

Abstract
A new cytotoxic macrocyclic glycoresin, ipomoeassin F (6), has been isolated from the leaves of Ipomoea squamosa. The structure was elucidated by the interpretation of spectral data. Compound 6 was strongly active in the A2780 (human ovarian cancer cell line) assay with an IC50 value of 0.036 μM.
Keywords: ipomoeassin F, glycoresin, Ipomoea squamosa, Convolvulaceae, cytotoxicity
1. Introduction
We have previously reported the isolation of the five cytotoxic macrocyclic glycoresins ipomoeassins A-E (1–5) from the leaves of Ipomoea squamosa Choisy (Convolvulaceae) from the Suriname rainforest [2]. Subsequent to this work we re-examined a fraction from the final HPLC separation which had eluted just after ipomoeassin A (1). This fraction had originally been assumed to consist of ipomoeassin A, based on its 1H NMR spectrum, which was very similar to that of 1, and its Rf value on TLC (silica-gel, CH2Cl2:CH3OH; 40:1), which was identical to that of 1. Further chromatography of the fraction by HPLC (C8, 75% CH3CN/H2O) indicated however that the fraction contained a new compound (6), with a different retention time (tR: 15.2 min) from that of compound 1 (tR: 11.8 min).
2. Results and Discussion
Compound 6 was obtained as a colorless oil, and its HR FABMS gave a pseudomolecular ion [M+1]+ at m/z 831.4211 (calcd for C44H63O15 831.4167), 28 amu more than that of ipomoeassin A (1). Its IR, UV, and 1H NMR spectra were nearly identical to those of compound 1. The major difference between their 13C NMR spectra was located in the high field region (10 to 50 ppm), and it was thus deduced that compound 6 was a homolog of 1 with a C16 rather than a C14 hydroxyacid side chain, with two more methylenes than in 1. The oxygenated methine must be located at position 11 since H-11 showed HMBC correlations to C-12 and C-13, and H3-16 to C-14, and C-15. The chemical shifts of C-1 to C-6 were close to those of 1, and the signals for C-7 to C-16 matched those of 11-hydroxyhexadecanoic acid [3], which confirmed the above deduction. The spin systems of 6 were determined from COSY and TOCSY spectra. The connectivities from H-1 of fucose to C-11, H-1 of glucose to C-2 of fucose, H2-6 of glucose to C-1 of the side chain, H-4 of fucose to the acetyl carbonyl, and H-3 and H-4 of glucose to the tigloyl and E-cinnamoyl carbonyls, respectively, were established from HMQC and HMBC spectra. The configurations at position-11 and the sugar moieties were determined by analysis of its ROESY spectrum (Figure 1). Hence, the structure of 6 was determined as shown.
Figure 1.
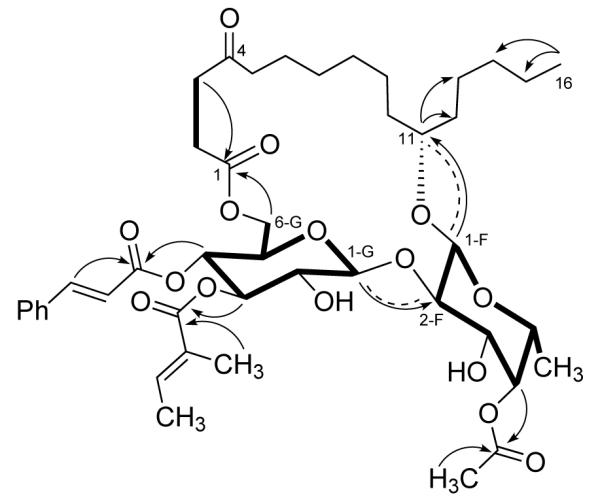
Key HMBC (arrows), ROESY (dashed), COSY and TOCSY (bold) correlations of ipomoeassin F (6)
Compound 6 exhibited potent cytotoxic activity against A2780 human ovarian cancer cell lines with an IC50 value of 0.036 μM, which was much more active than compound 1 (0.5 μM) [1]. Here we can see again that sometimes a slight modification of the structure will enhance the activity dramatically.
3. Experimental
3.1 General Procedures
General experimental procedures, cytotoxicity bioassays, plant material, and isolation procedures were as previously described [2].
3.2 Isolation
The crude extract of E940631 (10 g, IC50: 8.0 μg/mL) was separated into five fractions and fraction III was purified by HPLC over C18 using 75% MeCN/H2O to yield ipomoeassins A-E (1-5) as described previously [2]. A fraction collected immediately after compound 1 was examined by 1H NMR and normal phase TLC (silica-gel, CH2Cl2:CH3OH; 40:1, Rf = 0.3), and appeared to be identical with or very similar to compound 1. Further chromatography of this fraction was carried out using a semi-preparative C8 Varian Dynamax HPLC column (5 μ, 250×10 mm) with 75% CH3CN/H2O as eluents to afford compound 6 (1.4 mg, tR: 15.2 min), which had a different retention time from that of compound 1 (tR: 11.8 min).
3.3 Ipomoeassin F (6)
colorless oil; [α]D22 -54° (c 0.16, EtOH); IR (film) ψmax 3400, 2931, 2857, 1719, 1636, 1450, 1378, 1247, 1154, 1071, 1017; UV (EtOH) λmax (log ε) 278 (4.18) nm; 1H NMR (500 MHz, C6D6) and 13C NMR (125 MHz, C6D6) data, see Table 1; HRESIMS m/z 831.4211 (calcd for C44H63O15 831.4167).
Table 1.
1H and 13C NMR data of ipomoeassins F (6) and A (1)a
| no | 6 | 1 | 6 | 1 |
|---|---|---|---|---|
| 1 | 171.5 | 171.5 | ||
| 2 | 2.39 ddd (17.3, 9.6, 3.2) 2.14 ddd (17.3, 7.4, 3.4) |
2.40 ddd (17.4, 9.4, 3.4) 2.14 ddd (17.4, 7.7, 3.5) |
37.4b | 37.3b |
| 3 | 2.65 ddd (16.4, 7.4, 3.2) 2.52 ddd (16.4, 9.6, 3.4) |
2.65 ddd (16.1, 7.7, 3.4) 2.52 ddd (16.1, 9.4, 3.5) |
29.6c | 29.7c |
| 4 | 208.3 | 208.4 | ||
| 5 | 2.07 t (7.1) | 2.07 t (6.0) | 41.6 | 41.6 |
| 6 | 23.5 | 23.8 | ||
| 7 | 28.7 | 28.7 | ||
| 8 | 29.4c | 29.4c | ||
| 9 | 25.5 | 25.5 | ||
| 10 | 34.3 | 34.3 | ||
| 11 | 3.72b m | 3.72b m | 79.4 | 79.0 |
| 12 | 35.4b | 37.6b | ||
| 13 | 25.2 | 18.7 | ||
| 14 | 0.95 t (7.1) | 32.3 | 14.4d | |
| 15 | 23.1 | |||
| 16 | 0.92 t (7.1) | 14.3d | ||
| 1′ | 4.40 d (7.6) | 4.40 d (7.7) | 100.8 | 100.8 |
| 2′ | 3.96cdd (9.4, 7.6) | 3.96c dd (9.5, 7.7) | 84.0 | 84.0 |
| 3′ | 3.72b dd (9.4, 3.7) | 3.72b dd (9.5, 3.7) | 72.8e | 72.7e |
| 4′ | 5.15 br d (3.7) | 5.15 dd (3.7, 0.5) | 72.8e | 72.9e |
| 5′ | 3.11 br q (6.4) | 3.10 qd (6.4, 0.5) | 69.0 | 69.0 |
| 6′ | 1.11 d (6.4) | 1.10 d (6.4) | 14.1d | 14.1d |
| 1″ | 4.54 d (7.8) | 4.52 d (7.9) | 106.5 | 106.6 |
| 2″ | 3.91cdd (9.8, 7.8) | 3.91cdd (9.7, 7.9) | 74.8 | 74.8 |
| 3″ | 5.40 t (9.8) | 5.39 t (9.7) | 76.7 | 76.4 |
| 4″ | 5.70 t (9.8) | 5.69 t (9.7) | 67.8 | 67.8 |
| 5″ | 3.27 br d (9.8) | 3.24 ddd (9.7, 3.2, 1.6) | 73.0e | 73.0e |
| 6″ | 4.67 dd (12.6, 3.0) 4.13 br d (12.6) |
4.66 dd (12.6, 3.2) 4.11 dd (12.6, 1.6) |
61.5 | 61.5 |
| AA-1 | 170.9 | 171.0 | ||
| AA-2 | 1.84 s | 1.82 s | 20.5 | 20.5 |
| TA-1 | 168.7 | 168.5 | ||
| TA-2 | 128.0 | 128.0 | ||
| TA-3 | 6.95-7.00 m | 6.95 m | 139.4 | 139.2 |
| TA-4 | 1.26 d (7.1) | 1.23 d (7.1) | 16.6 | 16.6 |
| TA-5 | 1.71 br s | 1.68 br s | 12.0 | 12.1 |
| CA-1 | 165.6 | 165.6 | ||
| CA-2 | 6.39 d (16.0) | 6.39 d (15.9) | 117.6 | 117.6 |
| CA-3 | 7.82 d (16.0) | 7.81 d (15.9) | 146.1 | 146.1 |
| CA-4 | 134.4 | 134.5 | ||
| CA-5 | 6.92 d (7.1) | 6.89-7.07 | 128.5 | 128.5 |
| CA-6 | 7.05 d (7.1) | 6.89-7.07 | 128.9 | 128.9 |
| CA-7 | 6.95-7.00 m | 6.89-7.07 | 130.4 | 130.4 |
δ (ppm), in benzene-d6, 500 MHz for 1H NMR and 125 MHz for 13C NMR; multiplicities; J values (Hz) in parentheses. AA = acetoyl; TA = tigloyl; CA = cinnamoyl
overlapped
Structures 1-6.

Acknowledgments
This project was supported by the Fogarty International Center, the National Cancer Institute, the National Science Foundation, the National Heart Lung and Blood Institute, the National Institute of Mental Health, the Office of Dietary Supplements, and the Office of the Director of NIH, under Cooperative Agreement U01 TW000313 with the International Cooperative Biodiversity Groups, and this support is gratefully acknowledged. We also thank Mr. Bill Bebout for obtaining the HRFABMS spectrum, and Mr. Tom Glass for assistance obtaining NMR spectra.
Footnotes
See Ref. [1]
Reference
- [1].Biodiversity Conservation and Drug Discovery in Suriname, Part 18. For Part 17, see Adou E, Williams RB, Schilling JK, Malone S, Meyer J, Wisse JH, Frederik D, Koese D, Werkhoven MCM, Snipes CE, Werk TL, Kingston DGI. Bioorg. Med. Chem. 2005;13:6009. doi: 10.1016/j.bmc.2005.07.026. (this is erroneously listed as Part 16)
- [2].Cao SG, Guza RC, Wisse JH, Miller JS, Evans R, Kingston DGI. J. Nat. Prod. 2005;68:487. doi: 10.1021/np049629w. [DOI] [PubMed] [Google Scholar]
- [3].Fürstner A, Müller T. J. Am. Chem. Soc. 1999;121:7814. [Google Scholar]