

Abstract
Structures of truncated versions of the influenza A virus M2 proton channel have been determined recently by x-ray crystallography in the open conformation of the channel, and by NMR in the closed state. The structures differ in the position of the bound inhibitors. The x-ray structure shows a single amantadine molecule in the middle of the channel, whereas in the NMR structure four drug molecules bind at the channel's outer surface. To study this controversy we applied computational solvent mapping, a technique developed for the identification of the most druggable binding hot spots of proteins. The method moves molecular probes—small organic molecules containing various functional groups—around the protein surface, finds favorable positions using empirical free energy functions, clusters the conformations, and ranks the clusters on the basis of the average free energy. The results of the mapping show that in both structures the primary hot spot is an internal cavity overlapping the amantadine binding site seen in the x-ray structure. However, both structures also have weaker hot spots at the exterior locations that bind rimantadine in the NMR structure, although these sites are partially due to the favorable interactions with the interfacial region of the lipid bilayer. As confirmed by docking calculations, the open channel binds amantadine at the more favorable internal site, in good agreement with the x-ray structure. In contrast, the NMR structure is based on a peptide/micelle construct that is able to accommodate the small molecular probes used for the mapping, but has a too narrow pore for the rimantadine to access the internal hot spot, and hence the drug can bind only at the exterior sites.
Introduction
The integral membrane protein M2 of influenza virus forms pH-gated proton channels in the viral lipid envelope. The low pH of an endosome activates the M2 channel before hemagglutinin-mediated fusion. Conductance of protons acidifies the viral interior and thereby facilitates dissociation of the matrix protein from the viral nucleoproteins— a required process for unpacking of the viral genome (1). M2 is a 97-residue single-pass membrane protein that has its amino and carboxy termini directed toward the outside and inside of the virion, respectively, and forms a homotetramer in its native state (2). The four transmembrane helices yield a channel in which His37 is the pH sensor and Trp41 is the gate (3). M2 is the target of the anti-influenza drugs amantadine and its methyl derivative rimantadine; recently, resistance to these drugs has reached >90% (4).
The most complete structural information on the M2 transmembrane domain (M2TM) emerges from two studies published simultaneously in 2008. Stouffer et al. (5) used x-ray crystallography to determine the structure of residues 22–46 with and without amantadine in the detergent octyl-β-D-glucopyranoside. A crystal form that diffracts to 2.0 Å resolution was obtained at pH 7.3 in the absence of amantadine from a peptide in which Ile33 was changed to selenomethionine. The peptide crystallizes with six detergent molecules that form a bilayer-like environment. The structure shows a four-helix, cone-shaped bundle in which the helices are tilted by ∼35° with respect to the central axis. A second mutant, Gly34Ala, was crystallized at pH 5.3 in the presence of amantadine at 3.5 Å resolution. The two structures are very similar, with the primary differences lying near the carboxy-terminal region of the helices. Schnell and Chou (6) used NMR to determine the structure of the rimantadine-bound M2 peptide of residues 18–60 solubilized in dihexanoylphosphatidylcholine (DHPC) micelles at pH 7.5. The NMR results also show a four-helix bundle, but under the conditions of the experiment the channel is substantially less open than in the x-ray structure, and the tilt of each helix with respect to the central axis is <23°.
Despite the overall similarity of the channel structure, the x-ray and NMR studies show major disagreement in the position of the bound inhibitor (7). The x-ray structure shows a single amantadine molecule in the middle of the channel, surrounded by residues Val27, Ala30, Ser31, and Gly34. The finding of electron density in the pore of the channel in the presence of amantadine and the absence of density without amantadine is consistent with the drug being present in the pore of the channel, although at 3.5 Å resolution it cannot be proven that the density represents amantadine. However, the binding of amantadine at this position is supported by the fact that mutations of Val27, Ala30, Ser31, and Gly34 were observed in naturally occurring amantadine resistant strains (8,9). In addition, the Hill coefficient for amantadine inhibition was shown to be ∼1.0, consistent with a single amantadine molecule binding to the tetramer (10,11). In contrast to the x-ray structure, the NMR data show four rimantadine molecules bound to the lipid-exposed outer surface of the channel at the membrane boundary close to the cytoplasmic end of the helices. The binding includes interactions with residues 40–45, with a hydrogen bond between rimantadine and Asp44 (7). Residues Leu40, Ile42, and Leu43 form the hydrophobic walls of the binding pocket. Although the NMR structure is well defined in the TM region, the four rimantadine molecules display significant variability in their interactions. Based on the NMR structure, Schnell and Chou (6) proposed that the drug binds preferentially to, and thereby stabilizes, the closed state. They note that amantadine action is faster at neutral pH, where the channel is mostly closed, than at low pH, where the open state is favored (6), in accord with closed-state stabilization but not with open-pore block (10,11). However, a recent functional study (12) questions the significance of amantadine/rimantadine binding outside of the channel pore. In particular, using electrophysiological recordings in oocytes of Xenopus laevis and in mammalian cells it was shown that mutations of Asp44 and Arg45 to alanine do not alter the sensitivity of the channel to the drug, suggesting that the interactions of rimantadine with Asp44 and Arg45 are not important for inhibition.
The goal of this study is to investigate the potential origin of the different inhibitor binding modes in the two peptide-detergent constructs used in the x-ray and the NMR studies. Our main analysis tool is computational solvent mapping, a technique developed for the identification and characterization of “hot spots” in binding sites, i.e., regions of the protein surface that are major contributors to the binding free energy (13). Based on NMR (14) and x-ray (15) screening experiments with fragment-sized compounds, such hot spots bind a variety of small organic molecules, and hence the fraction of such molecules binding to a particular site is a good predictor of its druggability (14). Computational mapping is an analog of such screening experiments (16–18). The method moves molecular probes—small organic molecules containing various functional groups—around the protein surface, finds favorable positions using empirical free energy functions, clusters the conformations, and ranks the clusters on the basis of the average free energy (13,16–18). We have developed mapping algorithms that reproduce very well the results of the published NMR and x-ray screening studies (18). Applications to a variety of proteins show that the probes always cluster in major subsites of the binding site and the amino acid residues that interact with the probes also bind the specific ligands, suggesting that the number of different probes at a consensus site correlates with the importance of that site for ligand binding (13).
We have applied computational solvent mapping to both x-ray and NMR structures of M2TM to identify the most important binding sites. The mapping shows that both structures have binding hot spots both in the pore and on the lipid-exposed outer surface of the channel, but the internal site represents the main region of “druggability”, i.e., the potentially highest contribution to the binding free energy. However, the NMR structure is based on a peptide/micelle construct that is able to accommodate the small molecular probes used for the mapping, but appears to have a too narrow pore to bind adamantine-sized molecules inside the channel, and hence the drug can bind only at the exterior sites. Our results also suggest that in the open state of the channel the bound amantadine is more likely oriented with its amino group toward the N-terminal end of the channel rather than toward the C-terminal cytoplasmic end as shown in the x-ray structure.
Materials and Methods
The ligand-free x-ray structure (Protein Data Bank (PDB) code: 3bkd) and rimantadine bound NMR structure (PDB: code 2rlf, model 1) of the M2 protein were mapped using the FTMAP algorithm consisting of four steps as follows (18).
Step 1: soft rigid body docking of probe molecules
Protein structures are downloaded from the PDB (19). All bound ligands, ions, and water molecules are removed. For each structure, we use 16 small molecules as probes (ethanol, isopropanol, tert-butanol, acetone, acetaldehyde, dimethyl ether, cyclohexane, ethane, acetonitrile, urea, methylamine, phenol, benzaldehyde, benzene, acetamide, and N, N-dimethylformamide). For each probe, billions of docked conformations are sampled by soft rigid body docking based on Fast Fourier Transform (FFT) correlation approach (18). The method performs exhaustive evaluation of an energy function in the discretized 6D space of mutual orientations of the protein (receptor) and a small molecular probe (ligand). The center of mass of the receptor is fixed at the origin of the coordinate system. The translational space is represented as a grid of 0.8 Å displacements of the ligand center of mass, and the rotational space is sampled using 500 rotations. The energy expression includes a stepwise approximation of the van der Waals energy with attractive and repulsive contributions, and an electrostatics/solvation term based on Poisson-Boltzmann continuum calculation. The last term is approximated as an interaction of probe charges with an electrostatic potential of the protein-membrane system. This potential was calculated as a solution of a linearized Poisson-Boltzmann equation, using the dielectric constants of ɛ = 4 and ɛ = 80 for the protein and the solvent, respectively. The membrane was modeled as an infinite layer of low dielectric medium (ɛ = 4), approximately positioned with respect to the protein as shown in the original studies (5,6). A salt concentration was 0.15 M in the solution phase on both sides of the membrane. The potential was calculated by the Poisson-Boltzmann module PBEQ of CHARMM (20) on a 0.4 Å spaced grid. Note that mapping requires only the atomic coordinates of the two molecules, i.e., no a priori information on the binding site is used. The 2000 best poses for each probe are retained for further processing.
Step 2: minimization and rescoring
The 2000 complexes, generated in Step 1, are refined by off-grid energy minimization during which the protein atoms are held fixed whereas the atoms of the probe molecules are free to move. The energy function includes the bonded and van der Waals terms of the polar hydrogen CHARMM force field (20) and an electrostatic interaction term using the Poisson-Boltzmann potential generated at Step 1. The values of the potential were smoothly interpolated up to the first derivative from a 0.4 Å electrostatic grid using a tricubic algorithm (21).
Step 3: clustering and ranking
The minimized probe conformations from Step 2 are grouped into clusters using a simple greedy algorithm. The lowest energy structure is selected and the structures within 4 Å RMSD are joined in the first cluster. The members of this cluster are removed, and the next lowest energy structure is selected to start the second cluster. This step is repeated until the entire set is exhausted. Clusters with <10 members are excluded from consideration. The retained clusters are ranked on the basis of their Boltzman averaged energies. Six clusters with the lowest average free energies are retained for each probe.
Step 4: determination of consensus sites
To determine the hot spots, FTMAP finds consensus sites, i.e., regions on the protein where clusters of different probes overlap (14–17). Therefore the probe clusters are clustered again using the distance between the centers of mass of the cluster centers as the distance measure and 4 Å as the clustering radius. The consensus sites are ranked based on the number of their clusters. Duplicate clusters of the same type are considered in the count.
Docking of amantadine and rimantadine uses Steps 1–3 of the FTMAP algorithm, but all clusters (even with a single pose) are retained in Step 3.
Results and Discussion
Analysis of the x-ray structure
We have mapped the unbound and amantadine-bound structures of the transmembrane domain (PDB codes 3bkd and 3c9j, respectively). Because the differences are small, we report results for the higher resolution unbound structure. The structures were downloaded from the Protein Data Bank (19). All ligands, ions, and water molecules were removed before mapping. For each probe, the six lowest free energy clusters were superimposed (see Materials and Methods) to identify the consensus sites (CS) defined by overlapping probe clusters. The largest consensus site (defined as CS1) binds 28 probe clusters that form a very tight supercluster. Fig. 1 A shows the centers (i.e., the lowest energy structures) of these 28 clusters that include four clusters of isopropanol; three clusters of tert-butanol, phenol, and benzaldehyde; two clusters of N,N-dimethylformamide, acetone, and acetonitrile; and one cluster of each of the other nine probe compounds. The figure also includes amantadine (green), indicating that CS1 overlaps the amantadine binding site. The probes in the 28 clusters of CS1 primarily interact with the amino acid residues Ser31 (46.0%), Val27 (29.6%), Ala30 (22.8%), and Gly34 (1.2%), where the numbers in parentheses denote the percentage of nonbonded contacts between all probes and the protein. It is well known that each of these residues is mutated in some clinical isolates of amantadine-resistant viruses (4,8,9). We also note that in the lowest free energy clusters of most partially polar probes (ethanol, isopropanol, tert-butanol, acetone, acetaldehyde, dimethyl ether, acetonitrile, methylamine, phenol, benzaldehyde, acetamide, and N,N-dimethylformamide), the polar moiety is oriented toward the amino end of the channel, and forms a hydrogen bond with one of the Ser31 side chains. Indeed, the distributions of hydrogen bonds are as follows: Ser31 82.6%, Val27 12%, Ala30 5.3%, and His37 0.1%. Thus, the region of highest hydrogen bonding propensity is defined by the four Ser31 side chains, with some of the probes extending slightly further toward the extracellular end of the channel and interacting with the carbonyl oxygen of the Val27 backbone. Fig. 1 A shows that a number of the polar groups on the bound probes orient downward into the direction of the cytoplasmic end of the channel, but only ∼5% of all hydrogen bonds occur with the backbone of Ala30.
Figure 1.
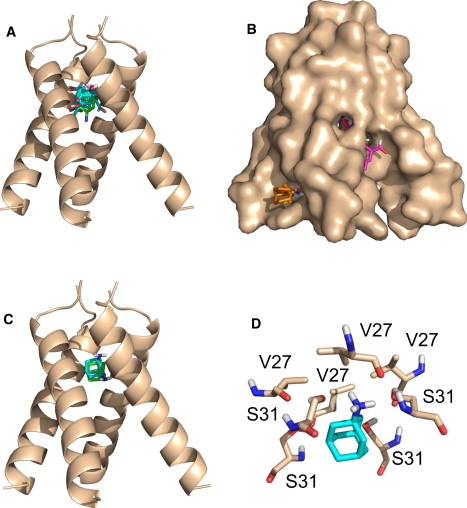
Analysis of the x-ray structure of the M2 channel (5). (A) Centers (i.e., lowest energy structures) of the 28 probe clusters in the largest consensus site (CS1). The color codes are as follows: oxygen, red; nitrogen, blue, and carbon, cyan. The figure also shows the bound amantadine with carbon atoms colored green, indicating that CS1 overlaps the amantadine binding site. (B). Surface representation of the channel with the four largest consensus site. CS1, shown in A, and CS2, a supercluster of 20 clusters between helices A and D, are hidden inside the pore. Some probes are visible for CS3, a supercluster of 17 probe clusters between helices B and C, shown in magenta. CS4, shown in orange, includes only eight probe clusters between helices C and D on the outside of the channel, interacting with residues 43–45 that bind rimantadine in the closed NMR structure. (C) Centers the lowest and second lowest energy docked amantadine cluster, shown in green and cyan, respectively. (D) The lowest energy amantadine conformation is oriented toward the N-terminal end of the channel, forming hydrogen bonds with two Ser31 side chains on two adjacent helices.
In addition to CS1 at the amantadine binding site, there are three locations where relatively large numbers of probe clusters overlap. CS2 with 20 probe clusters is located between helices A and D, and CS3 with 17 clusters is between helices B and C. Both CS2 and CS3 are elongated superclusters located inside the pore, extending from Ile33 (changed to selenomethionine in the x-ray structure) to the His37 side chain. The existence of these favorable binding regions suggests that the channel could accommodate more elongated molecules than amantadine. Fig. 1 B shows the surface representation of the x-ray structure with the four largest consensus sites. CS1 and CS2 are not visible inside the pore, and only a few probes of CS3 show (in magenta) through the openings between helices B and C. However, the figure shows (in orange) that the eight probe clusters of the fourth largest consensus site CS4 bind in a shallow pocket on the lipid-exposed outer surface of the channel. What makes CS4 interesting is that the probes interact with residues 43–45, i.e., the site that binds rimantadine in the NMR structure (6). Thus, the mapping shows a hot spot at this location even for the x-ray structure. However, binding occurs only between helices B and C, because the 35° tilt of helices yields large openings rather than pockets on the other three sides of the four-helix bundle. We also note (and will further discuss) that binding at CS4 is partially due to the favorable interactions between the probes and the interfacial region of the bilayer used in our model.
To further test the relative importance of the hot spot regions we have docked amantadine to the ligand-free x-ray structure using the FFT algorithm. The advantage of this approach is that there is no need for any a priori assumption on the location of the binding site, and the entire protein surface is considered in the docking calculation. Although Step 1 of the algorithm is rigid body docking, the smooth scoring function allows for partial atomic overlaps, and the flexible minimization in Step 2 can account for the limited flexibility of the amantadine molecule. Fig. 1 C shows the centers (i.e., lowest energy conformations) in the two lowest energy clusters. Cluster 1 (green) includes amantadine conformations with the NH2 group oriented upward, forming hydrogen bonds with two Ser31 side chains on two adjacent helices. In Cluster 2 (cyan) the NH2 group is oriented toward the cytoplasmic end of the channel and hydrogen bonds to the carbonyl group of Ala30. This second structure has better overlap with the amantadine pose given in the x-ray structure, but in the latter the orientation of the NH2 group could not be determined due to the 3.5 Å resolution. We note that docked amantadine molecules can also be observed at consensus site CS4 on the lipid-exposed outer surface of the channel, at the location that binds rimantadine in the NMR structure. However, the average energy of this cluster is much higher (−13.43 kcal/mol) than the average energy of Cluster 1 in the pore (−23.72 kcal/mol).
Both mapping and docking results imply that amantadine binding occurs in the pore at the CS1 site, in good agreement with the x-ray structure (5). However, three observations suggest that the amino group of amantadine may be preferentially oriented toward the N-terminal extracellular end of the channel rather than the C-terminus as deposited in the PDB. First, the mapping results show much higher propensity for hydrogen bonding in the region surrounded by the Ser31 side chains than anywhere else in the channel. Indeed, the amino group of amantadine in Cluster 1 overlaps with polar groups in the majority of partially polar probes, and forms hydrogen bonds with the Ser31 side chains. As an example, Fig. 1 D shows the lowest energy conformation of Cluster 1, with slightly higher energy members of the cluster interacting with Ser31 side chains on the other helices. Second, the average energy is somewhat lower in Cluster 1 than in Cluster 2 (−23.72 kcal/mol vs. −23.24 kcal/mol, respectively). We note that these values do not include contributions from changes in rotational, translational, and vibrational entropy, and hence do not represent valid estimates of the binding free energy. Third, in our model the bulky adamantane moiety is surrounded by small residues Ala30 and Gly34, in agreement with the observation that Ala30Thr and Gly34Glu result in resistance (22,23), and the amino group of the drug is in the polar environment of the Ser31 side chains. In contrast, in the x-ray structure the hydrophobic adamantane cage is coordinated to the hydroxyls of Ser31, making the model difficult to reconcile with the chemical properties of the drug (24). The upward orientation of the amantadine amino group is compatible with the observation that the dipolar splitting and chemical shift of Val27 are unaffected by inhibitor binding (25). A recent magic-angle-spinning solid-state NMR study of M2TMP bound to lipid bilayers also agrees with our model (26).
However, it is important to note that a number of models and observations support the opposite orientation of amantadine as seen in the deposited x-ray structure. An early model of the M2 channel correctly predicted that the Val27 side chains form the “lid” of the amantadine binding pocket (27), but placed the drug with its NH2 group toward the cytoplasmic end, forming hydrogen bonds with the Ser31 hydroxyls. This model was further supported by neutron diffraction data (28). Based on a more recent solid state NMR structure of the channel (29), Yi et al. (30) carried out a series of molecular dynamics (MD) simulations, initially placing amantadine in the channel pore around Ser31. According to the simulations the protein is quite flexible, and the drug molecule samples both the upward and the downward orientations, but the downward orientation toward the cytoplasmic end occurs for the majority of the time, with hydrogen bonds alternating among the Ser31 hydroxyls, the Ala30 backbone carbonyls, and water molecules (30). Thus, although the preference for the upward orientation seen in our rigid body docking removes the apparent incompatibility of surface properties, the MD simulations show that the drug molecule is quite mobile, and that the downward orientation can be favorable if the presence of water is taken into account explicitly. We recall that according to our docking results the two orientations of amantadine differ by <0.5 kcal/mol. In fact, the drug can turn over if conditions change: the docking of amantadine to the Ser31Ala mutant places the drug with its NH2 group downward, hydrogen bonding to the backbone carbonyl of Ala30. Because the mutant channel is known to be inhibited by the drug, hydrogen bonds with the Ser31 side chains are not required for inhibition.
Analysis of the NMR structure
The NMR structure of the closed channel shows rimantadine molecules bound at four external sites on the lipid-facing side of the channel, close to the membrane boundary. We have mapped all 15 conformers included in the PDB file 2rlf, but show detailed results only for the first. As for the open structure, all ligands were removed before mapping, but in some of the figures we show the bound rimantadine molecules for orientation. Interestingly, the largest consensus site (defined as CS1 and shown in cyan in Fig. 2 A) of 19 probe clusters is located close to the amantadine-binding site seen in the x-ray structure. The probes in CS1 interact with residues Ala30 (56%), Ser31 (19.5%), Gly34 (16.2%), and Ile33 (7.8%). Although three of these residues (Ala30, Ser31, and Gly34) are also part of the binding site in the x-ray structure, the consensus site in the NMR structure is slightly shifted toward the cytoplasmic end of the channel. In addition, >99% of hydrogen bonds are formed with the backbone carbonyl group of Ala30 rather than with the Ser31 side chains as in the x-ray structure. In fact, the channel is so narrow that even the smallest probes are unable to reach the Val27 side chains. In addition to the primary hot spot in the pore, Fig. 2 A shows the four rimantadine molecules bound to the membrane exposed outer surface of the channel as seen in the NMR structure (green).
Figure 2.
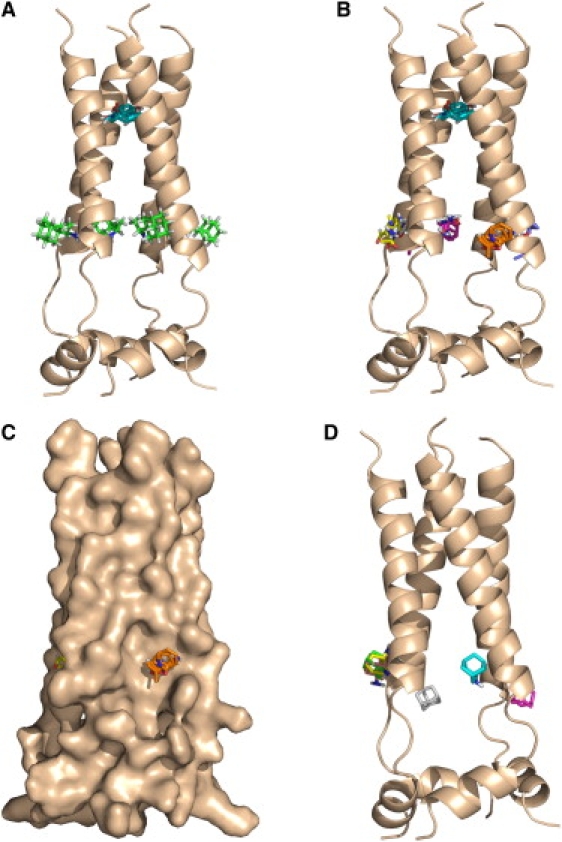
Analysis of the NMR structure of the M2 channel (6). (A) The largest consensus site (CS1, shown in cyan) of 19 probe clusters, located close to the amantadine-binding site seen in the x-ray structure. The four rimantadine molecules in the NMR structure are shown in green. (B). Consensus site CS1 in the pore (cyan), and consensus sites CS2 through CS5 at the four external sites. (C) Same as in B using surface representation of the M2 protein. (D) Centers of the six lowest energy amantadine clusters docked to the NMR structure. Note the lack of amantadine binding at the internal hot spot.
Fig. 2 B shows again the prime consensus site CS1 in the pore (cyan), and the next five largest (in terms of the number of probe clusters) consensus sites. Site CS2 (yellow) includes 17 probe clusters between helices B and C, CS3 (magenta) 15 clusters between helices C and D, CS4 (orange) 13 clusters between helices A and B, and CS5 (gray) 6 clusters between helices A and D. The consensus sites CS2 through CS5 are located at the four rimantadine binding sites, overlapping with the amino groups of the bound rimantadine molecules in the NMR structure. The probes in these four consensus sites interact with Asp44 (46.2%), Arg45 (25.3%), Leu43 (13.0%), Leu46 (5.6%), and Ile42 (4.6%) that define the rimantadine-binding pocket (6). Although the different sizes of the external consensus sites suggest substantial asymmetry of the channel, averaging the mapping results for the 15 structures shows that the four external sites are equally important. Based on the numbers of probe clusters, the external sites (on the average with 16 probe clusters) are still somewhat less important than the internal site with 19 probe clusters. We emphasize that probe binding at the exterior sites is largely due to the favorable interactions between the partially polar probes and the interfacial region of the lipid bilayer (31,32). In our model these sites are located on the low dielectric side of the dielectric boundary, and disappear in a homogeneous solvent environment modeled as a uniform high dielectric medium. In fact, as shown in Fig. 2 C using the surface representation of the protein, the external sites are in broad and shallow pockets.
To test why rimantadine binding is seen only at the external sites and not at the more important hot spot in the pore we have docked the slightly smaller amantadine to the NMR structure using the FFT approach used for the mapping. As already mentioned, the advantage of this approach is that a priori the entire protein surface, including the interior of the channel, is available for binding, and no a priori assumptions are made to constrain the search for the putative binding site. The most important result is that the docking does not place any drug molecule at the hot spot within the pore, despite the dense sampling and allowing for the rotation of the amantadine NH2 group. Fig. 2 D shows the centers of the six lowest energy amantadine clusters. The three lowest energy clusters, shown in green, cyan, and magenta, overlap with bound rimantadine molecules. In each cluster the NH2 group hydrogen bonds to the side chain of Asp44. We note that these results are based on the analysis of the first NMR conformer, and docking to the other 14 structures show comparable amantadine binding on all four sides of the channel.
To further test whether the NMR methodology may have missed inhibitor binding within the pore, in additional calculations we have placed amantadine molecules at the primary hot spot in both orientations, and refined the structures using energy minimization with a completely flexible ligand and allowing for the flexibility of the protein side chains. Although this procedure created a cavity that was able to accommodate rimantadine, the ligand was shifted toward the cytoplasmic side of the pore, and the docked rimantadine did not interact with Ser31, Val27, or Ala30, confirming that without allowing for changes in the backbone the pore in the NMR structure is too narrow to provide access for a bulky rimantadine-sized molecule, despite the existence of the internal binding hot spot.
Although the docked amantadine positions are in good agreement with the NMR results (6), the observed helical tilt of 23° with respect to the lipid bilayer normal is way out of the 32° to 38° range reported for the channel in the open state (26,33–36). It is possible that the open and closed states substantially differ, even in terms of the tilt angle. In fact, a study using 1D and 2D IR spectroscopies indicates a large conformational change at neutral pH when the channel is closing (37). In addition, recent MD simulations at high pH show the transmembrane helix to kink around Gly34 (38). The amantadine-bound form exhibits a single peak ∼10° in the distribution of helix kink angle, but the apo form exhibits two peaks ∼ 0° and 40°, and the corresponding structures have narrow and wide pores, respectively (38).
Although the difference between x-ray and NMR structures of M2 may be due to the difference in pH, it is known that detergent micelles may cause curvature stress to membrane proteins affecting their structure (39,40), or changing their functionality even when the structure is left largely intact (41), and thus one has to consider this possibility. In a relevant study, Poget and Girvin (41) determined solution NMR structures of Staphylococcal multidrug resistance transporter (Smr) in a number of detergents, including lysopalmitoylphospatidylglycerol, dodecylphosphocholine, n-dodecyl-β-D-maltopyranoside, and n-decyl-β-D-maltopyranosid. The Smr protein is known to be functional as a dimer, with four transmembrane helices per monomer and the chemical shift deviations were consistent with the presence of four individual α-helices in all four detergents. Poget and Girvin (41) also carried out a functional assay by monitoring the binding of the drug tetraphenylphosphonium to ascertain that Smr is in its native conformation. They found reproducible ligand binding in n-dodecyl-β-D-maltopyranoside (and to a lesser extent in n-decyl-β-D-maltopyranosid), but not in lysopalmitoylphospatidylglycerol or dodecylphosphocholine, indicating that although Smr formed a compact dimeric α-helical bundle in these detergents, this conformation did not fully correspond to the protein's native tertiary structure.
The Smr example obviously does not imply that the M2 channel is substantially affected by the DHPC micelle, but it shows that strong protein-micelle interactions are possible. In particular, Chou et al. (39) noted that in the absence of the peptide the DHPC micelle would adopt a spherical shape with the diameter of 36 Å, whereas the length of the M2 transmembrane channel is only ∼30 Å (6). Due to the symmetry of the governing forces it is plausible to assume that the channel extends straight through the middle of the micelle, from one side to the other. Therefore the channel appears to be stretched, and this may be responsible for the tilt angle that is substantially smaller than the one reported for the M2 channel in other biological membranes (33–36). However, little information is available on micelle structure in the presence of a protein. For example, Fernandez et al. (42) studied the interactions between DHPC and the integral membrane protein OmpX with a transmembrane height of 28 Å, thus similar to the M2 channel. They assumed that the DHPC molecules are oriented perpendicular to the hydrophobic protein surface as a distorted monolayer, with the polar headgroups forming the surface of a prolate ellipsoid. Thus, the environment of the protein substantially differs from the conditions seen in a natural lipid bilayer.
Conclusions
Computational solvent mapping of both x-ray and NMR structures of the influenza M2 proton channel shows the most important binding hot spot inside the pore, surrounded by the amino acid residues Ala30, Ser31, and Gly34. In the x-ray structure some probes also interact with the Val27 side chains, and most hydrogen bonds are formed with the Ser31 hydroxyls. In the much narrower pore of the NMR structure the probes cluster ∼2 Å closer to the cytoplasmic end of the channel, and do not interact with the Val27 side chains. In addition, the polar groups form hydrogen bonds with the backbone amino group of Ala30 rather than with the Ser31 side chains. In both x-ray and NMR structures, the mapping shows additional hot spots on the lipid exposed exterior of the channel at the location that is seen to bind rimantadine in the NMR structure. However, these hot spots are weaker (include fewer probe clusters) than the one in the pore, and are largely determined by polarization effects in the interfacial region of the lipid bilayer.
Docking of amantadine to the two structures confirms our mapping results and agrees well with experimental data. In the x-ray structure the docked poses of amantadine form low energy clusters only at the main hot spot in the pore. Combining the results of mapping and docking calculations suggest that the amino group of amantadine is oriented toward the N-terminal end of the channel, and forms hydrogen bonds with the Ser31 hydroxyls, whereas the adamantine ring primarily interacts with the Ala30 and Gly34. However, the energy difference between N-terminal and C-terminal orientations is small, and a number of arguments suggest that the bound drug is highly mobile, with both orientations feasible. In contrast to the x-ray structure, docking of amantadine to the NMR structure fails to place any inhibitor molecules inside the pore, suggesting that the M2 peptides in dihexanoyl-phosphatidyl-choline detergent micelles form a channel that is too narrow to provide access to the internal site for the bulky adamantane moiety. Thus, the drugs can bind only at the lipid-exposed external sites despite the hot spot in the middle of the channel. Although the narrow pore may be induced by high pH, the conditions in the DHPC micelle substantially differ from those seen in a natural lipid environment, and this also may affect the shape of the channel.
Acknowledgments
We thank Dr. Chris Miller for insightful discussion and Dr. Dagmar Ringe for suggesting the application of computational solvent mapping to the M2 channel.
This work has been supported by grant GM064700 from the National Institutes of Health.
Footnotes
Gwo-Yu Chuang, Dima Kozakov, Ryan Brenke, and Dmitri Beglov contributed equally to this work.
References
- 1.Helenius A. Unpacking the incoming influenza-virus. Cell. 1992;69:577–578. doi: 10.1016/0092-8674(92)90219-3. [DOI] [PubMed] [Google Scholar]
- 2.Sugrue R.J., Hay A.J. Structural characteristics of the M2protein of influenza A viruses: evidence that it forms a tetrameric channel. Virology. 1991;180:617–624. doi: 10.1016/0042-6822(91)90075-M. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tang Y., Zaitseva F., Lamb R.A., Pinto L.H. The gate of the influenza virus M2 proton channel is formed by a single tryptophan residue. J. Biol. Chem. 2002;277:39880–39886. doi: 10.1074/jbc.M206582200. [DOI] [PubMed] [Google Scholar]
- 4.Deyde V.M., Xu X., Bright R.A., Shaw M., Smith C.B. Surveillance of resistance to adamantanes among influenza A(H3N2) and A(H1N1) viruses isolated worldwide. J. Infect. Dis. 2007;196:249–257. doi: 10.1086/518936. [DOI] [PubMed] [Google Scholar]
- 5.Stouffer A.L., Acharya R., Salom D., Levine A.S., Di Costanzo L. Structural basis for the function and inhibition of an influenza virus proton channel. Nature. 2008;451:596–599. doi: 10.1038/nature06528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schnell J.R., Chou J.J. Structure and mechanism of the M2 proton channel of influenza A virus. Nature. 2008;451:591–595. doi: 10.1038/nature06531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miller C. Ion channels: coughing up flu's proton channels. Nature. 2008;451:532–533. doi: 10.1038/451532a. [DOI] [PubMed] [Google Scholar]
- 8.Bright R.A., Medina M.J., Xu X., Perez-Oronoz G., Wallis T.R. Incidence of adamantine resistance among influenza A (H3N2) viruses isolated worldwide from 1994 to 2005: a cause for concern. Lancet. 2005;366:1175–1181. doi: 10.1016/S0140-6736(05)67338-2. [DOI] [PubMed] [Google Scholar]
- 9.Bright R.A., Shay D.K., Shu B., Cox N.J., Klimov A.I. Adamantine resistance among influenza A viruses isolated early during the 2005–2006 influenza season in the United States. JAMA. 2006;295:891–894. doi: 10.1001/jama.295.8.joc60020. [DOI] [PubMed] [Google Scholar]
- 10.Wang C., Takeuchi K., Pinto L.H., Lamb R.A. Ion channel activity of influenza A virus M2 protein: characterization of the amantadine block. J. Virol. 1993;67:5585–5594. doi: 10.1128/jvi.67.9.5585-5594.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Czabotar P.E., Martin S.R., Hay A.J. Studies of structural changes in the M2 proton channel of influenza A virus by tryptophan fluorescence. Virus Res. 2004;99:57–61. doi: 10.1016/j.virusres.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 12.Jing X., Ma C., Ohigashi Y., Oliveira F.A., Jardetzky T.S. Functional studies indicate amantadine binds to the pore of the influenza A virus M2 proton-selective ion channel. Proc. Natl. Acad. Sci. USA. 2008;105:10967–10972. doi: 10.1073/pnas.0804958105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hajduk P.J., Huth J.R., Fesik S.W. Druggability indices for protein targets derived from NMR-based screening data. J. Med. Chem. 2005;48:2518–2525. doi: 10.1021/jm049131r. [DOI] [PubMed] [Google Scholar]
- 14.Mattos C., Ringe D. Locating and characterizing binding sites on proteins. Nat. Biotechnol. 1996;14:595–599. doi: 10.1038/nbt0596-595. [DOI] [PubMed] [Google Scholar]
- 15.Dennis S., Kortvelyesi T., Vajda S. Computational mapping identifies the binding sites of organic solvents on proteins. Proc. Natl. Acad. Sci. USA. 2002;99:4290–4295. doi: 10.1073/pnas.062398499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Landon M.R., Lancia D.R., Yu J., Thiel S.C., Vajda S. Identification of hot spots within druggable binding sites of proteins by computational solvent mapping. J. Med. Chem. 2007;50:1231–1240. doi: 10.1021/jm061134b. [DOI] [PubMed] [Google Scholar]
- 17.Silberstein M., Dennis S., Brown L., Kortvelyesi T., Clodfelter K. Identification of substrate binding sites in enzymes by computational solvent mapping. J. Mol. Biol. 2003;332:1095–1113. doi: 10.1016/j.jmb.2003.08.019. [DOI] [PubMed] [Google Scholar]
- 18.Brenke R., Kozakov D., Chuang G.Y., Beglov D., Mattos C. Fragment-based identification of druggable “hot spots” of proteins using Fourier domain correlation techniques. Bioinformatics. 2009;25:621–627. doi: 10.1093/bioinformatics/btp036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brooks B.R., Bruccoleri R.E., Olafson B.D., States D.J., Swaminathan S. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983;4:187–217. [Google Scholar]
- 21.Lekien F., Marsden J. Tricubic interpolation in three dimensions. Int. J. Numer. Methods Eng. 2005;63:455–471. [Google Scholar]
- 22.Hay A.J., Wolstenholme A.J., Skehel J.J., Smith M.H. The molecular basis of the specific anti-influenza action of amantadine. EMBO J. 1985;4:3021–3024. doi: 10.1002/j.1460-2075.1985.tb04038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pinto L.H., Lamb R.A. Controlling influenza virus replication by inhibiting its proton channel. Mol. Biosyst. 2007;3:18–23. doi: 10.1039/b611613m. [DOI] [PubMed] [Google Scholar]
- 24.Pielak R.M., Schnell J.R., Chou J.J. Mechanism of drug inhibition and drug resistance of influenza A M2 channel. Proc. Natl. Acad. Sci. USA. 2009;106:7379–7384. doi: 10.1073/pnas.0902548106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Song Z., Kovacs F.A., Wang J., Denny J.K., Shekar S.C. Transmembrane domain of M2 protein from influenza A virus studied by solid-state (15)N polarization inversion spin exchange at magic angle NMR. Biophys. J. 2000;79:767–775. doi: 10.1016/S0006-3495(00)76334-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cady S.D., Mishanina T.V., Hong M. Structure of amantadine-bound M2 transmembrane peptide of influenza a in lipid bilayers from magic-angle-spinning solid-state NMR: the role of Ser31 in amantadine binding. J. Mol. Biol. 2009;385:1127–1141. doi: 10.1016/j.jmb.2008.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sansom M.S., Kerr I.D. Influenza virus M2 protein: a molecular modeling study of the ion channel. Protein Eng. 1993;6:65–74. doi: 10.1093/protein/6.1.65. [DOI] [PubMed] [Google Scholar]
- 28.Duff K.C., Gilchrist P.J., Saxena A.M., Bradshaw J.P. Neutron diffraction reveals the site of amantadine blockade in the influenza A M2 ion channel. Virology. 1994;202:287–293. doi: 10.1006/viro.1994.1345. [DOI] [PubMed] [Google Scholar]
- 29.Hu J., Asbury T., Achuthan S., Li C., Bertram R. Backbone structure of the amantadine-blocked trans-membrane domain M2 proton channel from influenza A virus. Biophys. J. 2007;92:4335–4343. doi: 10.1529/biophysj.106.090183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yi M., Cross T.A., Zhou H.X. A secondary gate as a mechanism for inhibition of the M2 proton channel by amantadine. J. Phys. Chem. B. 2008;112:7977–7979. doi: 10.1021/jp800171m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chew C.F., Guy A., Biggin P.C. Distribution and dynamics of adamantanes in a lipid bilayer. Biophys. J. 2008;95:5627–5636. doi: 10.1529/biophysj.108.139477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li C., Yi M., Hu J., Zhou H.X., Cross T.A. Solid-state NMR and MD simulations of the antiviral drug amantadine solubilized in DMPC bilayers. Biophys. J. 2008;94:1295–1302. doi: 10.1529/biophysj.107.112482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kovacs F.A., Cross T.A. Transmembrane four-helix bundle of influenza A M2 protein channel: structural implications from helix tilt and orientation. Biophys. J. 1997;73:2511–2517. doi: 10.1016/S0006-3495(97)78279-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kovacs F.A., Denny J.K., Song Z., Quine J.R., Cross T.A. Helix tilt of the M2 transmembrane peptide from influenza A virus: an intrinsic property. J. Mol. Biol. 2000;295:117–125. doi: 10.1006/jmbi.1999.3322. [DOI] [PubMed] [Google Scholar]
- 35.Duong-Ly K.C., Nanda V., Degrado W.F., Howard K.P. The conformation of the pore region of the M2 proton channel depends on lipid bilayer environment. Protein Sci. 2005;14:856–861. doi: 10.1110/ps.041185805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cady S.D., Hong M. Amantadine-induced conformational and dynamical changes of the influenza M2 transmembrane proton channel. Proc. Natl. Acad. Sci. USA. 2008;105:1483–1488. doi: 10.1073/pnas.0711500105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Manor J., Mukherjee P., Lin Y.S., Leonov H., Skinner J.L. Gating mechanism of the influenza A M2 channel revealed by 1D and 2D IR spectroscopies. Structure. 2009;17:247–254. doi: 10.1016/j.str.2008.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yi M., Cross T.A., Zhou H.X. Conformational heterogeneity of the M2 proton channel: a structural model for channel activation. Proc. Natl. Acad. Sci. USA. 2009;106:13311–13316. doi: 10.1073/pnas.0906553106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chou J.J., Kaufman J.D., Stahl S.J., Wingfield P.T., Bax A. Micelle-induced curvature in a water-insoluble HIV-1 Env peptide revealed by NMR dipolar coupling measurement in stretched polyacrylamide gel. J. Am. Chem. Soc. 2002;124:2450–2451. doi: 10.1021/ja017875d. [DOI] [PubMed] [Google Scholar]
- 40.Chou J.J., Baber J.L., Bax A. Characterization of phospholipid mixed micelles by translational diffusion. J. Biomol. NMR. 2004;29:299–308. doi: 10.1023/B:JNMR.0000032560.43738.6a. [DOI] [PubMed] [Google Scholar]
- 41.Poget S.F., Girvin M.E. Solution NMR of membrane proteins in bilayer mimics: small is beautiful, but sometimes bigger is better. Biochim. Biophys. Acta. 2007;1768:3098–3106. doi: 10.1016/j.bbamem.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fernández C., Hilty C., Wider G., Wüthrich K. Lipid-protein interactions in DHPC micelles containing the integral membrane protein OmpX investigated by NMR spectroscopy. Proc. Natl. Acad. Sci. USA. 2002;99:13533–13537. doi: 10.1073/pnas.212515099. [DOI] [PMC free article] [PubMed] [Google Scholar]