: 10.1007/s00401-008-0400-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ala TA, Beh GO, Frey WH., 2nd Pure hippocampal sclerosis: a rare cause of dementia mimicking Alzheimer's disease. Neurology. 2000;54:843–848. doi: 10.1212/wnl.54.4.843. [DOI] [PubMed] [Google Scholar]
- 165.Amador-Ortiz C, Dickson DW. Neuropathology of hippocampal sclerosis. Handb Clin Neurol. 2008;89:569–572. doi: 10.1016/S0072-9752(07)01253-5. [DOI] [PubMed] [Google Scholar]
- 166.Attems J, Jellinger KA. Hippocampal sclerosis in Alzheimer disease and other dementias. Neurology. 2006;66:775. doi: 10.1212/01.wnl.0000200959.50898.26. [DOI] [PubMed] [Google Scholar]
- 167.Corey-Bloom J, Sabbagh MN, Bondi MW, Hansen L, Alford MF, Masliah E, Thal LJ. Hippocampal sclerosis contributes to dementia in the elderly. Neurology. 1997;48:154–160. doi: 10.1212/wnl.48.1.154. [DOI] [PubMed] [Google Scholar]
- 168.Dickson DW, Davies P, Bevona C, Van Hoeven KH, Factor SM, Grober E, Aronson MK, Crystal HA. Hippocampal sclerosis: a common pathological feature of dementia in very old (> or = 80 years of age) humans. Acta Neuropathol. 1994;88:212–221. doi: 10.1007/BF00293396. [DOI] [PubMed] [Google Scholar]
- 169.Josephs KA, Dickson DW. Hippocampal sclerosis in tau-negative frontotemporal lobar degeneration. Neurobiol Aging. 2006 doi: 10.1016/j.neurobiolaging.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 170.Hatanpaa KJ, Blass DM, Pletnikova O, Crain BJ, Bigio EH, Hedreen JC, White CL, 3rd, Troncoso JC. Most cases of dementia with hippocampal sclerosis may represent fronto-temporal dementia. Neurology. 2004;63:538–542. doi: 10.1212/01.wnl.0000129543.46734.c0. [DOI] [PubMed] [Google Scholar]
- 171.Lee EB, Lee VM, Trojanowski JQ, Neumann M. TDP-43 immunoreactivity in anoxic, ischemic and neoplastic lesions of the central nervous system. Acta Neuropathol. 2008;115:305–311. doi: 10.1007/s00401-007-0331-5. [DOI] [PubMed] [Google Scholar]
- 172.Zarow C, Sitzer TE, Chui HC. Understanding hippocampal sclerosis in the elderly: epidemiology, characterization, and diagnostic issues. Curr Neurol Neurosci Rep. 2008;8:363–370. doi: 10.1007/s11910-008-0057-3. [DOI] [PubMed] [Google Scholar]
- 173.Zhang YJ, Xu YF, Dickey CA, Buratti E, Baralle F, Bailey R, Pickering-Brown S, Dickson D, Petrucelli L. Progranulin mediates caspase-dependent cleavage of TAR DNA binding protein-43. J Neurosci. 2007;27:10530–10534. doi: 10.1523/JNEUROSCI.3421-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Dix MM, Simon GM, Cravatt BF. Global mapping of the topography and magnitude of proteolytic events in apoptosis. Cell. 2008;134:679–691. doi: 10.1016/j.cell.2008.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Hasegawa M, Arai T, Nonaka T, Kametani F, Yoshida M, Hashizume Y, Beach TG, Buratti E, Baralle F, Morita M, Nakano I, Oda T, Tsuchiya K, Akiyama H. Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann Neurol. 2008;64:60–70. doi: 10.1002/ana.21425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Zhang YJ, Xu YF, Cook C, Gendron TF, Roettges P, Link CD, Lin WL, Tong J, Castanedes-Casey M, Ash P, Gass J, Rangachari V, Buratti E, Baralle F, Golde TE, Dickson DW, Petrucelli L. Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc Natl Acad Sci USA. 2009;106:7607–7612. doi: 10.1073/pnas.0900688106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Lin WL, Dickson DW. Ultrastructural localization of TDP-43 in filamentous neuronal inclusions in various neurodegenerative diseases. Acta Neuropathol. 2008;116:205–213. doi: 10.1007/s00401-008-0408-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Dickson DW. TDP-43 immunoreactivity in neurodegenerative disorders: disease versus mechanism specificity. Acta Neuropathol. 2008;115:147–149. doi: 10.1007/s00401-007-0323-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Hutton M. Missense and splice site mutations in tau associated with FTDP-17: multiple pathogenic mechanisms. Neurology. 2001;56:S21–25. doi: 10.1212/wnl.56.suppl_4.s21. [DOI] [PubMed] [Google Scholar]
- 180.Baker M, Litvan I, Houlden H, Adamson J, Dickson D, Perez-Tur J, Hardy J, Lynch T, Bigio E, Hutton M. Association of an extended haplotype in the tau gene with progressive supranuclear palsy. Hum Mol Genet. 1999;8:711–715. doi: 10.1093/hmg/8.4.711. [DOI] [PubMed] [Google Scholar]
- 181.Maraganore DM, de Andrade M, Elbaz A, Farrer MJ, Ioannidis JP, Kruger R, Rocca WA, Schneider NK, Lesnick TG, Lincoln SJ, Hulihan MM, Aasly JO, Ashizawa T, Chartier-Harlin MC, Checkoway H, Ferrarese C, Hadjigeorgiou G, Hattori N, Kawakami H, Lambert JC, Lynch T, Mellick GD, Papapetropoulos S, Parsian A, Quattrone A, Riess O, Tan EK, Van Broeck-hoven C. Collaborative analysis of alpha-synuclein gene promoter variability and Parkinson disease. Jama. 2006;296:661–670. doi: 10.1001/jama.296.6.661. [DOI] [PubMed] [Google Scholar]
- 182.Rademakers R, Eriksen JL, Baker M, Robinson T, Ahmed Z, Lincoln SJ, Finch N, Rutherford NJ, Crook RJ, Josephs KA, Boeve BF, Knopman DS, Petersen RC, Parisi JE, Caselli RJ, Wszolek ZK, Uitti RJ, Feldman H, Hutton ML, Mackenzie IR, Graff-Radford NR, Dickson DW. Common variation in the miR-659 binding-site of GRN is a major risk factor for TDP43-positive frontotemporal dementia. Hum Mol Genet. 2008;17:3631–3642. doi: 10.1093/hmg/ddn257. [DOI] [PMC free article] [PubMed] [Google Scholar]