

Abstract
Tyrosine phosphorylation plays a critical role in growth regulation, and its aberrant regulation can be involved in carcinogenesis. The association of Shc (Src homolog and collagen homolog) adaptor protein family members in tyrosine phosphorylation signaling pathway is well recognized. Shc adaptor proteins transmit activated tyrosine phosphorylation signaling that suggest their plausible role in growth regulation including carcinogenesis and metastasis. In parallel, by sharing a similar mechanism of carcinogenesis, the steroids are involved in the early stage of carcinogenesis as well as the regulation of cancer progression and metastatic processes. Recent evidence indicates a cross-talk between tyrosine phosphorylation signaling and steroid hormone action in epithelial cells, including prostate and breast cancer cells. Therefore, the members of Shc proteins may function as mediators between tyrosine phosphorylation and steroid signaling in steroid-regulated cell proliferation and carcinogenesis. In this communication, we discuss the novel roles of Shc proteins, specifically p52Shc and p66Shc, in steroid hormone-regulated cancers and a novel molecular mechanism by which redox signaling induced by p66Shc mediates steroid action via a non-genomic pathway. The p66Shc protein may serve as an effective biomarker for predicting cancer prognosis as well as a useful target for treatment.
Introduction
Cancers in steroid hormone-responsive tissues presently account for more than 35% in men and more than 40% in women of all newly diagnosed cancers in the United States (Henderson & Feigelson 2000). Extensive research has clearly demonstrated that the abnormal changes in the levels, frequencies, and types of steroid hormones are important contributors to the development of major cancer types such as the cancers of the prostate (androgen, estrogen), testes (in utero estrogen), breast (estrogen, progesterone), ovary (FSH, estrogen, androgen), uterine endometrium (estrogen), and thyroid (TSH, estrogen; Henderson & Feigelson 2000). Thus, many studies have been focused on the involvement of steroids in the regulation of tumor development, cancer cell proliferation, progression, and metastatic processes. It is now evident that either in the early stage of carcinogenesis or in the advanced metastatic phenotype, steroid hormone action goes far beyond the classical receptor-mediated gene regulation. These steroid hormone-related cancers may share common mechanisms of carcinogenesis, such as DNA damage/mutation as well as the elevated levels of various growth factors induced by the excess of steroids, leading to aberrant growth regulation (Dickson & Lippman 1987, Dabrosin et al. 1997, Devanesan et al. 2001). Recent advances further indicate that these hormones could also induce rapid, non-genomic responses and a convoluted network of interactions with different intracellular signaling pathways.
Non-genomic actions by steroid hormones
Numerous studies have demonstrated the non-genomic action of steroid hormones, including androgens and estrogens, in cellular processes such as cell proliferation and motility (Berridge et al. 1998). The most intriguing facts on the non-genomic nature of steroids are that the effects depend on their rapid response and insisting no direct binding of nuclear receptors to gene expression, i.e., seconds to minutes and their insensitivity to the inhibitors of transcription and translation and/or the antagonists of the classical intracellular steroid hormone receptors. The non-genomic effects of steroids could be mediated by multiple pathways, and are discussed briefly as follows.
Direct and acute action of steroids
Steroids may have direct action on target molecules independent of steroid receptors. One such effect, for example, is the direct binding and activation of protein kinase C (PKC) isoforms such as PKCα and PKCδ by aldosterone and 17β-estradiol (E2) respectively via binding directly to their regulatory domains C2 that mediates calcium (Ca2+) binding and results in the autophosphorylation of these kinases (Alzamora & Harvey 2008). Furthermore, aldosterone and E2 rapidly and directly stimulate phospholipase A2 (PLA2) and cyclooxygenase (COX), which also result in the rapid increase in intracellular [Ca2+]i (Harvey et al. 2002). The other direct and acute effects of steroids are their rapid action on voltage-gated and calcium-activated ion channels. E2 activates the calcium-activated potassium channels via binding directly to its regulatory β-subunit (Valverde et al. 1999). Testosterone rapidly activates ATP-sensitive K+ channels (KATP) via opening KATP channels (Er et al. 2004) and inhibits the L-type and T-type calcium channels (ICa,L; Michels et al. 2006, Er et al. 2007).
Rapid action of steroids involving classical intracellular steroid receptors
Steroids may bind to the classical intracellular steroid receptors and activate the second messenger pathways such as c-Src kinase that rapidly stimulate the MAPK/ERK and PI3K/AKT kinase pathways (Migliaccio et al. 2000). Interestingly, an androgen receptor (AR)/Src/modulator of non-genomic action of estrogen receptor (MNAR) complex and the cooperative association of c-Src, estrogen receptors (ERs), and AR activates MAPK and c-Src kinase pathways respectively (Kousteni et al. 2001, Unni et al. 2004). Estrogens on binding to ERα may also serve as a transcriptional co-activator activating several transcriptional factors, such as activator protein 1 (AP-1), nuclear factor kappa B (NF-κB), and SP-1 in a non-genomic manner (Ray et al. 1997, Jakacka et al. 2001, Safe 2001). Steroids can also activate cAMP-dependent protein kinase A (PKA) via the transmembrane sex hormone-binding globulin (SHBG) receptor in association with transmembrane G-protein-coupled receptor (GPCR; Fortunati 1999, Rosner et al. 1999). The activation of PKA via the induction of cAMP by SHBG is observed in both prostate and breast cancer cells (Fortunati et al. 1996, Nakhla et al. 1997).
Rapid action of steroids involving non-classical membrane-bound steroid receptors
Steroids may undergo non-genomic action by binding to distinct non-classical membrane-bound steroid receptors. Several reports have presumed the presence of androgen- and estrogen-binding sites in a number of cells (Benten et al. 1999a,b, Armen & Gay 2000, Kampa et al. 2002). Interestingly, both the membrane androgen receptor (mAR) and the membrane ER (mER) are found to be associated with an integral membrane protein caveolin that facilitates the assembling of several signaling molecules, including phosphatidylinositol 3-kinase (PI3K), Ras, and Src kinase in their scaffold domain (Okamoto et al. 1998, Kim et al. 1999, Lu et al. 2001). Furthermore, mER perhaps exists as in a cytoplasmic pool and the rapid action requires their interaction with caveolin in association with MNAR, Shc and growth factor receptors, and striatin that translocated ER to the plasma membrane (Wong et al. 2002, Lu et al. 2004, Song et al. 2005).
Rapid action of membrane steroid receptors involving GPCR
The most preserved non-genomic action of steroid hormones is the rapid increase in intracellular calcium concentration [Ca2+] mediated via GPCR that constitutes α-, β-, and γ-subunits (Lieberherr & Grosse 1994, Benten et al. 1998), which ultimately results in the rapid activation of MAPK/ERK and PI3K/AKT pathways, leading to the activation of PKC and PKA (Kelly et al. 1999, Estrada et al. 2003). The interaction of mAR with GPCR results in the dissociation of Gα-subunit and the signal is transmitted from Gβγ through the activation of effector molecules including c-Src, Raf, and phospholipase C (PLC; Pierce et al. 2002). GPCR itself may also serve as the membrane receptor, i.e., binding of E2 to an orphan GPCR, termed GPR30, plays a critical role in the rapid signaling of E2-mediated stimulation of Ras-dependent MAPK activation through the phosphorylation of Shc (Luttrell et al. 1996).
Rapid action of membrane steroid receptors via trans-activation of growth factor receptors
The rapid non-genomic actions of membrane steroid receptors may function via trans-activation of the growth factor receptors (Levin 2005), and of all membrane steroid receptors, mER is the paramount and well studied. The phenomenon is further confirmed by the co-existence of endogenous membrane receptors, including AR and ER, G-proteins, GPCR, growth factor receptors (EGFR, IGFR), non-receptor tyrosine kinases (Src, Ras), and linker proteins such as MNAR and striatin in the plasma membrane termed as ‘signalosomes’ (Hammes & Levin 2007). Alternatively, steroids may activate growth factor receptor kinase activity by inhibiting the regulatory phosphatases (Meng et al. 2000).
Rapid non-transcriptional action of membrane steroid receptors
The other non-genomic action of steroids involving membrane receptors is the non-transcriptional effects of these receptors that provoke the posttranslational amendments including phosphorylation. By regulating kinases and phosphatases, steroids influence the cell functions such as cell motility via modifying actin cytoskeleton (Kampa et al. 2002, Meyer & Feldman 2002, Levin 2005).
Rapid action of steroids on membrane fluidity
Steroids may mediate the non-genomic fashion through changes in membrane flexibility. Androgens via interacting with phospholipids in the lipid bilayer decrease the membrane fluidity, and subsequently alter the function of Na+/K+ and Ca2+ ATPase systems and also influence cellular adhesion and cell–cell interaction (Duval et al. 1983, Van Bömmel et al. 1987).
In summary, we propose that though the non-genomic actions of steroids are mediated through multiple pathways, both genomic and non-genomic effects are interlinked as non-genomic actions of steroids ultimately influencing at least one of the classical genomic-mediated transcriptional activities. Nevertheless, the molecular mechanisms of steroids, especially androgens and estrogens, in both the development and progression of human endocrine-related cancers at the non-genomic levels need further investigations. Along with these observations, in this review, we emphasize a novel non-genomic action of steroids promoting various stages of carcinogenesis via Shc proteins.
Cross-talk of tyrosine phosphorylation signaling and steroids in carcinogenesis
Every step of carcinogenesis is essentially controlled by various growth factors and their receptors, either it is steroid regulated or not. Growth factors, including nerve growth factor (Engebraaten et al. 1993, Sachs et al. 1996), fibroblast growth factor (Mignatti et al. 1991, Engebraaten et al. 1993, Taylor et al. 1993), platelet-derived growth factor (Engebraaten et al. 1993, Choudhury et al. 1997), epidermal growth factor (EGF; Engebraaten et al. 1993, Hamada et al. 1995), keratin growth factor (Sachs et al. 1996), hepatocyte growth factor (Pelicci et al. 1995, Sachs et al. 1996), interleukin 2 (Ratner et al. 1992), insulin, and insulin-like growth factors (Stracke et al. 1989) are known to be involved in regulating cell proliferation, motility, invasion, and/or migration of various cell types. The activated receptors triggered by growth factors, cytokines, or adhesion molecules facilitate the docking of Src homology 2 (SH2) and phosphotyrosine-binding (PTB) domain-containing adaptor molecules that transduce signals via downstream intracellular cascades. Each of the receptors for the ligands described above activates the c-Src homology and collagen homolog (Shc) adaptor proteins for signal transduction, which suggests a conceivable role of the Shc proteins in various stages of carcinogenesis.
Recently, several lines of evidence indicate that Shc proteins mediate diverse biological activities; for example, they may mediate steroid actions other than serving as adaptors in tyrosine phosphorylation signaling. Since the role of Shc proteins in mediating tyrosine phosphorylation signaling and in regulating oxidative stress-induced apoptosis has received much attention (Migliaccio et al. 1997, 1999, Ravichandran 2001), in this communication, we will first briefly overview Shc proteins and then focus our efforts on discussing the novel roles of Shc proteins, specifically p52Shc and p66Shc, in steroid-regulated cancers.
Members of Shc family: structure and function in tyrosine phosphorylation signaling
Molecular structure of Shc isoforms
Shc proteins were first cloned using an SH2-coding sequence as a probe, and the Shc family includes three isoforms with molecular masses of 46, 52 and 66 kDa, which are encoded by the same gene at chromosome 1q21 (Pelicci et al. 1992). A promoter in the first intron of Shc locus transcribes the mRNA of p66Shc, whereas an alternate promoter and splicing generates the other two Shc isoforms, i.e., p46Shc and p52Shc (Ventura et al. 2002). These three isoforms of Shc protein contain overlapping amino acid sequences that contribute to a SH2 domain at the COOH-terminal and a PTB domain at the NH2-terminal, separated by a central region enriched in proline and glycine residues, i.e., collagen homology (CH1) domain (Ravichandran 2001), as shown in Fig. 1. The SH2 domain (∼100 amino acids) is the prototype for protein–protein interaction modules that mediate the formation of multiprotein complexes during signaling (Pawson & Scott 1997, Yoshida et al. 2004). Structurally, p66Shc differs from p52Shc and p46Shc by virtue of its unique NH2-terminal, a 110-amino acid CH2 region, which is also rich in proline and glycine residues (Migliaccio et al. 1997).
Figure 1.
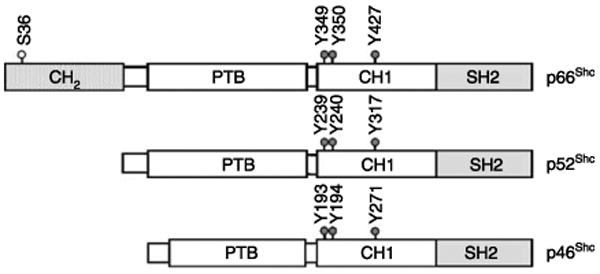
A schematic organization of Shc isoforms. The Shc proteins include three isoforms that are encoded by the same gene. They contain overlapping sequences. Three major tyrosine phosphorylation sites have been identified within the CH1 domain in all Shc isoforms. The unique CH2 domain of p66Shc isoform consists of 110 amino acids and contains a serine phosphorylation site (Ser-36). The PTB domain of p46Shc is deficient of the first 46 amino acids.
Subcellular localization of Shc isoforms
p66Shc is expressed primarily in epithelial cells, while p52Shc and p46Shc are expressed ubiquitously (Migliaccio et al. 1997). Most of p66Shc protein is distributed throughout the cytosol and a fraction of p66Shc localizes within the inner membrane and intermembrane spaces of mitochondria (Orsini et al. 2004, Ventura et al. 2004, Giorgio et al. 2005, Nemoto et al. 2006). p46Shc is found to be localized in the mitochondrial matrix (Orsini et al. 2004, Ventura et al. 2004, Nemoto et al. 2006). Unlike p46Shc and p66Shc, p52Shc is translocated to the plasma membrane from cytosol upon stimulation by growth factors, e.g., EGF (Migliaccio et al. 1997).
Shc isoforms in tyrosine phosphorylation signaling
p46Shc
Shc proteins are expressed in distinct patterns and exhibit diverse biological functions. Ventura et al. (2004) have reported specifically p46Shc as the first example to be localized in the mitochondrial matrix by means of a mitochondrion-targeting signal, which is inactive in p52Shc and p66Shc. Thus, p46Shc may play a role in the signal transduction pathways regulating mitochondrial physiology (Ventura et al. 2004). In addition to mediating tyrosine phosphorylation signaling (McGlade et al. 1992, Migliaccio et al. 1997), p46Shc may also modulate steroid action since steroids can regulate mitochondrial enzymatic activities (Ripple et al. 1997, 1999). Nevertheless, due to the limited studies on p46Shc in steroid action in carcinogenesis, it will not be discussed further in this communication.
p52Shc
p52Shc is responsible for transducing anchorage-dependent growth signaling (Pelicci et al. 1992). In general, when cells are stimulated by growth factors, p52Shc is recruited and binds to tyrosine kinase receptors through its PTB or SH2 domain, leading to its phosphorylation at tyrosine residues 239, 240, and 317 within the CH1 domain (Fig. 1; Rozakis-Adcock et al. 1992, van der Geer et al. 1996, Gotoh et al. 1996). Upon tyrosine phosphorylation, p52Shc recruits Grb2/SOS through a binding event between the SH2 domain of Grb2 and Shc phosphotyrosine residues (Pelicci et al. 1992, Rozakis-Adcock et al. 1992), which ultimately results in the activation of Ras and the MAPK cascade for mitogenesis (Fig. 2; Bonfini et al. 1996). The PTB domain of p52Shc binds to the phosphorylated tyrosine residues of receptor protein tyrosine kinases and functions similar to the SH2 domain. The CH1 and CH2 domains are putative SH3-binding regions (Lotti et al. 1996). Studies have also suggested that p52Shc mediates steroid action on cell proliferation as well as cell survival via tyrosine phosphorylation signal pathway (Kousteni et al. 2001, Lee et al. 2004a). Its aberrant expression and activation may lead to the dysregulation of one of the multi-pathways in carcinogenesis by steroids, a topic of focus in this review.
Figure 2.
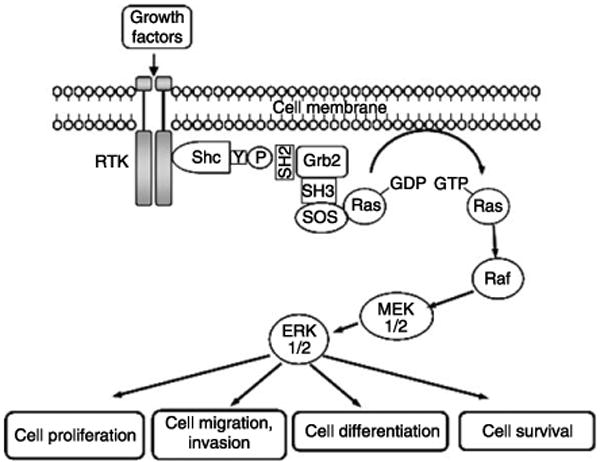
A schematic representation of Shc proteins activating the Ras and MAP kinase cascade. Upon stimulation by growth factors, RTK undergoes dimerization and tyrosine phosphorylation. Subsequently, Shc proteins are recruited and phosphorylated on tyrosine residue via forming a complex with RTK through the SH2 and/or PTB domain of Shc. The phosphorylated Shc proteins then associate with Grb2 adaptor protein through its tyrosine phosphorylation site to the SH2 domain of Grb2; the latter is constitutively complexed with SOS through its SH3 domains. These events result in the translocation of SOS to the plasma membrane and subsequently activate membrane-bound Ras in the exchange of GDP for GTP and trigger the activation of MAP kinase cascade, resulting in cell proliferation, differentiation, migration, invasion, and survival.
p66Shc
p66Shc is also phosphorylated at its tyrosine residues as p52Shc and p46Shc upon growth factor stimulation, e.g., EGF treatment, and forms complexes with Grb2. However, there are certain functional differences between p66Shc and the other two Shc members. Unlike p52Shc, p66Shc is unable to transform NIH3T3 mouse fibroblasts in culture (Migliaccio et al. 1999) and it could not augment EGF-induced extracellular signal-regulated kinases/mitogen-activated protein kinases (ERK/MAPK) activation in cell cultures such as HeLa, CHO, and COS-1 cells. One possible explanation is that increased expression of p66Shc has resulted in an elevated level of the basal activity of ERK/MAPK in the absence of stimulus, which thus minimizes the extent of further activation by growth factors (Migliaccio et al. 1997, Okada et al. 1997, Veeramani et al. 2005b). Recently, the task of p66Shc in mediating stress-induced apoptosis has received much attention (Migliaccio et al. 1999, Orsini et al. 2004, Giorgio et al. 2005). Once p66Shc is phosphorylated at Ser-36 in its CH2 domain in response to various stress factors, such as H2O2, UV radiation, and chemicals, e.g., Taxol, a fraction of cytosolic p66Shc associates with heat-shock proteins to mediate apoptotic response (Fig. 3; Orsini et al. 2004) and serves as an apoptotic sensitizer to those signals (Migliaccio et al. 1999). p66Shc also acts as a negative regulator of human and mouse T-cell survival and proliferation (Pacini et al. 2004). In parallel, p66Shc knockout mice exhibit a prolonged life span by 30% and those mouse embryo fibroblast (MEF) cells have increased resistance to oxidative and hypoxic stress (Migliaccio et al. 1999, Trinei et al. 2002, Zaccagnini et al. 2004). Thus, p66Shc may function as a longevity gene in mammals. Interestingly, p66Shc expression level in human dermal fibroblasts increases with aging, opposite to the knockout mouse model (Pandolfi et al. 2005). Collectively, these data indicate that p66Shc could function as a sensor and transduce signals in response to cellular stress while more studies are required for its role in human longevity.
Figure 3.
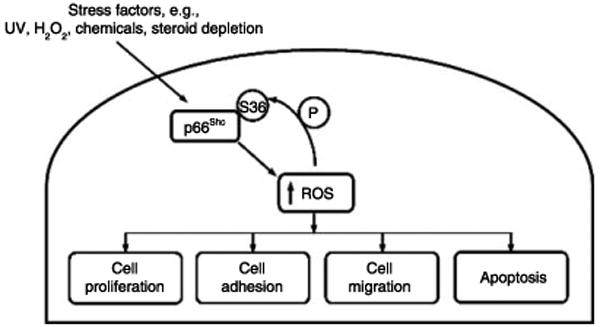
p66Shc acting as a stress sensor and increasing intracellular ROS level. Increased ROS induces the phosphorylation of serine 36 at p66Shc protein that promotes the generation of more ROS, leading to cell proliferation, adhesion, migration, or apoptosis.
Several lines of evidence suggest that aberrant expression of p66Shc could be involved in various stages of carcinogenesis (Jackson et al. 2000, Luzi et al. 2000, Ravichandran 2001, Davol et al. 2003, Lee et al. 2004b, De et al. 2005, Grossman et al. 2007). However, the role and the molecular mechanisms of p66Shc in this mode of regulation remain to be elucidated. In this review, we focus on discussing a novel functional role of p66Shc adaptor protein involved in steroid-related carcinogenesis, leading to its metastasis. This member of Shc protein family may serve as a new target for preventing tumor progression and metastasis.
Role of Shc proteins in steroid-regulated tumor progression and metastasis
Tumor progression and metastasis are the features of cancer. Cell proliferation, migration, and adhesion to the target tissues are the critical steps that allow tumor cells to obtain the metastatic phenotype. The process of metastasis requires the interaction of malignant cells with at least three distinct microenvironments, including the primary organ, the circulation or lymphatic channels, and the target organ where a metastatic lesion will develop (Radinsky & Fidler 1992, Mundy 1997, Cooper et al. 2003). Within these microenvironments, several factors are involved in the metastatic cascade (Gopalkrishnan et al. 2001). Tumor cells, after reaching to target organs and tissues, establish as successful foci in the conducible environments. Subsequently, tumor cells proliferate in the new, supportive microenvironment as micrometastasis where they induce angiogenesis for maintaining the growth of new lesion. Induction of angiogenesis is necessary for successful metastasis to meet the nutrient requirements when the tumor size becomes more than 2 mm in size (Ellis & Fidler 1996, Gopalkrishnan et al. 2001). Cell proliferation and migration depend on intracellular signals transmitted by the growth factors and adhesion proteins within the extracellular matrix (Pages et al. 1993). Both these processes employ many common intracellular signaling molecules, e.g., Rho family proteins and ERK cascades (Pages et al. 1993, Olson et al. 1995, Anand-Apte et al. 1997, Klemke et al. 1997). It is evident now that Shc isoforms, specifically p52Shc and p66Shc, play a crucial role in cell migration and adhesion, in addition to their roles in mediating cell proliferation induced by growth factor receptor signaling (Fig. 4; Klemke et al. 1994, Huttenlocher et al. 1995). These processes involve the rearrangement of actin cytoskeleton, the formation of new integrin substratum contacts, cell contraction, and the release of pre-existing cell-matrix contacts (Lauffenburger & Horwitz 1996). We will thus discuss the role of p52Shc and p66Shc in these processes and emphasize on steroid regulation.
Figure 4.
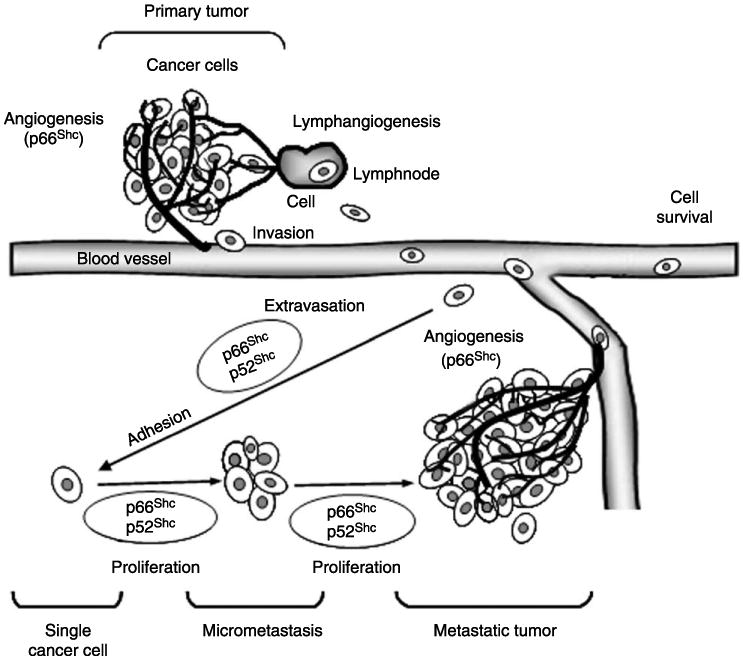
Involvement of p66Shc and p52Shc in tumor metastasis. Growing primary tumors attract new blood vessels, i.e., angiogenesis, and lymphatic vessels, i.e., lymphangiogenesis, to promote local tumor growth, involvement of regional lymph nodes, and finally distant metastasis. In the process of metastasis after degrading or remodeling the basement membrane, metastatic cells detach from the primary tumor mass, intravasate, survive the stress of vascular transportation, and then evade host defense mechanism, which are in part regulated by the adaptor proteins p52Shc and p66Shc. Furthermore, targeting via microvessels and cell adhesion molecules, the increased phosphorylation of p52Shc and p66Shc proteins mediate the proliferation of tumor cells that metastasize to their preferred sites after extravasation into the target organ parenchyma, and are permitted to reside in the target tissue in which cancer cells respond to transendothelial growth factors from that specific organ. (The figure is adapted from La Porta 2000).
Role of p52Shc in steroid-regulated cell proliferation and migration
In addition to the classical role in mediating tyrosine kinase-activated pathways, p52Shc functions as a primary adaptor protein for mediating the mitogenic signals of steroids at the non-genomic level in human breast and prostate cancer cells (Stevenson et al. 1999, Lee et al. 2004a). In prostate cancer, the data clearly showed that p52Shc is responsible for transducing androgen-activated ErbB-2 signaling, which leads to prostate cancer cell proliferation (Lee et al. 2004a). This androgen-stimulated cell proliferation requires Y317 phosphorylation since p52Shc Y317F mutant effectively abolishes androgen stimulation, and that inhibition occurs at certain phases of the cell cycle (Lee et al. 2004a). This observation on the role of p52Shc in androgen action on prostate cancer cells is in parallel to that of p52Shc in estrogen action on breast cancer cells, correlating with ErbB-2 activity (Stevenson & Frackelton 1998). In addition, vitamin D treatment caused a significant decrease in LNCaP cell growth, which is closely associated with the reduction in ErbB-2 activity and its downstream signaling mediator p52Shc via dephosphorylating Y317, thereby emphasizing the involvement of tyrosine phosphorylation of p52Shc in human prostate cancer cell proliferation (Stewart et al. 2005). Furthermore, the p52Shc Y317F mutant blocks estrogen-induced cell cycle progression at both the G0–G1 and G2–M junctions (Stevenson et al. 1999). Additionally, in the presence of steroids, ER α-transfected HeLa cells exhibit chemoresistance, where p52Shc mediates the anti-apoptotic activity of steroids and thus prevents those cells from etoposide-induced apoptosis (Kousteni et al. 2001). Clearly, p52Shc plays a critical role in mediating steroid action, at least in part, via tyrosine phosphorylation non-genomic signaling.
p52Shc may also be involved in the adhesion process of cancer cells. In rapidly adhering prostate cancer cells, e.g., LNCaP C-81 and PC-3 cells, the phosphorylation level of p52Shc protein at Y317 correlates with ErbB-2 activation, higher than in slow-adhering LNCaP C-33 cells (Lee et al. 2004a, Yuan et al. 2007). Yet, both the adhesion rate and the Y317 phosphorylation of p52Shc as well as ErbB-2 tyrosine phosphorylation level in slow-adhering LNCaP C-33 cells can be up-regulated by steroids. Thus, by correlating tyrosine phosphorylation and adhesion, p52Shc may play a crucial role in the adhesion of cancer cells during steroid hormone-induced metastasis. Furthermore, a direct involvement of p52Shc in breast cancer metastasis in transgenic mice that express polyomavirus middle T antigen with a mutated Shc-binding site has been demonstrated (Webster et al. 1998). Polyomavirus middle T antigen couples with and activates signaling molecules, such as Src, Shc, and phosphatidylinositol 3′-kinase (PI3K) for its oncogenic capacity. Importantly, in transgenic mice, which have metastatic tumors, the mutated p52Shc-binding site on middle T antigen had reverted to the wild type and regained its function, thus emphasizing the potential importance of the functional p52Shc in the process of metastasis in vivo (Webster et al. 1998). In addition, it has been revealed that in integrin signaling, Shc recruitment to the actin-associated cytoskeleton is important (McGlade et al. 1992, Schlaepfer et al. 1998, Wary et al. 1998). p52Shc potentiates integrin signaling, and integrin ligation results in the activation of non-receptor tyrosine kinases, such as Src, Fyn, and focal adhesion kinase (FAK), which phosphorylates p52Shc, leading to Ras activation and entering into the cell cycle (McGlade et al. 1992, Mainiero et al. 1995, Wary et al. 1996, 1998). Besides, the SH3 domain of Fyn interacts with the proline-rich region in the CH1 domain of p52Shc (Thomas & Bradshaw 1997) and the amino-terminal domain of p52Shc is shown to mediate the association of this adaptor protein to an actin-rich cellular fraction (Thomas & Bradshaw 1997). Additionally, a mutation of the PTB domain (S154P-p52Shc) abolishes integrin-induced p52Shc tyrosine phosphorylation where the SH2 domain of p52Shc is dispensable (Collins et al. 1999). p52Shc phosphorylation by c-Src can be augmented when the PTB domain binds to phospholipids (Zhou et al. 1995, Sato et al. 1997). These observations explain how the PTB domain localizes p52Shc to the membrane where it becomes phosphorylated by cytoskeleton-associated tyrosine kinases, which finally results in cell migration. It should be noted that these non-receptor tyrosine kinases, e.g., Src, closely interact with steroid hormone signaling pathway (Migliaccio et al. 2000, Guo et al. 2006). The molecular mechanism by which steroids induce cell adhesion and/or migration via p52Shc requires further investigation.
Role of p66Shc in steroid-regulated cell proliferation
In tyrosine phosphorylation signal transduction pathway, p66Shc conventionally known as an adaptor protein. Interestingly, p66Shc expression closely correlates with the growth rate of prostate cancer cells. For example, p66Shc protein levels in the rapidly growing cells, e.g., PC-3 and DU145, are approximately 4- to 13-fold higher than that in the slow-growing LNCaP C-33 cells and are over tenfold higher than that in even slower MDA PCa2b cells (Veeramani et al. 2005b). Recent data indicate that p66Shc may play a critical role in mediating steroid-stimulated cell proliferation. In the presence of steroid hormones (androgen and estrogen), p66Shc protein level as well as cell proliferation rate is increased in hormone-sensitive human prostate (LNCaP C-33 and MDA PCa2b), testicular (Tera-1 and Tera-2), and breast (MCF-7) cancer cells, higher than those cells cultured in the absence of steroids (Lee et al. 2004b). Thus, steroids increase p66Shc protein level and concurrently cell growth.
To elucidate directly the functional role of p66Shc protein in steroid-regulated cells, both cDNA and siRNA approaches were employed (Veeramani et al. 2005b). In both p66Shc cDNA, transiently transfected cell population and stable subclones of slow-growing LNCaP C-33 cells, elevated expression of p66Shc correlates with increased cell proliferation. On the contrary, a decreased cell growth rate is observed when p66Shc protein is knocked down by its siRNA in the rapidly growing LNCaP C-81 and PC-3 cells. The data clearly establish the causal relationship of p66Shc protein and cell growth. Furthermore, p66Shc mediates growth stimulation by androgens (Veeramani et al. 2008). The clinical relevance of these data is supported by the observations that in prostate cancer archival specimens, p66Shc protein level is significantly higher in prostate adenocarcinomatous cells than in adjacent benign glandular cells (Lee et al. 2004b). Similarly, the expression level of p66Shc is elevated in metastatic breast, ovarian, and thyroid tumors and may serve as a useful prognostic marker for stage-IIA colon cancer (Jackson et al. 2000, Abdollahi et al. 2003, Park et al. 2005, Grossman et al. 2007). Nevertheless, some studies showed that p66Shc protein is down-regulated in the primary tumors of breast cancers (Davol et al. 2003). Due to the potential importance of p66Shc in carcinogenesis, further studies are needed to clarify the correlation of p66Shc expression with breast cancer. In summary, cross-talks between tyrosine phosphorylation signaling and steroid hormones have been well established (Weigel 1996, Meng et al. 2000, Grossmann et al. 2001, Guo et al. 2006, Kraus et al. 2006, Migliaccio et al. 2006, Weigel & Moore 2007); it is thus reasonable to propose that p66Shc mediates steroid action in steroid-responsive epithelial cells. We therefore hypothesize that the elevated level of p66Shc protein in steroid-related cancer cells plays a critical role in up-regulating those cancer cell proliferation and thus contributes to the tumorigenicity of those cancers (Fig. 2). The role of p66Shc in this mode of regulation requires further investigation.
Role of p66Shc in metastasis
Several studies have shown the involvement of p66Shc in cellular invasion, motility, migration, and/or metastasis. Jackson et al. (2000) have shown that xenograft bone metastasis of breast cancer cell line, MDA-MB-231, expresses p66Shc and its metastatic variant F-11 cells have a threefold higher p66Shc expression level. Furthermore, increased expression of p66Shc in lymph node-positive breast cancers correlates with an increased number of positive lymph nodes (Jackson et al. 2000). As higher levels of p66Shc protein are observed in breast cancer specimens with higher metastatic potential, it suggests the possibility that p66Shc influences cell motility and invasion other than the MAPK pathway (Jackson et al. 2000). Furthermore, Northey et al. (2008) have demonstrated that decrease in the ShcA levels or the expression of a dominant-negative ShcA mutant blocked TGF-β-induced motility and the invasion of Neu/ErbB-2-expressing breast cancer cells, thus exploiting the crucial role of p66Shc in the migration and invasion of cancer cells. In addition, elevated expression of p66Shc by cDNA transfection in LNCaP C-33 cells is associated with increased motility and invasion (Yuan TC, Lin FF & Lin MF, unpublished data). In parallel, LNCaP C-81 and PC-3 prostate cancer cells express higher levels of p66Shc and exhibit higher metastatic potential than LNCaP C-33 cells in xenograft animals (Veeramani et al. 2005b, Sebeger J & Lin MF, unpublished observation). The observations on the increased expression of p66Shc, but not p52Shc or p46Shc, in cell lines with higher metastatic ability and in the node-positive primary breast cancers also imply that p66Shc functions in metastatic pathway.
Integrins play a vital role in cancer progression because of their ability to regulate various intracellular signaling molecules that are essential for cell motility, cell survival, and proliferation (Ruoslahti & Reed 1994, Hynes et al. 1999). It has been suggested that αvβ3 integrin plays a critical role in the metastasis of cancer cells to bone marrow (Cooper et al. 2002). αvβ3 integrin is expressed in breast and lung cancer cells that were originally derived from the bone marrow aspirates. αvβ3 is also expressed in highly tumorigenic, bone foci-derived human PC-3 prostate cancer cells, but not in low tumorigenic, lymph node foci-derived LNCaP cells (Zheng et al. 1999). The results of recent studies suggest that activated αvβ3 integrin regulates tumor growth in vivo by influencing VEGF expression. The up-regulation of VEGF expression depends on αvβ3 clustering where it promotes the recruitment of p66Shc and subsequently the phosphorylation of β3-associated p66Shc. Phosphorylation of p66Shc is a critical step for αvβ3-mediated potentiation of VEGF expression and tumor vascularization in vivo (De et al. 2005). These findings provide insights into the role of αvβ3 and p66Shc interaction as a regulator of tumor metastasis and angiogenesis. Thus, down-regulation of p66Shc inhibits VEGF expression as well as the tumor growth and angiogenesis in vivo (De et al. 2005). Although the molecular mechanism of p66Shc involvement in metastasis requires further investigations, it is hypothesized that p66Shc is involved in an early step of invasion or during cell motility (Jackson et al. 2000) and plays a role in steroid-regulated metastatic process.
Molecular mechanisms of p66Shc-mediated steroid action
In determining the prostate origin of metastatic cancers, cellular prostatic acid phosphatase (cPAcP) has been used as a biomarker, due to its cell-specific expression (Sakai et al. 1992, Chu & Lin 1998). The results of several studies collectively indicate that cPAcP exhibit the growth inhibitory activity by functioning as a PTPase (Lin & Meng 1996, Lin et al. 2001, Veeramani et al. 2005a). In parallel, the expression level of cPAcP negatively correlates with prostatic carcinogenesis, i.e., the level of cPAcP decreases in prostate cancer cells, lower than that in the adjacent non-cancerous cells (Reif et al. 1973, Foti et al. 1977, Loor et al. 1981, Chu & Lin 1998, Lin et al. 2001). Interestingly, in prostate cancer cells, the level of p66Shc protein shows an inverse correlation with cPAcP expression and has a positive correlation with Erb2 as well as ERK/MAPK activation (Veeramani et al. 2005b). In cPAcP cDNA-transfected stable subclone cells, p66Shc protein level is decreased and ErbB-2 as well as ERK/MAPK activity is diminished, correlating with decreased cell proliferation (Veeramani et al. 2005b). Conversely, elevated p66Shc protein level as well as ErbB-2 and ERK/MAPK activation is observed in cPAcP-inhibited LNCaP C-33 cells by inhibitors (Veeramani et al. 2005b). Similarly, in breast cancer cells, elevated expression of p66Shc protein correlates with ErbB-2 and/or MAPK1/MAPK activation (Stevenson & Frackelton 1998, Lee et al. 2004b). In addition, the up-regulation of p66Shc is shown in human ovarian, oral, and lung cancer cells expressing increased levels of ErbB-2 (Xie & Hung 1996). While the molecular mechanism of this inverse relationship between cPAcP and p66Shc protein remains to investigated further, it should be noted that the inverse correlation of cPAcP with p66Shc as well as ERK/MAPK activation is clinically relevant (Loor et al. 1981, Pontes et al. 1981, Solin et al. 1990, Sakai et al. 1993, Gioeli et al. 1999, Price et al. 1999, Lin et al. 2001, Lee et al. 2004b). Despite the fact that cPAcP may serve as a good prognostic marker for metastatic prostate cancers (Sakai et al. 1993), because of decreased cPAcP level upon tumor progression as well as increased p66Shc protein level in PCa cells, we contemplate that the p66Shc/cPAcP ratio may serve as a competitive surrogate biomarker for predicting the prognosis of advanced prostate carcinoma.
Furthermore, p66Shc may mediate steroid-stimulated cell proliferation via a non-genomic signaling pathway. In the rapidly growing cells, including steroid-stimulated cells, increased oxidative stress by the generation of reactive oxygen species (ROS) might contribute to the elevated p66Shc protein level. p66Shc protein can function as a stress sensor and is involved in regulating the intracellular level of ROS (Trinei et al. 2002). Activated metabolic reactions in the rapidly growing cells lead to increased production of ROS (Klaunig & Kamendulis 2004), which may in turn increase p66Shc protein levels to mediate oxidative stress signals (Fig. 3). Evidently, upon H2O2 treatment, in MEF cells or DLD-1 colorectal cancer cells, the expression level of p66Shc protein is increased (Trinei et al. 2002, Pacini et al. 2004). Additionally, ROS-induced phosphorylation of p66Shc protein at Ser-36 residue further promotes the generation of ROS (Nemoto & Finkel 2002, Orsini et al. 2004). Notably, recent studies reveal that p66Shc protein exhibits endogenous oxidase activity and its amino terminus contains a redox center, which is involved in electron transfer from cytochrome c to molecular oxygen and produces H2O2. This H2O2 mediates the opening of transition pores resulting in increased mitochondrial permeability and thus an abnormal high level of H2O2 can lead to apoptosis (Giorgio et al. 2005). Nevertheless, physiological levels of H2O2 function as growth stimuli. In addition to the above findings, p66Shc may also increase H2O2 production through Rac1-SOS-specific pathway (Khanday et al. 2006), possibly leading to androgen-independent cell proliferation (Knight-Krajewski et al. 2004). ROS therefore mediates diverse biological functions, including cell proliferation, cell adhesion, migration, and apoptosis (Fig. 3).
In prostate cancer archival specimens, the ROS level is higher in cancerous cells than in non-cancerous cells, correlating with the proliferation index (Lim et al. 2005). The functional role of ROS as a positive regulator of cell growth, including prostate cancer cells, is apparent in part by inhibiting the PTPase activity, and thus the corresponding RPTK can be activated (Finkel & Holbrook 2000, Liu et al. 2002, Lou et al. 2008, Veeramani et al. 2008). Furthermore, steroid hormones, e.g., androgens and estrogens, and growth factors, such as EGF, can up-regulate ROS production in cells, as such; cell proliferation is promoted (Sundaresan et al. 1996, Liu et al. 2002). This is consistent with our findings that androgenic treatment of LNCaP C-33 cells promotes cell proliferation via decreasing cellular PAcP and increasing p66Shc protein level and ROS production as well as ErbB-2 tyrosine phosphorylation (Meng et al. 2000, Veeramani et al. 2008). Therefore, an inverse correlation of p66Shc and cPAcP in prostate cancer cell is observed. Additionally, it has been demonstrated that ROS may play a critical role in the initiation and/or early progression of prostate cancer; as such, antioxidants are used in clinical trials for this cancer prevention (Veeramani & Lin 2007). Collectively, the data indicate that p66Shc can mediate non-genomic steroid action on cell proliferation and carcinogenesis including metastasis, while the molecular mechanisms require further investigations.
Conclusion and perspective
To discover the novel therapeutic targets for the prevention of tumor progression and metastasis, identifying the functional molecules that are involved in enhancing or suppressing these processes is one of the major challenges. The metastatic process in carcinogenesis is regulated dynamically by numerous factors, including growth factors, hormones, and extracellular matrices. To establish a metastatic lesion, tumor cells must complete all the steps in metastatic processes. Several studies have already shown that Shc proteins, including p52Shc and p66Shc, have marked effects on cell proliferation, invasion, and migration. In this review, we discuss a novel role of p52Shc and p66Shc in mediating steroid action on tumor proliferation and metastasis. We further present a novel non-genomic mechanism by which steroid signaling via p52Shc and p66Shc induces cancer cell proliferation, survival, migration and ultimately metastasis (Fig. 5). Nevertheless, the role of other key players engaged with p52Shc and/or p66Shc signaling pathway has yet to be identified. Thus, the molecular mechanisms by which p52Shc and p66Shc mediate steroid hormone-induced carcinogenesis require further investigations. Understanding the role of Shc adaptor proteins in cancer biology including the determination of the upstream regulators and downstream effectors of Shc functional pathways may lead to the development of novel anti-tumor strategies for targeting against steroid-induced epithelial cancers.
Figure 5.
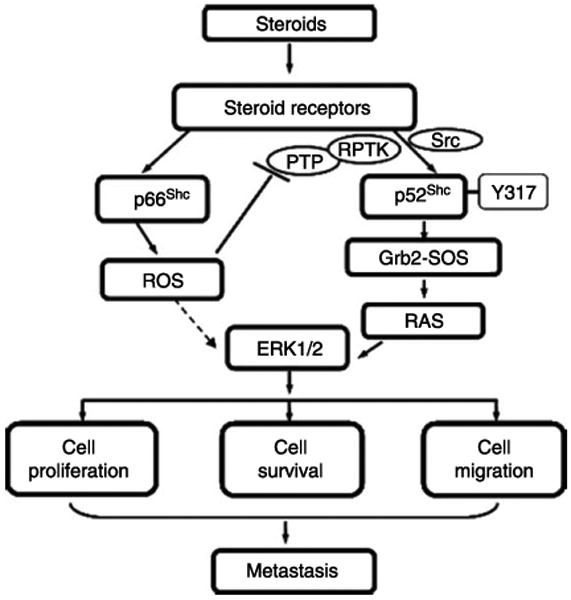
A proposed scheme of steroid-regulated cancer progression via Shc proteins. Upon steroid activation, p52Shc undergoes phosphorylation at Y317 and promotes tumor growth at least in part via transducing signals through the Grb2–Ras–MAPK pathway. Like p52Shc, p66Shc protein also promotes tumor progression, nevertheless, via mediating oxidative stress signals through the generation of ROS. Upon stimulation by steroids, p66Shc is translocated to mitochondria via Ser-36 phosphorylation-independent manner resulting in the generation of ROS. ROS may then inhibit PTP, resulting in RPTK activation, ERK/MAPK activation, and promote cell proliferation, survival, and migration, which collectively lead to metastasis.
Acknowledgments
We thank Ms Fen-Fen Lin for her tremendous help in our studies on Shc proteins.
Funding
This study was supported in part by the National Cancer Institute, National Institutes of Health (R01 CA88184), Department of Defense (W81XWH-06-1-0070 and W8lXWH-08-1-0459), Nebraska Research Initiative for Cancer Glycobiology, and Nebraska Cancer and Smoking Disease Research Program LB 506 (2008-20).
Footnotes
Declaration of interest
All authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
References
- Abdollahi A, Gruver BN, Patriotis C, Hamilton TC. Identification of epidermal growth factor-responsive genes in normal rat ovarian surface epithelial cells. Biochemical and Biophysical Research Communications. 2003;18:188–197. doi: 10.1016/s0006-291x(03)01140-9. [DOI] [PubMed] [Google Scholar]
- Alzamora R, Harvey BJ. Direct binding and activation of protein kinase C isoforms by steroid hormones. Steroids. 2008;73:885–888. doi: 10.1016/j.steroids.2008.01.001. [DOI] [PubMed] [Google Scholar]
- Anand-Apte B, Zetter BR, Viswanathan A, Qiu RG, Chen J, Ruggieri R, Symons M. Platelet-derived growth factor and fibronectin-stimulated migration are differentially regulated by the Rac and extracellular signal-regulated kinase pathways. Journal of Biological Chemistry. 1997;272:30688–30692. doi: 10.1074/jbc.272.49.30688. [DOI] [PubMed] [Google Scholar]
- Armen TA, Gay CV. Simultaneous detection and functional response of testosterone and estradiol receptors in osteoblast plasma membranes. Journal of Cellular Biochemistry. 2000;79:620–627. [PubMed] [Google Scholar]
- Benten WP, Lieberherr M, Giese G, Wunderlich F. Estradiol binding to cell surface raises cytosolic free calcium in T cells. FEBS Letters. 1998;422:349–353. doi: 10.1016/s0014-5793(98)00039-8. [DOI] [PubMed] [Google Scholar]
- Benten WP, Lieberherr M, Giese G, Wrehlke C, Stamm O, Sekeris CE, Mossmann H, Wunderlich F. Functional testosterone receptors in plasma membranes of T cells. FEBS Journal. 1999a;13:123–133. doi: 10.1096/fasebj.13.1.123. [DOI] [PubMed] [Google Scholar]
- Benten WP, Lieberherr M, Stamm O, Wrehlke C, Guo Z, Wunderlich F. Testosterone signaling through internalizable surface receptors in androgen receptor-free macrophages. Molecular Biology of the Cell. 1999b;10:3113–3123. doi: 10.1091/mbc.10.10.3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ, Bootman MD, Lipp P. Calcium – a life and death signal. Nature. 1998;395:645–648. doi: 10.1038/27094. [DOI] [PubMed] [Google Scholar]
- Van Bömmel T, Marsen T, Bojar H. Effects of high-dose medroxyprogesterone acetate and various other steroid hormones on plasma membrane lipid mobility in CAMA-1 mammary cancer cells. Anticancer Research. 1987;7:1217–1223. [PubMed] [Google Scholar]
- Bonfini L, Migliaccio E, Pelicci G, Lanfrancone L, Pelicci PG. Not all Shc's roads lead to Ras. Trends in Biochemical Sciences. 1996;21:257–261. [PubMed] [Google Scholar]
- Choudhury GG, Karamitsos C, Hernandez J, Gentilini A, Bardgette J, Abboud HE. PI-3-kinase and MAPK regulate mesangial cell proliferation and migration in response to PDGF. American Journal of Physiology. 1997;273:F931–938. doi: 10.1152/ajprenal.1997.273.6.F931. [DOI] [PubMed] [Google Scholar]
- Chu TM, Lin MF. PSA and acid phosphatase in the diagnosis of prostate cancer. Journal of Clinical Ligand Assay. 1998;21:24–34. [Google Scholar]
- Collins LR, Ricketts WA, Yeh L, Cheresh D. Bifurcation of cell migratory and proliferative signaling by the adaptor protein Shc. Journal of Cell Biology. 1999;147:1561–1568. doi: 10.1083/jcb.147.7.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper CR, Chay CH, Pienta KJ. The role of alpha(v)beta(3) in prostate cancer progression. Neoplasia. 2002;4:191–194. doi: 10.1038/sj.neo.7900224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper CR, Chay CH, Gendernalik JD, Lee HL, Bhatia J, Taichman RS, McCauley LK, Keller ET, Pienta KJ. Stromal factors involved in prostate carcinoma metastasis to bone. Cancer. 2003;97:739–747. doi: 10.1002/cncr.11181. [DOI] [PubMed] [Google Scholar]
- Dabrosin C, Ollinger K, Ungerstedt U, Hammar M. Variability of glutathione levels in normal breast tissue and subcutaneous fat during the menstrual cycle: an in vivo study with microdialysis technique. Journal of Clinical Endocrinology and Metabolism. 1997;82:1382–1384. doi: 10.1210/jcem.82.5.3957. [DOI] [PubMed] [Google Scholar]
- Davol PA, Bagdasaryan R, Elfenbein GJ, Maizel AL, Frackelton AR., Jr Shc proteins are strong, independent prognostic markers for both node negative and node positive primary breast cancer. Cancer Research. 2003;63:6772–6783. [PubMed] [Google Scholar]
- De S, Razorenova O, McCabe NP, O'Toole T, Qin J, Byzova TV. VEGF-integrin interplay controls tumor growth and vascularization. PNAS. 2005;102:7589–7594. doi: 10.1073/pnas.0502935102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devanesan P, Santen RJ, Bocchinfuso WP, Korach KS, Rogan EG, Cavalieri E. Catechol estrogen metabolites and conjugates in mammary tumors and hyperplastic tissue from estrogen receptor-alpha knockout (ERKO)/Wnt-1 mice: implications for initiation of mammary tumors. Carcinogenesis. 2001;22:1573–1576. doi: 10.1093/carcin/22.9.1573. [DOI] [PubMed] [Google Scholar]
- Dickson RB, Lippman ME. Estrogenic regulation of growth and polypeptide growth factor secretion in human breast carcinoma. Endocrine Reviews. 1987;8:29–43. doi: 10.1210/edrv-8-1-29. [DOI] [PubMed] [Google Scholar]
- Duval D, Durant S, Homo-Delarche F. Non-genomic effects of steroids. Interactions of steroid molecules with membrane structures and functions. Biochimica et Biophysica Acta. 1983;737:409–442. doi: 10.1016/0304-4157(83)90008-4. [DOI] [PubMed] [Google Scholar]
- Ellis LM, Fidler IJ. Angiogenesis and metastasis. European Journal of Cancer. 1996;32A:2451–2460. doi: 10.1016/s0959-8049(96)00389-9. [DOI] [PubMed] [Google Scholar]
- Engebraaten O, Bjerkvig R, Pedersen PH, Laerum OD. Effects of EGF, bFGF, NGF, and PDGF (bb) on cell proliferative, migratory, and invasive capacities of human brain-tumour biopsies in vitro. International Journal of Cancer. 1993;53:209–214. doi: 10.1002/ijc.2910530206. [DOI] [PubMed] [Google Scholar]
- Er F, Michels G, Gassanov N, Rivero F, Hoppe UC. Testosterone induces cytoprotection by activating ATP-sensitive K+ channels in the cardiac mitochondrial inner membrane. Circulation. 2004;110:3100–3107. doi: 10.1161/01.CIR.0000146900.84943.E0. [DOI] [PubMed] [Google Scholar]
- Er F, Michels G, Brandt MC, Khan I, Haase H, Eicks M, Lindner M, Hoppe UC. Impact of testosterone on cardiac L-type calcium channels and Ca2+ sparks: acute actions antagonize chronic effects. Cell Calcium. 2007;41:467–477. doi: 10.1016/j.ceca.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Estrada M, Espinosa A, Muller M, Jaimovich E. Testosterone stimulates intracellular calcium release and mitogen-activated protein kinases via a G protein-coupled receptor in skeletal muscle cells. Endocrinology. 2003;144:3586–3597. doi: 10.1210/en.2002-0164. [DOI] [PubMed] [Google Scholar]
- Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;9:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- Fortunati N. Sex hormone-binding globulin: not only a transport protein. What news is around the corner? Journal of Endocrinological Investigation. 1999;22:223–234. doi: 10.1007/BF03343547. [DOI] [PubMed] [Google Scholar]
- Fortunati N, Fissore F, Fazzari A, Becchis M, Comba A, Catalano MG, Berta L, Frairia R. Sex steroid binding protein exerts a negative control on estradiol action in MCF-7 cells (human breast cancer) through cyclic adenosine 30,50-monophosphate and protein kinase A. Endocrinology. 1996;137:686–692. doi: 10.1210/endo.137.2.8593818. [DOI] [PubMed] [Google Scholar]
- Foti AG, Cooper JF, Herschman H, Malvaez RR. Detection of prostatic cancer by solid-phase radioimmunoassay of serum prostatic acid phosphatase. New England Journal of Medicine. 1977;297:1357–1361. doi: 10.1056/NEJM197712222972501. [DOI] [PubMed] [Google Scholar]
- van der Geer P, Wiley S, Gish GD, Pawson T. The Shc adaptor protein is highly phosphorylated at conserved, twin tyrosine residues (Y239/240) that mediate protein-protein interactions. Current Biology. 1996;6:1435–1444. doi: 10.1016/s0960-9822(96)00748-8. [DOI] [PubMed] [Google Scholar]
- Gioeli D, Mandell JW, Petroni GR, Frierson HF, Jr, Weber MJ. Activation of mitogen-activated protein kinase associated with prostate cancer progression. Cancer Research. 1999;59:279–284. [PubMed] [Google Scholar]
- Giorgio M, Migliaccio E, Orsini F, Paolucci D, Moroni M, Contursi C, Pelliccia G, Luzi L, Minucci S, Marcaccio M, et al. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell. 2005;122:221–233. doi: 10.1016/j.cell.2005.05.011. [DOI] [PubMed] [Google Scholar]
- Gopalkrishnan RV, Kang DC, Fisher PB. Molecular markers and determinants of prostate cancer metastasis. Journal of Cellular Physiology. 2001;189:245–256. doi: 10.1002/jcp.10023. [DOI] [PubMed] [Google Scholar]
- Gotoh N, Tojo A, Shibuya M. A novel pathway from phosphorylation of tyrosine residues 239/240 of Shc, contributing to suppress apoptosis by IL-3. EMBO Journal. 1996;15:6197–6204. [PMC free article] [PubMed] [Google Scholar]
- Grossman SR, Lyle S, Resnick MB, Sabo E, Lis RT, Rosinha E, Liu Q, Hsieh CC, Bhat G, Frackelton AR, Jr, et al. p66 Shc tumor levels show a strong prognostic correlation with disease outcome in stage IIA colon cancer. Clinical Cancer Research. 2007;13:5798–5804. doi: 10.1158/1078-0432.CCR-07-0073. [DOI] [PubMed] [Google Scholar]
- Grossmann ME, Huang H, Tindall DJ. Androgen receptor signaling in androgen-refractory prostate cancer. Journal of the National Cancer Institute. 2001;93:1687–1697. doi: 10.1093/jnci/93.22.1687. [DOI] [PubMed] [Google Scholar]
- Guo Z, Dai B, Jiang T, Xu K, Xie Y, Kim O, Nesheiwat I, Kong X, Melamed J, Handratta VD, et al. Regulation of androgen receptor activity by tyrosine Phosphorylation. Cancer Cell. 2006;10:309–319. doi: 10.1016/j.ccr.2006.08.021. [DOI] [PubMed] [Google Scholar]
- Hamada J, Nagayasu H, Takayama M, Kawano T, Hosokawa M, Takeichi N. Enhanced effect of epidermal growth factor on pulmonary metastasis and in vitro invasion of rat mammary carcinoma cells. Cancer Letters. 1995;89:161–167. doi: 10.1016/0304-3835(95)03686-q. [DOI] [PubMed] [Google Scholar]
- Hammes SR, Levin ER. Extranuclear steroid receptors: nature and actions. Endocrine Reviews. 2007;28:726–741. doi: 10.1210/er.2007-0022. [DOI] [PubMed] [Google Scholar]
- Harvey BJ, Doolan CM, Condliffe SB, Renard C, Alzamora R, Urbach V. Non-genomic convergent and divergent signalling of rapid responses to aldosterone and estradiol in mammalian colon. Steroids. 2002;67:483–491. doi: 10.1016/s0039-128x(01)00169-6. [DOI] [PubMed] [Google Scholar]
- Henderson BE, Feigelson HS. Hormonal carcinogenesis. Carcinogenesis. 2000;21:427–433. doi: 10.1093/carcin/21.3.427. [DOI] [PubMed] [Google Scholar]
- Huttenlocher A, Sandborg RR, Horwitz AF. Adhesion in cell migration. Current Opinion in Cell Biology. 1995;7:697–706. doi: 10.1016/0955-0674(95)80112-x. [DOI] [PubMed] [Google Scholar]
- Hynes RO, Bader BL, Hodivala-Dilke K. Integrins in vascular development. Brazilian Journal of Medical and Biological Research. 1999;32:501–510. doi: 10.1590/s0100-879x1999000500002. [DOI] [PubMed] [Google Scholar]
- Jackson JG, Yoneda T, Clark GM, Yee D. Elevated levels of p66 shc are found in breast cancer cell lines and primary tumors with high metastatic potential. Clinical Cancer Research. 2000;6:1135–1139. [PubMed] [Google Scholar]
- Jakacka M, Ito M, Weiss J, Chien PY, Gehm BD, Jameson JL. Estrogen receptor binding to DNA is not required for its activity through the nonclassical AP1 pathway. Journal of Biological Chemistry. 2001;276:13615–13621. doi: 10.1074/jbc.M008384200. [DOI] [PubMed] [Google Scholar]
- Kampa M, Papakonstanti EA, Hatzoglou A, Stathopoulos EN, Stournaras C, Castanas E. The human prostate cancer cell line LNCaP bears functional membrane testosterone receptors that increase PSA secretion and modify actin cytoskeleton. FEBS Journal. 2002;16:1429–1431. doi: 10.1096/fj.02-0131fje. [DOI] [PubMed] [Google Scholar]
- Kelly MJ, Lagrange AH, Wagner EJ, Rønnekleiv OK. Rapid effects of estrogen to modulate G protein-coupled receptors via activation of protein kinase A and protein kinase C pathways. Steroids. 1999;64:64–75. doi: 10.1016/s0039-128x(98)00095-6. [DOI] [PubMed] [Google Scholar]
- Khanday FA, Santhanam L, Kasuno K, Yamamori T, Naqvi A, Dericco J, Bugayenko A, Mattagajasingh I, Disanza A, Scita G, et al. Sos-mediated activation of rac1 by p66shc. Journal of Cell Biology. 2006;172:817–822. doi: 10.1083/jcb.200506001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HP, Lee JY, Jeong JK, Bae SW, Lee HK, Jo I. Nongenomic stimulation of nitric oxide release by estrogen is mediated by estrogen receptor alpha localized in caveolae. Biochemical and Biophysical Research Communications. 1999;263:257–262. doi: 10.1006/bbrc.1999.1348. [DOI] [PubMed] [Google Scholar]
- Klaunig JE, Kamendulis LM. The role of oxidative stress in carcinogenesis. Annual Review of Pharmacology and Toxicology. 2004;44:239–267. doi: 10.1146/annurev.pharmtox.44.101802.121851. [DOI] [PubMed] [Google Scholar]
- Klemke RL, Yebra M, Bayna EM, Cheresh DA. Receptor tyrosine kinase signaling required for integrin alpha v beta 5-directed cell motility but not adhesion on vitronectin. Journal of Cell Biology. 1994;127:859–866. doi: 10.1083/jcb.127.3.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemke RL, Cai S, Giannini AL, Gallagher PJ, de Lanerolle P, Cheresh DA. Regulation of cell motility by mitogen-activated protein kinase. Journal of Cell Biology. 1997;137:481–492. doi: 10.1083/jcb.137.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight-Krajewski S, Welsh CF, Liu Y, Lyons LS, Faysal JM, Yang ES, Burnstein KL. Deregulation of the Rho GTPase, Rac1, suppresses cyclin-dependent kinase inhibitor p21(CIP1) levels in androgen-independent human prostate cancer cells. Oncogene. 2004;23:5513–5522. doi: 10.1038/sj.onc.1207708. [DOI] [PubMed] [Google Scholar]
- Kousteni S, Bellido T, Plotkin LI, O'Brien CA, Bodenner DL, Han L, Han K, DiGregorio GB, Katzenellenbogen JA, Katzenellenbogen BS, et al. Nongenotropic, sex-nonspecific signaling through the estrogen or androgen receptors: dissociation from transcriptional activity. Cell. 2001;104:719–730. [PubMed] [Google Scholar]
- Kraus S, Gioeli D, Vomastek T, Gordon V, Weber MJ. Receptor for activated C kinase 1 (RACK1) and Src regulate the tyrosine phosphorylation and function of the androgen receptor. Cancer Research. 2006;66:11047–11054. doi: 10.1158/0008-5472.CAN-06-0596. [DOI] [PubMed] [Google Scholar]
- Lauffenburger DA, Horwitz AF. Cell migration: a physically integrated molecular process. Cell. 1996;84:359–369. doi: 10.1016/s0092-8674(00)81280-5. [DOI] [PubMed] [Google Scholar]
- Lee MS, Igawa T, Lin MF. Tyrosine-317 of p52(Shc) mediates androgen-stimulated proliferation signals in human prostate cancer cells. Oncogene. 2004a;23:3048–3058. doi: 10.1038/sj.onc.1207451. [DOI] [PubMed] [Google Scholar]
- Lee MS, Igawa T, Chen SJ, Van Bemmel D, Lin JS, Lin FF, Johansson SL, Christman JK, Lin MF. p66Shc protein is upregulated by steroid hormones in hormone-sensitive cancer cells and in primary prostate carcinomas. International Journal of Cancer. 2004b;108:672–678. doi: 10.1002/ijc.11621. [DOI] [PubMed] [Google Scholar]
- Levin ER. Integration of the extranuclear and nuclear actions of estrogen. Molecular Endocrinology. 2005;19:1951–1959. doi: 10.1210/me.2004-0390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberherr M, Grosse B. Androgens increase intracellular calcium concentration and inositol 1,4,5-trisphosphate and diacylglycerol formation via a pertussis toxin-sensitive G-protein. Journal of Biological Chemistry. 1994;269:7217–7223. [PubMed] [Google Scholar]
- Lim SD, Sun C, Lambeth JD, Marshall F, Amin M, Chung L, Petros JA, Arnold RS. Increased Nox1 and hydrogen peroxide in prostate cancer. Prostate. 2005;62:200–207. doi: 10.1002/pros.20137. [DOI] [PubMed] [Google Scholar]
- Lin MF, Meng TC. Tyrosine phosphorylation of a 185 kDa phosphoprotein (pp185) inversely correlates with the cellular activity of human prostatic acid phosphatase. Biochemical and Biophysical Research Communications. 1996;226:206–213. doi: 10.1006/bbrc.1996.1334. [DOI] [PubMed] [Google Scholar]
- Lin MF, Lee MS, Zhou XW, Andressen JC, Meng TC, Johansson SL, West WW, Taylor RJ, Anderson JR, Lin FF. Decreased expression of cellular prostatic acid phosphatase increases tumorigenicity of human prostate cancer cells. Journal of Urology. 2001;166:1943–1950. [PubMed] [Google Scholar]
- Liu SL, Lin X, Shi DY, Cheng J, Wu CQ, Zhang YD. Reactive oxygen species stimulated human hepatoma cell proliferation via cross-talk between PI3-K/PKB and JNK signaling pathways. Archives of Biochemistry and Biophysics. 2002;406:173–182. doi: 10.1016/s0003-9861(02)00430-7. [DOI] [PubMed] [Google Scholar]
- Loor R, Wang MC, Valenzuela L, Chu TM. Expression of prostatic acid phosphatase in human prostate cancer. Cancer Letters. 1981;14:63–69. doi: 10.1016/0304-3835(81)90010-0. [DOI] [PubMed] [Google Scholar]
- Lotti LV, Lanfrancone L, Migliaccio E, Zompetta C, Pelicci G, Salcini AE, Falini B, Pelicci PG, Torrisi MR. Shc proteins are localized on endoplasmic reticulum membranes and are redistributed after tyrosine kinase receptor activation. Molecular and Cellular Biology. 1996;16:1946–1954. doi: 10.1128/mcb.16.5.1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou YW, Chen YY, Hsu SF, Chen RK, Lee CL, Khoo KH, Tonks NK, Meng TC. Redox regulation of the protein tyrosine phosphatase PTP1B in cancer cells. FEBS Journal. 2008;275:69–88. doi: 10.1111/j.1742-4658.2007.06173.x. [DOI] [PubMed] [Google Scholar]
- Lu ML, Schneider MC, Zheng Y, Zhang X, Richie JP. Caveolin-1 interacts with androgen receptor. A positive modulator of androgen receptor mediated transactivation. Journal of Biological Chemistry. 2001;276:13442–13451. doi: 10.1074/jbc.M006598200. [DOI] [PubMed] [Google Scholar]
- Lu Q, Pallas DC, Surks HK, Baur WE, Mendelsohn ME, Karas RH. Striatin assembles a membrane signaling complex necessary for rapid, nongenomic activation of endothelial NO synthase by estrogen receptor alpha. PNAS. 2004;101:17126–17131. doi: 10.1073/pnas.0407492101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luttrell LM, Hawes BE, van Biesen T, Luttrell DK, Lansing TJ, Lefkowitz RJ. Role of c-Src tyrosine kinase in G protein-coupled receptor- and Gbetagamma subunit-mediated activation of mitogen-activated protein kinases. Journal of Biological Chemistry. 1996;271:19443–19450. doi: 10.1074/jbc.271.32.19443. [DOI] [PubMed] [Google Scholar]
- Luzi L, Confalonieri S, Di Fiore PP, Pelicci PG. Evolution of Shc functions from nematode to human. Current Opinion in Genetics & Development. 2000;10:668–674. doi: 10.1016/s0959-437x(00)00146-5. [DOI] [PubMed] [Google Scholar]
- Mainiero F, Pepe A, Wary KK, Spinardi L, Mohammadi M, Schlessinger J, Giancotti FG. Signal transduction by the alpha 6 beta 4 integrin: distinct beta 4 subunit sites mediate recruitment of Shc/Grb2 and association with the cytoskeleton of hemidesmosomes. EMBO Journal. 1995;144:470–481. doi: 10.1002/j.1460-2075.1995.tb00126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlade J, Cheng A, Pelicci G, Pelicci PG, Pawson T. Shc proteins are phosphorylated and regulated by the v-Src and v-Fps protein-tyrosine kinases. PNAS. 1992;89:8869–8873. doi: 10.1073/pnas.89.19.8869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng TC, Lee MS, Lin MF. Interaction between protein tyrosine phosphatase and protein tyrosine kinase is involved in androgen-promoted growth of human prostate cancer cells. Oncogene. 2000;19:2664–2677. doi: 10.1038/sj.onc.1203576. [DOI] [PubMed] [Google Scholar]
- Meyer G, Feldman EL. Signaling mechanisms that regulate actin-based motility processes in the nervous system. Journal of Neurochemistry. 2002;83:490–503. doi: 10.1046/j.1471-4159.2002.01185.x. [DOI] [PubMed] [Google Scholar]
- Michels G, Er F, Eicks M, Herzig S, Hoppe UC. Long-term and immediate effect of testosterone on single T-type calcium channel in neonatal rat cardiomyocytes. Endocrinology. 2006;147:5160–5169. doi: 10.1210/en.2006-0186. [DOI] [PubMed] [Google Scholar]
- Migliaccio E, Mele S, Salcini AE, Pelicci G, Lai KM, Superti-Furga G, Pawson T, Di Fiore PP, Lanfrancone L, Pelicci PG. Opposite effects of the p52Shc/p46Shc and p66Shc splicing isoforms on the EGF receptor-MAP kinase-fos signaling pathway. EMBO Journal. 1997;16:706–716. doi: 10.1093/emboj/16.4.706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliaccio E, Giorgio M, Mele S, Pelicci G, Reboldi P, Pandolfi PP, Lanfrancone L, Pelicci PG. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature. 1999;402:309–313. doi: 10.1038/46311. [DOI] [PubMed] [Google Scholar]
- Migliaccio A, Castoria G, Di Domenico M, de Falco A, Bilancio A, Lombardi M, Barone MV, Ametrano D, Zannini MS, Abbondanza C, et al. Steroid-induced androgen receptor-oestradiol receptor beta-Src complex triggers prostate cancer cell proliferation. EMBO Journal. 2000;19:5406–5417. doi: 10.1093/emboj/19.20.5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliaccio A, Castoria G, Di Domenico M, Ciociola A, Lombardi M, De Falco A, Nanayakkara M, Bottero D, De Stasio R, Varricchio L, et al. Crosstalk between EGFR and extranuclear steroid receptors. Annals of the New York Academy of Sciences. 2006;1089:194–200. doi: 10.1196/annals.1386.006. [DOI] [PubMed] [Google Scholar]
- Mignatti P, Morimoto T, Rifkin DB. Basic fibroblast growth factor released by single, isolated cells stimulates their migration in an autocrine manner. PNAS. 1991;88:11007–11011. doi: 10.1073/pnas.88.24.11007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundy GR. Mechanisms of bone metastasis. Cancer. 1997;80:1546–1556. doi: 10.1002/(sici)1097-0142(19971015)80:8+<1546::aid-cncr4>3.3.co;2-r. [DOI] [PubMed] [Google Scholar]
- Nakhla AM, Romas NA, Rosner W. Estradiol activates the prostate androgen receptor and prostate-specific antigen secretion through the intermediacy of sex hormone-binding globulin. Journal of Biological Chemistry. 1997;272:6838–6841. doi: 10.1074/jbc.272.11.6838. [DOI] [PubMed] [Google Scholar]
- Nemoto S, Finkel T. Redox regulation of forkhead proteins through a p66shc-dependent signaling pathway. Science. 2002;29:2450–2452. doi: 10.1126/science.1069004. [DOI] [PubMed] [Google Scholar]
- Nemoto S, Combs CA, French S, Ahn BH, Fergusson MM, Balaban RS, Finkel T. The mammalian longevity-associated gene product p66shc regulates mitochondrial metabolism. Journal of Biological Chemistry. 2006;281:10555–10560. doi: 10.1074/jbc.M511626200. [DOI] [PubMed] [Google Scholar]
- Northey JJ, Chmielecki J, Ngan E, Russo C, Annis MG, Muller WJ, Siegel PM. Signaling through ShcA is required for transforming growth factor beta- and Neu/ErbB-2-induced breast cancer cell motility and invasion. Molecular and Cellular Biology. 2008;28:3162–3176. doi: 10.1128/MCB.01734-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada S, Kao AW, Ceresa BP, Blaikie P, Margolis B, Pessin JE. The 66-kDa Shc isoform is a negative regulator of the epidermal growth factor-stimulated mitogen-activated protein kinase pathway. Journal of Biological Chemistry. 1997;272:28042–28049. doi: 10.1074/jbc.272.44.28042. [DOI] [PubMed] [Google Scholar]
- Okamoto T, Schlegel A, Scherer PE, Lisanti MP. Caveolins, a family of scaffolding proteins for organizing ‘preassembled signaling complexes’ at the plasma membrane. Journal of Biological Chemistry. 1998;273:5419–5422. doi: 10.1074/jbc.273.10.5419. [DOI] [PubMed] [Google Scholar]
- Olson MF, Ashworth A, Hall A. An essential role for Rho, Rac, and Cdc42 GTPases in cell cycle progression through G1. Science. 1995;269:1270–1272. doi: 10.1126/science.7652575. [DOI] [PubMed] [Google Scholar]
- Orsini F, Migliaccio E, Moroni M, Contursi C, Raker VA, Piccini D, Martin-Padura I, Pelliccia G, Trinei M, Bono M, et al. The life span determinant p66shc localizes to mitochondria where it associates with mitochondrial heat shock protein 70 and regulates trans-membrane potential. Journal of Biological Chemistry. 2004;279:25689–25695. doi: 10.1074/jbc.M401844200. [DOI] [PubMed] [Google Scholar]
- Pacini S, Pellegrini M, Migliaccio E, Patrussi L, Ulivieri C, Ventura A, Carraro F, Naldini A, Lanfrancone L, Pelicci P, et al. p66SHC promotes apoptosis and antagonizes mitogenic signaling in T cells. Molecular and Cellular Biology. 2004;24:1747–1757. doi: 10.1128/MCB.24.4.1747-1757.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pages G, Lenormand P, L'Allemain G, Chambard JC, Meloche S, Pouyssegur J. Mitogen-activated protein kinases p42mapk and p44mapk are required for fibroblast proliferation. PNAS. 1993;90:8319–8323. doi: 10.1073/pnas.90.18.8319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandolfi S, Bonafe M, Di Tella L, Tiberi L, Salvioli S, Monti D, Sorbi S. Franceschi Cp66Shc is highly expressed in fibroblasts from centenarians. Mechanisms of Ageing and Development. 2005;126:839–844. doi: 10.1016/j.mad.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Park YJ, Kim TY, Lee SH, Kim H, Kim SW, Shong M, Yoon YK, Cho BY, Park DJ. p66shc expression in proliferating thyroid cells is regulated by thyrotropin receptor signaling. Endocrinology. 2005;146:2473–2480. doi: 10.1210/en.2004-1588. [DOI] [PubMed] [Google Scholar]
- Pawson T, Scott JD. Signaling through scaffold, anchoring, and adaptor proteins. Science. 1997;278:2075–2080. doi: 10.1126/science.278.5346.2075. [DOI] [PubMed] [Google Scholar]
- Pelicci G, Lanfrancone L, Grignani F, McGlade J, Cavallo F, Forni G, Nicoletti I, Pawson T, Pelicci PG. A novel transforming protein (SHC) with an SH2 domain is implicated in mitogenic signal transduction. Cell. 1992;70:93–104. doi: 10.1016/0092-8674(92)90536-l. [DOI] [PubMed] [Google Scholar]
- Pelicci G, Giordano S, Zhen Z, Salcini AE, Lanfrancone L, Bardelli A, Panayotou G, Waterfield MD, Ponzetto C, Pelicci PG, et al. The motogenic and mitogenic responses to HGF are amplified by the Shc adaptor protein. Oncogene. 1995;10:1631–1638. [PubMed] [Google Scholar]
- Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nature Reviews Molecular Cell Biology. 2002;3:639–650. doi: 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- Pontes JE, Rose NR, Ercole C, Pierce JM., Jr Immunofluorescence for prostatic acid phosphatase: clinical applications. Journal of Urology. 1981;126:187–189. doi: 10.1016/s0022-5347(17)54440-7. [DOI] [PubMed] [Google Scholar]
- La Porta CA. Links nPKCdelta a new therapeutic marker for melanoma metastasis. International Journal of Molecular Medicine. 2000;5:467–471. doi: 10.3892/ijmm.5.5.467. [DOI] [PubMed] [Google Scholar]
- Price DT, Della Rocca G, Guo C, Ballo MS, Schwinn DA, Luttrell LM. Activation of extracellular signal regulated kinase in human prostate cancer. Journal of Urology. 1999;162:1537–1542. [PubMed] [Google Scholar]
- Radinsky R, Fidler IJ. Regulation of tumor cell growth at organ-specific metastases. In Vivo. 1992;6:325–331. [PubMed] [Google Scholar]
- Ratner S, Patrick P, Bora G. Lymphocyte development of adherence and motility in extracellular matrix during IL-2 stimulation. Journal of Immunology. 1992;149:681–688. [PubMed] [Google Scholar]
- Ravichandran KS. Signaling via Shc family adapter proteins. Oncogene. 2001;20:6322–6330. doi: 10.1038/sj.onc.1204776. [DOI] [PubMed] [Google Scholar]
- Ray P, Ghosh SK, Zhang DH, Ray A. Repression of interleukin-6 gene expression by 17 beta-estradiol: inhibition of the DNA-binding activity of the transcription factors NF-IL6 and NF-kappa B by the estrogen receptor. FEBS Letters. 1997;409:79–85. doi: 10.1016/s0014-5793(97)00487-0. [DOI] [PubMed] [Google Scholar]
- Reif AE, Schlesinger RM, Fish CA, Robinson CM. Acid phosphatase isozymes in cancer of the prostate. Cancer. 1973;31:689–699. doi: 10.1002/1097-0142(197303)31:3<689::aid-cncr2820310331>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Ripple MO, Henry WF, Rago RP, Wilding G. Prooxidant-antioxidant shift induced by androgen treatment of human prostate carcinoma cells. Journal of the National Cancer Institute. 1997;89:40–48. doi: 10.1093/jnci/89.1.40. [DOI] [PubMed] [Google Scholar]
- Ripple MO, Henry WF, Schwarze SR, Wilding G, Weindruch R. Effect of antioxidants on androgen-induced AP-1 and NF-kappaB DNA-binding activity in prostate carcinoma cells. Journal of the National Cancer Institute. 1999;91:1227–1232. doi: 10.1093/jnci/91.14.1227. [DOI] [PubMed] [Google Scholar]
- Rosner W, Hryb DJ, Khan MS, Nakhla AM, Romas NA. Androgen and estrogen signaling at the cell membrane via G-proteins and cyclic adenosine monophosphate. Steroids. 1999;64:100–106. doi: 10.1016/s0039-128x(98)00108-1. [DOI] [PubMed] [Google Scholar]
- Rozakis-Adcock M, McGlade J, Mbamalu G, Pelicci G, Daly R, Li W, Batzer A, Thomas S, Brugge J, Pelicci PG, et al. Association of the Shc and Grb2/Sem5 SH2-containing proteins is implicated in activation of the Ras pathway by tyrosine kinases. Nature. 1992;360:689–692. doi: 10.1038/360689a0. [DOI] [PubMed] [Google Scholar]
- Ruoslahti E, Reed JC. Anchorage dependence, integrins, and apoptosis. Cell. 1994;77:477–478. doi: 10.1016/0092-8674(94)90209-7. [DOI] [PubMed] [Google Scholar]
- Sachs M, Weidner KM, Brinkmann V, Walther I, Obermeier A, Ullrich A, Birchmeier W. Motogenic and morphogenic activity of epithelial receptor tyrosine kinases. Journal of Cell Biology. 1996;133:1095–1107. doi: 10.1083/jcb.133.5.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safe S. Transcriptional activation of genes by 17 beta-estradiol through estrogen receptor-Sp1 interactions Vitamins. Hormones. 2001;62:231–252. doi: 10.1016/s0083-6729(01)62006-5. [DOI] [PubMed] [Google Scholar]
- Sakai H, Igawa T, Saha PK, Nomata K, Yushita Y, Kanetake H, Saito Y. A case of prostatic carcinoma presenting as a metastatic orbital tumor. Hinyokika Kiyo. Acta Urologica Japonica. 1992;38:77–80. [PubMed] [Google Scholar]
- Sakai H, Yogi Y, Minami Y, Yushita Y, Kanetake H, Saito Y. Prostate specific antigen and prostatic acid phosphatase immunoreactivity as prognostic indicators of advanced prostatic carcinoma. Journal of Urology. 1993;149:1020–1023. doi: 10.1016/s0022-5347(17)36285-7. [DOI] [PubMed] [Google Scholar]
- Sato K, Gotoh N, Otsuki T, Kakumoto M, Aoto M, Tokmakov AA, Shibuya M, Fukami Y. Tyrosine residues 239 and 240 of Shc are phosphatidylinositol 4,5-bisphosphate-dependent phosphorylation sites by c-Src. Biochemical and Biophysical Research Communications. 1997;240:399–404. doi: 10.1006/bbrc.1997.7667. [DOI] [PubMed] [Google Scholar]
- Schlaepfer DD, Jones KC, Hunter T. Multiple Grb2-mediated integrin-stimulated signaling pathways to ERK2/mitogen-activated protein kinase: summation of both c-Src- and focal adhesion kinase-initiated tyrosine phosphorylation events. Molecular and Cellular Biology. 1998;18:2571–2585. doi: 10.1128/mcb.18.5.2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solin T, Kontturi M, Pohlmann R, Vihko P. Gene expression and prostate specificity of human prostatic acid phosphatase (PAP): evaluation by RNA blot analyses. Biochimica et Biophysica Acta. 1990;1048:72–77. doi: 10.1016/0167-4781(90)90024-v. [DOI] [PubMed] [Google Scholar]
- Song RX, Zhang Z, Santen RJ. Estrogen rapid action via protein complex formation involving ERalpha and Src. Trends in Endocrinology and Metabolism. 2005;16:347–353. doi: 10.1016/j.tem.2005.06.010. [DOI] [PubMed] [Google Scholar]
- Stevenson LE, Frackelton AR., Jr Constitutively tyrosine phosphorylated p52 Shc in breast cancer cells: correlation with ErbB2 and p66 Shc expression. Breast Cancer Research and Treatment. 1998;49:119–128. doi: 10.1023/a:1006007227747. [DOI] [PubMed] [Google Scholar]
- Stevenson LE, Ravichandran KS, Frackelton AR., Jr Shc dominant negative disrupts cell cycle progression in both G0-G1 and G2-M of ErbB2-positive breast cancer cells. Cell Growth and Differentiation. 1999;10:61–71. [PubMed] [Google Scholar]
- Stewart LV, Lyles B, Lin MF, Weigel NL. Vitamin D receptor agonists induce prostatic acid phosphatase to reduce cell growth and HER-2 signaling in LNCaP-derived human prostate cancer cells. Journal of Steroid Biochemistry and Molecular Biology. 2005;97:37–46. doi: 10.1016/j.jsbmb.2005.06.011. [DOI] [PubMed] [Google Scholar]
- Stracke ML, Engel JD, Wilson LW, Rechler MM, Liotta LA, Schiffmann E. The type I insulin-like growth factor receptor is a motility receptor in human melanoma cells. Journal of Biological Chemistry. 1989;264:21544–21549. [PubMed] [Google Scholar]
- Sundaresan M, Yu ZX, Ferrans VJ, Sulciner DJ, Gutkind JS, Irani K, Goldschmidt-Clermont PJ, Finkel T. Regulation of reactive-oxygen-species generation in fibroblasts by Rac1. Biochemical Journal. 1996;318:379–382. doi: 10.1042/bj3180379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor WR, Greenberg AH, Turley EA, Wright JA. Cell motility, invasion, and malignancy induced by overexpression of K-FGF or bFGF. Experimental Cell Research. 1993;204:295–301. doi: 10.1006/excr.1993.1036. [DOI] [PubMed] [Google Scholar]
- Thomas D, Bradshaw RA. Differential utilization of ShcA tyrosine residues and functional domains in the transduction of epidermal growth factor-induced mitogen-activated protein kinase activation in 293T cells and nerve growth factor-induced neurite outgrowth in PC12 cells. Identification of a new Grb2.Sos1 binding site. Journal of Biological Chemistry. 1997;272:22293–22299. doi: 10.1074/jbc.272.35.22293. [DOI] [PubMed] [Google Scholar]
- Trinei M, Giorgio M, Cicalese A, Barozzi S, Ventura A, Migliaccio E, Milia E, Martin-Padura I, Raker VA, Maccarana M, et al. A p53-p66Shc signalling pathway controls intracellular redox status, levels of oxidation-damaged DNA and oxidative stress-induced apoptosis. Oncogene. 2002;30:3872–3878. doi: 10.1038/sj.onc.1205513. [DOI] [PubMed] [Google Scholar]
- Unni E, Sun S, Nan B, McPhaul MJ, Cheskis B, Mancini MA, Marcelli M. Changes in androgen receptor nongenotropic signaling correlate with transition of LNCaP cells to androgen independence. Cancer Research. 2004;64:7156–7168. doi: 10.1158/0008-5472.CAN-04-1121. [DOI] [PubMed] [Google Scholar]
- Velverde MA, Rojas P, Amigo J, Cosmelli D, Orio P, Bahamonde MI, Mann GE, Vergara C, Latorre R. Acute activation of Maxi-K channels (hSlo) by estradiol binding to the beta subunit. Science. 1999;285:1929–1931. doi: 10.1126/science.285.5435.1929. [DOI] [PubMed] [Google Scholar]
- Veeramani S, Lin MF. Role of reactive oxygen species in carcinogenesis. In: Banarjee R, editor. Redox Biochemistry. New Jercy: John Wiley & Sons Inc.; 2007. pp. 212–218. [Google Scholar]
- Veeramani S, Yuan TC, Chen SJ, Lin FF, Petersen JE, Shaheduzzaman S, Srivastava S, MacDonald RG, Lin MF. Cellular prostatic acid phosphatase: a protein tyrosine phosphatase involved in androgen-independent proliferation of prostate cancer. Endocrine-Related Cancer. 2005a;12:805–822. doi: 10.1677/erc.1.00950. [DOI] [PubMed] [Google Scholar]
- Veeramani S, Igawa T, Yuan TC, Lin FF, Lee MS, Lin JS, Johansson SL, Lin MF. Expression of p66(Shc) protein correlates with proliferation of human prostate cancer cells. Oncogene. 2005b;24:7203–7212. doi: 10.1038/sj.onc.1208852. [DOI] [PubMed] [Google Scholar]
- Veeramani S, Yuan TC, Lin FF, Lin MF. Mitochondrial redox signaling by p66Shc is involved in regulating androgenic growth stimulation of human prostate cancer cells. Oncogene. 2008;27:5057–5068. doi: 10.1038/onc.2008.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura A, Luzi L, Pacini S, Baldari CT, Pelicci PG. The p66Shc longevity gene is silenced through epigenetic modifications of an alternative promoter. Journal of Biological Chemistry. 2002;277:22370–22376. doi: 10.1074/jbc.M200280200. [DOI] [PubMed] [Google Scholar]
- Ventura A, Maccarana M, Raker VA, Pelicci PG. A cryptic targeting signal induces isoform-specific localization of p46Shc to mitochondria. Journal of Biological Chemistry. 2004;279:2299–2306. doi: 10.1074/jbc.M307655200. [DOI] [PubMed] [Google Scholar]
- Wary KK, Mainiero F, Isakoff SJ, Marcantonio EE, Giancotti FG. The adaptor protein Shc couples a class of integrins to the control of cell cycle progression. Cell. 1996;87:733–743. doi: 10.1016/s0092-8674(00)81392-6. [DOI] [PubMed] [Google Scholar]
- Wary KK, Mariotti A, Zurzolo C, Giancotti FG. A requirement for caveolin-1 and associated kinase Fyn in integrin signaling and anchorage-dependent cell growth. Cell. 1998;94:625–634. doi: 10.1016/s0092-8674(00)81604-9. [DOI] [PubMed] [Google Scholar]
- Webster MA, Hutchinson JN, Rauh MJ, Muthuswamy SK, Anton M, Tortorice CG, Cardiff RD, Graham FL, Hassell JA, Muller WJ. Requirement for both Shc and phosphatidylinositol 3′ kinase signaling pathways in polyomavirus middle T-mediated mammary tumorigenesis. Molecular and Cellular Biology. 1998;18:2344–2359. doi: 10.1128/mcb.18.4.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigel NL. Steroid hormone receptors and their regulation by phosphorylation. Biochemical Journal. 1996;319:657–667. doi: 10.1042/bj3190657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigel NL, Moore NL. Kinases and protein phosphorylation as regulators of steroid hormone action. Nuclear Receptor Signaling. 2007;5:e005. doi: 10.1621/nrs.05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong CW, McNally C, Nickbarg E, Komm BS, Cheskis BJ. Estrogen receptor-interacting protein that modulates its nongenomic activity-crosstalk with Src/Erk phosphorylation cascade. PNAS. 2002;99:14783–14788. doi: 10.1073/pnas.192569699. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Xie Y, Hung MC. p66Shc isoform down-regulated and not required for HER-2/neu signaling pathway in human breast cancer cell lines with HER-2/neu overexpression. Biochemical and Biophysical Research Communications. 1996;221:140–145. doi: 10.1006/bbrc.1996.0559. [DOI] [PubMed] [Google Scholar]
- Yoshida S, Masaki T, Feng H, Yuji J, Miyauchi Y, Funaki T, Yoshiji H, Matsumoto K, Uchida N, Watanabe S, et al. Enhanced expression of adaptor molecule p46 Shc in nuclei of hepatocellular carcinoma cells: study of LEC rats. International Journal of Oncology. 2004;25:1089–1096. [PubMed] [Google Scholar]
- Yuan TC, Lin FF, Veeramani S, Chen SJ, Earp HS, 3rd, Lin MF. ErbB-2 via PYK2 upregulates the adhesive ability of androgen receptor-positive human prostate cancer cells. Oncogene. 2007;26:7552–7559. doi: 10.1038/sj.onc.1210570. [DOI] [PubMed] [Google Scholar]
- Zaccagnini G, Martelli F, Fasanaro P, Magenta A, Gaetano C, Di Carlo A, Biglioli P, Giorgio M, Martin-Padura I, Pelicci PG, et al. p66ShcA modulates tissue response to hindlimb ischemia. Circulation. 2004;15:2917–2923. doi: 10.1161/01.CIR.0000129309.58874.0F. [DOI] [PubMed] [Google Scholar]
- Zheng DQ, Woodard AS, Fornaro M, Tallini G, Languino LR. Prostatic carcinoma cell migration via alpha(v)beta3 integrin is modulated by a focal adhesion kinase pathway. Cancer Research. 1999;59:1655–1664. [PubMed] [Google Scholar]
- Zhou MM, Ravichandran KS, Olejniczak EF, Petros AM, Meadows RP, Sattler M, Harlan JE, Wade WS, Burakoff SJ, Fesik SW. Structure and ligand recognition of the phosphotyrosine binding domain of Shc. Nature. 1995;378:584–592. doi: 10.1038/378584a0. [DOI] [PubMed] [Google Scholar]