

Abstract
Tripterygium wilfordii Hook F. has been used for centuries in traditional Chinese medicine to treat rheumatoid arthritis, an autoimmune disease associated with increased production of the pro-inflammatory cytokine, tumor necrosis factor (TNF)-α. Triptolide is a compound originally purified from T. wilfordii Hook F. and has potent anti-inflammatory and immunosuppressant activities. In this study, we investigated the effect of triptolide on the global gene expression patterns of macrophages treated with lipopolysaccharide (LPS). We found that LPS stimulation resulted in >5-fold increase in expression of 117 genes, and triptolide caused a >50% inhibition in 47 of the LPS-inducible 117 genes. A large portion of the genes that were strongly induced by LPS and significantly inhibited by triptolide were pro-inflammatory cytokine and chemokine genes, including TNF-α, IL-1β, and IL-6. Interestingly, LPS also induced the expression of micro-RNA-155 (miR-155) precursor, BIC, which was inhibited by triptolide. Confirming the cDNA array results, we demonstrated that triptolide blocked the induction of these pro-inflammatory cytokines as well as miR-155 in a dose-dependent manner. Profound inhibition of pro-inflammatory cytokine expression was observed at concentrations as low as 10–50 nM. However, triptolide neither inhibited the phosphorylation or degradation of IκBα after LPS stimulation, nor affected the DNA-binding activity of NF-κB. Surprisingly, we found that triptolide not only inhibited NF-κB-regulated reporter transcription, but also dramatically blocked the activity of other transcription factors. Our study offers a plausible explanation of the therapeutic mechanism of T. wilfordii Hook F.
Keywords: Inflammation, cytokines, transcription, Chinese medicine, rheumatoid arthritis, Tripterygium wilfordii
Introduction
The herb T. wilfordii Hook F., which is known as “Lei Gong Teng” in Chinese or “Thunder god vine” , has been used for centuries in traditional Chinese medicine to treat rheumatoid arthritis [1]. Although much of the clinical experience with this herb comes from uncontrolled studies and anecdotal reports, recent randomized double blind, placebo controlled clinical trials have confirmed the efficacy of the extracts of T. wilfordii Hook F. in the treatment of rheumatoid arthritis [2–4]. In addition to rheumatoid arthritis, extracts of T. wilfordii Hook F. have also shown efficacy in prolonging allograft survival, as well as treating several other autoimmune and inflammatory diseases including immune complex nephritis and systemic lupus erythematosus [5, 6]. Despite the remarkable clinical efficacy, the therapeutic mechanisms of T. wilfordii Hook F. extracts remain elusive. In addition to therapeutic properties, T. wilfordii Hook F. also exhibits a strong cellular toxicity [7, 8]. In fact, another name for T. wilfordii Hook F. in Chinese is “seven steps to death”, vividly describing its life-threatening severe toxicity [9, 10]. Because of its severe toxicity, wide spread medical application of this herb has been prohibited. Thus, identification of the active ingredients of this plant and understanding of the mode of action of these ingredients may facilitate the development of drugs that are highly efficacious but devoid of significant toxicity.
The extracts of T. wilfordii Hook F. contain more than 70 compounds including diterpenoids, triterpenoids, sesquiterpenoids, β-sitosterol, dulcitol, and glycosides [1]. Triptolide (C20H24O6), a diterpene triepoxide, is a major component of T. wilfordii Hook F. extracts, which has been shown to possess potent anti-inflammatory and immune-suppressive properties [1]. For example, triptolide has been shown to inhibit the expression of IL-2 and IFN-γ in T-cells [5, 11]. Moreover, it has been demonstrated that triptolide promotes apoptosis of T lymphocytes and dendritic cells [6, 12]. In addition to the anti-inflammatory and immunosuppressive activities, triptolide also exhibits potent anti-tumor and anti-leukemic activities [6, 13, 14]. Several studies have suggested that triptolide exerts its therapeutic activities by inhibiting the activity of several transcription factors, including NF-κB, AP-1, and NF-AT [13, 15]. In the present study, we examined the effects of triptolide on the inflammatory response of innate immune effector cells. cDNA array analysis revealed that triptolide acts as a selective transcriptional blocker predominantly affecting genes involved in the immune response. We also investigated the molecular mechanisms mediating the inhibition of cytokine expression by triptolide and demonstrated that triptolide inhibits gene transcription mediated by a number of transcription factors including NF-κB, VP16, and heat shock factor (HSF)-1 without affecting the transcription machinery.
Materials and methods
Animals
Pathogen-free female C57BL6 mice were maintained on Harlan Tecklad irradiated diet (Harlan) at 24°C with relative humidity between 30 and 70% on a 12-h day-night cycle. The experimental protocols were approved by the Institutional Animal Care and Use Committee of The Research Institute at Nationwide Children's Hospital.
Reagents
LPS (Escherichia coli 055:B5), tetracycline, and triptolide were purchased from Calbiochem (La Jolla, CA). Triptolide was dissolved in DMSO and added to the culture medium.
Cell culture
Thioglycollate-elicited peritoneal macrophages were isolated from female C57BL6 mice by peritoneal lavage and cultured as described previously [16]. RAW264.7 cells were cultured in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal bovine serum at 37°C in a humidified atmosphere containing 5% CO2. The CHO-AA8-Luc cells, purchased from Clontech (Mount View, CA) were cultured in Alpha Minimum Essential Medium (Cellgro, Herndon, VA) containing 10% Tet-approved fetal bovine serum (Clontech), 4 mM glutamine, 1% penicillin/streptomycin, and 100 μg/ml G418. The CHO-AA8-Luc cells harbor a stable luciferase reporter whose expression is under the control of a Tet-Off system (Clontech). Reporter gene transcription was turned off by the addition of tetracycline (2 μg/ml), and turned on by tetracycline withdrawal. Heat shock treatment of RAW264.7 cells were carried out by immersing cells in a 43°C water bath for 1 h.
cDNA microarray analysis
RAW264.7 cells were pre-treated with 50 nM triptolide or vehicle for 30 min, and then stimulated with 100 ng/ml LPS for 4 h. Total RNA was purified using Qiagen RNeasy kit (Qiagen, Valecia, CA). RNA integrity was confirmed with a Bioanalyzer 2100 capillary electrophoresis system and the Degradometer software [17]. Labeled cDNA probes were synthesized using 20 ng total RNA and a SPIA Biotin System [18]. Labeled cDNA was hybridized to 430A2.0 mouse GeneChips (Affymetrix, Santa Clara, CA) according to manufacturer's recommendations. The 430A2.0 mouse GeneChip contained sequences representing over 14,000 well defined gene transcripts. Scanned images (DAT files) were converted into CEL files using GCOS software (Affymetrix). Gene expression levels were calculated using RMAExpress [19]. Differentially expressed genes were analyzed for over-represented biological themes (Gene Ontology categories) using EASE software [20]. Cluster analyses were performed on two subset of genes, first those that are up-regulated 5-fold by LPS, and second those that are up-regulated 1-5-fold by LPS and also down-regulated at least 50% by triptolide.
Western blot analysis
Western blot analysis was carried out as previously described [16, 21–23]. A rabbit polyclonal antibody against mouse IL-1β was purchased from Chemicon (Temecula, CA). The antibodies against the p50 subunit of NF-κB, IKK, and IκBα were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The rabbit polyclonal IKK antibody recognizes both IKKα and IKKβ. The rabbit polyclonal antibody against the p65 subunit of NF-κB was a generous gift from Dr. Dennis Guttridge at the Ohio State University. The polyclonal antibody against phospho-IκBα (p-Ser 32) and an IκBα monoclonal antibody were purchased from Cell Signaling (Danvers, MA). To normalize for protein loading, blots were stripped and probed with a β-actin monoclonal antibody (Sigma, St. Louis, MO).
Northern blotting
Total RNA was isolated from cells using STAT-60 (Tel-Test, Friendswood, TX). Northern blot analysis was carried out using mouse IL-1β, TNF-α, luciferase, and Hsp70 cDNA probes as described previously [24, 25]. The membrane was stripped and reprobed with 18S rRNA for normalizing RNA loading.
Northern blotting was also used to assess miR-155 expression. RAW264.7 cells were pretreated with various doses of triptolide for 30 minutes followed by stimulation with LPS (100 ng/ml) for 4 h. Total RNA harvested using TRIzol reagent (Invitrogen, San Diego, CA) was resolved by electrophoresis on a 12% denaturing urea polyacrylamide gel. RNA was transferred onto a Hybond N+ membrane (Amersham, Piscataway, NJ), and crosslinked to the membrane using UV radiation. The miR-155 and U6 probes were labeled with [α-32P]dATP using StarFire Nucleic Acid labeling system (IDT DNA, Coralville, IA). Hybridization was carried out essentially as previously described [26]. The same membrane was stripped and rehybridized with a probe for the small nuclear RNA U6 for normalizing sample loading.
ELISA
RAW264.7 cells or primary peritoneal macrophages were plated into 6-well plates at a density of 3×105 cells per well. The next day, cells were pretreated either with triptolide (0.01–1 μM) or with equal volume of vehicle (DMSO) for 30 min, and then stimulated with 100 ng/ml LPS for 6 h. TNF-α and IL-6 concentrations in the media were determined by ELISA as previously described [22].
Animal studies
C57BL6 mice were first injected with triptolide intraperitoneally (0.15 or 0.25 mg/kg body weight) or an equal volume of vehicle (DMSO). Thirty min later, these mice were challenged with LPS intraperitoneally at a dose of 5 mg/kg body weight. Mice were euthanized 60 min post LPS challenge, and blood was collected by cardiac puncture. Concentrations of TNF-α and IL-6 in the serum were determined by ELISA as previously described [22].
Immunofluorescence
To examine the effect of triptolide on the nuclear translocation of NF-κB p65 subunit, we performed immunofluorescence. RAW264.7 cells were treated with LPS (100 ng/ml) in the presence or absence of triptolide (0.1 μM) for the indicated times, controls were treated with DMSO only. Immunofluorescence was essentially carried out as previously described [22, 27], using a rabbit polyclonal anti-p65 antibody (Santa Cruz) and Alexa488-conjugated goat anti-rabbit IgG secondary antibody (Invitrogen). Finally, the cells were stained with 4,6-diamidino-2-phenylindole (DAPI) to visualize the nuclei and were examined under a Zeiss Axioskop microscope (Carl Zeiss, Inc., New York). The fluorescent images were acquired with identical exposure time for all of the samples. The percentage of cells where p65 was localized in the nuclei was scored from at least 10 fields chosen randomly.
Electromobility gel-shift assay (EMSA)
RAW264.7 cells were pre-treated with 0.1 μM triptolide, and then stimulated with 100 ng/ml LPS. Nuclear extracts were prepared essentially as previously described [28]. Oligo-nucleotides (5'-AGTTGAGGGGACTTTCCCAGGC-3' and 5'-GCCTGGGAAAGTCCCCTCAACT-3') were annealed, labeled with [γ-32P]ATP using T4 polynucleotide kinase, and then purified using Sephadex G-25 spin columns. Nuclear extracts (5 μg protein) were incubated with the 32P-labeled double-stranded oligonucleotides for 30 min in a buffer containing 10 mM Tris-HCl (pH 8.0), 0.1% Triton X-100, 12.5% glycerol, 150 mM KCl, 1 mM DTT, 0.5 mM EDTA, and 2 μg of poly IC. Samples were resolved on a 5% nondenaturing polyacrylamide gel using 0.5xTBE (25 mM Tris-HCl pH8.0, 25 mM boric acid, 0.5 mM EDTA) as running buffer. Gels were dried, and subjected to PhosphoImaging.
Plasmids
Site-directed mutagenesis was carried out to remove the DNA region coding for the VP16 transactivation domain in the pTet-off plasmid (Clontech) while creating an unique Srf I site. The transactivation domain of NF-κB was amplified by PCR from a human p65 cDNA with high fidelity pfu DNA polymerase, using primers 5'-CCTCAGGCTGTGGCCCCACCTG-3' and 5'-TCATTAGGAGCTGATCTGACTCAG-3'. This blunt-end PCR product was then cloned into the Srf I site of the modified pTet-off plasmid. When expressed, this construct results in a fusion protein consisting of the Tet repressor DNA binding protein (TetR) and the transactivation domain of the p65 subunit of NF-κB, referred to hereafter as TetR-p65TA hereafter. The authenticity of the cloning was confirmed by sequencing.
Transfection and luciferase assays
To examine the effect of triptolide on reporter transcription mediated by the transactivation domain of NF-κB, RAW264.7 cells were transiently transfected with the plasmid (TetR-p65TA) and the pTRE2-hyg-luc reporter (Clontech), using the polyethyleneimine transfection reagent (Polysciences, Warrington, PA) in the presence of tetracycline. Twenty four h following transfection, cells were fed with medium containing tetracycline, or fed with tetracycline-free medium that contained either 0.1 μM triptolide or vehicle (DMSO) for 24 h. Cells were harvested and luciferase activity measured. Luciferase activity was normalized to lysate protein concentration.
CHO-AA8-Luc cells were grown in the presence of tetracycline until reaching a confluency of ∼50%. Then cells were washed with PBS twice and fed with fresh medium with or without tetracycline, in the absence or presence of triptolide. Cells were grown overnight, harvested, and luciferase activity in the lysates was measured using a luminometer with a Luciferase Assay System (Promega, Madison, WI).
In vitro transcription assay
Mixtures of transcription factors, DNA templates and α-32P CTP, were incubated with 100 nM of triptolide or vehicle (DMSO) at 30°C for 60 min. The reactions for the basal level transcription were performed with the transcription kit from ProteinOne (Bethesda, MD) according to the manufacturer's recommendations. In vitro assay for activated transcription was reconstituted with purified transcription factors (TFIID, TFIIA, GAL4-AH, PC4) in the presence of template pG5HM and pMLΔ53 essentially as described [29]. RNA was isolated by phenol chloroform extraction, resolved by electrophoresis on a 5% urea-polyacrylamide gel and visualized by autoradiography.
Statistics
The results from the experiments assessing the effects of triptolide on TNF-α and IL-6 production were analyzed by one-way ANOVA with LSD post hoc test. Statistical analysis was carried out using SPSS statistical software program (SPSS, Chicago, IL). Differences were considered significant when p < 0.05.
Results
Triptolide selectively inhibits the expression of inflammatory genes including proinflammatory cytokines and chemokines
To assess the effect of triptolide on the inflammatory response of innate immune effector cells, we performed microarray analysis. To this end, RAW264.7 macrophages were pre-treated with 50 nM triptolide or vehicle for 30 min, and then stimulated with LPS for 4 h. Total RNA was harvested and used to produce cDNA probes. Labeled cDNA was hybridized to 430A2.0 mouse GeneChips (Affymetrix), which contained sequences corresponding to over 14,000 well-defined gene transcripts. Gene expression levels were calculated using RMAExpress. Differentially expressed genes were analyzed for over-represented biological themes (Gene Ontology categories) using EASE software. LPS treatment resulted in >5-fold induction in the expression of 117 genes (Figure 1A). Among these 117 genes induced by LPS, 47 of them were inhibited by 50% or more by triptolide (50 nM). Many of these LPS-induced, triptolide-attenuated genes are involved in immune function/host defense, including pro-inflammatory cytokines (TNF-α, IL-1β, IL-6, colony stimulating factor (CSF)-2 and 3) and chemokines (CCL-1, 2, 5, 7, and 12 as well as CXCL-10 and 11). In addition to these cytokines and chemokines, triptolide also inhibited the expression of several adhesion molecules (laminin γ1, integrin α5, ICAM1), a number of transcription factors (ATF3, NF-κB, STAT5A), as well as BIC that is the precursor of miR-155. The global effects of triptolide on the expression of genes that are potently induced by LPS (>5-fold) are shown in Figure 2.
Figure 1.

Triptolide selectively inhibits the expression of a subset of genes involved in the immune response. RAW264.7 cells were either treated with vehicle or LPS in the presence or absence of triptolide, and microarray analyses were performed. Heat maps were generated representing cluster analyses of genes exhibiting 5-fold or greater induction by LPS (A) or 1–5-fold induction by LPS but inhibited by triptolide at least 50% (B).
Figure 2.
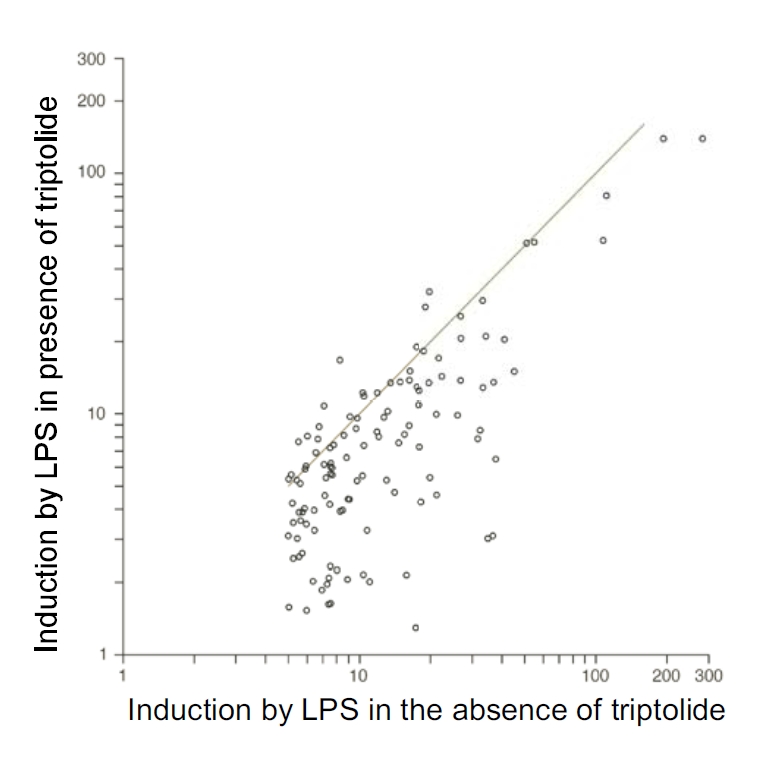
Triptolide potently inhibits the expression of genes that were potently induced by LPS. The graph depicts the effect of triptolide on the genes highly induced (>5-fold) by LPS. Dots above the line represent genes whose expression is enhanced by triptolide. Dots below the line represent genes whose expression is inhibited by triptolide.
We also analyzed the effects of triptolide on the expression of genes that are induced less potently by LPS or are relatively insensitive to LPS (1–5-fold induction by LPS relative to unstimulated control). Total of 10,600 genes fell into this category. Among these genes, the expression levels of 119 genes were decreased by triptolide pretreatment by at least 50% (Figure 1B). These results indicate that triptolide does not block global gene expression in a nondiscriminatory manner. Our analysis suggests that triptolide preferentially inhibits genes that are highly inducible by inflammatory stimuli.
Triptolide inhibits pro-inflammatory cytokine production in LPS-stimulated macrophages
To validate the results of the cDNA array analysis, we examined the effect of triptolide on the expression of a small set of pro-inflammatory cytokine genes. Since pro-inflammatory cytokines such as TNF-α and IL-1β are critical in the pathogenesis of inflammatory diseases, including rheumatoid arthritis, understanding of the mechanism underlying the inhibitory effect of triptolide on these cytokines may reveal important information on the mode of action of this novel compound. The effect of triptolide on the expression of IL-1β and TNF-α induced by LPS in RAW264.7 cells was examined by Northern blot analysis. While basal IL-1β and TNF-α mRNA levels were undetectable in control cells, these genes were dramatically induced upon LPS stimulation. Treatment with triptolide caused a dose-dependent inhibition in both IL-1β and TNF-α mRNA levels (Figure 3A). Substantial inhibition of IL-1β and TNF-α mRNA levels were observed in samples treated with triptolide at concentrations as low as 50 nM.
Figure 3.

Triptolide blocks the induction of pro-inflammatory cytokine mRNA in LPS-stimulated macrophages. RAW264.7 cells (A) and primary peritoneal macrophages (B) were pretreated with indicated doses of triptolide for 30 min, and then stimulated with LPS (100 ng/ml) for 4 h. Total RNA was harvested and analyzed by Northern blotting. The IL-1β and TNF-α mRNA signals were normalized to 18S rRNA signals, and expressed as fold increase relative to control. Numbers below the blots indicate fold of changes in gene expression relative to untreated controls.
The effect of triptolide on the induction of these cytokines in primary macrophages was also examined. As in the RAW264.7 cells, the induction of both TNF-α and IL-1β by LPS in primary macrophages was strongly inhibited by triptolide in a dose-dependent manner (Figure 3B). The inhibitory effects of triptolide on these cytokine mRNAs were clearly visible starting at 20 nM.
The effect of triptolide on the production of cytokine proteins was examined by ELISA. LPS potently induced the production of both TNF-α and IL-6 in RAW264.7 macrophages, and triptolide potently inhibited the cytokine production in a dose-dependent manner, with an IC50 of <30 nM for both TNF-α and IL-6 (Figure 4A). Impressively, treatment with 50 nM triptolide resulted in a greater than 80% decrease in both TNF-α and IL-6 production in LPS-stimulated cells. Triptolide also potently suppressed LPS-induced IL-1β production in a dose-dependent manner.
Figure 4.

Triptolide potently inhibits the production of pro-inflammatory cytokine proteins in macrophages treated with LPS. RAW264.7 cells (A) and primary peritoneal macrophages (B) were pretreated with indicated doses of triptolide for 30 min followed by stimulation with 100 ng/ml LPS for 6 h. The negative control was only treated with equal volume of vehicle (DMSO). Positive controls were those first treated with vehicle (DMSO) then stimulated with LPS. Upper panels represent the effect of triptolide on TNF-α and IL-6 secretion, which were assayed by ELISA. Results in the graph represent % relative to the mean value of the positive controls. Data were expressed as means ± S.E. from 3 independent experiments. *, p<0.05, compared to the group stimulated with LPS after pretreatment with vehicle. Lower panels demonstrate the effect of triptolide on IL-1βproduction detected by Western blot analysis. Presented are the representative results of at least three experiments.
Similar to what happened in RAW264.7 cells, triptolide potently inhibited IL-1β production in LPS-stimulated primary macrophages in a dose-dependent manner (Figure 4B). Likewise, production of both TNF-α and IL-6 production in primary macrophages were also inhibited by triptolide. Triptolide, at a concentration of 10 nM, inhibited TNF-α production by approximately 80%. Compared to TNF-α, IL-6 production appeared to be less sensitive to triptolide (Figure 4B). Triptolide, at concentrations of 10 and 50 nM, inhibited IL-6 production by ∼40% and ∼80%, respectively.
Triptolide inhibits the induction of miR-155 in LPS-stimulated macrophages
miR-155 is a key player in inflammatory response, our microarray results showed that the expression of the miR-155 precursor, BIC, was up regulated 7-fold following stimulation with LPS. Triptolide attenuates BIC expression by 80%. To validate these results, RAW264.7 cells were pre-treated cells with triptolide (0.01–0.5 μM) for 30 min followed by stimulation with LPS for 4 h, and miR-155 expression was assessed by Northern blot analysis. LPS treatment resulted in a robust increase in miR-155 level, and miR-155 induction by LPS was attenuated in a dose-dependent manner by triptolide (Figure 5).
Figure 5.

Triptolide inhibits the induction of miR-155 in LPS-stimulated macrophages. RAW264.7 cells were pretreated with the indicated doses of triptolide followed by stimulation with LPS for 4 h. Total RNA was harvested and Northern blot analysis was performed using a urea-PAGE denaturing gel. U6 was used as a loading control. Graph depicts the fold of changes in miR-155 expression in a representative experiment.
Triptolide attenuates the inflammatory response in vivo in LPS-challenged mice
To investigate the effects of triptolide on the inflammatory response in vivo, C57BL6 mice were injected intraperitoneally with either triptolide or the vehicle, and then challenged with LPS 30 min later. Blood was harvested 60 min post LPS challenge, and cytokine concentrations in the serum were measured. Challenge of C57BL6 mice with LPS caused a robust increase in TNF-α production as compared to un-challenged mice (Figure 6A). At a dose of 0.15 mg/kg body weight, triptolide decreased blood TNF-α levels by 64%. At a higher dose (0.25 mg/kg body weight), triptolide almost completely abolished TNF-α production in vivo. Likewise, triptolide markedly inhibited the LPS-stimulated increase in blood IL-6 levels in a dose-dependent manner (Figure 6B).
Figure 6.

Triptolide attenuates LPS-induced cytokine production in vivo in a dose-dependent manner. C57BL6 mice (5–6 weeks old) were given the indicated doses of triptolide, or equal volumes of vehicle i.p, and then challenged with LPS (5 mg/kg body weight). Cytokine concentrations in the serum were determined by ELISA. (A) Effects of triptolide on TNF-α biosynthesis in vivo. (B) Effects of triptolide on IL-6 production in vivo. Values represent the mean ± S.E. from 8–12 animals. †, p < 0.05, compared to control group (not stimulated with LPS). *, p<0.05, compared to the group received vehicle and then stimulated with LPS.
Triptolide has no effect on IκBα degradation and p65 nuclear translocation, and does not affect NF-κB DNA binding activity
Previously, it has been reported that NF-κB activity is inhibited by triptolide [15].
Furthermore, a study by Zhou et al. showed that IκB degradation and NF-κB DNA-binding activity are inhibited by a triptolide derivative [30]. To elucidate the mode of action of triptolide, RAW264.7 cells were treated with LPS in the absence or presence of triptolide, and kinetics of IκBα phosphorylation were determined by Western blot analysis using an anti-phospho-IκBα antibody (Figure 7A). Upon LPS stimulation, the levels of phospho-IκBα peaked at about 10 min, then rapidly plummeted. The decreases in phospho-IκB levels were associated with decreases in IκBα protein levels, detected by Western blotting using a monoclonal antibody recognizing total IκBα. At 60 min, IκBα protein levels increased again, likely due to de novo protein synthesis. This increase in newly synthesized IκBα coincided with increased phospho-IκBα levels, suggesting IκB kinases remained active at least up to 60 min post LPS stimulation (Figure 7A). Importantly, neither the kinetics of IκBα phosphorylation nor IκBα degradation was significantly affected by triptolide. The levels of p65, p50, and IKK proteins did not change regardless of whether the cells were pretreated with triptolide or not (Figure 7A). We then investigated the effect of triptolide on NF-κB translocation to the nucleus, by immunofluorescence staining of the p65 subunit of NF-κB (Figure 7B). p65 was almost exclusively localized in the cytoplasm in control cells, and was translocated to the nuclei upon LPS stimulation. Triptolide had no effect on the kinetics of nuclear translocation of NF-κB p65 subunit in LPS-stimulated RAW264.7 cells (Figure 7B). To examine if triptolide inhibits the DNA binding activity of NF-κB, RAW264.7 cells were pre-treated with triptolide for 30 min then stimulated with LPS. Nuclear extracts were collected at the indicated times and EMSA was performed. LPS stimulation resulted in an increase of NF-κB DNA-binding activity, which was not affected by triptolide (Figure 7C).
Figure 7.

Triptolide has no effect on IκBα degradation and p65 nuclear translocation, and does not affect NF-κB DNA-binding activity in LPS-stimulated macrophages. (A) Effects of triptolide on the components of the NF-κB pathway in LPS-stimulated macrophages. RAW264.7 cells were pretreated with 0.1 μM triptolide for 30 minutes, then stimulated with 100 ng/ml LPS for the indicated periods. Cells lysates were harvested and Western blotting was performed. Data shown were from a representative experiment. (B) Effect of triptolide on nuclear translocation of NF-κB p65 subunit. RAW264.7 cells were treated as in A, and immunofluorescence was performed with a p65 antibody (green), and then stained with DAPI (blue). The percentage of cells where p65 was localized in the nuclei was scored from at least 10 fields chosen randomly. Values in the graph on the right side represent mean ± S.E. from 3 independent experiments. (C) Effect of triptolide on NF-κB DNA-binding activity. RAW264.7 cells were treated as in A, and nuclear proteins were extracted. EMSA was performed using end-labeled double-stranded oligonucleotides containing a consensus NF-κB-binding element. Results were from a representative experiment.
Triptolide blocks gene transcriptional induction regulated by a variety of transcriptional factors
Previous studies have suggested that triptolide inhibits transactivation of NF-κB at a step after binding to DNA [15]. To gain insight into the inhibitory mechanism of NF-κB by triptolide, we examined the effect of triptolide on the activity of the transactivation domain of p65, a subunit of NF-κB, by taking advantage of the Tet-Off system. We modified the Tet-Off system by replacing the VP16 transactivation domain with the transactivation domain of p65. This modification yielded an artificial hybrid transcription factor TetR-p65TA, which consists of the DNA-binding domain of TetR and the transactivation domain of p65. This construct was transfected into RAW264.7 cells together with a tetracycline-regulated luciferase reporter gene (pTRE-Luc), and the effect of triptolide on reporter gene expression was assessed. TetR-p65TA efficiently turned on the transcription of the luciferase reporter in the absence of tetracycline and triptolide potently inhibited the reporter gene transcription (Figure 8A). As a control, we also assessed the effect of triptolide on reporter transcription mediated by the artificial transcription factor TetR-VP16, using a stable cell line (CHO-AA8-Luc) which harbors both the TetR-VP16 and the TRE-Luc reporter constructs. Surprisingly, triptolide showed a dose-dependent inhibition of the luciferase reporter transcription. Triptolide at concen-trations of 50–100 nM almost completely abrogated reporter expression elicited by tetracycline withdrawal (Figure 8B). Northern blot analysis further confirmed that triptolide, at a concentration of 20 nM, abolished luciferase mRNA expression induced by tetracycline withdrawal (Figure 8C).
Figure 8.

Triptolide blocks gene transcriptional induction regulated by a variety of transcriptional factors. Effect of triptolide on reporter expression mediated by the transactivation domains of p65 NF-κB (A) and VP16 (B). RAW264.7 (A) and CHO-AA8-Luc (B) cells were treated as described. Cells were then lysed and luciferase activity in the lysates was measured. Relative luciferase units were normalized to protein concentrations. Luciferase activities in both A and B were expressed in the graphs as fold change relative to cells kept in tetracycline-containing medium (controls). Values represent mean ± SE from 3 independent experiments. † different from control group (with tetracycline), p<0.05. *, different from the group without tetracycline but treated with vehicle (DMSO), p<0.05. (C) Effect of triptolide on luciferase reporter mRNA expression mediated by the VP16 transactivation domain. CHO-AA8-Luc cells were treated as in B. Cells were harvested to isolate total RNA, and Northern blot analysis was performed. Luciferase expression was normalized to 18S rRNA, and expressed as fold increase relative to control (signal of cells grown in medium containing only tetracycline). (D) Triptolide inhibits the induction of Hsp70 by heat shock. RAW264.7 cells were subjected to heat shock in the presence of indicated doses of triptolide, and Northern blot analysis was performed. (E) Triptolide does not inhibit gene transcription by RNA polymerase II in vitro. RAW264.7 cells were either left untreated (C) or treated with vehicle (V) or triptolide (T), and in vitro transcription assays were performed using the indicated templates.
To explore the specificity of triptolide on the transcriptional induction of endogenous genes, we examined the effect of triptolide on Hsp70, which is induced by heat shock through a completely different transcription factor, heat shock factor (HSF)-1 [31]. To this end, RAW264.7 cells were pre-treated with either various doses of triptolide or vehicle (DMSO), and then subjected to heat shock at 43°C for 1 h. Hsp70 mRNA was detected by Northern blot analysis. Hsp70 mRNA in control cells was undetectable, and its mRNA levels increased dramatically after heat shock. Triptolide pre-treatment caused a dose-dependent inhibition of Hsp70 mRNA induction (Figure 8D). These studies clearly indicate that triptolide not only inhibits the activity of NF-κB, but also has broad inhibitory effects on gene transcription mediated by other transcription factors.
To elucidate the mechanism by which triptolide blocks gene transcription, we examined the effect of triptolide on gene transcription using an in vitro gene transcription assay. The reaction mixtures containing RNA polymerase II complex, transcription factors and co-activators were incubated with 100 nM triptolide or DMSO. Basal activity of the transcription machinery was assayed using two plasmid DNA templates: pG5HM and pMLΔ53. Basal transcription requires a number of indispensable transcription factors (TBP, TFIIB, and TFIIE/F/H) while activated transcription relies on activators and cofactors that are not necessary for basal transcription (TFIIA, TFIID, GAL4-AH, and PC4). The pG5HM template contains 5 GAL4 binding sites and can support both basal and activated transcription. However, the pMLΔ53 plasmid has no GAL4 binding sites and can only be transcribed by the basal transcription machinery. As indicted in Figure 8E, triptolide at a concentration of 100 nM had little effect on in vitro gene transcription mediated by the basal transcriptional machinery. Likewise, triptolide also did not have an appreciable effect on gene transcription mediated by the GAL4-AH transcriptional factor in vitro. These results suggest that triptolide does not simply block the transcription machinery.
Discussion
Anti-inflammatory mechanism of triptolide
Triptolide has potent anti-inflammatory properties. It has been shown that triptolide inhibits experimental autoimmune uveoretinitis [32] and prolongs allograft survival [33]. Moreover, it has been demonstrated that a succinyl derivative of triptolide, PG490-88, can prevent graft-versus-host disease [34]. Although the immunosuppressive action of triptolide has been generally attributed to suppression of T-lymphocyte activation [1, 5], recent studies have also provided evidence that triptolide also inhibits innate immune functions. For example, Lu et al. have demonstrated that triptolide blocks the production of two chemokines, IL-8 and MCP-1, in cultured human corneal fibroblasts stimulated with proinflammatory cytokines [35]. These findings suggest that triptolide may exert its anti-inflammatory effects by limiting the infiltration of neutrophils and monocytes into the cornea [35]. Very recently, Zhu et al. and Chen et al. have shown that triptolide can inhibit the differentiation, maturation, trafficking, and function of dendritic cells, the professional antigen presenting cells [36, 37]. Moreover, triptolide has also been shown to suppress CD80 and CD86 expression and block IL-12 production in human monocytic THP-1 cells [38]. In the present report, we performed microarray analysis on LPS-treated RAW264.7 cells that have been pretreated with triptolide (Figures 1). Our results indicate that triptolide exerts its anti-inflammatory activity through selective transcriptional blockade of genes involved in the immune response (Figure 2). These results are in agreement with microarray results reported by Zhao et al. showing that triptolide is not a nonspecific inhibitor of transcription in bronchial epithelial cells [39]. The selectivity of transcriptional inhibition by triptolide suggests that it interferes with transcription factors regulating the transcription of a subset of genes, predominantly cytokines and chemokines. We have examined the effect of triptolide on the production of pro-inflammatory cytokines in macrophages, a major source of pro-inflammatory cytokines. We found that triptolide abrogated the production of pro-inflammatory cytokines both in vitro (Figure 4) and in vivo (Figure 6). Profound inhibition of pro-inflammatory cytokine expression was observed at concentrations as low as 10–50 nM. The inhibition of cytokine production by triptolide occurs at the mRNA level in both immortalized and primary macrophages (Figure 3), suggesting that triptolide blocks the transcription of pro-inflammatory cytokine genes. These results suggest that inhibition of pro-inflammatory cytokine expression by triptolide may play an important role in the anti-rheumatic/anti-inflammatory properties of T. wilfordii Hook F.
The mechanism by which triptolide inhibits the production of inflammatory cytokines
Triptolide has three highly reactive epoxide groups, which are capable of reacting with nucleophilic protein side chains such as those on histidine, cysteine, serine, and aspartic acid. Covalent modification of protein target(s) by an epoxide group(s) of triptolide is consistent with the exceptional potency of this compound. Very recently, it has been reported that triptolide binding to HeLa cells was saturable, reversible, and primarily localized to cell membrane proteins with molecular mass ranging from 75 to greater than 250 kDa [40]. The fact that triptolide binding to proteins can be detected after separation on denaturing SDS-polyacrylamide gels [40] strongly argues that triptolide interacts with its targets through covalent bonds. Due to the high reactivity of the epoxide rings, triptolide is expected to react with a variety of intracellular nucleophiles such as glutathione and have a relatively short half-life. This may explain the observed “reversibility” of the compound [40, 41], although it is also possible that the covalent linkage between triptolide and its protein target(s) is chemically reversible.
The observation that the transcription of several inflammatory cytokines is potently inhibited by triptolide suggests that triptolide may either disrupt the signaling process leading to cytokine transcription or directly interfere with the transcriptional process. NF-κB is a transcription factor that plays a critical role in the transcriptional induction of many inflammatory cytokines [42, 43]. Since it has been proposed that triptolide inhibits NF-κB activity at a step after NF-κB binding to DNA [15], we examined the effects of triptolide on the NF-κB pathway. We found that triptolide pretreatment neither affects the phosphorylation-mediated degradation of IκBα after LPS stimulation, nor decreases the DNA-binding activity of NF-κB in cell nuclear extracts (Figure 7). These observations are in agreement with the findings that DNA binding activity of NF-κB is not affected by triptolide [15, 39]. However, our findings contradict recent reports that triptolide inhibited LPS-induced increases in the DNA-binding activity of NF-κB in primary macrophages and RAW264.7 cells [44, 45].
The exact reasons for these discrepancies are unclear, but may represent differences in experimental methodologies.
Several earlier studies have examined the effect of triptolide on the regulation and activity of NF-κB [15, 46]. Using a luciferase reporter containing a NF-κB-binding element, Qiu et al. have shown that triptolide potently abolishes the reporter gene transcription elicited by TNF-α. Likewise, both Zhao et al. and Lee et al. have found that triptolide potently inhibits NF-κB-dependent reporter transcription induced by PMA or the inflammatory cytokines, TNF-α and IL-1β [39, 46]. More recently, Leuenroth and Crews have demonstrated that triptolide blocks NF-κB-dependent luciferase reporter transcription at a concentration greater than 50 nM [40]. Using a GAL4-NF-κB hybrid transcription factor construct together with a GAL4-dependent luciferase reporter system, Qiu et al. have shown that triptolide abolishes transcription activation mediated by the NF-κB transactivation domain [15]. These investigators have postulated that triptolide blocks NF-κB-mediated transcription by interfering with p65 modification or the recruitment of a transcriptional cofactor [15, 46]. In the present study, we have investigated the effects of triptolide on the transcription-enhancing property of the NF-κB transactivation domain, using a modified Tet-Off system (Figure 8A). Unlike the original Tet-Off system that uses the transactivation domain of the viral VP16 protein, the modified system described herein relies on the transactivation domain of NF-κB. Compared to the GAL4-NF-κB-coupled luciferase reporter system used by Qiu et al. [15], which is constitutively active when expressed, reporter gene expression driven by the NF-κB-based Tet-Off system described herein has the obvious advantage of being expressed only after tetracycline withdrawal. With this system, we found that triptolide, at a concentration of 100 nM, inhibited NF-κB-driven reporter gene transcription by ∼75% (Figure 8A).
Surprisingly, triptolide also dramatically inhibited the VP16-driven luciferase reporter activity (Figure 8B). To rule out the possibility that triptolide may directly react with luciferase and abolish its enzymatic activity, we performed Northern blot analysis (Figure 8C).
Our results indicate that luciferase transcription was completely abolished by triptolide at concentrations as low as 20 nM (Figure 8C). These results clearly indicate that the inhibition of gene transcription by triptolide is not only limited to genes transcriptionally regulated by NF-κB. This notion is further supported by the finding that induction of Hsp70 in response to heat shock, mediated by a distinct transcription factor HSF-1, is also strongly inhibited by triptolide (Figure 8D). The inhibitory effect of triptolide on Hsp70 induction in HeLa cells has been reported recently [41]. Similar to what was observed herein for NF-κB, Westerheide et al. found that triptolide abrogates the transactivation function of HSF-1 without interfering in the early events of trimer formation, hyperphos-phorylation, and DNA binding [41]. The fact that triptolide blocks the activities of three unrelated transcription factors (NF-κB, VP16, and HSF-1) suggests that triptolide inhibits a common step(s) of the transcripttional processes mediated by these transcripttion factors. While the mechanisms involved are currently unclear, we can make some speculations. First, it is possible that triptolide poisons the basal transcription machinery. However, we consider this scenario unlikely, since not all genes are inhibited equally by triptolide. Microarray experiments clearly indicate that some genes are more sensitive than others to inhibition by triptolide (Figure 1 and 2). For example, triptolide significantly inhibits TNF-α expression at concentrations between 20–100 nM. Although MKP-1 is also potently induced by LPS, significant inhibition was only observed at concentrations between 500–1,000 nM [21]. Second, a significant inhibition of basal gene transcription by triptolide was not observed in an in vitro assay. While we recognize the limitations of our in vitro transcription assays, we think that triptolide likely only inhibits the transcription mediated by a subset of transcription factors. This notion is consistent with a recent report that triptolide inhibits the interaction between p65 and the transcription coactivator CBP/p300 [47]. Indeed, the inhibition of a transcriptional mechanism utilized by a variety of transcription factors may explain the serious toxicity of triptolide and T. wilfordii Hook F. Further characterization of the interaction between triptolide and transcription factors or transcriptional coactivators may not only reveal novel targets for the development of anti-inflammatory and anti-rheumatic drugs, but also facilitate the development of less-toxic triptolide derivatives for therapeutic purposes.
Acknowledgments
This work was supported by grant R01AI57798 from NIAID, grant R01HL75261 from NHLBI, and funding from The Research Institute at Nationwide Children's Hospital. We are grateful to Dr. Lurong Zhang for helpful discussion. We thank Dr. Denis Guttridge for providing us with pCMV-Flag-p65 plasmid.
Abbreviations used
- LPS
lipopolysaccharide
- IL
interleukin
- TNF
tumor necrosis factor
- IFN-κ
interferon-κ
- NF-κB
nuclear factor κB
- IκB
inhibitor κB
- IKK
IκB kinase
- STAT
signal transducer and activator of transcription
- ATF
activating transcription factor
- AP-1
activating protein-1
- NF-AT
nuclear factor of activated T cells
- VP16
herpes simplex virus protein 16
- miR
microRNA
- DMSO
dimethyl sulfoxide
- PBS
phosphate-buffered saline
- Hsp
heat shock protein
- HSF
heat shock factor
- TetR-p65TA
fusion protein composed of TetR and p65 transactivation doman
- EMSA
electromobility gel-shift assay
- TBP
TATA-binding protein
- TFIIA, B, D, E, F, H
transcription factor IIA, IIB, IID, IIE, IIF, IIH
- CBP
cAMP response element-binding protein (CREB)-binding protein
- GAL4-AH
a fusion protein between N-terminal of GAL4 and an artificial 15 amino acid putative ampipathic hélix
- DAPI
4,6-diamidino-2-phenylindole
- ANOVA
analysis of variance
References
- 1.Chen BJ. Triptolide, a novel immunosuppressive and anti-inflammatory agent purified from a Chinese herb Tripterygium wilfordii Hook F. Leuk Lymphoma. 2001;42:253–265. doi: 10.3109/10428190109064582. [DOI] [PubMed] [Google Scholar]
- 2.Tao X, Cush JJ, Garret M, Lipsky PE. A phase I study of ethyl acetate extract of the chinese antirheumatic herb Tripterygium wilfordii hook F in rheumatoid arthritis. J Rheumatol. 2001;28:2160–2167. [PubMed] [Google Scholar]
- 3.Tao X, Younger J, Fan FZ, Wang B, Lipsky PE. Benefit of an extract of Tripterygium Wilfordii Hook F in patients with rheumatoid arthritis: a double-blind, placebo-controlled study. Arthritis Rheum. 2002;46:1735–1743. doi: 10.1002/art.10411. [DOI] [PubMed] [Google Scholar]
- 4.Cibere J, Deng Z, Lin Y, Ou R, He Y, Wang Z, Thorne A, Lehman AJ, Tsang IK, Esdaile JM. A randomized double blind, placebo controlled trial of topical Tripterygium wilfordii in rheumatoid arthritis: reanalysis using logistic regression analysis. J Rheumatol. 2003;30:465–467. [PubMed] [Google Scholar]
- 5.Qiu D, Kao PN. Immunosuppressive and anti-inflammatory mechanisms of triptolide, the principal active diterpenoid from the Chinese medicinal herb Tripterygium wilfordii Hook. f. Drugs R D. 2003;4:1–18. doi: 10.2165/00126839-200304010-00001. [DOI] [PubMed] [Google Scholar]
- 6.Liu Q, Chen T, Chen H, Zhang M, Li N, Lu Z, Ma P, Cao X. Triptolide (PG-490) induces apoptosis of dendritic cells through sequential p38 MAP kinase phosphorylation and caspase 3 activation. Biochem Biophys Res Commun. 2004;319:980–986. doi: 10.1016/j.bbrc.2004.04.201. [DOI] [PubMed] [Google Scholar]
- 7.Li R-L. Summary of the second symposium of Tripterygium wilfordii. Chin J Integr Trad West Med. 1991;11:758–760. [PubMed] [Google Scholar]
- 8.Chen JK. Lei Gong Teng (Radix Tripterygii Wilfordii): A Blessing or a Time Bomb? Acupuncture Today. 2004:5. [Google Scholar]
- 9.Zheng JR, Liu JH, Hsu LF, Gao JW, Jiang BL. Toxicity of total glycosides in Tripterygium wilfordii. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. 1983;5:73–78. [PubMed] [Google Scholar]
- 10.Chou WC, Wu CC, Yang PC, Lee YT. Hypovolemic shock and mortality after ingestion of Tripterygium wilfordii hook F.: a case report. Int J Cardiol. 1995;49:173–177. doi: 10.1016/0167-5273(95)02282-2. [DOI] [PubMed] [Google Scholar]
- 11.Chan MA, Kohlmeier JE, Branden M, Jung M, Benedict SH. Triptolide is more effective in preventing T cell proliferation and interferon-gamma production than is FK506. Phytother Res. 1999;13:464–467. doi: 10.1002/(sici)1099-1573(199909)13:6<464::aid-ptr483>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 12.Yang Y, Liu Z, Tolosa E, Yang J, Li L. Triptolide induces apoptotic death of T lymphocyte. Immunopharmacol. 1998;40:139–149. doi: 10.1016/s0162-3109(98)00036-8. [DOI] [PubMed] [Google Scholar]
- 13.Jiang XH, Wong BC, Lin MC, Zhu GH, Kung HF, Jiang SH, Yang D, Lam SK. Functional p53 is required for triptolide-induced apoptosis and AP-1 and nuclear factor-kappaB activation in gastric cancer cells. Oncogene. 2001;20:8009–8018. doi: 10.1038/sj.onc.1204981. [DOI] [PubMed] [Google Scholar]
- 14.Wang X, Matta R, Shen G, Nelin LD, Pei D, Liu Y. Mechanism of triptolide-induced apoptosis: effect on caspase activation and Bid cleavage and essentiality of the hydroxyl group of triptolide. J Mol Med 6 A.D. 84:405–415. doi: 10.1007/s00109-005-0022-4. [DOI] [PubMed] [Google Scholar]
- 15.Qiu D, Zhao G, Aoki Y, Shi L, Uyei A, Nazarian S, Ng JC, Kao PN. Immunosuppressant PG490 (triptolide) inhibits T-cell interleukin-2 expression at the level of purine-box/nuclear factor of activated T-cells and NF-kappaB transcriptional activation. J Biol Chem. 1999;274:13443–13450. doi: 10.1074/jbc.274.19.13443. [DOI] [PubMed] [Google Scholar]
- 16.Shepherd EG, Zhao Q, Welty SE, Hansen TN, Smith CV, Liu Y. The function of mitogen-activated protein kinase phosphatase-1 in peptidoglycan-stimulated macrophages. J Biol Chem. 2004;279:54023–54031. doi: 10.1074/jbc.M408444200. [DOI] [PubMed] [Google Scholar]
- 17.Auer H, Lyianarachchi S, Newsom D, Klisovic MI, Marcucci G, Kornacker K. Chipping away at the chip bias: RNA degradation in microarray analysis. Nat Genet. 2003;35:292–293. doi: 10.1038/ng1203-292. [DOI] [PubMed] [Google Scholar]
- 18.Barker CS, Griffin C, Dolganov GM, Hanspers K, Yang JY, Erle DJ. Increased DNA microarray hybridization specificity using sscDNA targets. BMC Genomics. 2005;6:57. doi: 10.1186/1471-2164-6-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 20.Hosack DA, Dennis G, Jr, Sherman BT, Lane HC, Lempicki RA. Identifying biological themes within lists of genes with EASE. Genome Biol. 2003;4:R70. doi: 10.1186/gb-2003-4-10-r70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen P, Li J, Barnes J, Kokkonen GC, Lee JC, Liu Y. Restraint of proinflammatory cytokine biosynthesis by mitogen-activated protein kinase phosphatase-1 in lipopolysaccharide-stimulated macrophages. J Immunol. 2002;169:6408–6416. doi: 10.4049/jimmunol.169.11.6408. [DOI] [PubMed] [Google Scholar]
- 22.Zhao Q, Shepherd EG, Manson ME, Nelin LD, Sorokin A, Liu Y. The role of mitogen-activated protein kinase phosphatase-1 in the response of alveolar macrophages to lipopolysaccharide: Attenuation of proinflammatory cytokine biosynthesis via feedback control of p38. J Biol Chem. 2005;280:8101–8108. doi: 10.1074/jbc.M411760200. [DOI] [PubMed] [Google Scholar]
- 23.Zhao Q, Wang X, Nelin LD, Yao Y, Matta R, Manson ME, Baliga RS, Meng X, Smith CV, Bauer JA, Chang CH, Liu Y. MAP kinase phosphatase 1 controls innate immune responses and suppresses endotoxic shock. J Exp Med. 2006;203:131–140. doi: 10.1084/jem.20051794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen W, Martindale JL, Holbrook NJ, Liu Y. Tumor promoter arsenite activates extracellular signal-regulated kinase through a signaling pathway mediated by epidermal growth factor receptor and Shc. Mol Cell Biol. 1998;18:5178–5188. doi: 10.1128/mcb.18.9.5178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li J, Gorospe M, Barnes J, Liu Y. Tumor promoter arsenite stimulates histone H3 phosphoacetylation of proto-oncogenes c-fos and c-jun chromatin in human diploid fibroblasts. J Biol Chem. 2003;278:13183–13191. doi: 10.1074/jbc.M300269200. [DOI] [PubMed] [Google Scholar]
- 26.Li J, Gorospe M, Hutter D, Barnes J, Keyse SM, Liu Y. Transcriptional induction of MKP-1 in response to stress is associated with histone H3 phosphorylation-acetylation. mole. 2001;21:8213–8224. doi: 10.1128/MCB.21.23.8213-8224.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen P, Hutter D, Yang X, Gorospe M, Davis RJ, Liu Y. Discordance between the binding affinity of mitogen-activated protein kinase subfamily members for MKP-2 and their ability to catalytically activate the phosphatase. J Biol Chem. 2001;276:29440–29449. doi: 10.1074/jbc.M103463200. [DOI] [PubMed] [Google Scholar]
- 28.Schreiber E, Matthias P, Muller MM, Schaffner W. Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nucleic Acids Res. 1989;17:6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ge H, Roeder RG. Purification, cloning, and characterization of a human coactivator, PC4, that mediates transcriptional activation of class II genes. Cell. 1994;78:513–523. doi: 10.1016/0092-8674(94)90428-6. [DOI] [PubMed] [Google Scholar]
- 30.Zhou R, Zheng SX, Tang W, He PL, Li XY, Yang YF, Li YC, Geng JG, Zuo JP. Inhibition of inducible nitric-oxide synthase expression by (5R)-5-hydroxytriptolide in interferon-gamma- and bacterial lipopolysaccharide-stimulated macrophages. J Pharmacol Exp Ther. 2006;316:121–128. doi: 10.1124/jpet.105.093179. [DOI] [PubMed] [Google Scholar]
- 31.Morimoto RI. Cells in stress: transcriptional activation of heat shock genes. Science. 1993;259:1409–1410. doi: 10.1126/science.8451637. [DOI] [PubMed] [Google Scholar]
- 32.Wu Y, Wang Y, Zhong C, Li Y, Li X, Sun B. The suppressive effect of triptolide on experimental autoimmune uveoretinitis by down-regulating Th1-type response. Int Immunopharmacol. 2003;3:1457–1465. doi: 10.1016/S1567-5769(03)00144-9. [DOI] [PubMed] [Google Scholar]
- 33.Kupchan SM, Court WA, Dailey RG, Jr, Gilmore CJ, Bryan RF. Triptolide and tripdiolide, novel antileukemic diterpenoid triepoxides from Tripterygium wilfordii. J Am Chem Soc. 1972;94:7194–7195. doi: 10.1021/ja00775a078. [DOI] [PubMed] [Google Scholar]
- 34.Chen BJ, Chen Y, Cui X, Fidler JM, Chao NJ. Mechanisms of tolerance induced by PG490–88 in a bone marrow transplantation model. Transplantation. 2002;73:115–121. doi: 10.1097/00007890-200201150-00022. [DOI] [PubMed] [Google Scholar]
- 35.Lu Y, Fukuda K, Nakamura Y, Kimura K, Kumagai N, Nishida T. Inhibitory effect of triptolide on chemokine expression induced by proinflammatory cytokines in human corneal fibroblasts. Invest Ophthalmol Vis Sci. 2005;46:2346–2352. doi: 10.1167/iovs.05-0010. [DOI] [PubMed] [Google Scholar]
- 36.Zhu KJ, Shen QY, Cheng H, Mao XH, Lao LM, Hao GL. Triptolide affects the differentiation, maturation and function of human dendritic cells. Int Immunopharmacol. 2005;5:1415–1426. doi: 10.1016/j.intimp.2005.03.020. [DOI] [PubMed] [Google Scholar]
- 37.Chen X, Murakami T, Oppenheim JJ, Howard Z. Triptolide, a constituent of immunosuppressive Chinese herbal medicine, is a potent suppressor of dendritic cell maturation and trafficking. Blood. 2005;106:2409–2416. doi: 10.1182/blood-2005-03-0854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu J, Wu QL, Feng YH, Wang YF, Li XY, Zuo JP. Triptolide suppresses CD80 and CD86 expressions and IL-12 production in THP-1 cells. Acta Pharmacol Sin. 2005;26:223–227. doi: 10.1111/j.1745-7254.2005.00035.x. [DOI] [PubMed] [Google Scholar]
- 39.Zhao G, Vaszar LT, Qiu D, Shi L, Kao PN. Anti-inflammatory effects of triptolide in human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2000;279:L958–L966. doi: 10.1152/ajplung.2000.279.5.L958. [DOI] [PubMed] [Google Scholar]
- 40.Leuenroth SJ, Crews CM. Studies on calcium dependence reveal multiple modes of action for triptolide. Chem Biol. 2005;12:1259–1268. doi: 10.1016/j.chembiol.2005.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Westerheide SD, Kawahara TL, Orton K, Morimoto RI. Triptolide, an inhibitor of the human heat shock response that enhances stress-induced cell death. J Biol Chem. 2006;281:9616–9622. doi: 10.1074/jbc.M512044200. [DOI] [PubMed] [Google Scholar]
- 42.Akira S, Kishimoto T. NF-IL6 and NF-kappa B in cytokine gene regulation. Adv Immunol. 1997;65:1–46. [PubMed] [Google Scholar]
- 43.Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25:280–288. doi: 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 44.Kim YH, Lee SH, Lee JY, Choi SW, Park JW, Kwon TK. Triptolide inhibits murine-inducible nitric oxide synthase expression by down-regulating lipopolysaccharide-induced activity of nuclear factor-kappa B and c-Jun NH2-terminal kinase. Eur J Pharmacol. 2004;494:1–9. doi: 10.1016/j.ejphar.2004.04.040. [DOI] [PubMed] [Google Scholar]
- 45.Wu Y, Cui J, Bao X, Chan S, Young DO, Liu D, Shen P. Triptolide attenuates oxidative stress, NF-kappaB activation and multiple cytokine gene expression in murine peritoneal macrophage. Int J Mol Med. 2006;17:141–150. [PubMed] [Google Scholar]
- 46.Lee KY, Chang W, Qiu D, Kao PN, Rosen GD. PG490 (triptolide) cooperates with tumor necrosis factor-alpha to induce apoptosis in tumor cells. J Biol Chem. 1999;274:13451–13455. doi: 10.1074/jbc.274.19.13451. [DOI] [PubMed] [Google Scholar]
- 47.Zhu W, Ou Y, Li Y, Xiao R, Shu M, Zhou Y, Xie J, He S, Qiu P, Yan G. A Small Molecule Triptolide Suppresses Angiogenesis and Invasion of Human Anaplastic Thyroid Carcinoma Cells via Down-regulation of NF-﹛kappa﹜B Pathway. Mol Pharmacol. 2009 doi: 10.1124/mol.108.052605. [DOI] [PubMed] [Google Scholar]