

Abstract
Rationale
Oxidants are important signaling molecules known to increase endothelial permeability, although the mechanisms underlying permeability regulation are not clear.
Objective
To define the role of caveolin-1 in the mechanism of oxidant-induced pulmonary vascular hyperpermeability and edema formation.
Methods and Results
Using genetic approaches, we show that phosphorylation of caveolin-1 Tyr14 is required for increased pulmonary microvessel permeability induced by hydrogen peroxide (H2O2). Caveolin-1 deficient mice (cav-1-/-) were resistant to H2O2-induced pulmonary vascular albumin hyperpermeability and edema formation. Furthermore, the vascular hyperpermeability response to H2O2 was completely rescued by expression of caveolin-1 in cav-1-/- mouse lung microvessels, but was not restored by the phosphorylation-defective caveolin-1 mutant. The increase in caveolin-1 phosphorylation induced by H2O2 was dose-dependently coupled to both increased 125I-albumin transcytosis and decreased transendothelial electrical resistance in pulmonary endothelial cells. Phosphorylation of caveolin-1 following H2O2 exposure resulted in the dissociation of vascular endothelial cadherin/β-catenin complexes, and resultant endothelial barrier disruption.
Conclusions
Caveolin-1 phosphorylation-dependent signaling plays a crucial role in oxidative stress-induced pulmonary vascular hyperpermeability via transcellular and paracellular pathways. Thus, caveolin-1 phosphorylation may be an important therapeutic target for limiting oxidant-mediated vascular hyperpermeability, protein-rich edema formation, and acute lung injury.
Keywords: vascular endothelial barrier, transcytosis, adherens junctions, caveolin-1, lung edema
Introduction
An increase in vascular permeability is a key hallmark of inflammation and has been implicated in the pathophysiology of many disease states including acute lung injury, ischemia-reperfusion injury, atherosclerosis, and diabetes.1 Under physiological conditions, adherens junctions predominate in endothelial cell-cell contacts and control pulmonary endothelial barrier integrity. Stabilization of adherens junctions is dependent on the association of vascular endothelial (VE)-cadherin, β-catenin, p120-catenin, and α-catenin proteins and their linkage to the actin cytoskeleton2 wherein VE-cadherin association with the actin cytoskeleton is thought to be dependent on the β-catenin linkage.3 Oxidants including superoxide and hydrogen peroxide (H2O2) generated by activated neutrophils and endothelial cells in response to inflammatory stimuli increase paracellular endothelial permeability by promoting the loss of cell–cell adhesion and activation of actin-myosin based cell retraction.1,4,5
We recently demonstrated that caveolae-mediated transendothelial transport (transcytosis) of macromolecules through the microvascular endothelial barrier is also an important mechanism responsible for inflammation-evoked pulmonary vascular hyperpermeability and protein-rich edema formation.6 We showed that an increase in transcellular (caveolae-mediated) permeability, triggered by the binding of neutrophils to endothelial cell surface intercellular adhesion molecule-1 (ICAM-1), was mediated by Src activation and phosphorylation of caveolin-1 (Cav-1)6 which thereby stimulates caveolae formation and trafficking.7-11 ICAM-1 expression and activation is mediated by oxidant signaling in lung endothelial cells.1 In addition, oxidants have also been shown to directly induce Src-dependent phosphorylation of Cav-1 at tyrosine 14 (Tyr14) in endothelial cells.12-14 Thus, oxidants generated by neutrophils and/or endothelial cells may serve as signal transduction mediators that regulate transcellular permeability through a Cav-1 dependent mechanism.
Cav-1 expression is required for caveolae-mediated endocytosis and transcytosis in endothelial cells.4,6,7-11,15-17 Recent evidence also points to the potential role of Cav-1 in cell-cell adhesion and thus paracellular permeability regulation. Cav-1 co-localizes with adherens junction proteins E-cadherin, β-catenin and γ-catenin in MDCK epithelial cells.18 Downregulation of Cav-1 leads to a loss and redistribution of tight junction proteins (occludin and ZO-1) in brain microvascular endothelial cells resulting in an increase in paracellular permeability.19 Furthermore, siRNA-mediated depletion of Cav-1 in the mouse lung induced an increase in the number of interendothelial gaps in pulmonary capillaries and veins.20 In contrast, deletion of Cav-1 attenuated protein kinase C (PKC)-induced inter-endothelial gap formation in myocardial microvascular endothelial cells.21 Whether and how Cav-1 regulates endothelial barrier function during oxidative stress remains an important question.
In the present study, using genetic approaches, we investigated the role of Cav-1 in pulmonary microvascular permeability regulation through both transcellular and paracellular pathways. We found that Cav-1 phosphorylation is required for H2O2-induced stimulation of transcytosis and destabilization of cell-cell junctions, and hence propose that tyrosine phosphorylation of Cav-1 plays an important role in the pathogenesis of oxidant-induced pulmonary vascular hyperpermeability.
Materials and Methods
An expanded Materials and Methods section is available in the online supplement.
Briefly, Cav-1 null (cav-1-/-) mice, wild-type B6/129SJ2 mice, and rat lung microvascular endothelial cells (RLMVECs) were used. Animal protocols received institutional review and committee approval. Endocytosis and transendothelial transport of 125I-albumin, fluorescent albumin uptake, transendothelial electrical resistance (TER), siRNA transfection, Western blotting and immunoprecipitation were performed as described previously.6 Rescue studies were made in mouse lungs from cav-1-/- mice by liposome-mediated plasmid cDNA transfection.6 125I-albumin permeability-surface area (PS) product was measured in lungs perfused with Krebs solution.
Results
H2O2 Induced Cav-1 Phosphorylation via Src and c-Abl Kinases
H2O2 in a concentration-dependent manner (0.05∼0.8 mmol/L) increased Cav-1 Tyr 14 phosphorylation (p(Y14)-Cav-1; Figure 1A). Quantitative analysis revealed that the level of phospho-Cav-1 was increased by 2- to 9-fold (Online Figure IA) following exposure to H2O2. Furthermore, Cav-1 phosphorylation increased within 5 min, peaked at 30 min, and then returned to basal levels 60 min after H2O2 treatment (Figure 1B and Online Figure IB). Coincident with the increase in p(Y14)-Cav-1 levels, we also observed activation of c-Src in endothelial cells following treatment with H2O2 as measured by p(Y418)-Src phospho-immunoblot (Figure 1A and Online Figure IA). Pretreatment of endothelial cells with the Src inhibitor PP2 (4-amino-5-(4-chlorophenyl)-7-(t-butyl) pyrazolo[3,4-d] pyrimidine) completely blocked Cav-1 phosphorylation following exposure of cells to 0.2 mmol/L H2O2, but it only blocked ∼ 65% of the effect of higher concentrations of H2O2 (0.6 mmol/L) (Figure 1C and Online Figure IC) suggesting Src-independent pathways may also be involved in the mechanism of Cav-1 phosphorylation induced by high concentration of H2O2.
Figure 1. H2O2-induced activation of Src and c-Abl and resultant phosphorylation of Cav-1.

(A) H2O2 induced the activation of Src and phosphorylation of Cav-1 in a concentration-dependent manner. RLMVECs were exposed to different concentrations of H2O2 (0.01-0.8 mmol/L) for 30 min. (B) Time course of H2O2-induced Cav-1 phosphorylation. Cells were stimulated with 0.6 mmol/L H2O2 for indicated times. (C) Effect of PP2 on Cav-1 phosphorylation induced by different concentrations of H2O2. (D) Dose-dependent c-Abl phosphorylation induced by H2O2. Cells were exposed to vehicle or H2O2 (0.1-0.6 mmol/L) for 30 min. c-Abl phosphorylation was measured by immunoprecipitation with c-Abl antibody and immunoblotting with phosphotyrosine antibody. Immunoblot of total c-Abl is shown as a loading control. (E) Effect of c-Abl siRNA on Cav-1 phosphorylation induced by the different concentrations of H2O2. (F) Effect of PP2 on H2O2-stimulated c-Abl activation. Cells were pretreated with PP2 or vehicle for 15 min and then stimulated with H2O2 (0.6 mmol/L) for 30 min. All blots are representative of 3 separate experiments.
H2O2 has been reported to induce Cav-1 phosphorylation via a c-Abl-dependent but Src-independent mechanism in fibroblasts.22 Therefore, we examined the potential role of c-Abl kinase in the regulation of Cav-1 phosphorylation in pulmonary endothelial cells. As shown in Figure 1D and Online Figure ID, H2O2 induced the phosphorylation (activation) of c-Abl kinase in a concentration-dependent manner. Downregulation of c-Abl with specific siRNA reduced Cav-1 phosphorylation induced by high but not low concentration of H2O2 (Figure 1E and Online Figure IE). To determine whether Src and c-Abl are activated by H2O2 independently of one another, we measured H2O2-induced c-Abl activation in the presence and absence of PP2. As shown in Figure 1F, PP2 significantly blocked H2O2 (0.6 mmol/L)-induced activation of c-Abl kinase (Online Figure IF) suggesting that c-Abl activation by oxidants occurs at least in part via activation of Src kinase.
Tyrosine Phosphorylation of Cav-1 Signals H2O2-Induced Transcellular Albumin Hyperpermeability
To evaluate whether H2O2 stimulates transcellular albumin permeability, we first addressed the possibility that H2O2 facilitates caveolae-mediated endocytosis of albumin in endothelial cells, the initial step in albumin transport via transcytosis.1,6,7-11,15-17 As shown in Figure 2A and 2B, H2O2 caused a concentration-dependent increase in Alexa 488 albumin and 125I-albumin endocytosis in endothelial cells. Confocal images (Figure 2A) indicated that albumin was internalized by endocytic vesicles and that exposure of endothelial cells to H2O2 induced a significant increase in albumin uptake. In parallel, tracer 125I-albumin uptake by endothelial cells following exposure to low dose H2O2 (0.05-0.2 mmol/L) was significantly increased (Figure 2B). We next determined the effect of H2O2 on transcytosis of 125I-albumin using a well-established technique.6,23 The concentration range of H2O2 (0.05-0.2 mmol/L) was chosen because at these levels, H2O2 did not disrupt the endothelial barrier (Figure 3A). Consistent with the activation of endocytosis, H2O2 induced a concentration-dependent increase in transendothelial 125I-albumin transport (Figure 2C). These data suggest that H2O2 stimulates transcellular albumin transport via a vesicular pathway.
Figure 2. Phosphorylation of caveolin-1 is required for H2O2-induced endocytosis and transendothelial albumin transport.

(A) Confocal images showing H2O2-induced concentration-dependent increase in the uptake of Alexa 488-labeled albumin (green). The nucleus (blue) was stained with DAPI. Scale bars = 10 μm. Results are typical of 3 experiments. (B) H2O2 increased 125I-albumin endocytosis in a concentration-dependent manner. (C) H2O2 induced a concentration-dependent increase in transendothelial transport of 125I-albumin. (D,E) Effect of H2O2 on 125I-albumin endocytosis (D) and transendothelial albumin permeability (E) in cells stably expressing WT and phosphorylation-defective Y14F-Cav-1 mutant. (F,G) Effect of Src inhibition and downregulation of c-Abl kinase on H2O2-induced increase in 125I-albumin endocytosis (F) and transcytosis (G). n = 4-6 for each group. The baseline permeability values for WT and control groups were 7.8±1.2 μl·min-1·cm-2×10-2 (E) and 6.1±1.1 μl·min-1·cm-2×10-2 (G), respectively. *P <0.05 compared with control (B, C, F, and G) and WT (D, E) groups, †P <0.05 compared with respective (D,E) and control (F,G) groups.
Figure 3. Phosphorylation of Cav-1 is required for signaling H2O2-induced endothelial barrier disruption.

RLMVECs were grown to confluence, treated with H2O2 at the concentration indicated, and TER was recorded. In the panels on the left, the original TER recordings are shown, and in the panels on the right, the mean value (±SEM) of the peak TER responses to H2O2 (relative to the starting value) is plotted. (A) Dose-response relationship of H2O2-induced decrease in TER; note there was no effect of ∼0.2 mmol/L H2O2. (B) Phospho-defective Y14F Cav-1 mutant blocks the effect of H2O2 on TER. Over-expression of WT-Cav-1 in RLMVECs reduced the threshold of H2O2 needed to decrease TER. (C) Effect of Src inhibitor PP2 and downregulation of c-Abl kinase on H2O2-induced decrease in TER. n = 4-6 for each group. *P < 0.05, compared to control (untreated) groups; †P < 0.05 compared with respective H2O2 groups.
To further assess the mechanism of vesicular albumin transport, cells were pretreated with the caveolae disrupting agent methyl-β-cyclodextrin and Cav-1 siRNA. As shown in Online Figure IIA and IIB, methyl-β-cyclodextrin prevented H2O2-induced increase in endocytosis and transcytosis of 125I-albumin. Furthermore, H2O2 increased albumin endocytosis and transendothelial 125I-albumin flux in endothelial cells transduced with scrambled siRNA, whereas Cav-1 knockdown by ∼90% abolished these effects.
To address the role of Cav-1 phosphorylation in H2O2-induced increase in transendothelial albumin permeability, we measured H2O2-induced endocytosis and transendothelial transport of 125I-albumin in RLMVEC lines overexpressing phosphorylation-defective Y14 F Cav-1 mutant (Y14F-Cav-1) and as a control, wild-type caveolin-1 (WT-Cav-1).6,24 H2O2 (0.2 mmol/L)-induced increase in endocytosis and transcytosis of 125I-albumin was significantly attenuated by Y14F-Cav-1 expression compared to WT-Cav-1 expressing cells (Figure 2D and 2E). Moreover, inhibition of Cav-1 phosphorylation by Src inhibitor PP2 reduced H2O2-stimulated endocytosis and transendothelial albumin permeability (Figure 2F and 2G) whereas downregulation of c-Abl kinase did not affect the endocytosis or transcytosis of albumin (Figure 2F and 2G).
Tyrosine Phosphorylation of Cav-1 Signals H2O2-Induced Paracellular Hyperpermeability
H2O2 at higher concentrations (0.4-0.8 mmol/L) induced inter-endothelial cell gap formation, detected as a reduction in TER, whereas H2O2 at lower concentrations (0.05-0.2 mmol/L) did not affect TER (Figure 3A). The effect of 0.4 and 0.6 mmol/L H2O2 was reversible in that TER returned to basal levels (4.7±0.7 Ω·cm2) within 5 hours, whereas endothelial cells treated with 0.8 mmol/L H2O2 did not fully recover (Figure 3A). Accordingly, 0.6 mmol/L H2O2 was chosen for subsequent experiments to explore the mechanism of H2O2-induced loss of monolayer integrity (increase in paracellular permeability).
To determine whether Cav-1 phosphorylation signals H2O2-induced increase in paracellular permeability, we measured H2O2-induced changes in TER in WT-and Y14F-Cav-1 expressing RLMVEC lines. The effect of low and high levels of Cav-1 phosphorylation on TER, induced by 0.2 and 0.6 mmol/L H2O2 respectively, was evaluated in endothelial monolayers. As shown in Figure 3A, 0.2 mmol/L H2O2 did not alter TER in native endothelial cells (non-transfected), but remarkably decreased TER in cells overexpressing WT-Cav-1 (Figure 3B). Importantly, 0.2 mmol/L H2O2 had no effect on TER in cells expressing the same level of mutant Y14F-Cav-1. The decrease in TER induced by 0.6 mmol/L H2O2 in endothelial cells expressing WT-Cav-1 was also significantly greater than the response observed in native cells and endothelial cells expressing Y14F-Cav-1 (Figure 3A and 3B). Inhibition of Cav-1 phosphorylation by Src inhibitor PP2 or c-Abl siRNA similarly reduced the magnitude of the H2O2 (0.6 mmol/L)-induced decrease in TER and resembled the response observed in cells expressing the phosphorylation-defective Cav-1 mutant. One difference noted was that the response was more transient in cells treated with c-Abl siRNA compared to PP2-treated cells (Figure 3C).
Cav-1 Phosphorylation Mediates H2O2-Induced Dissociation of VE-cadherin and β-catenin
To gain further insight into the regulatory mechanism of oxidant-induced endothelial barrier disruption and increase in paracellular permeability, we investigated the effect of H2O2-induced Cav-1 phosphorylation on the stability of adherens junctions by assessing VE-cadherin/β-catenin complexes using confocal microscopy and immunoprecipitation analysis. Confocal images showed significant co-localization of Cav-1 and β-catenin at cell-cell borders (Figure 4A) and immunoprecipitation studies also demonstrated an association between Cav-1 with β-catenin at baseline (Figure 4B). However, the association between Cav-1 and β-catenin at the cell borders observed by immunostaining and as well as the co-immunoprecipitated proteins measured in the immunoblots were reduced by ∼ 25% and 75% following exposure of endothelial cells to 0.2 and 0.6 mmol/L H2O2, respectively (Figure 4B).
Figure 4. Caveolin-1 phosphorylation mediates H2O2-induced dissociation of caveolin-1 from β-catenin.
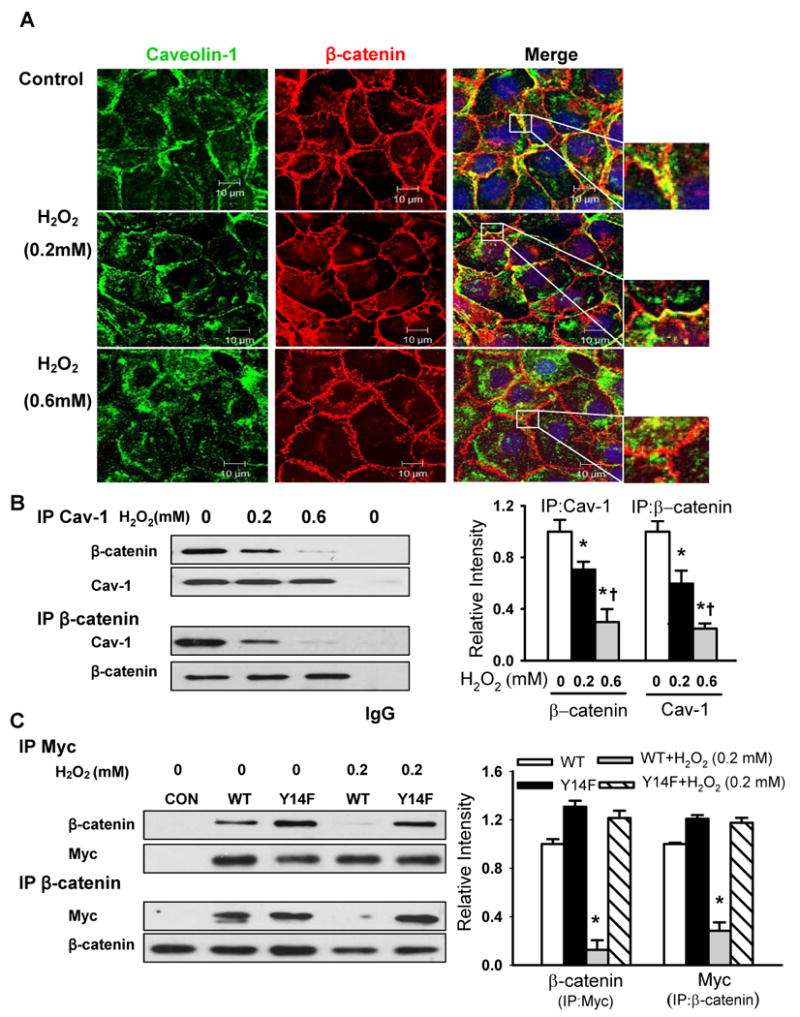
(A) Effects of H2O2 on the co-localization of caveolin-1 (green) and β-catenin (red) in naïve RLMVECs. Cells were exposed to different concentrations of H2O2 for 30 min. The nucleus (blue) was stained with DAPI. Scale bars =10 μm. (B, C) The association of endogenous caveolin-1 (B) or over-expressed Myc-tagged WT-Cav-1 or Y14F-Cav-1 mutant (C) with β-catenin in naïve endothelial cells and stable endothelial cell lines was determined by immunoprecipitation and immunoblot (IP/IB) with anti-caveolin-1, anti-Myc, or anti-β-catenin antibodies. Left, representative Western blots for β-catenin and caveolin-1 (or Myc); Right, protein quantification by densitometry. The density of proteins in each untreated control group was used as a standard (1 arbitrary unit) to compare the relative density of the other groups. *P < 0.05 compared to control (untreated) groups; † P < 0.05 compared with H2O2 (0.2 mmol/L) groups.
To assess the role of Cav-1 phosphorylation in the mechanism of H2O2-induced dissociation of Cav-1 and β-catenin, RLMVEC lines expressing Myc-tagged WT-Cav-1 or Y14F-Cav-1 were stimulated, lysed, and immunoprecipitated with anti-Myc or anti-β-catenin antibodies. As shown in Figure 4C, β-catenin was associated with WT-Cav-1 and to a greater extent with Y14F-Cav-1 in untreated cells, suggesting that β-catenin binds Cav-1 in the non-phosphorylated state. Stimulation of cells with 0.2 mmol/L H2O2 led to a decrease in association between β-catenin and WT-Cav-1 whereas H2O2 had no effect on the association between β-catenin and Y14F-Cav-1 (Figure 4C). These data strongly argue that phosphorylation of Cav-1 leads to its dissociation from β-catenin.
We next determined whether the dissociation of Cav-1 from β-catenin induced by Cav-1 phosphorylation also led to the disruption of VE-cadherin/β-catenin complexes. As shown in Figure 5A, association between VE-cadherin and β-catenin was reduced by 15% and 80% upon exposure to 0.2 and 0.6 mmol/L H2O2 compared to untreated endothelial cells. Furthermore, H2O2- induced dissociation of VE-cadherin and β-catenin observed in WT-Cav-1 expressing cells was abolished in Y14F-Cav-1 expressing cells (Figure 5B).
Figure 5. Caveolin-1 phosphorylation mediates H2O2-induced dissociation of VE-cadherin and β-catenin.
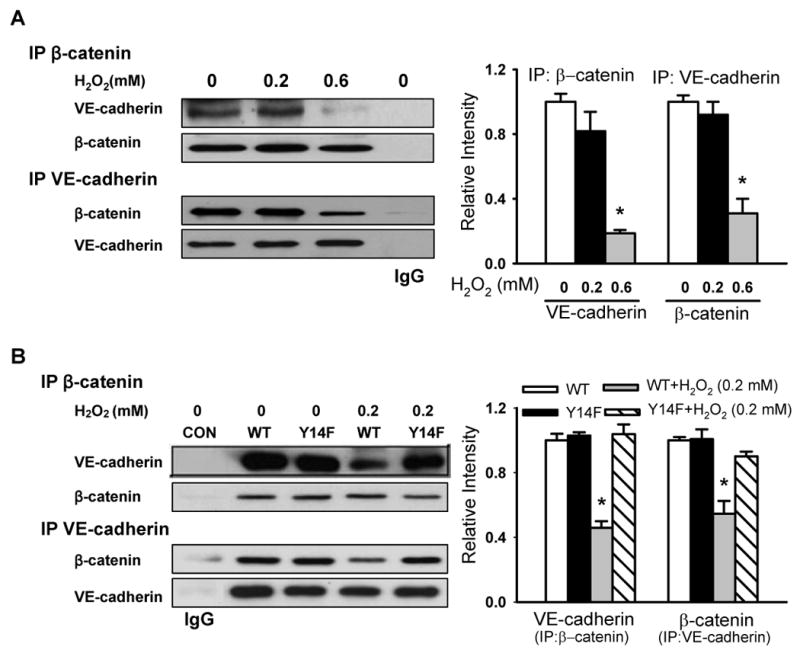
The association of VE-cadherin with β-catenin was determined by immunoprecipitation and immunoblot analysis using anti-β-catenin or anti-VE-cadherin antibodies. Left, representative Western blots for VE-cadherin and β-catenin; Right, protein quantification by densitometry. The density of proteins in each untreated (control) group was used as a standard (1 arbitrary unit) to compare the relative density in the other groups. (A) Effect of H2O2 on the association of VE-cadherin with β-catenin in naïve endothelial cells. Note dissociation upon exposure to 0.6 mmol/L H2O2. (B) Effect of H2O2 on the association of VE-cadherin with β-catenin in cells stably expressing WT and phosphorylation-defective Y14F-Cav-1 mutant. *P < 0.05, compared to control (untreated) group (A) or WT alone group (B); n = 3/each group.
To further assess whether β-catenin translocates from the membrane following H2O2 stimulation at the higher concentration (0.6 mmol/L), cells were treated, lysed, fractionated, and immunoblotted. Figure 6A shows β-catenin predominantly localized in the membrane basally and then in the cytosolic compartment after treatment of endothelial cells with 0.6 mmol/L H2O2. Moreover, we observed H2O2-induced β-catenin translocation in WT-Cav-1 expressing cells but not in Y14F-Cav-1 expressing cells (Figure 6B). Similarly, confocal imaging showed wide-spread gap formation, increased β-catenin translocation into the cytosol, and a loss of β-catenin staining in membranes at sites of cell-cell contact in WT-Cav-1 expressing cells whereas β-catenin localization was unaffected and gaps in the monolayer did not form in Y14F-Cav-1 mutant expressing cells following exposure to 0.2 mmol/L H2O2 (Online Figure III). Thus, Cav-1 phosphorylation leads to its dissociation from β-catenin, disruption of β-catenin/VE-cadherin complexes, and β-catenin translocation from the membrane to cytosolic compartments.
Figure 6. Caveolin-1 phosphorylation is required for H2O2-induced β-catenin redistribution.
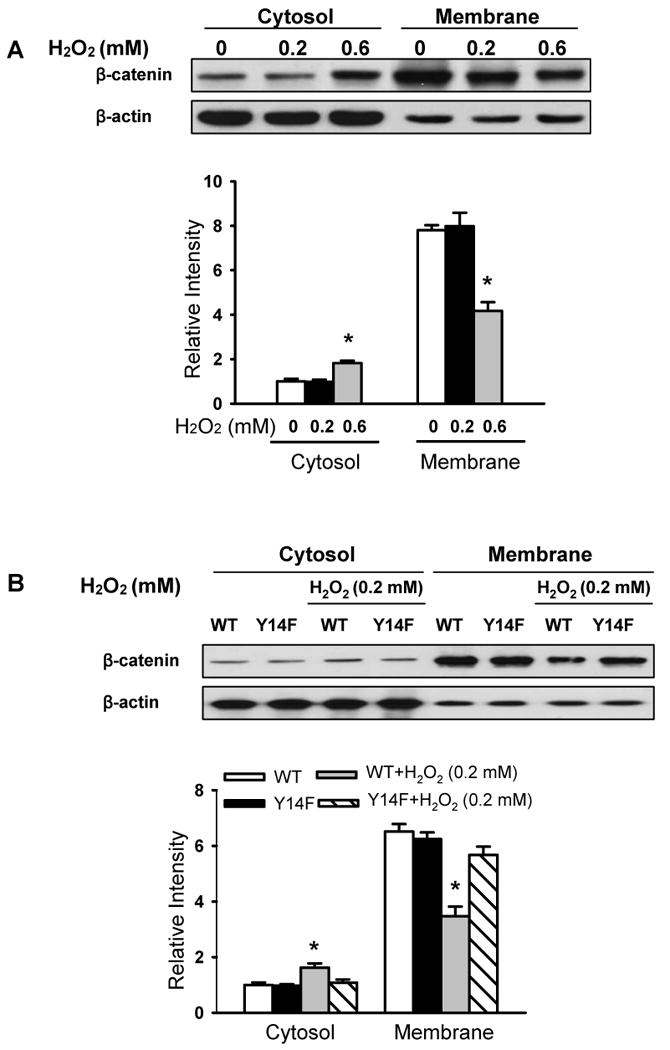
Cells were exposed to the indicated concentrations of H2O2 for 30 min and lysates separated into cytosolic and membrane fractions followed by Western blotting with β–catenin antibody to determine β–catenin localization. Top panel shows a representative Western blot for β-catenin and β-actin loading control; bottom panels show protein quantification by densitometry. The density of proteins in each untreated control group was used as a standard (1 arbitrary unit) to compare relative densities in the other groups. (A) H2O2 at 0.6 mmol/L concentration induced β-catenin redistribution from the membrane to cytosol in RLMVECs. (B) H2O2 (0.2 mmol/L) induced the translocation of β-catenin from cytosol to membrane in a manner dependent on Cav-1 phosphorylation in Cav-1 over-expressing cells. The redistribution of β-catenin was observed in cells stably expressing WT but not the phosphorylation-defective Y14F-Cav-1 mutant. n = 3. *P < 0.05, compared to control (untreated) group (A) or WT alone group (B).
H2O2-Induced Vascular Albumin Hyperpermeability and Lung Edema Formation Requires Cav-1 Phosphorylation in Mouse Lungs
To address whether phosphorylation of Cav-1 mediates H2O2-induced increase in pulmonary vascular hyperpermeability and edema formation, we infused H2O2 into isolated, perfused mouse lungs. As shown in Figure 7A, H2O2 (0.5 mmol/L) induced a robust increase in Cav-1 phosphorylation in wild type (WT) mouse lungs. We chose 0.5 mmol/L H2O2 as this dose reliably induced an increase in 125I-albumin permeability (PS product) (Figure 7B) and lung edema formation (wet/dry ratio) (Figure 7C) in the isolated lung. In contrast, in cav-1-/- mouse lungs, which exhibit reduced basal albumin PS (Figure 7B), H2O2 did not induce an increase in albumin and fluid permeability. To assess the role of Cav-1 phosphorylation in these permeability enhancing-effects of H2O2 in vivo, we used liposome-mediated Cav-1 cDNA delivery to rescue vascular endothelial Cav-1 expression in cav-1-/- mouse lungs6,25,26 with either WT-Cav-1 or the phosphorylation-defective Y14F-Cav-1 mutant (Figure 7A). Consistent with the in vitro data, H2O2 induced an increase in albumin PS product and lung wet/dry weight ratio in WT-Cav-1 expressing mouse lungs, whereas these effects were not reconstituted upon expression of Y14F-Cav-1 in cav-1-/- lungs (Figure 7B and 7C).
Figure 7. H2O2-induced increase in lung microvessel albumin permeability and pulmonary edema formation requires caveolin-1 phosphorylation.
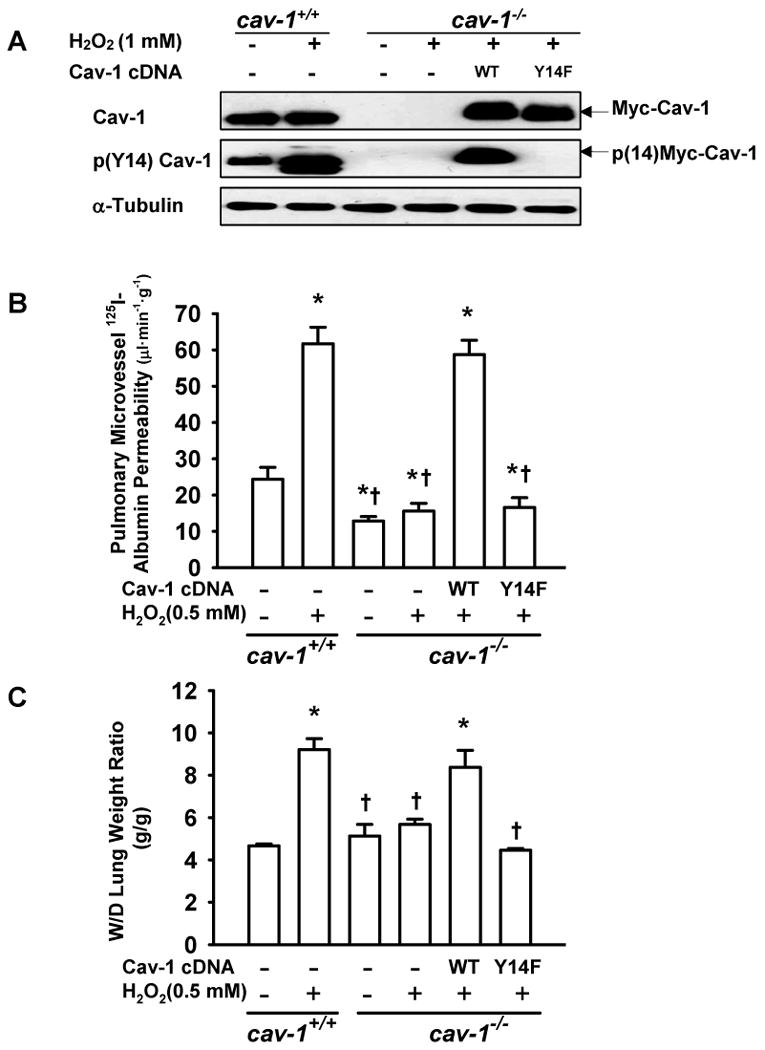
Cav-1-/- mice were injected intravenously with liposomes containing WT- or Y14F-Cav-1 cDNA. After 48 h, lungs were isolated and perfused with H2O2 (0.5 mmol/L) for 30 min. (A) Western blots show increase in Cav-1 phosphorylation in whole lung homogenates induced by H2O2, the absence of Cav-1 in null mice, and exogenous expression of Myc-tagged WT-Cav-1 and Y14F-Cav-1 in cav-1-/- lungs. Note that H2O2 induced phosphorylation of reconstituted WT-Cav-1 in the isolated mouse lung. (B) Effect of H2O2 on pulmonary microvessel 125I-albumin permeability (PS product) in wild-type (cav-1+/+) and cav-1-/- lungs with or without rescue with WT-Cav-1 or Y14F-Cav-1 mutant cDNA. (C) H2O2 induced and increase in lung wet/dry (W/D) weight ratio in cav-1+/+ lungs but not in cav-1-/- lungs. Rescue of Cav-1 expression with WT-Cav-1 but not Y14F-Cav-1 cDNA restored the lung edema response to H2O2. n = 6/each group. * P < 0.05 compared with control groups (cav-1+/+ mouse without H2O2 treatment); †P < 0.05 compared with cav-1+/+ mouse with H2O2 treatment group.
Discussion
This study provides strong evidence indicating that Cav-1 phosphorylation in endothelial cells plays a fundamental role in the mechanism of oxidant-induced pulmonary vascular hyperpermeability. Oxidant-induced increase in caveolae-mediated albumin transport and decrease in endothelial barrier integrity were both dependent on tyrosine phosphorylation of Cav-1, suggesting Cav-1 phosphorylation is a common signal regulating transcellular and paracellular permeability pathways in lung microvessels. The molecular mechanisms regulating transcellular and paracellular permeability induced by Cav-1 phosphorylation following oxidant stress appear to be different. The increase in transcellular permeability in response to low concentration of oxidant is entirely mediated by Src-dependent Cav-1 phosphorylation, whereas increased paracellular permeability induced by high concentration of oxidant depends on both Src- and c-Abl-mediated Cav-1 phosphorylation and subsequent regulation of β-catenin localization.
We established that exposure of pulmonary microvascular endothelial cells to pathophysiological concentrations of H2O2 (0.05-0.8 mmol/L)27 stimulated Cav-1 phosphorylation in a concentration-dependent manner. Our results are distinct from previous reports which showed that higher concentrations of H2O2 (≥ 1 mmol/L) were required to induce an increase in Cav-1 phosphorylation in cultured bovine pulmonary artery,12 aortic,13,14 and human umbilical vein14 endothelial cells.
Cav-1 Tyr14 is a principal target for Src kinase phosphorylation in response to oxidative stress,12-14,24,28 and thus we predicted that treatment of endothelial cells with Src inhibitor PP2 to inactivate Src kinase6 would block H2O2-induced Cav-1 Tyr14 phosphorylation. However, our results demonstrated that the level of phospho-Cav-1 following PP2 treatment was only decreased by approximately 65%, suggesting the existence of a Src-independent pathway, particularly when higher concentrations of H2O2 were used to stimulate Cav-1 phosphorylation. Depletion of c-Abl with specific siRNA demonstrated that c-Abl also contributed to H2O2-induced Cav-1 phosphorylation in pulmonary microvascular endothelial cells. In contrary to previous findings that c-Abl phosphorylates Cav-1 independently of Src,22,29 our results showed that c-Abl activation and subsequent Cav-1 phosphorylation was in part dependent on Src activity. It is likely that Src family kinases directly phosphorylate tyrosine residues in the kinase domain of c-Abl, leading to enhanced activity.30
Studies from our laboratory6,7,9-11 and others16,17 have suggested that Cav-1 phosphorylation plays an essential role in the mechanisms regulating caveolae formation and caveolae-mediated endocytosis and transcytosis of albumin in microvascular endothelial cells. Using 125I-albumin and Alexa 488-albumin tracers, we observed that H2O2 (0.05-0.2 mmol/L) increased both the endocytosis and transcytosis of albumin. Cholesterol-depleting agent methyl-β-cyclodextrin and siRNA-induced depletion of Cav-1 prevented H2O2-induced endocytosis and transcytosis of albumin, indicative of a caveolae-dependent mechanism. Furthermore, the H2O2-induced increase in endocytosis and transcytosis of 125I-albumin in WT-Cav-1 expressing cells was abolished in phospho-defective Cav-1 mutant over-expressing endothelial cell line. Noteworthy is that Src inhibitor PP2 but not c-Abl siRNA inhibited endocytosis and transcytosis of albumin induced by H2O2. These findings provide further support for the concept that Src-dependent phosphorylation of Cav-1 mediates oxidant-induced transcellular transport via caveolae in pulmonary microvascular endothelial cells.
The present data for the first time show that Cav-1 phosphorylation, which is an important mechanism regulating caveolae-mediated transcellular permeability,7-11 is also a crucial regulator of oxidant-induced paracellular hyperpermeability. Expression of a Cav-1 mutant lacking the tyrosine phosphorylation site resulted in the inhibition of H2O2-induced decrease in TER, a measure of loss of endothelial junctional integrity. Furthermore, overexpression of wild type Cav-1 in endothelial cells reduced the threshold concentration of H2O2 needed to disrupt endothelial cell-cell junctions, presumably due to an increase in the total amount of phosphorylated Cav-1.6,22 In agreement with these findings, inhibition of Cav-1 phosphorylation by either Src inhibitor PP2 or c-Abl siRNA partially attenuated H2O2-induced decrease in TER. These results highlight the importance of Cav-1 Tyr14 phosphorylation in the regulation of H2O2-induced endothelial barrier disruption. A recent study showed that oxidized phospholipid-induced Cav-1 phosphorylation is associated with sphingosine 1-phosphate receptor signaling in caveolae that leads to endothelial barrier enhancement.31 However, it remains unclear whether Cav-1 phosphorylation per se plays an important signaling role in strengthening the endothelial barrier in this scenario.
In cav-1-/- 32 and Cav-1 siRNA-treated20 mice, an increase in the number of open inter-endothelial junctions in pulmonary capillaries and veins was observed, indicating there is a compensatory mechanism that increases basal paracellular permeability in the absence of the vesicular (transcellular) permeability pathway.33 In our study, however, both transcellular and paracellular permeability pathways were stimulated upon Cav-1 phosphorylation, suggesting these two pathways are affected in a coordinate-manner in response to oxidative stress. Consistent with previous findings,6,34 the present study demonstrated that the increase in transendothelial permeability occurred prior to an increase in paracellular permeability. These data suggest that the mechanism mediating increased transcellular permeability, namely Src phosphorylation of Cav-1, is also a trigger signal for increasing paracellular permeability. Thus, it is likely that the cooperative mechanism between the two pathways via Cav-1 phosphorylation may play an important role in the development and progression of vascular hyperpermeability and inflammatory lung injury.
Recent studies indicate the important role of Cav-1 in stabilization of adherens junctions.19,35 Knockdown of Cav-1 was shown to be associated with a significant decrease in both VE-cadherin and β-catenin localized at inter-endothelial junctions.19 In our studies with confluent endothelial cell monolayers, we observed that Cav-1 co-localized with adherens junction-associated protein β-catenin, consistent with a previous study conducted in MDCK cells.18 Confocal imaging and immunoprecipitation studies indicated that exposure of endothelial cells to higher concentrations of H2O2 caused a marked dissociation of β-catenin from Cav-1 as well as VE-cadherin. H2O2-induced Src activation may also promote direct phosphorylation of VE-cadherin and/or β-catenin, inducing their dissociation from cytoskeletal anchors.1,4,36 However, using a Cav-1 overexpressing endothelial cell line, we showed that dissociation of β-catenin from Cav-1 and VE-cadherin following exposure to H2O2 was dependent on Cav-1 Tyr14 phosphorylation as these changes did not occur in endothelial cells expressing the phosphorylation-defective Cav-1 mutant. Furthermore, Cav-1 phosphorylation promoted the translocation of β-catenin from the membrane to cytosol. These data support a model in which, when phospho-Cav-1 is present in cell-cell junctions at significant levels, it negatively regulates adherens junction stability by decreasing the association between VE-cadherin and β-catenin. At this stage, we cannot rule out the possibility that Cav-1-mediated endocytosis of VE-cadherin and/or β-catenin may lead to the disassembly of adherens junction complexes and thereby contribute to the increase in paracellular permeability.37 While Cav-1 phosphorylation is known to regulate caveolae-mediated endocytosis, as also shown herein, this explanation of β-catenin internalization is less plausible knowing that the co-immunoprecipitation of β-catenin with Cav-1 decreased upon Cav-1 phosphorylation.
The significance of Cav-1 phosphorylation as a molecular signal in endothelial permeability regulation is not clear. Phosphorylation of Cav-1 may reduce interactions between Cav-1 and other signaling proteins that are negatively regulated by association with Cav-1.38 Increased Cav-1 phosphorylation may also be necessary for translocation of proteins normally associated with Cav-1 at the plasma membrane.39 Our data support these hypotheses, as indicated by the dissociation of Cav-1 and β-catenin and subsequent translocation of β-catenin from the membrane to the cytosol in a manner dependent on increased Cav-1 phosphorylation induced by high concentration of H2O2.
We also found that H2O2-induced increase in pulmonary vascular permeability was dependent on Cav-1 Tyr14 phosphorylation in vivo, consistent with our findings from pulmonary endothelial monolayers. Mice lacking Cav-1 expression were resistant to H2O2-induced increase in pulmonary vascular hyperpermeability and edema formation. The pulmonary vascular response to H2O2 was rescued by adding back WT-Cav-1 to the cav-1-/- pulmonary vasculature, but not by expression of the phospho-defective Cav-1 mutant. These findings, together with those from pulmonary endothelial cells, strongly suggest that Cav-1 phosphorylation plays a pivotal role in the mechanism of increased pulmonary vascular permeability and edema formation evoked by oxidants.
In summary, our findings implicate Cav-1 phosphorylation as a critical mechanism mediating oxidant-induced pulmonary vascular hyperpermeability. H2O2 stimulated caveolae-mediated transcellular transport and the opening of inter-endothelial junctions in a manner dependent on Cav-1 phosphorylation. Although these studies focused on oxidative stress as a means of increasing endothelial permeability and inducing acute lung injury, it is possible that our findings have broader applicability in understanding the mechanisms and development of inflammatory pulmonary vascular hyperpermeability. Therefore, therapeutic inhibition of Cav-1 phosphorylation may be an effective means of limiting lung vascular injury by preventing increased transcellular albumin permeability and stabilizing the endothelial junctional barrier.
Supplementary Material
Legends to Supplemental Figures
Online Figure I. H2O2 induced Src and c-Abl activation and caveolin-1 (Cav-1) phosphorylation. Densitometric analysis of phosphorylated caveolin-1 (p(Y14) Cav-1), Src (p(Y418) Src) and c-Abl (c-Abl-pY) in RLMVEs after different treatments (n = 3 samples). The density of proteins in each control (untreated) group was used as a standard (1 arbitrary unit) to compare relative densities in the other groups. *P < 0.05, compared to control groups (untreated); †P < 0.05, compared to H2O2 (0.6 mM) groups. (A) Relative densities of the bands of p(Y14) Cav-1and p(Y418) Src expression, as shown in Figure 1A. (B) Relative densities of the bands of p(Y14) Cav-1 expression, as shown in Figure 1B. (C) Relative densities of the bands of p(Y14) Cav-1 expression, as shown in Figure 1C. (D) Relative densities of the bands of c-Abl-pY expression, as shown in Figure 1D. (E) Relative densities of the bands of p(Y14) Cav-1 expression, as shown in Figure 1E. (F) Relative densities of the bands of c-Abl-pY expression, as shown in Figure 1F.
Online Figure II. H2O2 increases caveolae-mediated transendothelial 125I-albumin permeability in Rat lung microvascular endothelial cell (RLMVEC) monolayers. (A) Effects of pretreatment of RLMVECs with methyl-β-cyclodextrin (MβCD) and downregulation of caveolin-1 (Cav-1) with siRNA on transendothelial 125I-albumin permeability. (B) Effects of pretreatment of RLMVECs with MβCD and downregulation of Cav-1 with siRNA on endothelial endocytosis of 125I-albumin. (C) Western blots showing Cav-1 knockdown. Endothelial cells were transfected with Cav-1 siRNA or scrambled (Sc) siRNA. Results are typical of 3 experiments. n = 6-8/group (A and B). *P < 0.05, compared to control (untreated) groups; †P < 0.05 compared with respective H2O2 (0.2 mM) groups.
Online Figure III. Immunofluorescent staining of β-catenin and F-actin in RLMVECs over-expressing WT vs. Y14F-Cav-1. Cells were grown to confluence and treated with H2O2 (0.2 mM) for 30 min. After fixation and permeabilization, cells were incubated with anti-β-catenin primary antibody followed by FITC-conjugated secondary antibody (green). The F-actin and nucleus were stained with Alexa 546 phalloidin (red) and DAPI (blue), respectively. In the third panel (WT + 0.2 mM H2O2), note gap formation (arrow), increased β-catenin translocation into the cytosol (arrow head), and absence of β-catenin in areas where gaps in the monolayer have formed. The fourth panel (Y14F-Cav-1 + 0.2 mM H2O2) shows no gap formation as well as β-catenin translocation in response to H2O2. Scale bars =10 μm. WT = wild type-caveolin-1 expressing cells; Y14F = Y14F-caveolin-1 mutant expressing cells
Acknowledgments
The authors thank David J. Visintine and Maricela Castellon for technical assistance and Richard Ye for critical reading of the manuscript.
Sources of Funding: This work was supported by American Heart Association Scientist Development Grant 0730331N (GH) and NIH NHLBI grants 5R01 HL071626 and P01 HL060678 (RDM).
Non-standard Abbreviations and Acronyms
- Cav-1
caveolin-1
- H2O2
hydrogen peroxide
- ICAM-1
intercellular adhesion molecule-1
- MDCK
Madin-Darby canine kidney
- PKC
protein kinase C
- PP2
4-amino-5-(4-chlorophenyl)-7-(t-butyl) pyrazolo[3,4-d] pyrimidine
- RLMVEC
rat lung microvascular endothelial cells
- TER
transendothelial electrical resistance
- Tyr
tyrosine
- VE
vascular endothelial
- WT
wild type
Footnotes
Disclosures: None.
References
- 1.Lum H, Roebuck KA. Oxidant stress and endothelial cell dysfunction. Am J Physiol. 2001;280:C719–741. doi: 10.1152/ajpcell.2001.280.4.C719. [DOI] [PubMed] [Google Scholar]
- 2.Dejana E. Endothelial cell-cell junctions: happy together. Nat Rev Mol Cell Biol. 2004;(5):261–270. doi: 10.1038/nrm1357. [DOI] [PubMed] [Google Scholar]
- 3.Lampugnani MG, Corada M, Caveda L, Breviario F, Ayalon O, Geiger B, Dejana E. The molecular organization of endothelial cell-to-cell junctions: differential association of plakoglobin, β-catenin, and α-catenin with vascular endothelial cadherin (VE-cadherin) J Cell Biol. 1995;129:203–217. doi: 10.1083/jcb.129.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86:279–367. doi: 10.1152/physrev.00012.2005. [DOI] [PubMed] [Google Scholar]
- 5.Cai H. Hydrogen peroxide regulation of endothelial function: origins, mechanisms, and consequences. Cardiovasc Res. 2005;68:26–36. doi: 10.1016/j.cardiores.2005.06.021. [DOI] [PubMed] [Google Scholar]
- 6.Hu G, Vogel SM, Schwartz DE, Malik AB, Minshall RD. Intercellular adhesion molecule-1-dependent neutrophil adhesion to endothelial cells induces caveolae-mediated pulmonary vascular hyperpermeability. Circ Res. 2008;102:e120–131. doi: 10.1161/CIRCRESAHA.107.167486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Minshall RD, Tiruppathi C, Vogel SM, Malik AB. Vesicle formation and trafficking in endothelial cells and regulation of endothelial barrier function. Histochem Cell Biol. 2002;117:105–112. doi: 10.1007/s00418-001-0367-x. [DOI] [PubMed] [Google Scholar]
- 8.Minshall RD, Sessa WC, Stan RV, Anderson RG, Malik AB. Caveolin regulation of endothelial function. Am J Physiol. 2003;285:L1179–1183. doi: 10.1152/ajplung.00242.2003. [DOI] [PubMed] [Google Scholar]
- 9.Shajahan AN, Timblin BK, Sandoval R, Tiruppathi C, Malik AB, Minshall RD. Role of Src-induced dynamin-2 phosphorylation in caveolae-mediated endocytosis in endothelial cells. J Biol Chem. 2004;279:20392–20400. doi: 10.1074/jbc.M308710200. [DOI] [PubMed] [Google Scholar]
- 10.Minshall RD, Tiruppathi C, Vogel SM, Niles WD, Gilchrist A, Hamm HE, Malik AB. Endothelial cell-surface gp60 activates vesicle formation and trafficking via Gi-coupled Src kinase signaling pathway. J Cell Biol. 2000;150:1057–1070. doi: 10.1083/jcb.150.5.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shajahan AN, Tiruppathi C, Smrcka AV, Malik AB, Minshall RD. Gβγ activation of Src induces caveolae-mediated endocytosis in endothelial cells. J Biol Chem. 2004;279:48055–48062. doi: 10.1074/jbc.M405837200. [DOI] [PubMed] [Google Scholar]
- 12.Vepa S, Scribner WM, Natarajan V. Activation of protein phosphorylation by oxidants in vascular endothelial cells: identification of tyrosine phosphorylation of caveolin. Free Radic Biol Med. 1997;22:25–35. doi: 10.1016/s0891-5849(96)00241-9. [DOI] [PubMed] [Google Scholar]
- 13.Parat MO, Stachowicz RZ, Fox PL. Oxidative stress inhibits caveolin-1 palmitoylation and trafficking in endothelial cells. Biochem J. 2002;361:681–688. doi: 10.1042/0264-6021:3610681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aoki T, Nomura R, Fujimoto T. Tyrosine phosphorylation of caveolin-1 in the endothelium. Exp Cell Res. 1999;253:629–636. doi: 10.1006/excr.1999.4652. [DOI] [PubMed] [Google Scholar]
- 15.Drab M, Verkade P, Elger M, Kasper M, Lohn M, Lauterbach B, Menne J, Lindschau C, Mende F, Luft FC, Schedl A, Haller H, Kurzchalia TV. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science. 2001;293:2449–2452. doi: 10.1126/science.1062688. [DOI] [PubMed] [Google Scholar]
- 16.Parton RG, Joggerst B, Simons K. Regulated internalization of caveolae. J Cell Biol. 1994;127:1199–1215. doi: 10.1083/jcb.127.5.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rothberg KG, Heuser JE, Donzell WC, Ying YS, Glenney JR, Anderson RG. Caveolin, a protein component of caveolae membrane coats. Cell. 1992;68:673–682. doi: 10.1016/0092-8674(92)90143-z. [DOI] [PubMed] [Google Scholar]
- 18.Galbiati F, Volonte D, Brown AM, Weinstein DE, Ben-Ze'ev A, Pestell RG, Lisanti MP. Caveolin-1 expression inhibits Wnt/beta-catenin/Lef-1 signaling by recruiting beta-catenin to caveolae membrane domains. J Biol Chem. 2000;275:23368–23377. doi: 10.1074/jbc.M002020200. [DOI] [PubMed] [Google Scholar]
- 19.Song L, Ge S, Pachter JS. Caveolin-1 regulates expression of junction-associated proteins in brain microvascular endothelial cells. Blood. 2007;109:1515–1523. doi: 10.1182/blood-2006-07-034009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miyawaki-Shimizu K, Predescu D, Shimizu J, Broman M, Predescu S, Malik AB. siRNA-induced caveolin-1 knockdown in mice increases lung vascular permeability via the junctional pathway. Am J Physiol. 2006;290:L405–413. doi: 10.1152/ajplung.00292.2005. [DOI] [PubMed] [Google Scholar]
- 21.Waschke J, Golenhofen N, Kurzchalia TV, Drenckhahn D. Protein kinase C-mediated endothelial barrier regulation is caveolin-1-dependent. Histochem Cell Biol. 2006;126:17–26. doi: 10.1007/s00418-005-0140-7. [DOI] [PubMed] [Google Scholar]
- 22.Sanguinetti AR, Mastick CC. c-Abl is required for oxidative stress-induced phosphorylation of caveolin-1 on tyrosine 14. Cell Signal. 2003;15:289–298. doi: 10.1016/s0898-6568(02)00090-6. [DOI] [PubMed] [Google Scholar]
- 23.John TA, Vogel SM, Tiruppathi C, Malik AB, Minshall RD. Quantitative analysis of albumin uptake and transport in the rat microvessel endothelial monolayer. Am J Physiol. 2003;284:L187–196. doi: 10.1152/ajplung.00152.2002. [DOI] [PubMed] [Google Scholar]
- 24.Li S, Seitz R, Lisanti MP. Phosphorylation of caveolin by Src tyrosine kinases. The α-isoform of caveolin is selectively phosphorylated by v-Src in vivo. J Biol Chem. 1996;271:3863–3868. [PubMed] [Google Scholar]
- 25.Zhou MY, Lo SK, Bergenfeldt M, Tiruppathi C, Jaffe A, Xu N, Malik AB. In vivo expression of neutrophil inhibitory factor via gene transfer prevents lipopolysaccharide-induced lung neutrophil infiltration and injury by aβ2 integrin-dependent mechanism. J Clin Invest. 1998;101:2427–2437. doi: 10.1172/JCI407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Broman MT, Kouklis P, Gao X, Ramchandran R, Neamu RF, Minshall RD, Malik AB. Cdc42 regulates adherens junction stability and endothelial permeability by inducing alpha-catenin interaction with the vascular endothelial cadherin complex. Circ Res. 2006;98:73–80. doi: 10.1161/01.RES.0000198387.44395.e9. [DOI] [PubMed] [Google Scholar]
- 27.Ward PA. Mechanisms of endothelial cell killing by H2O2 or products of activated neutrophils. Am J Med. 1991;91:89S–94S. doi: 10.1016/0002-9343(91)90290-e. [DOI] [PubMed] [Google Scholar]
- 28.Volonté D, Galbiati F, Pestell RG, Lisanti MP. Cellular stress induces the tyrosine phosphorylation of caveolin-1 (Tyr14) via activation of p38 mitogen-activated protein kinase and c-Src kinase. Evidence for caveolae, the actin cytoskeleton, and focal adhesions as mechanical sensors of osmotic stress. J Biol Chem. 2001;276:8094–8103. doi: 10.1074/jbc.M009245200. [DOI] [PubMed] [Google Scholar]
- 29.Galvagni F, Anselmi F, Salameh A, Orlandini M, Rocchigiani M, Oliviero S. Vascular endothelial growth factor receptor-3 activity is modulated by its association with caveolin-1 on endothelial membrane. Biochemistry. 2007;46:3998–4005. doi: 10.1021/bi061400n. [DOI] [PubMed] [Google Scholar]
- 30.Plattner R, Kadlec L, DeMali KA, Kazlauskas A, Pendergast AM. c-Abl is activated by growth factors and Src family kinases and has a role in the cellular response to PDGF. Genes Dev. 1999;13:2400–2411. doi: 10.1101/gad.13.18.2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Singleton PA, Chatchavalvanich S, Fu P, Xing J, Birukova AA, Fortune JA, Klibanov AM, Garcia JG, Birukov KG. Akt-mediated transactivation of the S1P1 receptor in caveolin-enriched microdomains regulates endothelial barrier enhancement by oxidized phospholipids. Circ Res. 2009;104:978–986. doi: 10.1161/CIRCRESAHA.108.193367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schubert W, Frank PG, Woodman SE, Hyogo H, Cohen DE, Chow CW, Lisanti MP. Microvascular hyperpermeability in caveolin-1 (-/-) knock-out mice. Treatment with a specific nitric-oxide synthase inhibitor, L-NAME, restores normal microvascular permeability in Cav-1 null mice. J Biol Chem. 2002;277:40091–40098. doi: 10.1074/jbc.M205948200. [DOI] [PubMed] [Google Scholar]
- 33.Van Driessche W, Kreindler JL, Malik AB, Margulies S, Lewis SA, Kim KJ. Interrelations/cross talk between transcellular transport function and paracellular tight junctional properties in lung epithelial and endothelial barriers. Am J Physiol. 2007;293:L520–524. doi: 10.1152/ajplung.00218.2007. [DOI] [PubMed] [Google Scholar]
- 34.Childs EW, Udobi KF, Hunter FA, Dhevan V. Evidence of transcellular albumin transport after hemorrhagic shock. Shock. 2005;23:565–570. [PubMed] [Google Scholar]
- 35.Miotti S, Tomassetti A, Facetti I, Sanna E, Berno V, Canevari S. Simultaneous expression of caveolin-1 and E-cadherin in ovarian carcinoma cells stabilizes adherens junctions through inhibition of Src-related kinases. Am J Pathol. 2005;167:1411–1427. doi: 10.1016/S0002-9440(10)61228-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aberle H, Schwartz HR, Kemler R. Cadherin-catenin complex: protein interactions and their implications for cadherin function. J Cell Biochem. 1996;61:514–523. doi: 10.1002/(SICI)1097-4644(19960616)61:4%3C514::AID-JCB4%3E3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 37.Lu Z, Ghosh S, Wang Z, Hunter T. Downregulation of caveolin-1 function by EGF leads to the loss of E-cadherin, increased transcriptional activity of beta-catenin, and enhanced tumor cell invasion. Cancer Cell. 2003;4:499–515. doi: 10.1016/s1535-6108(03)00304-0. [DOI] [PubMed] [Google Scholar]
- 38.Klinge CM, Wickramasinghe NS, Ivanova MM, Dougherty SM. Resveratrol stimulates nitric oxide production by increasing estrogen receptor alpha-Src-caveolin-1 interaction and phosphorylation in human umbilical vein endothelial cells. FASEB J. 2008;22:2185–2197. doi: 10.1096/fj.07-103366. [DOI] [PubMed] [Google Scholar]
- 39.Khan EM, Heidinger JM, Levy M, Lisanti MP, Ravid T, Goldkorn T. Epidermal growth factor receptor exposed to oxidative stress undergoes Src- and caveolin-1-dependent perinuclear trafficking. J Biol Chem. 2006;281:14486–14493. doi: 10.1074/jbc.M509332200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Legends to Supplemental Figures
Online Figure I. H2O2 induced Src and c-Abl activation and caveolin-1 (Cav-1) phosphorylation. Densitometric analysis of phosphorylated caveolin-1 (p(Y14) Cav-1), Src (p(Y418) Src) and c-Abl (c-Abl-pY) in RLMVEs after different treatments (n = 3 samples). The density of proteins in each control (untreated) group was used as a standard (1 arbitrary unit) to compare relative densities in the other groups. *P < 0.05, compared to control groups (untreated); †P < 0.05, compared to H2O2 (0.6 mM) groups. (A) Relative densities of the bands of p(Y14) Cav-1and p(Y418) Src expression, as shown in Figure 1A. (B) Relative densities of the bands of p(Y14) Cav-1 expression, as shown in Figure 1B. (C) Relative densities of the bands of p(Y14) Cav-1 expression, as shown in Figure 1C. (D) Relative densities of the bands of c-Abl-pY expression, as shown in Figure 1D. (E) Relative densities of the bands of p(Y14) Cav-1 expression, as shown in Figure 1E. (F) Relative densities of the bands of c-Abl-pY expression, as shown in Figure 1F.
Online Figure II. H2O2 increases caveolae-mediated transendothelial 125I-albumin permeability in Rat lung microvascular endothelial cell (RLMVEC) monolayers. (A) Effects of pretreatment of RLMVECs with methyl-β-cyclodextrin (MβCD) and downregulation of caveolin-1 (Cav-1) with siRNA on transendothelial 125I-albumin permeability. (B) Effects of pretreatment of RLMVECs with MβCD and downregulation of Cav-1 with siRNA on endothelial endocytosis of 125I-albumin. (C) Western blots showing Cav-1 knockdown. Endothelial cells were transfected with Cav-1 siRNA or scrambled (Sc) siRNA. Results are typical of 3 experiments. n = 6-8/group (A and B). *P < 0.05, compared to control (untreated) groups; †P < 0.05 compared with respective H2O2 (0.2 mM) groups.
Online Figure III. Immunofluorescent staining of β-catenin and F-actin in RLMVECs over-expressing WT vs. Y14F-Cav-1. Cells were grown to confluence and treated with H2O2 (0.2 mM) for 30 min. After fixation and permeabilization, cells were incubated with anti-β-catenin primary antibody followed by FITC-conjugated secondary antibody (green). The F-actin and nucleus were stained with Alexa 546 phalloidin (red) and DAPI (blue), respectively. In the third panel (WT + 0.2 mM H2O2), note gap formation (arrow), increased β-catenin translocation into the cytosol (arrow head), and absence of β-catenin in areas where gaps in the monolayer have formed. The fourth panel (Y14F-Cav-1 + 0.2 mM H2O2) shows no gap formation as well as β-catenin translocation in response to H2O2. Scale bars =10 μm. WT = wild type-caveolin-1 expressing cells; Y14F = Y14F-caveolin-1 mutant expressing cells