

Abstract
The nature of specific RNA-RNA and protein-RNA interactions involved in the process of genome dimerization and isomerization in HIV-1, which is mediated in vitro by the stemloop 1 (SL1) of the packaging signal and by the nucleocapsid (NC) domain of the viral Gag polyprotein, was investigated by using archetypical nucleic acid ligands as non-covalent probes. Small-molecule ligands make contact with their target substrates through complex combinations of H-bonds, salt bridges, and hydrophobic interactions. Therefore, their binding patterns assessed by electrospray ionization (ESI) mass spectrometry can provide valuable insights into the factors determining specific recognition between species involved in biopolymer assemblies. In the case of SL1, dimerization and isomerization create unique structural features capable of sustaining stable interactions with classic nucleic acid ligands. The binding modes exhibited by intercalators and minor groove binders were adversely affected by the significant distortion of the duplex formed by palindrome annealing in the kissing-loop (KL) dimer, whereas the modes observed for the corresponding extended duplex (ED) confirmed a more regular helical structure. Consistent with the ability to establish electrostatic interactions with highly negative pockets typical of helix anomalies, polycationic aminoglycosides bound to the stem-bulge motif conserved in all SL1 conformers, to the unpaired nucleotides located at the hinge between kissing hairpins in KL, and to the exposed bases flanking the palindrome duplex in ED. The patterns afforded by intercalators and minor groove binders did not display detectable variations when the corresponding NC-SL1 complexes were submitted to probing. In contrast, aminoglycosides displayed the ability to compete with the protein for overlapping sites, producing opposite effects on the isomerization process. Indeed, displacing NC from the stem-bulges of the KL dimer induced inhibition of stem melting and decreased the efficiency of isomerization. Competition for the hinge region, instead, eliminated the NC stabilization of a grip motif formed by nucleobases of opposite strands, thus facilitating the strand-exchange required for isomerization. These non-covalent probes provided further evidence that the structural context of the actual binding sites has significant influence on the chaperone activities of NC, which should be taken in account when developing potential drug candidates aimed at disrupting genome dimerization and isomerization in HIV-1.
Keywords: Non-covalent probes, nucleic acid ligands, aminoglycoside antibiotics, electrospray ionization mass spectrometry, RNA dimerization and isomerization, HIV-1 nucleocapsid, chaperone activity
Introduction
Despite recent advances by high-resolution techniques in the structural elucidation of nucleic acids and protein-nucleic acid assemblies, methods based on chemical probing are still very widely used in the pursuit of substrates that are available in limited amounts, exceed practical size limitations, or do not possess favorable crystallization properties. Traditional protocols involve labeling the ends of the nucleic acid components with radioactive or fluorescent tags, applying selected reagents to covalently modify functional groups exposed to the medium, performing probe-specific reactions to induce strand cleavage at the modified sites, and finally completing product analysis by polyacrylamide gel electrophoresis (PAGE) to reveal the position of probed nucleotides [1-5]. With the goals of streamlining the process and increasing the information afforded by structural probes, we have replaced PAGE with electrospray ionization (ESI)[6, 7] Fourier transform ion cyclotron resonance (FTICR)[8, 9] mass spectrometry. Thanks to the unique mass signatures exhibited by the different probes, this analytical platform dispenses with end-labeling procedures, allows for the utilization of a broad range of nucleases in place of probe-specific hydrolysis, and enables multiplexed probe application [10-12]. The potential of this concerted approach for structural elucidation was substantiated by the demonstration that the spatial constraints afforded by mono- and bifunctional reagents could be readily employed for generating all-atom structures through established molecular modeling techniques [13, 14].
Traditional structural probes consist of mono- and bifunctional alkylating agents that possess very specific points of attack in the substrate structure, defined by stringent rules of chemical reactivity [3-5]. When the biopolymer of interest presents susceptible functional groups exposed on its surface, or located within the span of crosslinkers reactive centers, the detection of specific covalent adducts reveals the structural context of the target groups. Unlike typical alkylating agents used as structural probes, small-molecule ligands do not possess a unique attack point, but rather make contact with cognate substrates through complex combinations of H-bonds, salt bridges, and hydrophobic interactions [15, 16]. In this case, the structural context conducive to binding must present multiple functional groups with favorable chemical and steric characteristics. This observation provided a powerful rationale for testing small-molecule ligands as non-covalent probes for investigating complex features of biopolymer structures, as illustrated by the recent elucidation of the factors determining specific recognition between selected nucleic acid substrates and their cognate proteins in retroviral systems [17, 18].
Archetypical nucleic acid binders constitute ideal non-covalent probes for these types of substrates because of the extensive knowledge of their binding properties and selectivity. Due to their well-characterized tropism for specific structural features, general intercalators, minor groove binders, mixed-mode intercalator/groove binders, and multifunctional polycationic aminoglycosides can provide valuable information about the base stacking pattern, the general groove depth and geometry, and the presence of electronegative pockets associated with bulging nucleobases, kinks, and other helix anomalies. Direct infusion ESI mass spectrometry constitutes an excellent readout for these types of probes, due to its ability to transfer non-covalent complexes of nucleic acids to the gas phase without affecting their solution association state [19-21]. The binding stoichiometry, interaction strength, and position of putative binding sites can be interpreted on the basis of the known binding properties of the selected probe to infer the sought-after structural and functional information. This approach was employed, for example, to investigate the features of the HIV-1 polypurine tract (PPT), which are responsible for the specific recognition of this RNA:DNA hybrid by the viral reverse transcriptase (RT) during plus-strand synthesis [18]. In this case, the binding patterns exhibited by intercalators and groove binders indicated that the PPT hybrid did not present any unique groove geometries or stacking arrangements that may contribute to the specificity of RT recognition. Instead, the preferential binding of aminoglycoside antibiotics to well-defined sites of the hybrid structure served to highlight helix anomalies that could be specifically recognized by the thumb subdomain and the ribonuclease H region of RT. These data were corroborate by NMR and isothermal titration calorimetry, which provided validation to the non-covalent probing approach [18].
In this report, we have employed non-covalent probes to investigate the structure and dynamics of the dimeric complexes formed by RNA constructs corresponding to the stemloop 1 (SL1) of the HIV-1 packaging signal (Lai variant, Scheme 1), which includes the palindromic sequence necessary to initiate genome dimerization [22-25]. In vitro, this two-step process involves annealing of the self-complementary sequences of homologous constructs to form a metastable kissing-loop dimer (KL) [26-28], followed by isomerization into a more stable extended duplex (ED) [29-32], which is facilitated by the chaperone activity of the retroviral nucleocapsid (NC) protein [31, 33-36]. Using ad hoc mutants that isolated individual motifs, or locked dimeric complexes in either conformation, we identified the NC binding sites [37] and elucidated the effects of individual binding events on the outcome of the RNA remodeling process [38]. Now, the non-covalent probes have presented us with the opportunity of investigating the significance of distinctive structural features in stabilizing the alternative dimeric forms and the role of the different interactions in the mechanism of RNA dimerization and stabilization mediated by NC.
Scheme 1.

Sequences and secondary structures of the ribonucleic acid constructs and nucleocapsid protein included in the study. Nucleotides are numbered according to the subtype B sequence (Lai variant) of HIV-1. The self-complementary sequences are highlighted in gray. For each species, the monoisotopic masses observed experimentally and calculated from sequence are included. Structures of nucleic acid-active agents incorporated in the study, calculated from monoisotopic masses and experimentally determined by FTICR.
Results and Discussion
A close examination of the high-resolution structures available for the monomeric/dimeric forms assumed in vitro by SL1 provides a glimpse of the salient motifs expected to undergo major rearrangements during the two-step process of RNA dimerization and isomerization. In monomeric SL1, a 1×3 bulge interrupts the regular helical structure of the double-stranded stem [39, 40], which is capped by a flexible loop consisting of nine unpaired nucleotides (Scheme 2). In the KL dimer, the stem and bulge regions are conserved in the bound hairpins, but the six palindromic nucleotides of the apical loop are now paired with those of a homologous copy to form an inter-molecular duplex (Scheme 2). The other loop nucleotides remain unpaired at the hinge between the kissing hairpins and assume inward/outward orientations depending on the technique employed for structural elucidation, suggesting a rather dynamic situation [41-44]. In the ED dimer, extensive inter-molecular pairing provides a double-helical structure interrupted by bulge motifs homologous to those of the hairpin forms, but comprising nucleotides contributed by the opposite strands, and by unpaired bases flanking the palindrome duplex, which are subjected to significant dynamics [45-48]. In the absence of high-resolution structures for the corresponding NC complexes, we employed non-covalent ligands to reveal the effects induced by specific protein interactions. For this purpose, we first probed selected RNA structures to establish baseline behaviors for the ligands included in the study, then we repeated the experiments under identical conditions after addition of NC to obtain well-characterized ribonucleoprotein assemblies [37].
Scheme 2.
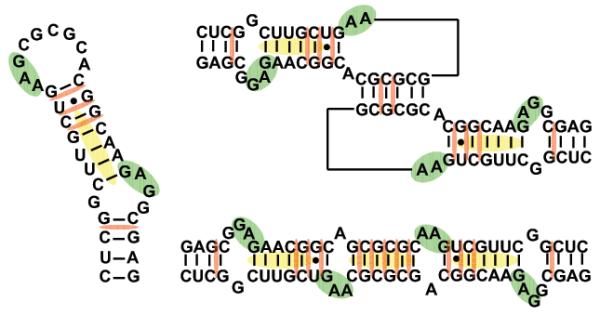
Sequences and secondary structures of the monomer, kissing-loop, and extended duplex conformations of SL1. Shaded regions indicate proposed binding sites of the aminoglycosides (green), intercalators (red), and minor-groove binders (yellow).
Probing monomeric SL1
The binding properties of monomeric RNA were established by using a dimerization-deficient mutant with disrupted palindrome (SL1G259A, Scheme 1). Titration experiments were performed by adding increasing amounts of selected probe to a 5 μM solution of RNA in 150 mM ammonium acetate (pH adjusted to 7.0), followed by equilibration for 15 min at room temperature (see Material and Methods). The formation of non-covalent complexes was monitored directly by ESI-FTICR mass spectrometry to obtain an unambiguous determination of the composition and stoichiometry of any species at equilibrium in solution. In this way, stable complexes with distinctive binding modes were readily observed for the different classes of ligands employed in the study (Figure 1a). Complexes with increasing stoichiometries were detected for intercalative agents, such as ethidium bromide, acridine, and the mixed-mode mitoxantrone (Scheme 1), which bound in stepwise fashion at increasing concentrations with no sign of cooperativity. In the presence of excess ligand, these probes shared the same maximum stoichiometry of four equivalents per RNA (Table 1). This stoichiometry exceeds the number of sites comprised between contiguous GC pairs in the stem structure (red marks in Scheme 2), which maximize hydrophobic stabilization between stacked aromatic systems [49]. Additional sites could be represented by those sandwiched between GC pairs and the GU wobble located in the upper stem section (red marks in Scheme 2), which would account for the remaining binding events observed in these experiments. The fact that minor groove ligands (distamycin A, Hoechst 33258 and 33342, Scheme 1) afforded only 1:1 stoichiometries is consistent with the composition and geometry of the stem’s helical sections (Figure 1b and Table 1). In fact, this class of molecules binds preferentially to the narrow groove of B-type DNA duplexes, with higher affinity for stretches of four or five consecutive AT base-pairs [50, 51]. Due to the wider and shallower dimensions exhibited by A-type helices in RNA, the minor groove can support only weaker interactions. In SL1G259A, the upper stem presents two contiguous AU pairs that may afford sufficient stabilization to sustain the binding of one equivalent only in the presence of excess ligand (yellow mark in Scheme 2).
Figure 1.

Nanospray-FTICR mass spectra of 5 μM solutions of dimerization-deficient SL1G259A in 150mM ammonium acetate after a 5-fold addition of: (a) mitoxantrone; (b) distamycin A; (c) neomycin B (see Materials and Methods).
Table 1.
Summary of dissociation constants (KD’s) determined for SL1 complexes with individual ligands. All values of KD are expressed in μM units. The two binding sites of aminoglycosides were determined utilizing mutant SL1 substrates, which eliminated the loop and bulge region respectively (Scheme 1). The maximum observed stoichiometry for each ligand were obtained with a 10x fold excess over the RNA substrate (see Materials and Methods).
| Stoichiometry | |||||
|---|---|---|---|---|---|
| Ligand | Kd SL1 loop | Kd SL1 bulge | Monomer | KL | ED |
| Neomycin B | 3.3 ± 0.7 | 4.1 ± 0.6 | 2 | 4 | 4 |
| Paromomycin | 7.2 ± 0.5 | 7.5 ± 0.5 | 2 | 4 | 4 |
| Kanamycin A | 8.8 ± 0.3 | 8.2 ± 0.7 | 2 | 4 | 4 |
| Kanamycin B | 7.7 ± 0.4 | 7.1 ± 0.5 | 2 | 4 | 4 |
| Tobramycin | 6.9 ± 0.8 | 7.6 ± 0.6 | 2 | 4 | 4 |
| Ethidium Br- | 2.1 ± 0.1 | NA | 3 | 10 | 13 |
| Acridine | 2.5 ± 0.2 | NA | 3 | 10 | 13 |
| Mitoxantrone | 3.3 ± 0.9 | NA | 3 | 10 | 13 |
| Distamycin A | 27.6 ± 6.6 | NA | 1 | 3 | 3 |
| Hoechst 33342 | 34.1 ± 7.6 | NA | 1 | 3 | 3 |
| Hoechst 33258 | 38.2 ± 3.6 | NA | 1 | 3 | 3 |
In the case of aminoglycosides such as neomycin B (NB), up to two equivalents bound the monomeric RNA during the titration experiments (Figure 1c and Table 1). Due to their polycationic nature and relatively flexible scaffold, these molecules can adapt to substrate surfaces through multiple electrostatic interactions and hydrogen bonds (Scheme 1) [52]. As demonstrated by assemblies of NC with other stemloop domains of the HIV-1 packaging signal (i.e., SL3 and SL4), the electronegative pockets favored by aminoglycosides constitute excellent binding sites for highly basic proteins [17]. With the goal of locating such sites onto the SL1 structure, we submitted the ligand-RNA complexes to tandem mass spectrometry [53, 54], which was performed by isolating the precursor ion of interest in the FTICR cell, and then activating it by sustained off-resonance irradiation collision-induced dissociation (SORI-CID)[55] (see Materials and Methods). We have previously shown that direct contacts with ligand molecules introduce characteristic gaps in the sequence coverage afforded by fragmentation of the precursor ion [56], which are ascribable to putative protection effects [18, 57]. This observation provided the basis for a gas-phase footprinting approach that enables the identification of nucleotides involved in specific interactions with cognate ligands. Applied to the SL1G259A•2NB, this technique revealed reduced fragmentation at G254:G257 and G270:G272, which are located in double-stranded regions near the internal bulge and immediately below the apical loop (Figure 2 and green marks in Scheme 2). These findings were corroborated by docking the aminoglycoside molecule onto electrostatic plots generated from the high-resolution coordinates of wild-type SL1 (see Materials and Methods). In spite of the fact that the ligand had been allowed to sample the entire hairpin surface without constraints, the cluster of hits with the lower binding energies consisted of complexes with the aminoglycoside docked to either of the regions identified by tandem mass spectrometry (Figure 2).
Figure 2.

(a) Product ion spectrum and (b) fragmentation map of the SL1G259A•2NB assembly. The fragmentation pattern obtained by SORI-CID displayed protection of G254:G257 and G270:G272, which are located in double-stranded regions near the internal bulge and immediately below the apical loop, as indicated on the hairpin secondary structure (c). These results were corroborated by docking the ligand molecule onto the electrostatic surfaces of SL1 (d). Docking simulations were performed using Autodock 3.0 [67] (see Materials and Methods), using electrostatic surfaces calculated by Delphi [68] from the high-resolution coordinates of SL1 (PDB:2D1B) in a 150 mM salt environment. The results were visualized in Pymol [69], with red color marking highly electronegative regions.
Probing SL1 dimers
The binding profiles of dimeric RNA were obtained by using wild-type and mutant constructs that were fully capable of undergoing the two-step process of dimerization and isomerization (Scheme 1). For this reason, each desired conformer was obtained by performing heat denaturation followed by either fast cooling in ice slurry, which refolds the kinetic product KL, or slow cooling completed at room temperature, which produces the thermodynamic product ED (see Materials and Methods, ref. [37, 38] and references therein). In each experiment, addition of probe induced the formation of ligand-RNA complexes with distinctive stoichiometries, but did not result in significant shifts of the initial monomer-dimer equilibrium observed in its absence. In particular, titration of KL samples with intercalating and mixed-mode ligands produced complexes with a maximum stoichiometry of ten equivalents per RNA dimer (Table 1), which exceeded by two units the total calculated by doubling the stoichiometry afforded by each individual hairpin. Up to thirteen ligands per dimer were instead observed for the ED conformer (Table 1), which suggested the creation of additional viable sites upon RNA isomerization. Mutant constructs with guanines replacing the palindrome-flanking adenines (Scheme 1) did not display detectable deviations from the binding modes obtained from wild-type SL1.
When probe-RNA complexes were submitted to tandem mass spectrometry under activation conditions that were insufficient to induce fragmentation of the covalent backbones (see Materials and Methods), the different conformers displayed characteristic dissociation patterns consistent with their spatial organization [38]. In agreement with the respective number of inter-molecular base pairs, intercalator-KL complexes underwent symmetric dissociation into individual ligand-hairpin components, whereas no strand dissociation was observed for the corresponding ED complexes (Figure 3). Each hairpin product retained four ligand units, as observed also for the dimerization-deficient mutant SL1G259A, thus indicating that the loop-loop duplex responsible for stabilizing the KL structure accounted for the remaining two binding sites afforded by this conformer (red marks in Scheme 2). In the ED dimer, at least eight binding events were ascribable to the double-stranded structures replicating the stems of individual hairpins, which would leave the palindrome duplex as the only possible region responsible for the remaining five units. The high-resolution structure of KL shows that the helix established by the loop-loop duplex deviates significantly from the canonical A-type form [41]. The participation of the palindrome nucleotides in a loop structure introduces mechanical strain at the ends of the duplex, which causes sizeable distortion of the normal base stacking pattern and reduces its ability to stabilize intercalative binding. In contrast, the palindrome sequence is not part of a loop in the ED structure, therefore the resulting duplex is not subjected to strain [46]. Despite the unpaired flanking purines, the annealed nucleotides form a nearly canonical A-helix with a regular base stacking arrangement that could readily stabilize up to five intercalators, matching the number of contiguous GC pairs with our experimental observation (red marks in Scheme 2).
Figure 3.

Product ion spectra obtained by submitting to mild SORI-CID (a) the 2SL1•10MT complex folded via the KL-inducing protocol and (b) the 2SL1•13MT folded via the ED-inducing protocol. The former results in the dissociation of the dimer coupled with the loss of two mitoxantrone units, while the latter remains intact under the same energy regime. The precursor ion is marked by the  symbol.
symbol.
The clearly different structural contexts created by palindrome annealing were reflected also by the binding patterns obtained from minor groove probes (Table 1). Indeed, while the KL construct was found capable of binding only two minor groove ligands, the ED counterpart bound up to three (Scheme 2). While two units were accounted for by the upper stem sites identified earlier on the monomeric structure, which are conserved in KL and are reconstituted in identical form in ED, the additional binding afforded by the latter could be sustained by the palindrome helix itself (yellow mark in Scheme 2). This is consistent the more regular geometry afforded by this duplex than by the corresponding loop-loop structure in the KL dimer, as highlighted by the intercalative probes.
Treated with aminoglycoside antibiotics, the dimeric conformers shared the same stoichiometry doubling the units bound by each monomeric component (Table 1). In this case, two binding events were accounted for by the stem-bulge sites conserved in all monomeric/dimeric forms (green marks in Scheme 2). The remaining events were ascribable to sites comprising the unpaired purines at the hinge between kissing hairpins, or flanking the palindrome duplex in ED, which represent structural anomalies that are readily recognizable by these types of ligands [17, 57-59]. Indeed, crystal structures obtained from truncated KL and ED dimers lacking the stem-bulge motifs displayed uncanny similarity with the natural target of aminoglycoside antibiotics, the eubacterial 16S ribosomal A site [58]. In particular, the KL conformer was shown to bind NB in a pocket lined by the major groove of the loop-loop helix, which was made accessible by a bulged-out conformation of the hinge purines. In the corresponding ED structure, access to an analogous pocket was controlled by the orientation of the same unpaired purines, which was decidedly influenced by the type of aminoglycoside employed in the experiment [60]. Significantly, when we compared the binding activities of these pockets with that of the more freely accessible stem-bulge site, the latter was found to possess a noticeably greater affinity for NB, as demonstrated by competition experiments pitting truncated KL/ED structures against a duplex that isolated the stem-bulge motif (SL1TR dimers and SL1BG, respectively, Scheme 1).
Probing ribonucleoprotein complexes
Possible structural effects induced by NC on monomeric RNA were investigated by using assemblies of the dimerization-deficient mutant SL1G259A. Earlier ESI-FTICR analyses had unambiguously demonstrated that this construct can bind up to two protein units to form a stable 2NC•SL1G259A complex [37]. Treated with intercalators and groove binders, these assemblies showed no signs of dissociation, while ternary complexes including ligand molecules were readily detected (Figure 4a and b). The maximum stoichiometries observed in these titration experiments matched those obtained from the RNA substrate in the absence of protein (Table 1), which suggested that NC interactions did not induce significant distortion of the original hairpin structure and did not reduce accessibility to the salient structural features targeted by the probes. In contrast, the addition of progressively larger amounts of aminoglycosidic antibiotics appeared to undermine the stability of the protein-RNA interactions. In particular, when preformed 2NC•SL1G259A was treated with NB, a NC•SL1G259A•NB assembly was initially observed, which was readily transformed into SL1G259A•2NB by further ligand addition (Figure 4c). The possibility of direct competition between NC and aminoglycosides for the same structures was supported by the observation that the ligand sites identified by tandem mass spectrometry matched regions involved in specific protein contacts described in earlier studies [37]. Furthermore, these results paralleled those provided by the SL3 and SL4 domains of the packaging signal, which include motifs capable of supporting specific interactions with both NC and aminoglycosides and, consequently, are subjected to significant competitive effects [17].
Figure 4.

Nanospray-FTICR mass spectra of a 5 μM solution of 2NC•SL1 G259A assembly in 150mM ammonium acetate after addition of 5-fold excess of (a) mitoxantrone, (b) distamycin A, and (c) neomycin B. The latter induced dissociation of the 2NC•SL1 G259A complex, while the two former classes of ligands create ternary complexes with 444.20 and 481.24 Da increments, respectively.
The binding profiles afforded by dimeric species were investigated by using wild-type SL1, as well as selected mutants varying the palindrome-flanking purines, which were heat-refolded to provide almost exclusively the KL or the ED conformer, as described above. The ability of NC to mediate isomerization required that addition of protein to form the desired ribonucleoprotein substrates be followed with only short delay by the titration with ligand to minimize the incidence of KL-ED transition (see Materials and Methods). In earlier studies, we have shown that KL dimers are capable of binding up to three NC units at the stem-bulges and at the hinge region, with the latter interaction being reinforced by replacing the flanking adenines with guanines (e.g., SL1A255G, Scheme 1) [38]. ED conformers, instead, bound up to four NC units at the stem-bulges and palindrome-flanking purines [38]. Addition of intercalators and groove binders did not affect the stability of these assemblies, but induced binding to the initial species with stoichiometries equivalent to those obtained from the same RNA samples in the absence of protein (Table 1). This outcome suggested that NC interactions did not induce major variations of the original RNA structures, nor constituted a significant hindrance to establishing stable contacts with these types of ligands.
In analogy with the behavior displayed by complexes of monomeric RNA, treating dimeric assemblies with increasing amounts of aminoglycosides promoted competition for overlapping sites. In particular, starting from preformed 3NC•2SL1A255G assemblies folded in the KL conformation, NB induced facile displacement of one protein unit to form a distinctive 2NC•2SL1A255G•NB complex that was further displaced only in the presence of larger amounts of ligand. This observation is consistent with the greater affinity exhibited by NC for the stem-bulge motifs than for the hinge site [37, 38], which makes the latter more susceptible to aminoglycoside competition. In similar fashion, preformed 4NC•2SL1A255G assemblies in the ED conformation produced a relatively stable 2NC•2SL1A255G•2NB intermediate that was transformed into 2SL1A255G •4NB upon addition of excess probe. Also in this case, the probing results agree with the observation that the G-rich stem-bulges afford higher protein affinity than the palindrome-flanking sites [37, 38].
Effects of ligand binding on NC chaperoning activity
The fact that different non-covalent probes had clearly distinctive effects on the stability of the ribonucleoprotein assemblies, consistent with the nature of their specific interactions with the RNA substrates, prompted the investigation of the possible influence of such ligands on the ability of the KL dimer to undergo isomerization catalyzed by NC. These experiments were carried out by adding NC to heat-refolded KL dimers at 37°C, followed by a 10 min interval before ligand titration. The partitioning between KL and ED assemblies was assessed by tandem mass spectrometry immediately after addition of probe and after 3 h incubation at 37°C. Under the selected conditions, control experiments showed that 89% of total RNA in solution was folded in the ED conformation after incubation in the absence of ligand (Table 2). In the presence of intercalating probes, instead, the percentage of ED decreased to 70-76%, thus indicating that ligand interactions induced detectable inhibitory effects on isomerization (Table 2). Considering that these molecules do not interfere with NC binding, the observed outcome cannot be attributed to direct disruption of its chaperone activity. A possible explanation can be found, instead, in the ability of intercalators to hamper the melting of double-stranded regions by increasing the stability of stacked base pairs. For the KL-ED transition to take place, intra-molecular base pairs in the stems of the kissing hairpins have to dissociate and then reconstitute in inter-molecular fashion to produce the final ED structure [38]. Therefore, stabilization of the initial stemloops poses an obstacle to the strand-exchange process mediated by NC. The same explanation is valid also for the less significant, but still detectable, inhibition induced by mixed-mode and minor groove binders (Table 2), which also act without competing with NC (Table 1).
Table 2.
Partitioning between monomeric, kissing-loop (KL), and extended duplex (ED) forms determined from solutions containing ribonucleoprotein assemblies in the presence of a 5-fold excess ligand per RNA dimer. The percentage of each species in solution was obtained before and after incubation to assess the effects of ligands on isomerization (see Materials and Methods). Each determination was repeated in triplicate and the final values carried an average standard deviation of ±2.5%. The SL1A255G mutant, which exhibits stronger NC-hinge interaction [38], was probed with a 3-fold (*) and a 5-fold (**) excess ligand per RNA dimer to distinguish the effects induced by NC-neomycin B competition at the hinge and stem-bulge sites, respectively (see Results and Discussion).
| t = 0 h | t = 3 h | ||
|---|---|---|---|
| SL1 No ligand |
Mono % | 5 | 1 |
| KL% | 95 | 10 | |
| ED% | 0 | 89 | |
| Total dimer % | 95 | 99 | |
|
| |||
| Ethidium bromide | Mono % | 5 | 5 |
| KL% | 95 | 25 | |
| ED% | 0 | 70 | |
| Total dimer % | 95 | 95 | |
|
| |||
| Acridine | Mono % | 8 | 8 |
| KL% | 92 | 16 | |
| ED% | 0 | 76 | |
| Total dimer % | 92 | 99 | |
|
| |||
| Mitoxantrone | Mono % | 4 | 1 |
| KL% | 96 | 20 | |
| ED% | 0 | 79 | |
| Total dimer % | 96 | 99 | |
|
| |||
| Neomycin B | Mono % | 5 | 2 |
| KL% | 95 | 51 | |
| ED% | 0 | 47 | |
| Total dimer % | 95 | 98 | |
|
| |||
| Paromomycin | Mono % | 3 | 3 |
| KL% | 97 | 36 | |
| ED% | 0 | 61 | |
| Total dimer % | 97 | 97 | |
|
| |||
| Tobramycin | Mono % | 9 | 4 |
| KL% | 91 | 40 | |
| ED% | 0 | 65 | |
| Total dimer % | 91 | 99 | |
|
| |||
| Kanamycin A | Mono % | 5 | 5 |
| KL% | 95 | 24 | |
| ED% | 0 | 71 | |
| Total dimer % | 95 | 95 | |
|
| |||
| Kanamycin B | Mono % | 9 | 5 |
| KL% | 91 | 36 | |
| ED% | 0 | 59 | |
| Total dimer % | 91 | 95 | |
|
| |||
| Distamycin A | Mono % | 5 | 5 |
| KL% | 95 | 14 | |
| ED% | 0 | 81 | |
| Total dimer % | 95 | 95 | |
|
| |||
| H33352 | Mono % | 4 | 5 |
| KL% | 96 | 15 | |
| ED% | 0 | 80 | |
| Total dimer % | 96 | 95 | |
|
| |||
| H33258 | Mono % | 7 | 2 |
| KL% | 93 | 15 | |
| ED% | 0 | 80 | |
| Total dimer % | 93 | 95 | |
|
| |||
| SL1A255G No ligand |
Mono % | 5 | 2 |
| KL% | 95 | 20 | |
| ED% | 0 | 78 | |
| Total dimer % | 95 | 98 | |
|
| |||
| Neomycin B* | Mono % | 5 | 5 |
| KL% | 95 | 13 | |
| ED% | 0 | 82 | |
| Total dimer % | 95 | 95 | |
|
| |||
| Neomycin B** | Mono % | 4 | 5 |
| KL% | 96 | 50 | |
| ED% | 0 | 45 | |
| Total dimer % | 96 | 95 | |
Interpreting the more sizeable inhibitory effects observed for aminoglycosides under the selected experimental conditions is complicated by their ability to disrupt protein interactions that cover different roles in the isomerization process (Table 2). In fact, NC binding to the stem-bulge motif induces destabilization of the surrounding base pairs, which facilitates stem melting and strand-exchange. In contrast, hinge binding stabilizes a base grip formed by the stacking of unpaired purines from the opposite stemloops, which requires instead destabilization to enable strand-exchange [38]. Consistent with these activities, aminoglycoside concentrations that displaced hinge-bound NC from the 3NC•2SL1A255G assembly provided a total of ~82% ED in solution after incubation (Figure 5b), up from 76% recorded for the same sample in the absence of ligand (Figure 5a). Concentrations that displaced the units bound to the bulge motifs allowed for the detection of only ~45% ED under the same test conditions (Figure 5c), thus inhibiting the NC chaperoning activity at this site. The significance of bulge interactions was further proved by control experiments using the stem-bulge duplex SL1BG, which underwent prompt dissociation into its single-stranded components in the presence of excess NC, but remained intact when aminoglycosides were added to the mixture. Finally, it is important to note that although aminoglycosides shared NC’s binding activity and specificity, they did not exhibit chaperoning capabilities of their own. This observation is consistent with the fact that these ligands possess a flexible backbone and a polycationic nature, which enables them to replicate the nucleic acid binding properties of highly basic proteins, but they do not possess the zinc-finger structures that are responsible for the unique RNA-remodeling activities of NC [61, 62].
Figure 5.

Nanospray-FTICR mass spectrum of a sample containing 30 μM NC (○) and 10 μM SL1A255G (◆) in 150 mM ammonium acetate (pH 7.0) after 3 hour incubation at 37°C in the (a) absence of ligand, (b) in the presence of 30 μM and (c) 50μM neomycin B (•). The percentages in parentheses indicate the proportions of KL/ED within each protein-RNA assembly obtained by tandem mass spectrometry under mild activation conditions (see Materials and Methods). From the resulting spectra, dimeric products were assigned to the ED conformer, whereas monomeric products were assigned to KL (as shown in Figure 3). The percentages were then summed to determine the partitioning between the two conformers within each precursor ion population, as reported in the box [38].
Conclusions
In the absence of high-resolution structures for the assemblies of NC with the different forms assumed in vitro by SL1, non-covalent probes can provide valuable information about their organization and significance in the two-step process of RNA dimerization and isomerization. Following this approach requires establishing baseline behaviors in the absence of protein, which should be subsequently employed to interpret the data obtained in its presence. For all ligands included in the study, the stoichiometries determined experimentally matched very closely the number of suitable sites inferred from the high-resolution structures of the respective RNA substrates. Although the initial stoichiometries were conserved in the presence of NC, the effects exerted by the various probes on the protein’s ability to catalyze structure remodeling were clearly influenced by their distinctive binding modes.
Intercalators, mixed-mode, and minor groove ligands did not interfere directly with NC interactions, but reduced the efficiency of isomerization by increasing the stability of double-stranded structures operated upon by the protein to facilitate strand-exchange. In contrast, aminoglycoside probes manifested the ability to compete with NC for specific sites of the SL1 conformers, thus preventing the protein from performing its chaperone activities. As shown by the high-resolution structures of complexes with other hairpin domains of the HIV-1 packaging signal, the highly basic N-terminal region of NC folds into a 310 helix that makes specific contacts with electronegative patches on the substrates upper stems, while the more hydrophobic finger domains can sandwich unpaired guanines exposed on their loop regions [63, 64]. We have previously demonstrated that highly charged aminoglycosides are capable of displacing the N-terminal tail from its electrostatic interactions, thus destabilizing protein-RNA binding [17, 57]. In similar fashion, neomycin B competed here with the positively-charged NC for the electronegative surfaces presented by the stem-bulge, hinge, and palindrome flanking sites. Given that aminoglycosides lack the ability to establish hydrophobic interactions with exposed guanines, which are critical for the chaperone activity of NC [61, 62], displacing the protein from the stem-bulge motifs eliminated its participation to the melting of the double-stranded stems necessary for KL-ED isomerization. Conversely, inhibiting NC interactions with the hinge region eliminated its active stabilization of the base grip motif created by the stacking of the unpaired purines, which would stand in the way of strand-exchange [38], thus having the opposite effect on isomerization efficiency. These results provide further evidence that the structural context of the actual binding sites has significant influence on the chaperone activities of NC, which should be taken in account when developing potential drug candidates aimed at disrupting genome dimerization and isomerization in HIV-1.
In conclusion, this application of non-covalent probes exemplifies the potential, but also the possible pitfalls of this approach. Unlike covalent probes reporting on the accessibility of susceptible functional groups, which can highlight the interface between bound biopolymers, non-covalent ligands can also reveal the nature of their specific binding interactions. The type and extent of information are still limited by the fact that available reagents were developed for purposes other than probing. Compiling a large database of reagents for targeting a wide variety of interactions and structural contexts would provide a very versatile tool for the investigation of biomolecular assemblies, which could be employed not only to tackle substrates that are not readily amenable to established techniques, but also to drive the development of novel therapeutic strategies in pharmacologically relevant systems.
Materials and Methods
RNA, NC, and ribonucleoprotein complexes
All RNA constructs in the study (Scheme 1) were purchased from IDT (Coralville, IA), desalted by ultrafiltration, and quality-controlled by ESI-FTICR. Each stock concentration was determined by UV absorbance using molar extinction coefficients calculated from the respective sequences. Each RNA sample was heat-refolded to obtain the desired dimeric conformation (see [37, 38] and references therein). Briefly, RNA solutions were heated to 95°C for 5 minutes in a water-bath, then fast-cooled in ice slurry to obtain predominantly the kinetic product KL. The cooling process was completed slowly at room temperature to produce the thermodynamic product ED. The conformational state of each dimeric form was verified by using tandem mass spectrometry to selectively induce dissociation of the KL dimer in the corresponding hairpin components [38]. Recombinant NC was expressed in E. coli BL21 (DE3)-pLysE, purified under non-denaturing conditions, and extensively desalted by ultrafiltration against 150 mM ammonium acetate with pH adjusted to 7.0 (see [20] and references therein). The purity and integrity of the protein were confirmed directly by ESI-FTICR. Protein-RNA assemblies were obtained by mixing appropriate amounts of each component in 150 mM ammonium acetate (pH 7.0). Although a wide range of protein-RNA ratios was explored, typical samples consisted of 30 μM and 5 μM final concentrations of NC and RNA, respectively, for a final 6:2 molar ratio. After 15 min at room temperature, complex composition and conformation were verified by ESI-FTICR.
Non-covalent probing
All ligands used in the study were obtained from Sigma Chemical Co. (St. Louis, MO) and utilized without further purification. Stock solutions were prepared by weighing each compound and dissolving it in 150 mM ammonium acetate (pH 7.0). For each step of the titration series, aliquots of substrate and ligand solution were mixed together and added with an appropriate volume of 150 mM ammonium acetate (pH 7.0) to obtain a fixed final 5 μM concentration of RNA. Total ligand concentrations ranged between 5 and 50 μM in subsequent steps, corresponding to a 1-10 fold excess of total ligand. Each sample was incubated at room temperature for 15 min to ensure that a binding equilibrium was established in solution before ESI-FTICR analysis. Isomerization experiments were carried out by incubating samples in a water bath at 37°C for up to 3 h. More extensive incubation periods did not result in significant differences in the observed outcome.
Mass spectrometry
All analyses were performed on a Bruker Daltonics (Billerica, MA) Apex III FTICR mass spectrometer equipped with a 7T actively-shielded superconducting magnet and a nano-ESI source built in house [37]. Desolvation temperature, skimmer voltage, and other source parameters known to control the ionization process were optimized to allow for the observation of the RNA-RNA and protein-RNA noncovalent complexes, as previously described [20, 37]. Analyte solutions were mixed with iso-propanol immediately before analysis to a final concentration of 10% in volume to assist desolvation. Typically, 5 μL samples were loaded into the nano-electrospray needle and a spray voltage of less than 1 kV was applied to the solution through a stainless steel wire inserted into back. No solvent pumps were necessary, as the solution flow-rate was dictated by the applied voltage and the size of the nano-ESI needle tip (typically ~1-2 μm). Spectra were acquired in negative ionization mode and processed with ApexControl 2.0 (Bruker Daltonics, Billerica, MA). Scans were performed in broadband mode that allowed for a typical 150,000 resolving power at m/z 2000. The spectra were externally calibrated using a 1 mg/ml solution of CsI, which produced a series of peaks throughout the mass range of 1000-7000 m/z and enabled to achieve a typical mass accuracy of 20 ppm or better across this range. Each analysis was performed a minimum of three times and only representative spectra were shown.
In tandem mass spectrometry experiments, the precursor ion of interest was isolated in the FTICR cell using correlated rf sweeps (CHEF)[65], followed by activation through sustained off-resonance irradiation-collision induced dissociation (SORI-CID)[55]. Frequency offsets below and above the resonant frequency of the precursor ion were sampled to avoid possible “blind spots” in the product spectra [66]. Best results were achieved by using irradiation frequencies that were 600 to 2000 Hz below that of the precursor ion. In experiments designed to assess the conformation of dimeric SL1, mild activation regimes were obtained by applying off-resonant pulses for 250 msec using 26 to 31 dB attenuation of the maximum power output allowed by the hardware. In sequencing experiments designed to locate the ligand sites, more energetic dissociation conditions were achieved by activating the precursor ion of interest for the same time interval, but employing only17 to 20 dB attenuation. Argon was used as the collision gas in pulsed bursts of 100-250 ms, which resulted in momentary increases from 1×10−11 to 1×10−7 mbar of the background pressure measured by the instrument gauge located underneath the ion optics. No attempt was made to determine the actual pressure within the FTICR cell. Twenty-five to fifty scans were typically averaged for each spectrum.
Data analysis
Concentrations of free and bound species obtained upon ligand addition were calculated from the known initial concentrations of RNA by using the molar fractions of the different species in solution, which was directly determined from the intensity of the respective signals divided by their charge state. This method relies on the reasonable assumption that any effect induced upon binding of a small ligand to a much larger and highly charged substrate is likely to result in a minor shift of the overall charge state distribution, rather than in complete signal suppression. Therefore, using all the signals detected for each species divided by their charge state can account for such a shift. In our case, the validity of such assumption was confirmed by the fact that no significant changes of charge distribution could be detected during the binding experiments (see Results and Discussion).
Solution concentrations obtained from the respective molar fractions were employed to plot binding curves and calculate the respective dissociation constant (KD) using the data-fitting algorithm included in Origin 7 (Silverdale Scientific, Buckinghampshire, UK). All titration experiments were performed at least in triplicate, thus the reported precision reflects the overall uncertainty of each KD determination.
The different protein-RNA and RNA-RNA species observed in solution by ESI-FTICR, or obtained from their gas-phase activation by SORI-CID, were unambiguously identified from the observed molecular masses. The typical accuracy afforded by these determinations can be appreciated by comparing the experimental masses with the theoretical ones calculated from sequence. The partitioning among the different forms assumed by SL1 in solution was estimated in a semi-quantitative fashion from the signal intensity of each species divided by its respective charge state. The use of normalized intensities is legitimated by the fact that NC binding did not induce complete neutralization of the nucleic acid component, but rather produced a mere shift in charge distribution (see also [20, 37] and references therein).
Docking calculations
Docking simulations were performed using Autodock 3.0, with additional help from the Autodocktools package [67]. Neomycin B was docked into SL1 (PDB:2D1B) using the Lamarckian genetic algorithm (LGA) included in the package. The Marsili-Gasteiger and the Kollman United Atom algorithms were used to add hydrogens and assign charges to the ligand and receptors. A total of 50 docked structures for each stem loop were generated, 10 at a time, using varying grid sizes ranging from 90×90×90 to 126×126×126 and a grid spacing of 0.150-0.375 Å. Typical settings employed for this procedure include a population size of 150, a maximum number of 250,000 energy evaluations, a maximum number of 27,000 generations, a mutation rate of 0.02, and a crossover rate of 0.8 [67].
Acknowledgments
This research was funded by the National Institutes of Health (GM064328) and by the National Science Foundation (CHE-0439067).
References
- 1.Peattie DA, Gilbert W. Chemical probes for higher-order structure in RNA. Proc. Natl. Acad. Sci. U.S.A. 1980;77(8):4679–82. doi: 10.1073/pnas.77.8.4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walker TA, et al. Enzymatic and chemical structure mapping of mouse 28S ribosomal ribonucleic acid contacts in 5.8S ribosomal ribonucleic acid. Biochemistry. 1982;21:2320–29. doi: 10.1021/bi00539a008. [DOI] [PubMed] [Google Scholar]
- 3.Ehresmann C, et al. Probing the structure of RNAs in solution. Nucleic Acids Res. 1987;12(22):9109–9128. doi: 10.1093/nar/15.22.9109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krol A, Carbon P. A guide for probing native small nuclear RNA and ribonucleoprotein structures. Methods Enzymol. 1989;180:212–27. doi: 10.1016/0076-6879(89)80103-x. [DOI] [PubMed] [Google Scholar]
- 5.Brunel C, Romby P. Probing RNA structure and RNA-ligand complexes with chemical probes. Methods Enzymol. 2000;318:3–21. doi: 10.1016/s0076-6879(00)18040-1. [DOI] [PubMed] [Google Scholar]
- 6.Yamashita M, Fenn JB. Electrospray ion source. Another variation on the free-jet theme. J. Phys. Chem. 1984;88:4671–4675. [Google Scholar]
- 7.Aleksandrov ML, et al. Extraction of ions from solutions under atmospheric pressure: a method of mass spectrometric analysis of bioorganic compounds. Doklady Akademii Nauk. 1984;277:379–383. [Google Scholar]
- 8.Comisarow MB, Marshall AG. Fourier transform ion cyclotron resonance. Chem. Phys. Lett. 1974;25(2):282–283. [Google Scholar]
- 9.Hendrickson CL, Emmett MR, Marshall AG. Electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry. Annu. Rev. Phys. Chem. 1999;50:517–536. doi: 10.1146/annurev.physchem.50.1.517. [DOI] [PubMed] [Google Scholar]
- 10.Yu E, Fabris D. Direct probing of RNA structures and RNA-protein interactions in the HIV-1 packaging signal by chemical modification and electrospray ionization Fourier transform mass spectrometry. J. Mol. Biol. 2003;330(2):211–223. doi: 10.1016/s0022-2836(03)00589-8. [DOI] [PubMed] [Google Scholar]
- 11.Yu ET, Fabris D. Toward multiplexing the application of solvent accessibility probes for the investigation of RNA three-dimensional structures by electrospray ionization - Fourier transform mass spectrometry. Anal. Biochem. 2004;344:356–366. doi: 10.1016/j.ab.2004.07.034. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Q, et al. Toward building a database of bifunctional probes for the MS3D investigation of nucleic acids structures. J Am Soc Mass Spectrom. 2006;17:1570–1581. doi: 10.1016/j.jasms.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 13.Yu ET, Zhang Q, Fabris D. Untying the FIV frameshifting pseudoknot structure by MS3D. J. Mol. Biol. 2005;345:69–80. doi: 10.1016/j.jmb.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 14.Yu ET, et al. MS3D structural elucidation of the HIV-1 packaging signal. Proc Natl Acad Sci U S A. 2008;105:12248–12253. doi: 10.1073/pnas.0800509105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Probst CL, Perun TJ. Nucleic acid targeted drug design. Marcel Dekker, Inc.; New York, NY: 1992. [Google Scholar]
- 16.Hermann T. Chemical and functional diversity of small molecule ligands for RNA. Biopolymers. 2003;70:4–18. doi: 10.1002/bip.10410. [DOI] [PubMed] [Google Scholar]
- 17.Turner KB, Hagan NA, Fabris D. Inhibitory effects of archetypical nucleic acid ligands on the interactions of HIV-1 nucleocapsid protein with elements of Ψ-RNA. Nucl. Acids Res. 2006;34(5):1305–1316. doi: 10.1093/nar/gkl004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Turner KB, et al. Structural probing of HIV-1 polypurine tract RNA:DNA hybrid using classic nucleic acid ligands. Nucleic Acids Res. 2008;36:2799–2810. doi: 10.1093/nar/gkn129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ganem B, Li Y-T, Henion JD. Detection of oligonucleotide duplex forms by ion-spray mass spectrometry. Tetrahedron Lett. 1993;34:1445–1448. [Google Scholar]
- 20.Hagan N, Fabris D. Direct mass spectrometric determination of the stoichiometry and binding affinity of the complexes between HIV-1 nucleocapsid protein and RNA stem-loops hairpins of the HIV-1 Ψ-recognition element. Biochemistry. 2003;42(36):10736–10745. doi: 10.1021/bi0348922. [DOI] [PubMed] [Google Scholar]
- 21.Hofstadler SA, Griffey RH. Analysis of noncovalent complexes of DNA and RNA by mass spectrometry. Chem. Rev. 2001;101(2):377–390. doi: 10.1021/cr990105o. [DOI] [PubMed] [Google Scholar]
- 22.Laughrea M, Jetté L. A 19-nucleotide sequence upstream of the 5′ major splice donor is part of the dimerization domain of human immunodeficiency virus 1 genomic RNA. Biochemistry. 1994;33:13464–13474. doi: 10.1021/bi00249a035. [DOI] [PubMed] [Google Scholar]
- 23.Skripkin E, et al. Identification of the primary site of the human immunodeficiency virus type 1 RNA dimerization in vitro. Proc Natl Acad Sci U S A. 1994;91(11):4945–9. doi: 10.1073/pnas.91.11.4945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paillart JC, et al. Dimerization of retroviral genomic RNAs: structural and functional implications. Biochimie. 1996;78(7):639–53. doi: 10.1016/s0300-9084(96)80010-1. [DOI] [PubMed] [Google Scholar]
- 25.Berkhout B, van Wamel JL. Role of the DIS hairpin in replication of human immunodeficiency virus type 1. J. Virol. 1996;70(10):6723–32. doi: 10.1128/jvi.70.10.6723-6732.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muriaux D, et al. Dimerization of HIV-1Lai RNA at low ionic strength. An autocomplementary sequence in the 5′ leader region is evidenced by an antisense oligonucleotide. J Biol Chem. 1995;270(14):8209–16. doi: 10.1074/jbc.270.14.8209. [DOI] [PubMed] [Google Scholar]
- 27.Clever J, Wong ML, Parslow TG. Requirements for kissing-loop-mediated dimerization of human immunodeficiency virus RNA. J. Virol. 1996;70(9):5902–5908. doi: 10.1128/jvi.70.9.5902-5908.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paillart JC, et al. A loop-loop “kissing” complex is the essential part of the dimer linkage of genomic HIV-1 RNA. Proc Natl Acad Sci U S A. 1996;93(11):5572–7. doi: 10.1073/pnas.93.11.5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muriaux D, Fossé P, Paoletti J. A kissing complex together with a stable dimer is involved in the HIV-1Lai RNA dimerization process in vitro. Biochemistry. 1996;35(15):5075–82. doi: 10.1021/bi952822s. [DOI] [PubMed] [Google Scholar]
- 30.Laughrea M, Jetté L. Kissing-loop model of HIV-1 genome dimerization: HIV-1 RNAs can assume alternative dimeric forms, and all sequences upstream or downstream of hairpin 248-271 are dispensable for dimer formation. Biochemistry. 1996;35(5):1589–98. doi: 10.1021/bi951838f. [DOI] [PubMed] [Google Scholar]
- 31.Takahashi KI, et al. Structural requirement for the two-step dimerization of human immunodeficiency virus type 1 genome. RNA. 2000;6(1):96–102. doi: 10.1017/s1355838200991635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bernacchi S, et al. Mechanism of hairpin-duplex conversion for the HIV-1 dimerization initiation site. J Biol Chem. 2005;280(48):40112–21. doi: 10.1074/jbc.M503230200. [DOI] [PubMed] [Google Scholar]
- 33.Feng YX, et al. HIV-1 nucleocapsid protein induces “maturation” of dimeric retroviral RNA in vitro. Proc. Natl. Acad. Sci. U S A. 1996;93(15):7577–81. doi: 10.1073/pnas.93.15.7577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muriaux D, et al. NCp7 activates HIV-1Lai RNA dimerization by converting a transient loop-loop complex into a stable dimer. J. Biol. Chem. 1996;271(52):33686–33692. doi: 10.1074/jbc.271.52.33686. [DOI] [PubMed] [Google Scholar]
- 35.Rist MJ, Marino JP. Mechanism of nucleocapsid protein catalyzed structural isomerization of the dimerization initiation site of HIV-1. Biochemistry. 2002;41(50):14762–70. doi: 10.1021/bi0267240. [DOI] [PubMed] [Google Scholar]
- 36.Takahashi K, et al. Two basic regions of NCp7 are sufficient for conformational conversion of HIV-1 dimerization initiation site from kissing-loop dimer to extended-duplex dimer. J. Biol. Chem. 2001;276(33):31274–8. doi: 10.1074/jbc.M104577200. [DOI] [PubMed] [Google Scholar]
- 37.Hagan NA, Fabris D. Dissecting the protein-RNA and RNA-RNA interactions in the nucleocapsid-mediated dimerization and isomerization of HIV-1 stemloop 1. J. Mol. Biol. 2007;365:396–410. doi: 10.1016/j.jmb.2006.09.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Turner KB, Hagan NA, Fabris D. Understanding the isomerization of the HIV-1 dimerization initiation domain by the nucleocapsid protein. J. Mol. Biol. 2007;369:812–828. doi: 10.1016/j.jmb.2007.03.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yuan Y, et al. Stem of SL1 RNA in HIV-1: structure and nucleocapsid protein binding for a 1 × 3 internal loop. Biochemistry. 2003;42(18):5259–69. doi: 10.1021/bi034084a. [DOI] [PubMed] [Google Scholar]
- 40.Lawrence DC, et al. Structure of the intact stem and bulge of HIV-1 Psi-RNA stem-loop SL1. J. Mol. Biol. 2003;326(2):529–42. doi: 10.1016/s0022-2836(02)01305-0. [DOI] [PubMed] [Google Scholar]
- 41.Mujeeb A, et al. Structure of the dimer initiation complex of HIV-1 genomic RNA. Nat. Struct. Biol. 1998;5(6):432–436. doi: 10.1038/nsb0698-432. [DOI] [PubMed] [Google Scholar]
- 42.Baba S, et al. Solution RNA Structures of the HIV-1 Dimerization Initiation Site in the Kissing-Loop and Extended-Duplex Dimers. J. Biochem. (Tokyo) 2005;138(5):583–92. doi: 10.1093/jb/mvi158. [DOI] [PubMed] [Google Scholar]
- 43.Ennifar E, et al. Crystal structures of coaxially stacked kissing complexes of the HIV-1 RNA dimerization initiation site. Nat Struct Biol. 2001;8(12):1064–8. doi: 10.1038/nsb727. [DOI] [PubMed] [Google Scholar]
- 44.Ennifar E, Dumas P. Polymorphism of bulged-out residues in HIV-1 RNA DIS kissing complex and structure comparison with solution studies. J Mol Biol. 2006;356(3):771–82. doi: 10.1016/j.jmb.2005.12.022. [DOI] [PubMed] [Google Scholar]
- 45.Ennifar E, et al. The crystal structure of the dimerization initiation site of genomic HIV-1 RNA reveal an extended duplex with two adenine bulges. Struct. Fold. Des. 1999;7(11):1439–1449. doi: 10.1016/s0969-2126(00)80033-7. [DOI] [PubMed] [Google Scholar]
- 46.Mujeeb A, et al. NMR structure of the mature dimer complex of HIV-1 genomic RNA. FEBS Lett. 1999;458(3):387–392. doi: 10.1016/s0014-5793(99)01183-7. [DOI] [PubMed] [Google Scholar]
- 47.Girard F, et al. Dimer initiation sequence of HIV-1Lai genomic RNA: NMR solution structure of the extended duplex. J. Biomol. Struct. Dyn. 1999;16(6):1145–57. doi: 10.1080/07391102.1999.10508323. [DOI] [PubMed] [Google Scholar]
- 48.Ulyanov NB, et al. NMR structure of the full-length linear dimer of stem-loop-1 RNA in the HIV-1 dimer initiation site. J Biol Chem. 2006;281(23):16168–77. doi: 10.1074/jbc.M601711200. [DOI] [PubMed] [Google Scholar]
- 49.Sun J-S, et al. Sequence-specific intercalating agents. Intercalation at specific sequences on duplex DNA via major groove recognition by oiligonucleotide-intercalator conjugates. Proc. Nat. Acad. Sci. USA. 1989;86:9198–9202. doi: 10.1073/pnas.86.23.9198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Spink N, et al. Sequence-dependent effects in drug-DNA interaction: the crystal structure of Hoechst 33258 bound to the d(CGCAAATTTGCG)2 duplex. Nucl. Acids Res. 1994;22(9):1607–1612. doi: 10.1093/nar/22.9.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tanious FA, et al. Effects of compound structure on carbazole dication-DNA complexes: tests of the minor-groove complex models. Biochemistry. 2000;39:12091–12101. doi: 10.1021/bi001236i. [DOI] [PubMed] [Google Scholar]
- 52.Busscher GF, Rutjes FPJT, van Delft FL. 2-Deoxystreptamine: central scaffold of aminoglycoside antibiotics. Chem. Rev. 2005;105(3):775–791. doi: 10.1021/cr0404085. [DOI] [PubMed] [Google Scholar]
- 53.McLafferty FW. Tandem mass spectrometry. Science. 1981;214:280–287. doi: 10.1126/science.7280693. [DOI] [PubMed] [Google Scholar]
- 54.Cooks RG. Collision-induced Dissociation: Readings and Commentary. J Mass Spectrom. 1995;30:1215–1221. [Google Scholar]
- 55.Gauthier JW, Trautman TR, Jacobson DB. Sustained off-resonance irradiation for collision-activated dissociation involving Fourier transform mass spectrometry. Collision-activated dissociation technique that emulates infrared multiphoton dissociation. Anal. Chim. Acta. 1991;246:211–25. [Google Scholar]
- 56.McLuckey SA, Habibi-Goudarzi S. Decompositions of multiply charged oligonucleotide anions. J. Am. Chem. Soc. 1993;115:12085–95. [Google Scholar]
- 57.Turner KB, et al. Mapping noncovalent ligand binding to stemloop domains of the HIV-1 packaging signal by tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 2006;17:1401–1411. doi: 10.1016/j.jasms.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 58.Ennifar E, et al. HIV-1 RNA dimerization initiation site is structurally similar to the ribosomal A site and binds aminoglycoside antibiotics. J Biol Chem. 2003;278(4):2723–30. doi: 10.1074/jbc.M205726200. [DOI] [PubMed] [Google Scholar]
- 59.Fourmy D, et al. Structure of the A-site of E. coli 16S rRNA complexed with an aminoglycoside antibiotic. Science. 1996;274:1367–1371. doi: 10.1126/science.274.5291.1367. [DOI] [PubMed] [Google Scholar]
- 60.Freisz S, et al. Binding of aminoglycoside antibiotics to the duplex form of the HIV-1 genomic RNA dimerization initiation site. Angew. Chem. Int. Ed. 2008;47:1–5. doi: 10.1002/anie.200800726. [DOI] [PubMed] [Google Scholar]
- 61.Rein A, Henderson LE, Levin JG. Nucleic-acid-chaperone activity of retroviral nucleocapsid proteins: significance for retroviral replication. Trends Biochem. Sci. 1998;23(8):297–301. doi: 10.1016/s0968-0004(98)01256-0. [DOI] [PubMed] [Google Scholar]
- 62.Levin JG, et al. Nucleic acid chaperone activity of HIV-1 nucleocapsid protein: critical role in reverse transcription and molecular mechanism. Prog. Nucleic Acid Res. Mol. Biol. 2005;80:217–86. doi: 10.1016/S0079-6603(05)80006-6. [DOI] [PubMed] [Google Scholar]
- 63.De Guzman RN, et al. Structure of the HIV-1 nucleocapsid protein bound to the SL-3 Ψ-RNA recognition element. Science. 1998;279(5349):384–388. doi: 10.1126/science.279.5349.384. [DOI] [PubMed] [Google Scholar]
- 64.Amarasinghe GK, et al. NMR structure of the HIV-1 nucleocapsid protein bound to stem-loop SL2 of the Ψ-RNA packaging signal. Implications for genome recognition. J. Mol. Biol. 2000;301(2):491–511. doi: 10.1006/jmbi.2000.3979. [DOI] [PubMed] [Google Scholar]
- 65.de Koning LJ, et al. Mass selection of ions in a Fourier transform ion cyclotron resonance trap using correlated harmonic excitation fields (CHEF) Int. J. Mass Spectrom. Ion Proc. 1997;165/166:209–219. [Google Scholar]
- 66.Senko MW, Speir JP, McLafferty FW. Collisional activation of large multiply charged ions using Fourier transform mass spectrometry. Anal Chem. 1994;66(18):2801–8. doi: 10.1021/ac00090a003. [DOI] [PubMed] [Google Scholar]
- 67.Morris GM, et al. Automated docking using a Lamarckian genetic algorithm and empirical binding free energy function. J. Comp. Chem. 1998;19:1639–1662. [Google Scholar]
- 68.Honig B, Nicholls A. Classical electrostatics in biology and chemistry. Science. 1995;268:1144–1149. doi: 10.1126/science.7761829. [DOI] [PubMed] [Google Scholar]
- 69.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA, USA: 2002. [Google Scholar]