

Abstract
Histone H3 lysine 56 acetylation (H3K56Ac) has recently been identified and shown to be important for genomic stability in yeast. However, whether or not H3K56 acetylation occurs in mammals is not clear. Here, we report that H3K56Ac exists in mammals. Mammalian H3K56Ac requires the histone chaperone Asf1 and occurs mainly at the S phase in unstressed cells. Moreover, SIRT1, which is a mammalian member of sirtuin family of NAD+-dependent deacetylases, regulates the deacetylation of H3K56. We further showed that proper H3K56 acetylation is critical for genomic stability and DNA damage response. These results establish the existence and functional significance of H3K56Ac in mammals and identify two regulators of this modification.
Keywords: histone H3, K56, acetylation, SIRT1, ASF1, replication, genomic stability
Introduction
In eukaryotes, genetic information is packed in a higher order structure called chromatin. The nucleosome is the basic unit of chromatin and consists of 147 bp of DNA wrapped around a histone octamer that contains two copies of each of the four core histones: H3, H4, H2A and H2B.1,2 Each of these four histones is a small basic protein with a globular core domain and less-ordered N-terminus and C-terminus called histone tails. Histones are modified by phosphorylation, methylation, ubiquitination and acetylation at both tails and globular domains.3 Post-translational modifications of histones are linked to different states of chromatin and regulate distinct processes such as DNA replication, gene transcription and DNA repair.4–6
Histone H3 lysine 56 (K56), a core domain residue that localizes at both the entry and exit points of a nucleosome, has recently been shown to be acetylated in yeast.7–12 In budding yeast, the residue is acetylated predominantly during the S phase, but disappears rapidly when cells enter the G2/M phase of the cell cycle.9,11–13 Strains lacking an acetylatable lysine at this position were genetically unstable and sensitive to a subset of genotoxic agents, such as camptothecin (CPT).7–9 These phenotypes are possibly due to an important role of this modification in nucleosome assembly following DNA replication and DNA repair.14,15 Together, these data suggest that the appropriate acetylation of H3 K56 in budding yeast is important for genome stability.
Although H3 K56 acetylation play an important role in genomic stability and is conserved in P. falciparum and Drosophila, previous studies failed to detect the acetylation of H3 K56 in mammalian cells.7,10 A recent study suggest possible K56 acetylation in mammalian cells using mass spectrometry,16 however the signal was weak and no conclusive immunological evidence were shown. Furthermore, the function of this modification has not been explored. In this report, we investigated whether H3 K56 was acetylated in mammals and if so, the possible functions of this modification.
Result
In budding yeast histone H3 K56 was found to be acetylated by mass spectrometry7–12 and this modification regulates replication or repair coupled nucleosome assembly, a process that is important for the inheritance of epigenetic information as well as maintenance of genome stability. In mammals, however, H3 K56Ac has not been well established, let alone the function of this modification. We found that H3 K56 is acetylated in human, mouse and rat cells using an antibody that we generated against yeast H3K56Ac (Fig. 1A), although the signals in mammalian cells were weaker than those of yeast. The specificity of this antibody has been vigorously tested toward yeast H3 K56Ac.12,17 To confirm the specificity of this antibody in human cells, we expressed wild type H3 tagged with the FLAG epitope as well as mutant forms of H3 in which human H3 K56 was mutated to Gln (K56Q) or Arg (K56R). As shown in Figure 1B, anti-H3K56Ac antibodies no longer recognize mutated H3 K56. Furthermore, H3K56Ac peptides, but not H3K56Me peptides, could block the signals by H3K56Ac antibody (Suppl. Fig. 1A). H3K56Ac antibody from another source also generates similar signals as our antibody (Suppl. Fig. 1B). These results confirm the specificity of the antibodies recognizing H3K56Ac in mammalian cell extracts.
Figure 1.
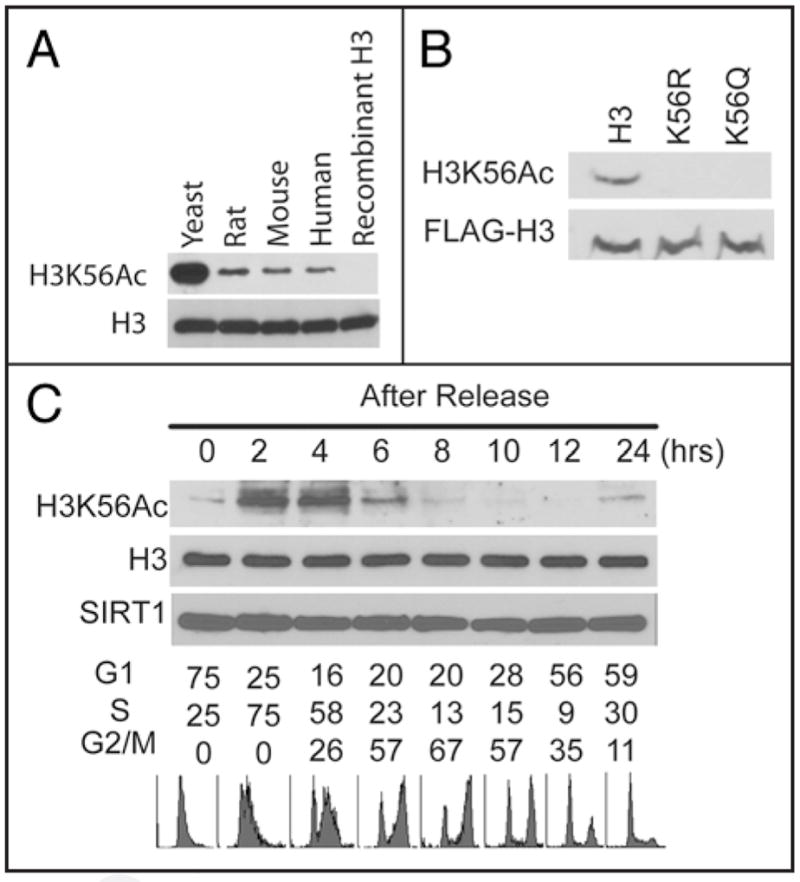
H3K56 acetylation was present in mammalian cells. (A) Yeast, H1299 (Human), mouse embryonic fibroblast (Mouse) and Rat1a (Rat) cells were lysed and cell lysates were blotted with antibodies against H3K56Ac or H3. GST-H3 purified from bacteria cells were used as negative control. (B) Cells expressing FLAG-tagged wild-type H3 or K56 mutants were blotted with antibodies against H3K56Ac or the FLAG epitope. (C) H1299 cells were synchronized by double thymidine block and released into the cell cycle. Cells were harvested at indicated time points and cell extracts analyzed by western blot using indicated antibodies against H3K56Ac and H3. Cell cycle distribution at each time point was determined by fluorescence-activated cell sorting (FACS).
In yeast, H3K56Ac is an abundant modification that marks newly synthesized histone H3 molecules before they are incorporated into nucleosomes during the S phase and this modification largely disappears during the G2/M phase.9,11–13 To investigate whether H3K56Ac is regulated in a cell cycle-dependent manner in mammalian cells, we synchronized cells at the G1/S interface by double-thymidine block, and then released cells into the cell cycle. As shown in Figure 1C, H3K56Ac occurred during the S phase and disappeared at the late stages of the cell cycle. These results suggest that H3 K56Ac occurs preferentially in S-phase of the cell cycle and may play a role in during DNA replication in mammalian cells.
We next investigated the regulation of H3K56Ac in mammalian cells. In yeast, the acetyltransferase Rtt109 as well as histone chaperone Asf 1 are required for H3K56Ac.11,17–21 While it is currently unclear whether there are mammalian homologs of Rtt109, two mammalian Asf isoforms Asf1a and Asf1b have been identified.22 Knockdown of Asf1a expression significantly reduced H3K56Ac, whereas knockdown Asf1b had only a slight effect on H3K56Ac (Fig. 2A). Since H3K56Ac preferentially occur in the S phase, the affect of Asf1a toward H3K56Ac could be the result an abnormal cell cycle profile in cells depleted of Asf1a. However, we found that cells accumulated in the S phase in these cells (Fig. 2A), which could not explain the decreased H3K56Ac. These results suggest that Asf1, like its yeast counterpart, is required for H3 K56 acetylation, with Asf1a the prevalent isoform in this process in HeLa cells.
Figure 2.

Asf1 and SIRT1 regulate H3K56 acetylation in mammalian cells. (A) Asf1a and Asf1b were knocked down separately or together in HeLa cells for 72 hr. Cells were lysed and cell lysates were analyzed for H3K56Ac by western blot. A fraction of cells were fixed and examined for cell cycle distribution by FACS. (B) Cell lysates from SIRT1+/+, SIRT1−/− or SIRT1−/− MEFs expressing WT SIRT1 were blotted with H3 K56Ac or H3 antibodies. Cell cycle distribution of these cells was determined by FACS.
The deacetylation of H3 K56 is mediated by sirtuin-family histone deacetylases, Hst3 and Hst4 in yeast.13,23,24 SIRT1, a mammalian member of sirtuins, has been shown to regulate epigenetic modification of histones in mammals,25 such as K14 of H3 and K16 of H4. A recent study also showed that loss of SIRT1 causes increased H3K9ac, decreased H3K9me3, and delocalization of HP1 from heterochromatin.26 We found that SIRT1−/− cells showed H3K56 hyperacetylation compared to SIRT1+/+ cells (Fig. 2B); expression of WT SIRT1 in SIRT1−/− cells reduced H3 K56 acetylation (Fig. 2B). On the other hand, downregulation of SIRT6, a sirtuin implicated in regulating H3K9 acetylation,27 had no obvious effect on H3K56 acetylation (Suppl. Fig. 1D). These results suggest a specific role of SIRT1 in deacetylating H3K56 in mammalian cells.
The H3K56Ac is dynamically controlled to maintain genomic stability in yeast. Strains lacking either H3K56 histone acetyltransferase Rtt109 or H3K56 deacetylase Hst3/Hst4 are sensitive to genotoxic agents and exhibit increased levels of spontaneous DNA damage as well as synthetic phenotypes with strains lacking genes involved in DNA repair and replication.13,17,21,24,28 Similar to Hst3/4-deficient cells, SIRT1−/− MEFs showed more spontaneous DNA damage as these cells displayed more γH2AX foci, which are markers of DNA damage (Fig. 3A and B). Reconstitution of SIRT1−/− cells with WT SIRT1, but not catalytically inactive H363Y mutant,29 decreased γ-H2AX foci. Many of the γH2AX foci colocalize with PCNA foci, suggesting that the spontaneous DNA breaks in SIRT1−/− cells occur during the S phase (Suppl. Fig. 2A). γH2AX levels were also increased in SIRT1−/− MEFs cells (Fig. 3C). Reconstitution of SIRT1−/− cells with WT SIRT1 decreased γ-H2AX levels (Fig. 3C). These results suggest that SIRT1 maintains genomic stability.
Figure 3.

SIRT1 deficiency induces spontaneous DNA damages. (A) SIRT1+/+, SIRT1−/− or SIRT1−/− MEFs reconstituted with WE SIRT1 or SIRT1H363Y were fixed and stained with indicated antibody and DAPI. Bars, 10 μm. (B) Quantification of the percentage of cells that display γ-H2Ax foci in (A). 3 independents and 100 cells per sample were counted for deriving the bar graphs. **p < 0.01; *p < 0.05. (C) Cell lysates from SIRT1+/+, SIRT1−/− MEFs or SIRT1−/− MEFs reconstituted with SIRT1 were blotted with γ-H2AX antibody and PCNA antibody.
To directly test the functional significance of H3K56Ac, we generated cells stably expressing H3.1 shRNA and at the same time, stably expressing shRNA-resistant WT H3.1, H3.1K56A and H3.1K56Q (Fig. 4A). As shown in Figure 4B and C, the K56R and K56Q mutations increased phosphorylated-H2AX (γ-H2AX) foci and levels. These results suggest that mutations at H3 K56 induce spontaneous DNA breaks. Since H3 K56Ac preferentially occurs at the S phase, the spontaneous DNA breaks in H3 K56 mutants are probably due to a defect in DNA replication process, likely nucleosome assembly. Consistent with this notion, cells expressing H3K56R or H3K56Q mutant have higher S phase population accumulation than cells expressing WT H3 (Suppl. Fig. 1E). Furthermore, the γ-H2AX foci in H56K mutants occur in high percentage in cells with PCNA foci, which are markers of replicating cells (Suppl. Fig. 2B).
Figure 4.

Histone H3 K56 contributes to genomic stability and DNA damage response. (A) H1299 stably transfected with H3 shRNA together with FLAG-tagged H3 WT, H3 K56R or H3 K56Q were lysed and the expression of H3 was examined. The right panel is the quantification of H3 level normalized to β-actin (B) Cells from (A) were fixed and stained with indicated antibody and DAPI. The right panel represented % of cells with γ-H2Ax foci. 3 independent experiments and 100 cells per sample were counted to derive the bar graphs. **p < 0.01; Bars: 10 μm. (C) Cells from (A) were lysed and cell lysates were blotted with indicated antibodies. (D) HeLa cells were treated with CPT and HU, and H3K56 acetylation was examined. (E) Cells from (A) were treated with HU for 16 hrs, then released into the cell cycle. Cell cycle progression of these cells was then examined by FACS. (F) Cells from (A) were treated with CPT, MMS or HU at indicated dosage, cell viability were detected by the MTS assay.
We also found that H3K56 acetylation was increased in response to genotoxic stress (Fig. 4D). In yeast, high level of H3K56Ac was suggested to create a favorable chromatin environment for DNA damage response.9 We used cells expressing K56R and K56Q mutants to test whether it is the case in mammalian cells. K56R and K56Q mutants had a slower progression through the S-phase in recovering from hydroxyurea (HU) induced cell cycle arrest (Fig. 4E and Suppl. Fig. 2C). The moderate effect on cell cycle progression could be an effect of H3 K56 acetylation on nucleosome assembly. Alternatively, the increased DNA damage occurs in cells expressing H3K56 mutants could trigger cell cycle checkpoints, which could temporarily arrest cell cycle. Furthermore, cells expressing K56R and K56Q mutants were more sensitive to CPT, HU and MMC, which cause DNA damage (Fig. 4F). Overall, these results suggest that mutations at H3K56 not only result in spontaneous DNA damage, but also render cells hypersensitive to DNA damage agents.
Discussion
In S. cerevisiae acetylation of H3K56 regulates nucleosome assembly, DNA damage response and genomic stability.30,31 However, whether H3K56Ac is present in human cells is controversial. Two previous studies did not detect H3 K56 acetylation in mammals,7,10 leaving open the question as whether H3K56 acetylation as well as the function of this modification are conserved from yeast to mammals. In this study, we detected H3K56 acetylation in human, mouse and rat cells, establishing that H3K56 is indeed acetylated in mammals. We further show that similar to yeast studies, H3K56 is acetylated preferentially during S-phase, and deacetylated when cells enter into the later stages of the cell cycle. In addition, we show that H3K56 acetylation is important for genomic stability and DNA damage response. These results suggest that the function of H3K56 acetylation is likely to be conserved from yeast to mammals.
In yeast cells, histone chaperone functions to present newly-synthesized H3 acetylation by Rtt109-Vps75 HAT complex and Asf1 is essential for H3K56Ac in yeast cells.11,17,18,21,24 In mammalian cells, there are two Asf1 isoforms, Asf1a and Asf1b. We have shown that Asf1 is required for efficient H3K56Ac in mammalian cells. Surprisingly, Asf1a appears to be more important in the regulation of H3K56Ac than Asf1b. This differential effect of Asf1a and 1b could mean that Asf1b is not as important as Asf1a in regulating H3K56 acetylation. Alternatively, Asf1a could be more abundant than 1b in HeLa cells, making them more dependent on Asf1a. However, a previous publication did show almost equivalent levels of Asf1a and 1b in HeLa cells.22 To date, no obvious sequence homologs of Rtt109 have been identified in mammalian cells. Recent structural studies on Rtt109 suggest that Rtt109 shares sequence homology with the catalytic domain of p300/CBP. Therefore, it is possible that p300 and CBP is the H3K56 HAT in mammalian cells. Future studies will be needed to test this hypothesis.
We also demonstrate that SIRT1 is involved, either directly or indirectly, in the deacetylation of H3K56 in mammalian cells. In yeast, the deacetylation of H3 K56 at G2/M phase is due to the increased expression of Hst3/4.13,24 However, SIRT1 expression remains constant throughout the cell cycle (Fig. 1C). It remains to be determined how H3 K56Ac is specifically deacetylated during the G2/M phase. Previous studies have shown cells deficient of SIRT1 display genomic instability26,32,33 through several potential mechanisms. SIRT1 is important for NBS1’s function in DNA damage response.32 Furthermore, SIRT1 relocalizes to the sites of DNA damage and promotes DNA repair.26,33 Here we report another potential mechanism by which SIRT1 regulates genomic stability through its effect toward H3K56. A previous report showed that although SIRT1−/− cells show genomic instability, the formation of γH2AX foci is defection.26 In contrast to this previous report, we found increased γH2AX foci in SIRT1−/− cells. Currently, it is not clear how to reconcile this difference.
We have shown that cells expressing H3 K56R and K56Q mutants have increased levels of spontaneous DNA damage and are more sensitive to genotoxic stress compared to WT H3. We only could substitute a subpopulation of H3 with H3K56R and K56Q (Fig. 4A), however, it is sufficient to induce many spontaneous DNA breaks. This suggests that proper modification at H3K56 is important for genomic stability. Interestingly, in yeast, the K56Q mutant that supposedly to mimic K56 acetylation, show less sensitivity to CPT than the K56R mutant.9 In our hand, the K56Q and K56R mutants behave in a similar fashion. One explanation is that the K56Q mutant might not behave as a acetylation-mimic mutant in mammalian system.
Together, our results answered an unresolved question and established that H3K56 acetylation is conserved from yeast to mammals.
Methods
Cell culture, plasmids and antibodies
HeLa and H1299 cells were cultured in RPMI supplemented with 10% FBS. SIRT1 MEFs were kindly provided by Dr. Chuxia Deng (NIH). MEFs cells were cultured in DMEM supplemented with 15% FBS.
FLAG tagged histone H3.1 (pOZ-FH-N vector) was kindly provided by Dr. Yoshihiro Nakatani (Harvard Medical School). pBabe retrovirus vectors encoding SIRT1 were constructed according to the manufacturer’s directions (Invitrogen Gateway System). Mutants were generated by site-directed mutagenesis (Stratagene). H3.1cK56R primer: 5′-CGA AAT CCG TCG CTA CCA GAG GTC CAC CGA GCT GCT GAT CC-3′ and 5′-GGA TCA GCA GCT CGG TGG ACC TCT GGT AGC GAC GGA TTT CG-3′; H3.1cK56Q primer: 5′-CGA AAT CCG TCG CTA CCA GCA GTC CAC CGA GCT GCT GAT CC-3′ and 5′-GGA TCA GCA GCT CGG TGG ACT GCT GGT AGC GAC GGA TTT CG-3′; H3.1c shRNA-resistant Mutant primer: 5′-CCG CCC GGG CAC CGT GGC CCT CCG CGA AAT CCG TCG CTA C-3′ and 5′-GTA GCG ACG GAT TTC GCG GAG GGC CAC GGT GCC CGG GCG G-3′.
SIRT1 antibodies were made as described previously.34 H3 and H3K56Ac antibodies were made as described previously.17 Another H3K56Ac antibody (07–677) was purchased from Millipore. PCNA antibody (SC-56) was purchased form Santa Cruz. Rabbit©-H2AX antibody (A300-081A) was purchased from Bethyl. FLAG antibody (M2) were purchased from Sigma. Asf1a and Asf1b were generated by immunizing rabbits with peptides (Asf1a: LEDAESSNPN; Asf1b: NCTPIKGLGL). The antisera were affinity-purified with AminoLink Plus immobilization and purification kit (Pierce).
Preparation and infection of retrovirus
Packaging cell line BOSC23 was transfected with the retrovirus plasmid pBabe-FLAG-SIRT1. Media was changed 24 h later and collected at 48 or 72 h after transfection. The media were filter-sterilized (0.45 μm filter) and used to infect SIRT−/− MEFs cell. Infected cells were selected with 2 μg/ml puromycin (Sigma). Resistant clones were picked and expanded for further analysis.
RNAi
Asf1a shRNA (RHS4531-NM_014034#V2LHS_60027) were purchased from Openbiosystems. Asf1b siRNA was purchased from Dharmacon (SmartPool M-020553-01). Transfection was performed twice at 24 h interval with 200 nM of siRNA using Oligofectamine reagent according to the manufacturer’s instruction (Invitrogen). Histone H3.1c shRNAs were purchased from Sigma (NM_003531), the target sequence is CCT TGC GCG AAA TCC GTC GCT, this sequence only target to H3.1c. Lentivirus vector pLK0.1 encoding H3.1c shRNAs were made according to the protocol shown on Sigma website (http://www.sigmaaldrich.com/life-science/functional-genomics-and-rnai/learning-center/mission-protocols.html).
Histone H3 stable cell lines generation
H1299 cells were cotransfected with H3.1c shRNA (puromycin resistent) and shRNA-resistent FLAG-tagged wild type H3.1c or mutant H3.1c (K56R or K56Q) by using lipofectamine 2000 according to the manufacturer’s instruction (Invitrogen). After 48 hrs, cells were selected with 2 μg/ml puromycin (Sigma). After 6 days, resistant clones were screened with anti-H3 and anti-FLAG antibodies. Clones which express equal levels of endogenous H3 and recombinant H3 were picked and expanded for further analysis.
Immunoblotting and antibody blocking
Histone H3 used for anti-Ac-K56 immunoblotting was isolated by acid extraction. Cells were lysed in ice-cold NETN lysis buffer (20 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5% Nonidet P-40) containing 50 mM β-glycerophosphate, 10 mM NaF, and 1 mg/ml each of pepstatin A and aprotinin. 1 M HCl was added to a final concentration of 0.2 M, followed by incubation on ice for 30 min and centrifugation at 13,000 rpm for 10 min. The supernatant was retained and adjusted to pH 7 by adding 5 M NaOH. Cell extracts were separated by 15% SDS-PAGE and subjected to immunoblot analysis with the indicated antibodies.
Cell synchronization and FACS analysis
H1299 cells were synchronized by double thymidine block and release. Briefly, cells were treated with thymidine (2 mM) for 16 h. Cells were then washed three times, replaced with fresh medium and grown for 8h prior to a second treatment with thymidine (2 mM). 16 h after the second thymidine treatment, cells were washed, refed with fresh medium, and collected at the indicated time points. H1299 cells were synchronized by HU (1 mM) for 16 hrs. cell were then washed with three times, replaced with fresh medium and collected at the indicated time points. Cells were stained with propidium iodine (final concentration is 20 μg/ml) and subjected to FACS analysis and data were analyzed using the ModFit program.
Immunofluorescence
Immunofluorescence staining was performed using standard procedure. Cells were plated on dishes containing glass coverslips. After 24 hrs, cells were fixed in 4% paraformaldehyde for 10 min at room temperature and permeabilized with 0.2% Triton X-100 for 10 min. The fixed cells were then incubated in 1% BSA/PBS solution for 30 min. Anti-γH2AX and PCNA antibodies were added in 1% BSA/PBS for 25 min at 37°C. After washing with PBS, Rhodamine-conjugated anti-mouse IgG and FITC-conjugated anti-rabbit IgG antibody was added and incubated for 20 min at 37°C. Cells were then washed three times with PBS, and the stained cells were mounted with mounting medium and sealed with nail polish. Immunofluorescence was recorded using an Nikon Elipse 80i microscope.
Cell viability assay
We used the MTS assay to measure cell viability. The MTS cell proliferation assay is a colorimetric method to identify the cytotoxic potential of a test item. The assay measures the formation of a soluble formazan product (absorbance maximum at 490–500 nm), which is directly proportional to the number of live cells in culture. Cells were plated on 96-well dishes at a density of 3 × 103 cells/well. After overnight incubation, cells were treated with capmtothecin (CPT), methyl methanesulfonate (MMS) and hydroxyurea (HU) at the indicated dosages. After 24 hrs (MMS) or 48 hrs (CPT and HU) incubation, CellTiter 96® Aqueous One Solution Reagent (MTS assay, Promega, Cat.#: G3582) was added to each well according to the manufacturer’s instructions. After 1 hr incubation, cell viability was determined by measuring the absorbance at 490 nm using a 550 BioRad plate-reader (Bio-Rad, Hertfordshire, UK).
Supplementary Material
Acknowledgments
We thank Dr. Yoshihiro Nakatani for histone H3 constructs, Dr. Wei Gu for SIRT1 and SIRT1H363Y constructs, and Dr. Chuxia Deng for providing SIRT1+/+ and SIRT1−/− MEFs. This work is supported by NIH RO1 grant GM71729 (Z.Z.) and CA 129344 (Z.L.) and Richard Schulze Family Foundation (Z.L.).
Footnotes
Note
Supplementary materials can be found at: www.landesbioscience.com/supplement/YuanCC8-11-Sup.pdf
References
- 1.White CL, Suto RK, Luger K. Structure of the yeast nucleosome core particle reveals fundamental changes in internucleosome interactions. EMBO J. 2001;20:5207–18. doi: 10.1093/emboj/20.18.5207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Luger K. Structure and dynamic behavior of nucleosomes. Curr Opin Genet Dev. 2003;13:127–35. doi: 10.1016/s0959-437x(03)00026-1. [DOI] [PubMed] [Google Scholar]
- 3.Peterson CL, Laniel MA. Histones and histone modifications. Curr Biol. 2004;14:546–51. doi: 10.1016/j.cub.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 4.Margueron R, Trojer P, Reinberg D. The key to development: interpreting the histone code? Curr Opin Genet Dev. 2005;15:163–76. doi: 10.1016/j.gde.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 5.Narlikar GJ, Fan HY, Kingston RE. Cooperation between complexes that regulate chromatin structure and transcription. Cell. 2002;108:475–87. doi: 10.1016/s0092-8674(02)00654-2. [DOI] [PubMed] [Google Scholar]
- 6.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–80. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 7.Ozdemir A, Spicuglia S, Lasonder E, Vermeulen M, Campsteijn C, Stunnenberg HG, et al. Characterization of lysine 56 of histone H3 as an acetylation site in Saccharomyces cerevisiae. J Biol Chem. 2005;280:25949–52. doi: 10.1074/jbc.C500181200. [DOI] [PubMed] [Google Scholar]
- 8.Hyland EM, Cosgrove MS, Molina H, Wang D, Pandey A, Cottee RJ, et al. Insights into the role of histone H3 and histone H4 core modifiable residues in Saccharomyces cerevisiae. Molecular and cellular biology. 2005;25:10060–70. doi: 10.1128/MCB.25.22.10060-10070.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Masumoto H, Hawke D, Kobayashi R, Verreault A. A role for cell cycle-regulated histone H3 lysine 56 acetylation in the DNA damage response. Nature. 2005;436:294–8. doi: 10.1038/nature03714. [DOI] [PubMed] [Google Scholar]
- 10.Xu F, Zhang K, Grunstein M. Acetylation in histone H3 globular domain regulates gene expression in yeast. Cell. 2005;121:375–85. doi: 10.1016/j.cell.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 11.Recht J, Tsubota T, Tanny JC, Diaz RL, Berger JM, Zhang X, et al. Histone chaperone Asf1 is required for histone H3 lysine 56 acetylation, a modification associated with S phase in mitosis and meiosis. Proc Natl Acad Sci USA. 2006;103:6988–93. doi: 10.1073/pnas.0601676103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou H, Madden BJ, Muddiman DC, Zhang Z. Chromatin assembly factor 1 interacts with histone H3 methylated at lysine 79 in the processes of epigenetic silencing and DNA repair. Biochemistry. 2006;45:2852–61. doi: 10.1021/bi0521083. [DOI] [PubMed] [Google Scholar]
- 13.Maas NL, Miller KM, DeFazio LG, Toczyski DP. Cell cycle and checkpoint regulation of histone H3 K56 acetylation by Hst3 and Hst4. Mol Cell. 2006;23:109–19. doi: 10.1016/j.molcel.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 14.Li Q, Zhou H, Wurtele H, Davies B, Horazdovsky B, Verreault A, et al. Acetylation of histone H3 lysine 56 regulates replication-coupled nucleosome assembly. Cell. 2008;134:244–55. doi: 10.1016/j.cell.2008.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen CC, Carson JJ, Feser J, Tamburini B, Zabaronick S, Linger J, et al. Acetylated lysine 56 on histone H3 drives chromatin assembly after repair and signals for the completion of repair. Cell. 2008;134:231–43. doi: 10.1016/j.cell.2008.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garcia BA, Hake SB, Diaz RL, Kauer M, Morris SA, Recht J, et al. Organismal differences in post-translational modifications in histones H3 and H4. J Biol Chem. 2007;282:7641–55. doi: 10.1074/jbc.M607900200. [DOI] [PubMed] [Google Scholar]
- 17.Han J, Zhou H, Horazdovsky B, Zhang K, Xu RM, Zhang Z. Rtt109 acetylates histone H3 lysine 56 and functions in DNA replication. Science. 2007;315:653–5. doi: 10.1126/science.1133234. [DOI] [PubMed] [Google Scholar]
- 18.Tsubota T, Berndsen CE, Erkmann JA, Smith CL, Yang L, Freitas MA, et al. Histone H3-K56 acetylation is catalyzed by histone chaperone-dependent complexes. Mol Cell. 2007;25:703–12. doi: 10.1016/j.molcel.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rufiange A, Jacques PE, Bhat W, Robert F, Nourani A. Genome-wide replication-independent histone H3 exchange occurs predominantly at promoters and implicates H3 K56 acetylation and Asf1. Mol Cell. 2007;27:393–405. doi: 10.1016/j.molcel.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 20.Han J, Zhou H, Li Z, Xu RM, Zhang Z. Acetylation of lysine 56 of histone H3 catalyzed by RTT109 and regulated by ASF1 is required for replisome integrity. J Biol Chem. 2007;282:28587–96. doi: 10.1074/jbc.M702496200. [DOI] [PubMed] [Google Scholar]
- 21.Driscoll R, Hudson A, Jackson SP. Yeast Rtt109 promotes genome stability by acetylating histone H3 on lysine 56. Science. 2007;315:649–52. doi: 10.1126/science.1135862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sillje HH, Nigg EA. Identification of human Asf1 chromatin assembly factors as substrates of Tousled-like kinases. Curr Biol. 2001;11:1068–73. doi: 10.1016/s0960-9822(01)00298-6. [DOI] [PubMed] [Google Scholar]
- 23.Yang B, Miller A, Kirchmaier AL. HST3/HST4-dependent deacetylation of lysine 56 of histone H3 in silent chromatin. Mol Biol Cell. 2008;19:4993–5005. doi: 10.1091/mbc.E08-05-0524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Celic I, Masumoto H, Griffith WP, Meluh P, Cotter RJ, Boeke JD, et al. The sirtuins hst3 and Hst4p preserve genome integrity by controlling histone h3 lysine 56 deacetylation. Curr Biol. 2006;16:1280–9. doi: 10.1016/j.cub.2006.06.023. [DOI] [PubMed] [Google Scholar]
- 25.Michan S, Sinclair D. Sirtuins in mammals: insights into their biological function. Biochem J. 2007;404:1–13. doi: 10.1042/BJ20070140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang RH, Sengupta K, Li C, Kim HS, Cao L, Xiao C, et al. Impaired DNA damage response, genome instability and tumorigenesis in SIRT1 mutant mice. Cancer Cell. 2008;14:312–23. doi: 10.1016/j.ccr.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Michishita E, McCord RA, Berber E, Kioi M, Padilla-Nash H, Damian M, et al. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature. 2008;452:492–6. doi: 10.1038/nature06736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brachmann CB, Sherman JM, Devine SE, Cameron EE, Pillus L, Boeke JD. The SIR2 gene family, conserved from bacteria to humans, functions in silencing, cell cycle progression and chromosome stability. Genes Dev. 1995;9:2888–902. doi: 10.1101/gad.9.23.2888. [DOI] [PubMed] [Google Scholar]
- 29.Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, et al. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137–48. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- 30.Ozdemir A, Masumoto H, Fitzjohn P, Verreault A, Logie C. Histone H3 lysine 56 acetylation: a new twist in the chromosome cycle. Cell Cycle. 2006;5:2602–8. doi: 10.4161/cc.5.22.3473. [DOI] [PubMed] [Google Scholar]
- 31.Downs JA. Histone H3 K56 acetylation, chromatin assembly, and the DNA damage checkpoint. DNA Repair (Amst) 2008;7:2020–4. doi: 10.1016/j.dnarep.2008.08.016. [DOI] [PubMed] [Google Scholar]
- 32.Yuan Z, Zhang X, Sengupta N, Lane WS, Seto E. SIRT1 regulates the function of the Nijmegen breakage syndrome protein. Mol Cell. 2007;27:149–62. doi: 10.1016/j.molcel.2007.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oberdoerffer P, Michan S, McVay M, Mostoslavsky R, Vann J, Park SK, et al. SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell. 2008;135:907–18. doi: 10.1016/j.cell.2008.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim JE, Chen J, Lou Z. DBC1 is a negative regulator of SIRT1. Nature. 2008;451:583–6. doi: 10.1038/nature06500. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.