

Catalytic RNA molecules can be found at the heart of many essential biological reactions.1-3 It has been posited that such ribozymes can achieve rate acceleration by shifting base pKa values,4 by providing functional groups for general acid/base catalysis,5,6 by coordinating water molecules for specific acid/base catalysis,7,8 and by providing electrostatic stabilization of high-energy intermediates.9-11 Although these chemical transformation strategies are similar to those utilized by protein enzymes, the limited chemical repertoire of RNA would appear to place it at a significant disadvantage. A major challenge to the field is to elucidate how ribozymes gain biological functionality by forging structure-function relationships.
The hairpin ribozyme (HPRZ) is a catalytic RNA motif derived from the negative polarity strand of the 359-nt satellite RNA of the tobacco ringspot virus. In vivo the HPRZ generates unit-length circular transcripts from concatenated replication intermediates produced during rolling-circle replication.12 Cleavage of the complementary RNA substrate occurs when a 2′-hydroxyl is activated by a localized base responsible for transient deprotonation.13 The activated nucleophile attacks the scissile phosphorus between residues A-1 and G+1 generating a trigonal-bipyramidal transition state (Figure 1A) that ultimately yields a free 5′-hydroxyl group and a cyclic-2′,3′-phosphate. The HPRZ is an ideal system for understanding how RNA nucleobases contribute to rate enhancement because it does not rely on a metal cofactor,14 thus implicating the nucleobases themselves in reaction chemistry. At present, the definitive mechanism of action by which such bases contribute to catalysis remains an area of intense focus.
Figure 1.
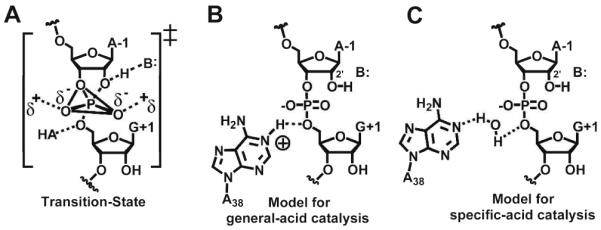
Small ribozyme transition state and models of HPRZ catalysis. (A) The phosphorane transition-state of small ribozymes. (B) A model of general-acid catalysis, where A38 is HA in Figure 1A. (C) A model of specific-acid catalysis, where water is HA in Figure 1A.
The preponderance of biochemical data suggests that the N1-imine atoms of nucleobases A38 and G8 facilitate proton transfer in the HPRZ reaction.6,7,9 Crystal structures revealed both residues are located within hydrogen-bonding distance to functional groups involved in phosphoryl transfer.10,15 Both biochemical and structural analyses have concluded that A38 is critical for catalysis and structural integrity.7,16 However, the function of A38 remains uncertain, which has led to several disparate mechanistic proposals. Simulated pH-rate profiles, abasic probing, and computational analyses have suggested that A38 participates in catalysis by nucleobase protonation or by orienting a water molecule (Figure 1B or 1C, respectively).4,7
Experimental pH-rate profiles for the HPRZ show titration points at 5.4 and 9.6.13,14 Although the more basic of the two values compares well with the imino pKa for G or U, the pKa for A or C is closer to 4.0 (Ref. 17), thus suggesting a base whose imino group pKa is shifted toward neutrality. The use of abasic variants at positions 8 and 38 provided insight into this observation. Whereas the position 8 variant showed a 350-fold loss in cleavage, its pH-rate profile was unaltered.6 By contrast, abasic substitution at position 38 reduced cleavage 14,000-fold, and shifted the pH-rate profile three log units into the basic.7 These results suggested A38 is primarily responsible for the pKa at 5.4, although its microscopic pKa has not been reported.
Recently, our lab probed the chemical contribution of the N1 imine of A38 in relation to cleavage and active site integrity. To accomplish this, we synthesized and incorporated N1-deazaadenosine (N1dA) - an inert isostere of adenosine - at position 38 of the HPRZ.18 The N1dA38 variant reduced cleavage by ~107-fold relative to wild-type, but revealed only nominal changes to the active site in the context of pre-cleavage and transition-state analogues. This investigation supported the existence of an A38-N1(H+) state (Figure 1B). However, the question of whether the lower titration point of 5.4 in the pH-rate profile arose directly from A38, and if so, whether the pKa of the N1-imine is shifted in pre-catalytic, as well as the transition-state conformations remained unresolved.
To address these questions, we directly measured the A38 N1-imino pKa using Raman crystallography, which allows analysis of group-specific RNA properties in single crystals.19 Herein, we examined crystals of the HPRZ whose structure was reported previously to 2.05 Å resolution.20 To prevent cleavage, the 2′-nucleophile was capped by an -O-methyl moiety. Although the non-bridging oxygens of the scissile bond do not occupy the active site pocket,10,21 this structure has been dubbed ‘pre-catalytic’ due to its near in-line geometry (Figure S1).15
First we established the Raman marker bands required to measure the pKa of an N1-imino group of adenine. We initially subjected a concentrated solution of AMP to Raman spectroscopy titration analysis. We chose to monitor the 734 cm-1 band, which corresponds to an adenine ring mode that occurs in a spectral window and thus is readily distinguishable from other functional groups.19 Using the Raman spectrum at pH 6.5 as the subtrahend and the pH invariant PO2- peak as the internal standard, a series of difference spectra were obtained from pH 2.0 to 6.0. The results provided a differential feature that reported on the N1-protonation state (Figure S2A). The peak areas were plotted as a function of pH and fit to a sigmoidal function revealing a solution pKa of 3.68 ± 0.06 for AMP (Figure S3), which is comparable to literature values.17,22 A similar approach was taken for single crystals of the HPRZ (Figure 2 and S2B). By contrast, the pKa was 5.46 ± 0.05, which corresponds closely to the macroscopic pKa of the HPRZ in solution.13
Figure 2.
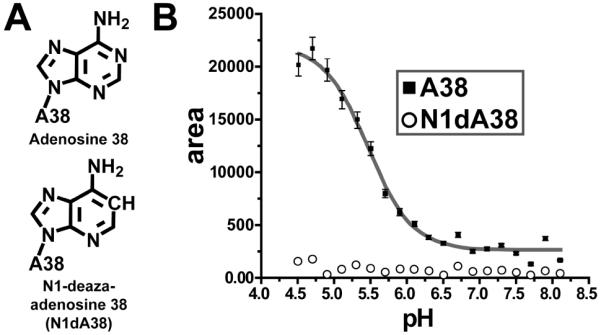
Position 38 variants and pH titration profiles for pre-catalytic HPRZ crystals. (A) The adenosine and N1-deazaadenosine (N1dA) bases investigated at position 38. (B) pH titration profile for A38 and N1dA38. The Y-axis plots the area of the positive 731 cm-1 feature in the difference spectra.
Although the measured microscopic pKa appeared consistent with a single proton transfer event, as many as eleven adenine bases in our HPRZ construct do not participate in Watson-Crick pairing (Figure S1A). To pinpoint the site of titration, we used N1dA38, which is devoid of a titratable N1-imine (Figure 2A). Prior crystal structures of this variant revealed that N1dA38 does not disrupt the active site fold,18 thus providing a control experiment with single-atom resolution. The resulting Raman titration profile for N1dA38 showed no transition over the pH range recorded for A38 (Figure 2B), suggesting that the observed pKa corresponds directly to the N1-imine of A38.
A general acid/base mechanism for the HPRZ has long been considered viable since the A38 and G8 bases directly flank the scissile bond. However, when one considers the microscopic pKa of 5.46 at A38 observed herein, and the recently reported value of 9.5 for G8,23 the results suggest that A38 is solely responsible for the rate-limiting proton transfer step in the reaction (Figure 1B). Prior analysis of N1dA38 supported both essential chemical and structural roles for the A38 N1-imine.18 Moreover, our finding that the pKa of A38 is shifted toward neutrality in crystals representative of a pre-catalytic conformation implies that the base is protonated prior to the transition-state, which would promote phosphorane formation and stabilization.17 A similar scenario has been proposed for the HDV ribozyme.24 Although the determinants contributing to the elevated A38 pKa are unknown, a plausible electrostatic source is the proximity of the N1-imine to the oxygen-rich environment of the scissile bond, which includes a 3.1 Å contact distance to the O3′-atom of A-1 (Figure S1B, inset).
Prior work on the HPRZ suggested a water molecule positioned between the N1 imino of A38 and the O5′-leaving group of G+1 could be operative in proton transfer (Figure 1C).7 However, crystal structures of the HPRZ representative of the reaction coordinate were inconsistent with the binding of such a water molecule.18,20,21 Given that our crystals are in a pre-catalytic conformation, and that the A38 pKa therein has excellent agreement with the macroscopic pKa of 5.4, a role for water as a specific acid in cleavage does not appear viable.
MD simulations have led to the hypothesis that A38 serves as both a general acid and base in cleavage.25 Our data suggest that A38 is protonated in the pre-cleavage state, which precludes a general base role. However, MD simulations also place the A38-N1 group in close proximity to the 2′-nucleophile,25 consistent with several crystal structures.18,20 As such, we hypothesize that proximity of A38-N1(H+) in the pre-catalytic state decreases the pKa of the 2′-nucleophile, thereby facilitating proton transfer to oriented waters observed near A9 and A10 (refs. 20, 21) or to other unidentified groups in the local environment. Electrostatic coupling of reactive groups has been described for C75-(N3+) and Mg2+ in the context of the HDV ribozyme active site.24
In summary, our data present direct evidence for an elevated pKa at residue A38 of the HPRZ. This observation suggests that the A38-N1(H+) species could serve to: (i) promote and stabilize the transition state, (ii) directly protonate the 5′-leaving group of G+1 in cleavage, and (iii) lower the pKa of the 2′-nucleophile of A-1 for deprotonation prior to cleavage. Our results provide clear evidence that the fold of an RNA catalyst can perturb the biochemical properties of nucleobases to impart a level of functionality more often associated with a protein catalyst.
Supplementary Material
Acknowledgment
We thank Philip Bevilacqua for helpful discussions. Funding was provided in part by grants from the NIH (GM63162) and donors of the Petroleum Research Fund (45534 AC4) to JEW, and by NIH GM84120 to PRC. RCS was supported by an Elon Huntington-Hooker graduate fellowship.
References
- (1).Zaug AJ, Grabowski PJ, Cech TR. Nature. 1983;301:578–83. doi: 10.1038/301578a0. [DOI] [PubMed] [Google Scholar]
- (2).Guerrier-Takada C, Gardiner K, Marsh T, Pace N, Altman S. Cell. 1983;35:849–57. doi: 10.1016/0092-8674(83)90117-4. [DOI] [PubMed] [Google Scholar]
- (3).Nissen P, Hansen J, Ban N, Moore PB, Steitz TA. Science. 2000;289:920–30. doi: 10.1126/science.289.5481.920. [DOI] [PubMed] [Google Scholar]
- (4).Bevilacqua PC. Biochemistry. 2003;42:2259–65. doi: 10.1021/bi027273m. [DOI] [PubMed] [Google Scholar]
- (5).Das SR, Piccirilli JA. Nat Chem Biol. 2005;1:45–52. doi: 10.1038/nchembio703. [DOI] [PubMed] [Google Scholar]
- (6).Kuzmin YI, Da Costa CP, Fedor MJ. J Mol Biol. 2004;340:233–51. doi: 10.1016/j.jmb.2004.04.067. [DOI] [PubMed] [Google Scholar]
- (7).Kuzmin YI, Da Costa CP, Cottrell JW, Fedor MJ. J Mol Biol. 2005;349:989–1010. doi: 10.1016/j.jmb.2005.04.005. [DOI] [PubMed] [Google Scholar]
- (8).Walter NG. Mol Cell. 2007;28:923–9. doi: 10.1016/j.molcel.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Lebruska LL, Kuzmine, Fedor MJ. Chem Biol. 2002;9:465–73. doi: 10.1016/s1074-5521(02)00130-8. [DOI] [PubMed] [Google Scholar]
- (10).Rupert PB, Massey AP, Sigurdsson ST, Ferre-D’Amare AR. Science. 2002;298:1421–4. doi: 10.1126/science.1076093. [DOI] [PubMed] [Google Scholar]
- (11).Nam K, Gao J, York DM. RNA. 2008;14:1501–7. doi: 10.1261/rna.863108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Hampel A, Tritz R. Biochemistry. 1989;28:4929–33. doi: 10.1021/bi00438a002. [DOI] [PubMed] [Google Scholar]
- (13).Fedor MJ. J. Mol. Biol. 2000;297:269–91. doi: 10.1006/jmbi.2000.3560. [DOI] [PubMed] [Google Scholar]
- (14).Nesbitt S, Hegg LA, Fedor MJ. Chem Biol. 1997;4:619–30. doi: 10.1016/s1074-5521(97)90247-7. [DOI] [PubMed] [Google Scholar]
- (15).Rupert PB, Ferre-D’Amare AR. Nature. 2001;410:780–6. doi: 10.1038/35071009. [DOI] [PubMed] [Google Scholar]
- (16).MacElrevey C, Salter JD, Krucinska J, Wedekind JE. RNA. 2008;14:1600–16. doi: 10.1261/rna.1055308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Bevilacqua PC, Brown TS, Nakano S, Yajima R. Biopolymers. 2004;73:90–109. doi: 10.1002/bip.10519. [DOI] [PubMed] [Google Scholar]
- (18).Spitale RC, Volpini R, Heller MG, Krucinska J, Cristalli G, Wedekind JE. J Am Chem Soc. 2009;131:6093–5. doi: 10.1021/ja900450h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Gong B, Chen JH, Yajima R, Chen Y, Chase E, Chadalavada DM, Golden BL, Carey PR, Bevilacqua PC. Methods. 2009 doi: 10.1016/j.ymeth.2009.04.016. PMID: 19409996. (doi: 10.1016/j.ymeth.2009.04.016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Salter J, Krucinska J, Alam S, Grum-Tokars V, Wedekind JE. Biochemistry. 2006;45:686–700. doi: 10.1021/bi051887k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Torelli AT, Krucinska J, Wedekind JE. RNA. 2007;13:1052–70. doi: 10.1261/rna.510807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Moody EM, Brown TS, Bevilacqua PC. J Am Chem Soc. 2004;126:10200–1. doi: 10.1021/ja047362h. [DOI] [PubMed] [Google Scholar]
- (23).Liu L, Cottrell JW, Scott LG, Fedor MJ. Nat Chem Biol. 2009;5:351–7. doi: 10.1038/nchembio.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Gong B, Chen JH, Chase E, Chadalavada DM, Yajima R, Golden BL, Bevilacqua PC, Carey PR. J Am Chem Soc. 2007;129:13335–42. doi: 10.1021/ja0743893. [DOI] [PubMed] [Google Scholar]
- (25).Ditzler MA, Sponer J, Walter NG. RNA. 2009;15:560–75. doi: 10.1261/rna.1416709. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.