

Abstract
Self-assembled monolayers (SAMs) on gold prepared from amine-terminated alkanethiols have long been employed as model positively charged surfaces. Yet in previous studies significant amounts of unexpected oxygen containing species are always detected in amine terminated SAMs. Thus, the goal of this investigation was to determine the source of these oxygen species and minimize their presence in the SAM. The surface composition, structure, and order of amine-terminated SAMs on Au were characterized by X-ray photoelectron spectroscopy (XPS), time-of-flight secondary ion mass spectroscopy (ToF-SIMS), sum frequency generation (SFG) and near edge X-ray absorption fine structure (NEXAFS) spectroscopy. XPS determined compositions of amine-terminated SAMs in the current study exhibited oxygen concentrations of 2.4 ± 0.4 atomic %, a substantially lower amount of oxygen than reported in previously published studies. High-resolution XPS results from the S2p, C1s and N1s regions did not detect any oxidized species. Angle-resolved XPS indicated that the small amount of oxygen detected was located at or near the amine head group. Small amounts of oxidized nitrogen, carbon and sulfur secondary ions, as well as ions attributed to water, were detected in the ToF-SIMS data due to the higher sensitivity of ToF-SIMS. The lack of N-O, S-O, and C-O stretches in the SFG spectra are consistent with the XPS and ToF-SIMS results and together show that oxidation of the amine-terminated thiols alone can only account for, at most, a small fraction of the oxygen detected by XPS. Both the SFG and angle-dependent NEXAFS indicated the presence of gauche defects in the amine SAMs. However, the SFG spectral features near 2865 cm−1, assigned to the stretch of the methylene group next to the terminal amine unit, demonstrate the SAM is reasonably ordered. The SFG results also show another broad feature near 3200 cm−1 related to hydrogen-bonded water. From this multi-technique investigation it is clear that the majority of the oxygen detected within these amine-terminated SAMs arises from the presence of oxygen containing adsorbates such as tightly bound water.
1. Introduction
Self-assembled monolayers (SAMs) of alkanethiol molecules on metal surfaces (e.g., Au and Ag) have been extensively employed to control the composition and charge of surfaces1–3. These charged substrates have been used to drive the absorption of various biological materials (proteins, cells, etc.)4–9. In particular, amine-terminated SAMs (NH2-SAMs) have been used to model positively charged surfaces. However, unlike simple methyl terminated SAMs (CH3-SAMs), the quality of NH2-SAMs is more difficult to control. This difficulty is illustrated by the previously reported x-ray photoelectron spectroscopy (XPS) results from NH2-SAMs, all of which describe an unusually high and unexpected concentration of oxygen within the monolayer (a pure amine-terminated thiol should not contain oxygen). Across a range of protocols, the amount of oxygen detected by XPS varies from 5–8 atomic %4,5,10. In contrast, XPS does not detect the presence of oxygen in solution assembled CH3-SAMs11.
There are two possible hypotheses that can explain the presence of oxygen within the NH2-SAMs. The first is that the SAM is covered with oxygen containing coadsorbates12,13, while the second assumes the nitrogen, carbon, or sulfur species within the NH2-SAMs are oxidizing10. Even though there is a large body of work focused on various protocols that try to improve the quality of an NH2-SAMs (to avoid double layers, control the charge of the terminal group, etc), no study has directly addressed the validity of either of these two hypotheses 4,10,14–16. The inability to control the composition of these SAMs has implications for the studies where these substrates are used. The formation of oxidized carbon, nitrogen or sulfur species within the SAM will affect stability, while a layer of contamination could adversely affect the absorption of proteins or cells12.
In this investigation these two hypotheses were tested using several complementary surface analysis techniques to characterize the NH2-SAMs. The composition and oxidation state of each component within the monolayer was probed with XPS and static time-of-flight secondary mass spectroscopy (ToF-SIMS). Monolayer order and identification of potential coadsorbates was explored via angle resolved XPS (ARXPS) in conjunction with both sum frequency generation (SFG) and near edge x-ray absorption fine structure (NEXAFS) spectroscopy.
2. Experimental
2.1 Substrate preparation and assembly of amine-terminated thiols
Silicon substrates (Microelectronics Inc., San Jose, CA) were cleaned by sequential sonications in DI water, dichloromethylene, acetone, and methanol. In a high vacuum thermal evaporator (pressure below 1 × 10−6 Torr) 1 cm × 1 cm silicon substrates were first coated with a thin layer of Ti (10nm) followed by a high purity (99.99%) Au layer (80nm).
The Au coated substrates were then submerged into a 0.5mM solution of 11-amino-1-undecanthiol, hydrochloride (Asemblon, Redmond, WA) in 200 proof ethanol (Decon Labs, King of Prussia, PA). The substrates were allowed to soak in this ethanol-thiol solution for at least 16 hrs. Following assembly, the substrates were removed from solution, rinsed with pure ethanol, and then vortexed within a 10% v/v NH4OH-ethanol solution. Previous investigations have shown that a basic, 10% v/v NH4OH-ethanol rinse reduces the presence of unbound 11-amino-1-undecanthiol molecules that tend to be susceptible to oxidation4,14. Following the rinsing, the samples were dried and stored under pure N2. Unbound 11-amino-1-undecanthiol controls were created by pressing the pure thiolated powder onto double sided sticky tape (Scotch Double Linerless 665, St. Paul, MN) attached onto clean Si substrates.
CH3-SAMs were assembled with a protocol similar to that used to assemble the NH2-SAMs. Briefly, the Au substrates were soaked in a 1mM 1-dodecanethiol (Aldrich, St. Louis, MO) – ethanol solution for at least 16 hrs and then rinsed within pure ethanol.
2.2 X-ray photoelectron spectroscopy
The XPS determined NH2-SAM composition was an average from three spots on each of eight distinct samples (a total of 24 analysis spots). The data was acquired on a Kratos AXIS Ultra DLD instrument (Kratos, Manchester, England) in the hybrid mode using a 0° take-off angle (TOA) and a monochromatic Al Kα1,2 X-ray source (hν=1486.6eV). Angle-resolved measurements were conducted at 0°, 55°, and 70° TOAs using the electrostatic mode. Here, the photoelectron TOA is defined as the angle between the surface normal and the axis of the analyzer lens. Atomic compositions were calculated from C1s and Au4f peak areas obtained from survey (0–1100eV), detail O1s (524–544eV), detail N1s (390–410eV), and detail S2p (155–173eV) spectra (analyzer pass energy=80eV) collected in the hybrid mode. Molecular environments of the samples were probed by collecting high-resolution (analyzer pass energy= 20eV) spectra from the N1s, C1s, and S2p regions. Energy scales were calibrated by normalizing the large CHx peak in the C1s region to 285.0eV and a linear background was subtracted for all peak quantifications. The peak areas were normalized by the manufacturer supplied sensitivity factors and surface concentrations were calculated using the Kratos Vision software.
The oxidation state of the monomer powder was explored via high-resolution scans from a SSI S-Probe XPS system (Surface Science Instruments, Mountain View, CA) using a monochromatic Al Kα1,2 X-ray source (hν=1486.6eV). Charge neutralization was accomplished via an electron flood gun. High-resolution scans (analyzer pass energy= 50eV) of the N1s, C1s, and S2p regions were collected at a TOA of 55°.
2.3 Time-of-Flight Secondary Ion Mass Spectrometry
Negative and positive secondary ion spectra were collected with an ION-TOF TOF.SIMS 5–100 instrument (ION-TOF, Münster, Germany), using a pulsed 25keV Bi3+ primary ion beam under static conditions (primary ion dose < 1013 ions/cm2). Spectra were collected from a 100μm ×100μm region and five spots on three replicate samples (15 spots total) were examined. Positive spectra were mass calibrated using the CH3+, NH4+, C3H5+ and Au+ peaks. Negative spectra were mass calibrated using the CH−, OH−,, and Au− peaks. Secondary ions were collected over a range of 0–400 m/z at a mass resolution (m/Δm) between 4000–8000. In addition, mass calibration corrections were typically kept below 20 ppm. Secondary ion peak intensities, within each spectrum, were normalized by the total ion intensity of that spectrum.
2.4 Near-Edge X-ray Absorption Fine Structure Spectroscopy
NEXAFS spectra were taken at the National Synchrotron Light Source (NSLS) U7A beamline at Brookhaven National Laboratory, using an elliptically polarized beam with ~85% p-polarization. This beam line uses a monochromator with a 600 l/mm grating that provides a full-width at half-maximum (FWHM) resolution of ~0.15 eV at the carbon K-edge (285 eV). The monochromator energy scale was calibrated using the 285.35 eV C1s → π* transition17 on a graphite transmission grid placed in the path of the X-rays. To eliminate the effect of incident beam intensity fluctuations and monochromator absorption features, nitrogen K-edge NEXAFS spectra were normalized by the signal from a dodecanethiol SAM and C K-edge spectra were normalized by the spectrum of a clean gold surface prepared by in vacuo evaporation of gold. Both reference and signal channels were divided by the beam flux during each respective data acquisition18. Partial electron yield was monitored by a detector with the bias voltage maintained at −150 V and −360 V for the C K-edge and N K-edge, respectively. Samples were mounted to allow rotation about the vertical axis to change the angle between the sample surface and the incident X-ray beam. The NEXAFS angle is defined as the angle between the incident X-ray beam and the sample surface.
2.5 SFG Setup and Data Acquisition
The SFG vibrational spectra were acquired on an EKSPLA instrument (EKSPLA, Vilnius, Lithuania) by overlapping visible and tunable IR laser pulses (25 ps) in time and space at incidence angles of 60° and 54°, respectively. The visible beam with a wavelength of 532 nm was delivered by an EKSPLA Nd:YAG laser operating at 50 Hz, which was also used to pump an EKSPLA optical parametric generation/amplification and difference frequency unit based on barium boarate and AgGaS2 crystals to generate tunable IR laser radiation from 1000 – 4000 cm−1. The bandwidth was 1 cm−1 for the visible pump pulses and 1 – 6 cm−1 for the IR laser radiation (1 cm−1 for 2750 – 3000 cm−1 and 6 cm−1 for higher and lower wave numbers). Both beams were unfocussed and had a diameter of approximately 2 mm at the sample. The energy for both beams was 160 μJ per pulse. The SFG signal generated at the sample was then analyzed by filters and a monochromator, detected with a gated photomultiplier tube and stored in a computer. The spectra were collected with 400 shots per data point in 4 cm−1 increments. All spectra were recorded in the ppp (sum, visible, and infrared) polarization combination and were normalized by a reference SFG signal generated in a ZnS crystal.
The fitting routine for the SFG data19 used the following expression for the SFG intensity:20.
| (1) |
Here, is the second order nonlinear susceptibility of the nonresonant background, Av is the strength of the νth vibrational mode, φ denotes the phase of the respective mode and ωIR refers to the frequency of the incident IR field. The integral is over Lorentzian lines, centered at ωL with a width ÃL having a Gaussian distribution. In the fits, the Lorentzian line widths were set to 2 cm−1 and Γν was allowed to vary since the two contributions to the total line width could not be separated within the accuracy of the measurements. Phase values are reported as relative values to the nonresonant gold background and identical phases were assumed for all methyl and methylene related stretches.
3. Results and Discussion
3.1 XPS analysis
Theoretically, XPS data from an NH2(CH2)11SH monolayer on gold should only contain carbon from the alkylchain (84.6%), nitrogen from the amine head group (7.7%), and sulfur of the thiol group at the tail (7.7%), as well as gold from the substrate. However, several groups have reported anomalies with amine-terminated monolayers, including XPS determined compositions that vary from these ideal values. Previously, Wang and coworkers reported a method to interrupt double layer formation between amine-terminated thiols. This protocol incorporated tri-ethylamine into the ethanol-thiol solution followed by a basic wash after assembly. However, they also reported XPS oxygen concentrations of 5.5 atomic % for these NH2-SAMs10. Other studies, in an effort to reduce the amount of oxygen, have adjusted the pH of both the assembly and rinsing solution, yet they still reported O/C atomic ratios of >0.1 4,5.
Average XPS determined compositions of NH2-SAMs from this investigation are reported in Table 1. As typically seen for amine-terminated SAMs, the surface composition showed the presence of carbon, nitrogen, sulfur, oxygen, and gold (from the underlying substrate). Omitting the Au contribution (Table 1), two large differences between the theoretical and experimental compositions become clear. First, the atomic % of S is lower than expected and secondly, there is a significant amount of O within the SAM. The lower percentage of sulfur can be explained by attenuation of the sulfur signal due to inelastic scattering from the long hydrocarbon over layer1. This result supports the conclusion that a relatively ordered monolayer was formed. With regards to the oxygen concentration, in comparison to the previously published compositions (a reported range of 5–8 atomic % when gold is included in the composition), the compositions reported here contain a substantially lower concentration of oxygen (2.4 ± 0.4 atomic %).
Table 1.
Summary of XPS determined elemental compositions.
| Au | C | O | N | S | |
|---|---|---|---|---|---|
| Theoretical Composition | -- | 84.6 | -- | 7.7 | 7.7 |
| Experimental Composition* | 39.2 | 53.0 | 2.4 | 3.6 | 1.8 |
| Std dev | 1.9 | 2.0 | 0.4 | 0.3 | 0.2 |
| Experimental Composition w/o Au | - | 87.2 | 4 | 5.9 | 2.9 |
| Std dev | - | 0.9 | 0.6 | 0.6 | 0.3 |
Average experimental composition from eight distinct samples (three spots per sample).
All data were collected at a 0° TOA in the hybrid mode.
High-resolution C1s spectra collected from the NH2-SAMs, yield peak envelopes made up of two distinct peaks, one at 285eV corresponding to the CH2 alkyl chain backbone and a small shoulder at 286.6eV (see Figure 1 for a typical spectrum). This small peak is at a binding energy that can be attributed to either a N-C or C-O species21.
Figure 1.

High-resolution XPS C1s spectra of the Cl−NH3+(CH2)11SH powder (bottom) and the NH2-SAM on Au (top). In both cases the spectra have peaks at 285eV (CHx) and 286.6eV (C-N/C-O).
Close examination of the high-resolution C1s spectrum from the thiol-monomer powder produces results that are identical to those from the SAM on Au (Figure 1). Again there are two peaks within the C1s region, one corresponding to the CHx species and the other corresponding to a C-N or C-O species. Theoretically, the C-N species should account for only 1/11th or 9% of the total carbon species within the SAM. In actuality, this small shoulder accounts for 14 ± 2 % of the total area of the C1s peak envelope (Table 2) from the SAM on Au and 10% for the monomer powder. The difference between the theoretical and experimental percentage of the NH2-SAM C-N peak could be due to the fact that the C-N species is at the top of the monolayer and its emitted photoelectron would be less attenuated than those emitted from the underlying methylene carbons. It also should be noted that no C=O peak at ~ 288.5eV corresponding to a primary amide group was detected in any of the high-resolution C1s spectra.
Table 2.
High-resolution XPS C1s peak fit results for the NH2-SAM on Au.
| C-Hx (285eV) | C-N/C-O (286.6eV) | |
|---|---|---|
| Theory | 91% | 9% |
| Experimental Composition* | 86% | 14% |
| Std dev | 2% | 2% |
Average experimental composition from eight distinct samples (three spots per sample).
All data were collected at a 0° TOA in the hybrid mode.
The high-resolution N1s spectra taken from the NH2-SAMs on Au exhibited two distinct peaks (see Figure 2 for a typical spectrum) located at 399.5 eV and 401.4 eV. The first peak at 399.5 eV corresponds to the C-N species of the amine head group. The 401.4 eV binding energy of the other peak is consistent with that of a protonated amine group22. In contrast, the N1s spectrum of the thiol powder shows only a single peak at 401.4 eV. This is not unexpected since the thiol powder is in the salt form (Cl− NH3+(CH2)11SH). After assembly no Cl is detected in the XPS survey spectra. Thus, the Cl is removed from the NH2-SAM surface during the rinsing step. However, the existence of two distinct peaks within the N1s spectrum of the NH2-SAM illustrates the fact that a substantial portion of the amine head groups were converted to a neutral state during the assembly and rinsing process. After assembly, the area of the peak assigned to the protonated head group varied from sample to sample, ranging from 24 –35% of the total area of the N1s peak envelope.
Figure 2.
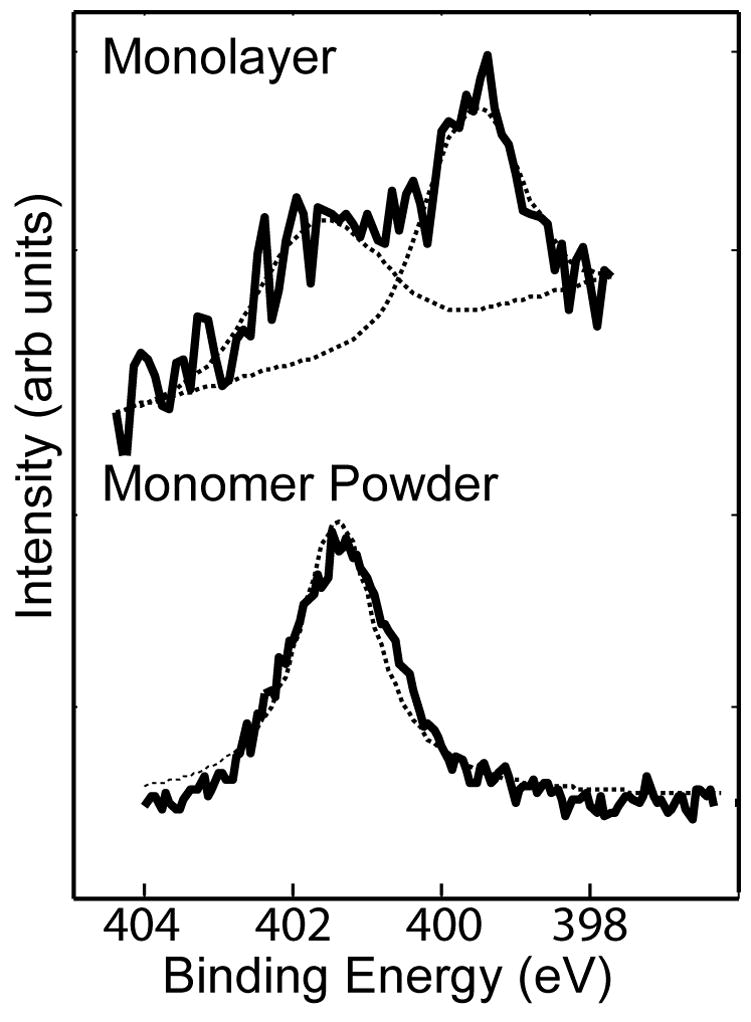
High-resolution XPS N1s spectra of the Cl−NH3+(CH2)11SH powder (bottom) and the NH2-SAM on Au (top). The powder spectrum has a single peak at 401.4eV (protonated amine). The NH2-SAM on Au spectrum has peaks at 399.5eV (C-N) and 401.4 eV (protonated amine).
The S2p high resolution spectra from the NH2-SAM on Au shown in Figure 3 exhibits a single doublet with peaks at 162.0 and 163.5eV due to the presence of bound thiols23. No unbound or oxidized sulfur species were detected. Angle-resolved XPS was performed to determine the location of the oxygen within the NH2-SAM. Data acquired at three different photoelectron TOAs (0°, 55°, and 75°) are summarized in Table 3. A TOA of 0° is normal to the surface and will have the largest sampling depth. As expected, the Au signal from the substrate is increasingly attenuated by the NH2-SAM with increasing TOA. The atomic ratio of C to Au increases three-fold over this 75° span. At a TOA of 55°, the atomic ratio of C/Au is equal to 2.16. Previously, Graham described a linear relationship between chain length and the C/Au attenuation ratio for CH3-SAMs of various chain lengths24. For a 12-carbon chain length CH3-SAM, at a TOA of 55°, Graham reported an atomic ratio of C/Au of 1.9. The slightly larger Au attenuation observed from the NH2-SAM is consistent with the presence of an ad-layer on that SAM. The ratio of N and O to Au in the NH2-SAM is also seen to increase with increasing TOA. It should also be noted that the ratio of O/N remains constant at all three TOAs. This suggests that the N and O are co-located in the monolayer.
Figure 3.
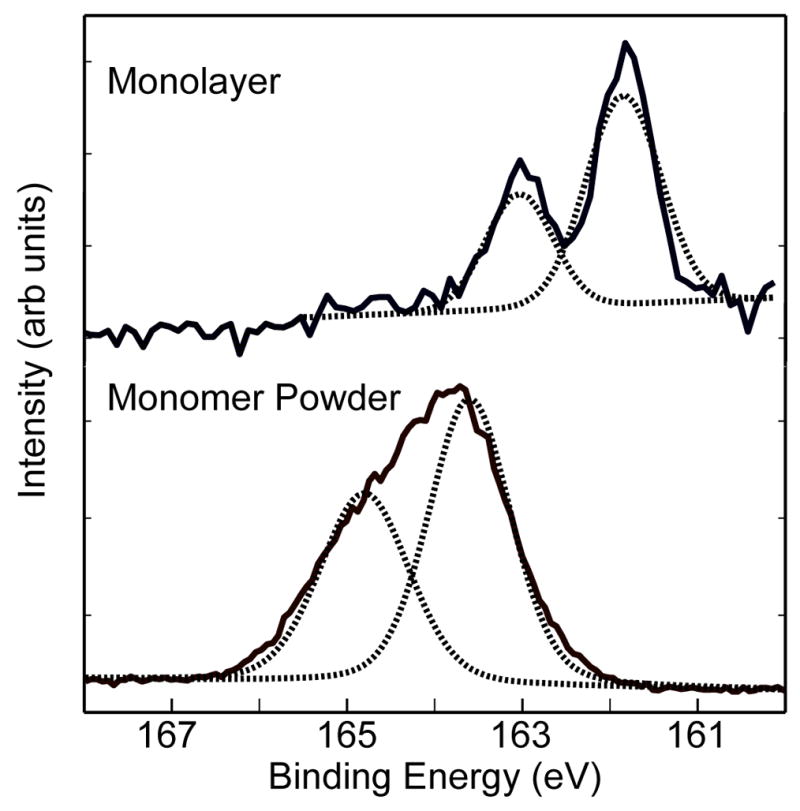
High-resolution XPS S2p spectra of the Cl−NH3+(CH2)11SH powder (bottom) and the NH2-SAM on Au (top). Each spectrum was fit using one S2p doublet with a splitting of 1.2eV.
Table 3.
Angle-resolved XPS determined elemental compositions of NH2-SAMs on Au.
| Atomic % (StdDev)* | |||||
|---|---|---|---|---|---|
| TOA | Au | C | O | N | S |
| 0 | 41.1 (2.5) | 49.9 (2.5) | 3.1 (0.3) | 4.2 (0.5) | 1.7 (0.1) |
| 55 | 28.5 (2.4) | 61.7 (2.7) | 3.4 (0.6) | 4.7 (0.4) | 1.8 (0.1) |
| 75 | 17.3 (1.3) | 70.2 (1.4) | 4.7 (0.6) | 5.9 (0.6) | 1.8 (0.1) |
| Atomic % Ratios (StdDev)* | |||||
|---|---|---|---|---|---|
| TOA | C/Au | N/Au | S/Au | O/Au | O/N |
| 0 | 1.21 (0.10) | 0.10 (0.01) | 0.04 (0.00) | 0.08 (0.01) | 0.75 (0.11) |
| 55 | 2.16 (0.21) | 0.16 (0.02) | 0.06 (0.01) | 0.12 (0.02) | 0.71 (0.13) |
| 75 | 4.05 (0.31) | 0.34 (0.04) | 0.10 (0.01) | 0.27 (0.04) | 0.79 (0.12) |
Average experimental composition from three distinct samples (one spot per sample). All data were taken at 0°, 55°, and 75° takeoff angles in the electrostatic mode. Note: the takeoff angle is defined as the angle between the surface normal and the analyzer.
3.2 ToF-SIMS analysis
ToF-SIMS is a versatile tool that probes complex surfaces with a high degree of sensitivity and molecular specificity25. As a consequence of the increased sensitivity of ToF-SIMS relative to XPS, small peaks from oxidized carbon, nitrogen, and sulfur secondary species were detected in both the positive and negative secondary ion spectra. The detection of oxidized secondary ions from SAMs on Au is common26–28 and a list of these secondary ions can be found in Table 4. To investigate the source of the oxygen contamination within the NH2-SAMs, we compared the normalized intensities of each oxidized fragment to those from a CH3-SAM of the same chain length (Table 4). CH3-SAMs were chosen for comparison because XPS does not detect oxygen in freshly prepared CH3-SAMs.
Table 4.
ToF-SIMS peak intensity ratios for selected oxygen containing peaks detected on NH2- and CH3-SAMs.*
| Ion | Mass | RATIO NH2-SAM/CH3-SAM |
|---|---|---|
| O− | 15.995 | 6.7 |
| OH− | 17.0029 | 3.2 |
| CHO− | 29.0034 | 7.9 |
| C2O− | 39.9959 | 6.5 |
| C2HO− | 41.0043 | 5.9 |
| CNO− | 41.9999 | 30.5 |
| CHNO− | 43.0027 | 29.5 |
| C2HO− | 43.0207 | 4.6 |
| CO2− | 43.9917 | 3.2 |
| CHO2− | 45.0002 | 6.2 |
| SO− | 47.9702 | 2.5 |
| SOH− | 48.9777 | 1.9 |
| H2O3− | 50.0045 | 13.2 |
| C3O− | 51.9977 | 8.4 |
| C3HO− | 53.0055 | 6.0 |
| C3H3O− | 55.0223 | 10.2 |
| C3H5O− | 57.038 | 6.7 |
| CHSO− | 60.9778 | 3.2 |
| SO2− | 63.9656 | 2.0 |
| SO3− | 79.9625 | 2.7 |
| SO4− | 95.9617 | 0.8 |
| S2O3− | 111.9358 | 2.6 |
| AuSO− | 244.9287 | 1.3 |
| H3O+ | 19.0185 | 3.4 |
| CHO+ | 29.0027 | 5.1 |
| CH3O+ | 31.0191 | 5.4 |
| C2H2O+ | 42.008 | 2.2 |
| CH2NO+ | 44.0144 | 7.7 |
| CH4NO+ | 46.029 | 40.3 |
| C3H3O+ | 55.0317 | 2.7 |
| C2H6NO+ | 60.046 | 16.5 |
| SO2H+ | 64.9708 | 1.8 |
| H2SO4+ | 97.9758 | 5.5 |
Intensities for each peak were normalized to the total intensity of the corresponding spectrum. Ratios presented here are calculated from average normalized peak intensities taken from five different spots on three samples of each SAM.
The biggest variation within the ToF-SIMS data from the two types of SAMs stems from the differences in head group. This is illustrated by the large differences in peak intensities (16–40 fold) of oxygen and nitrogen containing secondary ions from the two SAMs. In addition, the peak intensities of oxidized sulfur secondary ions from the NH2-SAMs are two times larger than those from the CH3-SAMs. While these oxidized sulfur secondary ions are always observed in ToF-SIMS data from thiol based SAMs, one possible explanation for the increased intensities in the NH2-SAMs is that these negatively charged oxidized sulfur species interact with the amine head group making it difficult to wash them away during the rinsing step13. Another possible explanation is that the rate of photooxidation of the NH2-SAM could be higher due to the polarity of the amine head group and a lower degree of ordering of the SAM. Cooper and Leggett, in their studies of SAM photooxidation rates, proposed that a high degree of ordering would limit access of reactive oxygen species to the thiol group, thus limiting the rate of sulfur, nitrogen, or carbon photooxidation28. While the polarity of the head group or the lower degree of ordering could drive a higher rate of photoxidation, compared to CH3-SAMs, these oxidized species are not detected by XPS and thus we believe cannot be the major source of the oxygen contamination
At low masses there are O− and OH− peaks in the negative spectra as well as H3O+ in the positive spectra that can be attributed to the presence of water. The relative peak intensities of these secondary ions from the NH2-SAMs are 3–7 times larger than the peak intensities observed for the hydrophobic CH3-SAM, suggesting a greater amount of water is present on the NH2-SAM.
3.3 NEXAFS Spectroscopy
NEXAFS spectroscopy can provide structural information about the molecular alignment in SAMs by measuring the angle dependence of the absorption peak intensities in spectra acquired at x-ray incidence angles from normal to glancing. The peaks present in the NEXAFS spectra also provide information about the chemical identity and bonding of surface species. N K-edge spectra of NH2-SAMs acquired at NEXAFS angles of 70°, 55° and 20° are presented in Figure 4 along with the difference between the 70° and 20° spectra. The spectra are dominated by a broad σ* resonance near 406 eV related to the N-C bond. This peak is accompanied by an intense absorption resonance at 400.6 eV which can be assigned either to amide or primary amine (−NH2) moieties.29–31 The latter possibility is more likely here since there is no evidence for C=O bonds in the NEXAFS C K-edge spectra (see below) or XPS C1s spectra (see section 3.1). The low-intensity pre-edge feature visible around 398 eV has been hypothesized to result from a hybridized or dissociated state of ammonia32. Other groups have proposed that a positively charged form of amine may be found at a different energy than the neutral form of the amine31. Since XPS results have shown a significant portion of the NH2-SAM monolayer has a −NH3+ character, this peak may be due to the positively charged form of the amine group (−NH3+). Since radiation damage from the X-rays also becomes visible in this spectral range, a careful beam damage study was performed to rule out this possibility33. The spectra exhibit a weak but discernible negative dichroism for the amine transition, indicating a small degree of orientational order for this species. In addition, the negative sign of the observed difference peak suggests an upright orientation of the C-N bond for this group18.
Figure 4.

NEXAFS spectra of the nitrogen K-edge for amine SAMs acquired at angles of 70°, 55° and 20° along with the 70 and the 20° difference spectrum.
The C K-edge NEXAFS spectra are consistent with conclusions determined from the N K-edge spectra. Figure 5 shows the 55° x-ray incidence angle spectrum (“magic angle” spectrum) and a difference spectrum (70° - 20°) for the NH2-SAM as well as corresponding reference CH3-SAM spectra. The 55° spectra of the amine and the methyl SAMs are very similar. Both exhibit the expected resonances at ~287.7 eV (R*/C-H σ*), ~293.3 eV (C-C σ*) and 301.3 eV (C-C′ σ*)18,34–36. A very weak peak at ~285.2 eV is also observed and is tentatively assigned to an excitation into the alkane-metal orbitals, small amounts of contaminants present on the upstream gold grid used for beam current normalization, or a combination of both effects37,38. We performed a radiation damage study to rule out the possibility of beam damage as a cause for this feature33. Note, that a C=O related resonance expected near 288.5 eV18,37,39 is not visible in the NH2-SAM spectrum, consistent with no detectable C=O related peak in the XPS NH2-SAM C1s spectrum. Thus, the C K-edge NEXAFS spectrum provides further evidence that conversion of the amine group to an amide group can’t account for the O contamination detected by XPS. Also the lack of C=O resonance in the C K-edge spectrum is consistent with the interpretation that the 400.6 eV feature observed in the N K-edge spectrum is due to an amine species.
Figure 5.

NEXAFS spectra of the carbon K-edge for the NH2- and CH3-terminated SAMs acquired at 55° along with their respective difference spectra (70°-20°).
The polarization dependence displayed as the 70° - 20° difference spectra (Figure 5) is very similar in both the NH2- and CH3-SAMs, with the R* and σ* resonances exhibiting positive and negative anisotropy, respectively. The NH2-SAM shows a weaker dichroism compared to the CH3-SAM. The molecular orbitals related to the R* transition are oriented relatively perpendicular to the alkyl chain axis while those related to transitions into the C-C and C-C′ σ* orbitals are oriented more parallel to the chain axis. Consequently, the data indicates that both the NH2- and the CH3-SAMs are oriented upright (i.e., away from the Au substrate)18. The weaker dichroism of the NH2-SAM can be explained either by a stronger inclination of the alkyl chains, a lower degree of structural order, or a combination of both.
3.4 SFG Spectroscopy
SFG spectra of the NH2-SAM and a reference CH3-SAM along with the corresponding fits are shown in Figure 6. The CH3-SAM spectrum exhibits the expected resonances for the three peaks assigned to the terminal methyl unit at 2969 cm−1 (r−), 2936 cm−1 (r−FR), and 2882 cm−1 (r+). These three resonances are observed on well ordered and densely packed SAMs. There are no CH2 related stretching resonances visible in the spectrum, indicating that the alkyl chains are predominantly in an all-trans conformation40. Conversely, the NH2-SAM spectrum is dominated by CH2 related stretching vibrations in the region below 3000 cm−1. A strong resonance observed near 2929 cm−1 is commonly assigned to asymmetric stretching vibrations of methylene (d−) and it indicates a significant amount of trans-gauche defects in the alkyl chains7,40. This agrees well with the NEXAFS C K-edge data where the NH2-SAM exhibits a lower linear dichroism compared to the CH3-SAM. Thus, both NEXAFS and SFG suggest the alkyl chains in the NH2-SAM have a lower degree of structural order. The spectral feature near 2865 cm−1 has been observed in NH2-SAMs previously and assigned to the methylene group next to the terminal NH2 unit (d+w)7,40. This peak indicates that the SAM surface is still reasonably ordered in spite of the trans-gauche defects in the chain since an SFG peak would not be observed if this group was randomly oriented.
Figure 6.

SFG spectrum of NH2- (upper trace) and CH3-terminated (lower trace) SAMs on gold. Solid lines are best fits based on equation 1.
The region above 3000 cm−1 is dominated by a broad OH resonance near 3200 cm−1 related to strongly hydrogen-bonded water (“ice-like” water)41, and is also observed on peptide films where NH2 terminated lysine side chains are exposed to water42. The presence of bound water in the SFG spectrum is consistent with the observation of water related fragments in ToF-SIMS data. Spectral features at 3333 cm−1 and 3247 cm−1 are assigned to the asymmetric (d−) and symmetric (d+) N-H stretches of the terminal NH2 unit, respectively. Previous SFG studies have observed NH stretches from amine groups in this spectral range42,43. The mode observed near 3020 cm−1 is more difficult to assign. It could be related to symmetric or asymmetric stretching modes of NH3+.
4. Concluding Remarks
The aim of this investigation was to use complementary surface analysis techniques to investigate the source of the oxygen contamination found within NH2-SAMs and minimize its presence. NEXAFS and SFG experiments did indicate the presence of gauche defects in the alkyl chains, yet, other spectral features pointed to a reasonably ordered surface. While it has been shown that these defects could allow oxygen to penetrate the SAM and interact/oxidize with the thiol molecule, XPS did not detect the presence of any oxidized sulfur or nitrogen species. ToF-SIMS, with its higher sensitivity, did detect the presence of some fragments containing both oxygen and nitrogen, but XPS, NEXAFS and SFG did not detect any peaks that could be attributed to amide groups. Thus, as the concentration of oxidized sulfur and amine species were below the XPS detection limits this indicates that oxidized sulfur and amine species are not the major contributors to the oxygen contamination observed for the NH2-SAMs in this study. Angle-resolved XPS experiments show that the oxygen is co-located with the amine head group. In addition, ToF-SIMS and SFG results suggest that there is a measurable amount of water bound to the surface. Thus, we believe that the majority of the oxygen detected within these amine SAMs most likely arises from the presence of oxygen containing coadsorbates such as tightly bound water.
Acknowledgments
This research was supported by NIH grants EB-002027, EB-006163 and GM-074511. TW thanks the Deutsche Forschungsgemeinschaft for a research fellowship. We thank Daniel Fischer (NIST) and Cherno Jaye (Hunter College) for providing us with the experimental equipment for NEXAFS spectroscopy and their help at the synchrotron. NEXAFS studies were performed at the NSLS, Brookhaven National Laboratory, which is supported by the U.S. Department of Energy, Division of Materials Science and Division of Chemical Sciences.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bain CD, Evall J, Whitesides GM. Journal of the American Chemical Society. 1989;111:7155–7164. [Google Scholar]
- 2.Ostuni E, Yan L, Whitesides GM. Colloids and Surfaces B-Biointerfaces. 1999;15:3–30. [Google Scholar]
- 3.Wink T, vanZuilen SJ, Bult A, vanBennekom WP. Analyst. 1997;122:R43–R50. [Google Scholar]
- 4.Wang H, Castner DG, Ratner BD, Jiang SY. Langmuir. 2004;20:1877–1887. doi: 10.1021/la035376f. [DOI] [PubMed] [Google Scholar]
- 5.Chuang WH, Lin JC. Journal of Biomedical Materials Research Part A. 2007;82A:820–830. doi: 10.1002/jbm.a.31193. [DOI] [PubMed] [Google Scholar]
- 6.Faucheux N, Schweiss R, Lutzow K, Werner C, Groth T. Biomaterials. 2004;25:2721–2730. doi: 10.1016/j.biomaterials.2003.09.069. [DOI] [PubMed] [Google Scholar]
- 7.Palyvoda O, Bordenyuk AN, Yatawara AK, McCullen E, Chen CC, Benderskii AV, Auner GW. Langmuir. 2008;24:4097–4106. doi: 10.1021/la7032675. [DOI] [PubMed] [Google Scholar]
- 8.Schilp S, Ballav N, Zharnikov M. Angewandte Chemie-International Edition. 2008;47:6786–6789. doi: 10.1002/anie.200801858. [DOI] [PubMed] [Google Scholar]
- 9.Ballav N, Schilp S, Zharnikov M. Angewandte Chemie-International Edition. 2008;47:1421–1424. doi: 10.1002/anie.200704105. [DOI] [PubMed] [Google Scholar]
- 10.Wang H, Chen SF, Li LY, Jiang SY. Langmuir. 2005;21:2633–2636. doi: 10.1021/la046810w. [DOI] [PubMed] [Google Scholar]
- 11.Graham DJ, Price DD, Ratner BD. Langmuir. 2002;18:1518–1527. [Google Scholar]
- 12.Sprik M, Delamarche E, Michel B, Rothlisberger U, Klein ML, Wolf H, Ringsdorf H. Langmuir. 1994;10:4116–4130. [Google Scholar]
- 13.Sahoo RR, Patnaik A. Applied Surface Science. 2005;245:26–38. [Google Scholar]
- 14.Li LY, Chen SF, Jiang SY. Langmuir. 2003;19:3266–3271. [Google Scholar]
- 15.Fears KP, Creager SE, Latour RA. Langmuir. 2008;24:837–843. doi: 10.1021/la701760s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wallwork ML, Smith DA, Zhang J, Kirkham J, Robinson C. Langmuir. 2001;17:1126–1131. [Google Scholar]
- 17.Batson PE. Physical Review B. 1993;48:2608. doi: 10.1103/physrevb.48.2608. [DOI] [PubMed] [Google Scholar]
- 18.Stöhr J. NEXAFS Spectroscopy. Vol. 25 Springer-Verlag; Berlin: 1992. [Google Scholar]
- 19.Moore FG, Becraft KA, Richmond GL. Applied Spectroscopy. 2002;56:1575–1578. [Google Scholar]
- 20.Bain CD. J Chem Soc, Faraday Trans. 1995;91:1281–1296. [Google Scholar]
- 21.Michel R, Subramaniam V, McArthur SL, Bondurant B, D’Ambruoso GD, Hall HK, Brown MF, Ross EE, Saavedra SS, Castner DG. Langmuir. 2008;24:4901–4906. doi: 10.1021/la800037r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ratner BD, Castner DG. Biomaterials. 1990;11:143–146. [Google Scholar]
- 23.Castner DG, Hinds K, Grainger DW. Langmuir. 1996;12:5083–5086. [Google Scholar]
- 24.Graham DJ. PhD Dissertation. University of Washington; 2001. [Google Scholar]
- 25.Belu AM, Graham DJ, Castner DG. Biomaterials. 2003;24:2625–2653. doi: 10.1016/s0142-9612(03)00159-5. [DOI] [PubMed] [Google Scholar]
- 26.Graham DJ, Ratner BD. Langmuir. 2002;18:5861–5868. [Google Scholar]
- 27.Hutt DA, Leggett GJ. Journal of Physical Chemistry. 1996;100:6657–6662. [Google Scholar]
- 28.Cooper E, Leggett GJ. Langmuir. 1998;14:4795–4801. [Google Scholar]
- 29.Cooper G, Gordon M, Tulumello D, Turci C, Kaznatcheev K, Hitchcock AR. Journal of Electron Spectroscopy and Related Phenomena. 2004;137:795–799. [Google Scholar]
- 30.Gordon ML, Cooper G, Morin C, Araki T, Turci CC, Kaznatcheev K, Hitchcock AP. Journal of Physical Chemistry A. 2003;107:6144–6159. [Google Scholar]
- 31.Zubavichus Y, Zharnikov M, Schaporenko A, Grunze M. Journal of Electron Spectroscopy and Related Phenomena. 2004;134:25–33. [Google Scholar]
- 32.Ozawa K, Hasegawa T, Edamoto K, Takahashi K, Kamada M. J Phys Chem B. 2002;106:9380–9386. [Google Scholar]
- 33.For the beam damage study at the N K-edge, the 388 eV to 410 eV energy range was repeatedly scanned and analyzed. In addition, the partial electron yield at 398 eV was monitored over time to rule out damage related changes in the first seconds. No changes in the spectra were observed within 15 minutes, which is more than the exposure time in the longest nitrogen scans performed within this work. An analogous radiation damage study for the C K-edge proved that the pre-edge feature observed near 285.2 eV is not caused by beam damage.
- 34.Outka DA, Stöhr J, Rabe JP, Swalen JD. Journal of Chemical Physics. 1988;88:4076. [Google Scholar]
- 35.Weiss K, Bagus PS, Wöll C. Journal of Chemical Physics. 1999;111:6834. [Google Scholar]
- 36.Väterlein P, Fink R. Journal of Chemical Physics. 1998;108:3313. [Google Scholar]
- 37.Cabarcos OM, Shaporenko A, Weidner T, Uppili S, Dake LS, Zharnikov M, Allara DL. J Phys Chem C. 2008;112:10842–10854. [Google Scholar]
- 38.Witte G, Weiss K, Jakob P, Braun J, Kostov KL, Wöll C. Physical Review Letters. 1998;80:121. [Google Scholar]
- 39.Ballav N, Schupbach B, Dethloff O, Feulner P, Terfort A, Zharnikov M. J Am Chem Soc. 2007;129:15416–15417. doi: 10.1021/ja0751882. [DOI] [PubMed] [Google Scholar]
- 40.Himmelhaus M, Eisert F, Buck M, Grunze M. J Phys Chem B. 2000;104:576–584. [Google Scholar]
- 41.Du Q, Freysz E, Shen YR. Science. 1994;264:826–828. doi: 10.1126/science.264.5160.826. [DOI] [PubMed] [Google Scholar]
- 42.Mermut O, Phillips DC, York RL, McCrea KR, Ward RS, Somorjai GA. J Am Chem Soc. 2006;128:3598–3607. doi: 10.1021/ja056031h. [DOI] [PubMed] [Google Scholar]
- 43.Jung SY, Lim SM, Albertorio F, Kim G, Gurau MC, Yang RD, Holden MA, Cremer PS. J Am Chem Soc. 2003;125:12782. doi: 10.1021/ja037263o. [DOI] [PubMed] [Google Scholar]