

Abstract
PDZ domains have well known binding preferences for distinct C-terminal peptide motifs. For most PDZ domains, these motifs are of the form [S/T]-W-[I/L/V]. Although the preference for S/T has been explained by a specific hydrogen bond interaction with a histidine in the PDZ domain and the (I/L/V) is buried in a hydrophobic pocket, the mechanism for Trp specificity at the second to last position has thus far remained unknown. Here, we apply a method to compute the free energies of explicit water molecules and predict that potency gained by Trp binding is due to a favorable release of high-energy water molecules into bulk. The affinities of a series of peptides for both wild-type and mutant forms of the PDZ domain of Erbin correlate very well with the computed free energy of binding of displaced waters, suggesting a direct relationship between water displacement and peptide affinity. Finally, we show a correlation between the magnitude of the displaced water free energy and the degree of Trp-sensitivity among subtypes of the HTRA PDZ family, indicating a water-mediated mechanism for specificity of peptide binding.
Keywords: PDZ domain, peptide binding affinity, water thermodynamics, molecular dynamics, WaterMap
Introduction
PDZ domains are small modular protein domains of around 70–90 amino acids that play an important role in the assembly of complexes of proteins at the plasma membrane.1 PDZ domains have been shown to interact with G-protein coupled receptors (GPCRs), ion channels, transporters, and receptor tyrosine kinases, as well as with cytoskeletal proteins, extra-cellular matrix proteins, and components of tight junctions (claudins, occludins). Biological processes involving PDZ domains include the establishment and maintenance of epithelial polarity2,3; the organization of signaling complexes at the synapse4 and trafficking of proteins to the plasma membrane.5 Many different types of PDZ domain-containing proteins have been identified, with as few as one and as many as 10 copies of the domain, often in combination with other regulatory domains, including SH2, SH3, WW, guanylate kinase, PTB, or LRR domains. The combination of several domains in these proteins enables them to act as molecular switchboards, bringing together components of signaling complexes.6 In total, more than 500 PDZ domains in over 300 different proteins have been identified in the human genome.7 Known PDZ domain-mediated protein–protein interactions have been compiled in a PDZ specific database, named PDZBase.8
Insight into the mechanism underlying affinity and specificity of these interactions has come from X-ray crystallography,9 NMR spectroscopy,10 sequence analysis,11 computational biophysics,12 and combinatorial peptide experiments.13–15 One particularly attractive experimental technique to study the binding properties of these domains is phage display, whereby large collections of peptides are screened and amplified in an in vivo selection process, revealing the optimal binding motifs of each PDZ domain. The research group of Sidhu and co-workers has determined optimal binding profiles for a large number of PDZ domains, initially for Erbin,16 ZO-1,17,18 and the HTRA (high-temperature requirement A) family.19,20 More recently, they profiled as many as 100 human and C. elegans PDZ domains.21 Together with the experimentally detected endogenous PDZ interactions, these experiments have enabled a comprehensive analysis of determinants of specificity and promiscuity of the entire PDZ domain family.
C-terminal PDZ binding motifs are often classified as Class I (S/T-X-φ) or Class II (φ-X-φ), where φ is any hydrophobic residue (the terminal residue in the ligand interacting with the PDZ domain is termed the P-0 position, and the next residue towards the N-terminus is designated as the P-1 position, etc.). Correspondingly, PDZ domains have been classified as Class I binding or Class II binding based on the observed correlation between binding specificity and the occurrence of specific residues in the peptide binding site.13,22 For example, most PDZ domains have a His residue at the αB1 position in the PDZ domain (see Fig. 1), and these domains prefer peptides with a Thr/Ser at the P-2 position.9 The specific size and shape of the P-0 hydrophobic pocket, formed by residues in the αB and βB structural elements, has been correlated with the observed preference for L or V at the P-0 position in NHERF and PSD-95 PDZ domains.23 More distal interactions with P-3 and P-4 sites appear to be mediated by the βB-βC loop, and the variability of this loop in terms of length and sequence appears to modulate specificity through these positions.24 Large scale phage display experiments have significantly extended the dual classification scheme, showing that these domains can be subdivided in as many as 16 different classes and can recognize features of up to seven peptide residues.21
Figure 1.
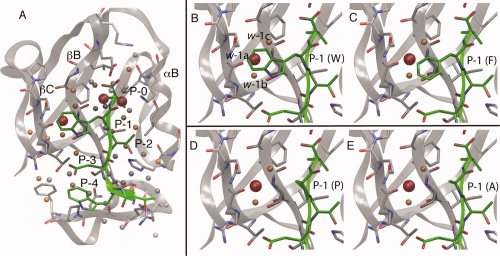
(A) WaterMap of the Erbin PDZ domain (gray) complex with the WETWV peptide (green). The αB helix and the βB and βC strands form the peptide binding site. Peptide residues are numbered P-0 to P-4, with the numbering starting at the C-terminal valine residue. All identified water sites in the peptide binding site are shown as spheres. Water sites with ΔG > 3.5 kcal/mol are shown in red, sites with 1.5 > ΔG > 3.5 kcal/mol are shown in orange, and sites with ΔG < 1.5 kcal/mol in gray; The three highest energy water sites overlap with side-chains at the P-0 and P-1 positions. (B) Close-up of the P-1 pocket with three predicted water-sites, named w-1a (orange), w-1b (red), and w-1c (orange). The w-1b site is more unfavorable (−TΔS 2.6 kcal/mol, ΔH 3.2 kcal/mol, ΔG 5.8 kcal/mol) than the w-1a (−TΔS 1.6 kcal/mol, ΔH −0.1 kcal/mol, ΔG 1.5 kcal/mol) and w-1c sites (−TΔS 1.7 kcal/mol, ΔH 0.6 kcal/mol, ΔG 2.3 kcal/mol). (C) WETFV-Erbin model. (D) WETPV-Erbin model. (E) WETAV-Erbin model. Note that while the wild-type Trp almost completely displaces the unfavorable waters, the Phe only has partial displacement and both Ala and Pro do not overlap with any of the three high-energy water sites, in good agreement with the experimentally measured differences in affinity.
Interestingly, the P-1 (second to last) position was originally thought to be relatively unimportant for specificity, based on the variability of amino acid types at this position in known ligands of PDZ domains,13 and the exposure to solvent of the P-1 side chain in crystal structures. However, more recent experiments, most notably a phage-display study of the Erbin PDZ domain,16 showed that certain side chains at P-1 can have a major contribution to affinity. For example, the Erbin phage display profile indicated that a WETWV peptide was the most optimal binding motif. The affinity of this super-binding peptide was determined to be 20 nM, a 1500-fold increase in potency compared to the peptide in which the Trp P-1 residue is replaced by Ala (i.e., WETAV). The NMR structure of Erbin with the WETWV peptide revealed that a pocket formed by Ser26, Arg51, and Gln49 residues accommodated the Trp side chain. However, specific side-chain interactions appeared to not be important, as Ala mutants at position 26, 49, and 51 had little effect on binding affinity of the peptide. Thus, the authors suggested that non-specific, backbone-mediated interactions were important for the high affinity of the Trp containing peptide.
We have recently developed a method, called WaterMap, for calculating the location and thermodynamics of water sites in proteins.25,26 This method is based on molecular dynamics (MD) simulation and computes both the enthalpy and entropy of water sites around a protein, relative to bulk water, from an analysis of explicit water molecules from the MD trajectory (see Materials and Methods). It has been shown that WaterMap can be used as a tool for qualitatively understanding the properties of binding sites,25 and as a quantitative tool for predicting small molecule binding affinities.26 Here, we have used WaterMap to investigate the mechanism of high-affinity binding of peptides to PDZ domains. In particular, the method was used to investigate peptide binding affinity of the structurally well-characterized PDZ domains of Erbin and the HTRA family. Erbin (ERB-interacting protein) is a member of the LAP (leucine-rich repeat and PDZ containing) family, a class of proteins involved in the maintenance of epithelial polarity.27 The PDZ domain of Erbin was originally identified through yeast two-hybrid experiments as an interacting partner of the ERB-B2 receptor,28 and has been shown to interact in vivo with p120-like catenin proteins.29 The HTRA family of proteins are characterized by a combination of a serine protease domain and a PDZ domain.30 The best characterized member of the family is HTRA2 (also known as Omi), which is released from mitochondria in cellular stress and participates in the regulation of apoptosis.31
Using WaterMap, we predict the presence of high-energy water sites on the surface of several PDZ domains, including Erbin and the HTRA family. In Erbin, the high-energy water site at the P-1 position is formed by the hydrophobic parts of the side chains Ser26, Arg49, and Gln51, as well as the surface of the βB-βC β-sheet. We show that the Trp dependence of high-affinity peptides can be correlated to the ability of Trp to displace this high-energy water, thus releasing it into the bulk and resulting in a net free energy gain. Additional predicted high-energy water sites on the PDZ domain, including those in the P-0 pocket, are also important for affinity, and we show that the reported affinities of a series of peptides for Erbin correlate well with the total energy of released water, suggesting a direct relationship between water displacement and peptide binding affinity. Finally, we demonstrate a correlation between the degree of Trp dependence and the magnitude of the water free energy among subtypes of the HTRA family, linking water displacement to the specificity of PDZ domains.
Results
Prediction of high-energy water sites in Erbin
The results from running WaterMap on the PDZ domain of Erbin are shown in Figure 1. Water sites are predicted along the entire peptide binding site and have been classified into low-energy (i.e., relatively stable with ΔG < 1.5 kcal/mol, gray spheres), medium-energy (1.5 < ΔG < 3.5 kcal/mol, orange spheres), and high-energy sites (highly unstable with ΔG > 3.5 kcal/mol, large red spheres). High-energy water sites are found only in the P-0 and P-1 pockets. This is consistent with the experimental data, which shows that the P-0 and P-1 sites are most important for high-affinity binding of the WETWV peptide (see Table I). Substitutions of the WETWV peptides at the P-1 and P-0 sites lead to large reductions in affinity; at P-1, Trp to Ala (WTEAV) leads to a 1500-fold loss of binding affinity (corresponding to a ΔΔG of 4.1 kcal/mol), while at P-0, Val to Ala (WETWA) results in 3700-fold lower affinity (ΔΔG 4.6 kcal/mol.) Ala substitutions at positions P-2, P-3, and P-4 only affect affinity 100-fold (2.5 kcal/mol), 140-fold (2.7 kcal/mol), and 50-fold (2.2 kcal/mol), respectively. Even a conservative substitution at the P-1 site of Trp to Phe causes a significant loss of affinity (170-fold, 2.8 kcal/mol). Finally, mutation to Pro at this position leads to the largest loss in affinity (3300-fold, 4.5 kcal/mol).
Table I.
Erbin-Peptide Experimental and Computational Binding Affinities
| Peptide | Affinity, IC50 (μM) | Relative affinity | RTln (IC50) | WaterMap -TΔS (kcal/mol) | WaterMap ΔH (kcal/mol) | WaterMap ΔG (kcal/mol) | MW | MM–GB/SA ΔG (kcal/mol) |
|---|---|---|---|---|---|---|---|---|
| WETWV | 0.02 ± 0.002 | 1 | −8.6 | −30.4 | −6.8 | −37.2 | 718 | −23.9 |
| FETWV | 0.16 ± 0.01 | 8.0 | −7.5 | −28.1 | −6.6 | −34.7 | 678 | −22.7 |
| YETWV | 0.16 ± 0.01 | 8.0 | −7.5 | −28.1 | −6.6 | −34.7 | 694 | −29.4 |
| WDTWV | 0.41 ± 0.06 | 21 | −6.9 | −28.9 | −7.5 | −36.4 | 704 | −21.3 |
| WESWV | 0.16 ± 0.01 | 8.0 | −7.5 | −30.0 | −6.3 | −36.3 | 704 | −26.2 |
| WEVWV | 0.26 ± 0.01 | 13 | −7.2 | −30.5 | −6.9 | −37.4 | 716 | −23.2 |
| WETFV | 3.3 ± 0.2 | 170 | −5.8 | −27.1 | −4.2 | −31.3 | 679 | −23.5 |
| WETPV | 66 ± 4 | 3300 | −4.1 | −24.5 | −3.3 | −27.8 | 630 | −14.6 |
| WETWL | 1.8 ± 0.1 | 90 | −6.1 | −28.8 | −3.9 | −32.7 | 732 | −29.0 |
| WETWI | 1.7 ± 0.1 | 85 | −6.2 | −30.4 | −6.8 | −37.2 | 732 | −29.4 |
| AETWV | 1.0 ± 0.1 | 50 | −6.4 | −27.5 | −6.6 | −34.1 | 603 | −22.0 |
| WATWV | 2.8 ± 0.7 | 140 | −5.9 | −28.1 | −7.7 | −35.8 | 661 | −19.8 |
| WEAWV | 2.0 ± 0.1 | 100 | −6.1 | −29.8 | −6.1 | −35.9 | 688 | −23.3 |
| WETAV | 30 ± 7 | 1500 | −4.5 | −24.5 | −3.3 | −27.8 | 603 | −15.4 |
| WETWA | 74 ± 4 | 3700 | −4.0 | −28.0 | −2.6 | −30.6 | 690 | −21.3 |
Experimental affinities were determined as IC50 using a glutathione S-transferase-based chemiluminescence assay.16 WaterMap affinities were calculated from the overlap of the peptides with the water-sites shown in Figure 1, according to the protocol described in Abel et al.26 MM–GB/SA energies were calculated using Prime,32,33,35 employing a brief minimization of the protein/peptide structure; peptide strain energies were not included in the total energy. MW, molecular weight.
Inspection of the WaterMap in the P-1 pocket suggests a reason why Trp is a much better binder than Phe, Ala, or Pro. Three water sites are predicted in the pocket formed by the Ser26, Arg49, and Gln51 side chains and the β-sheet backbone (see Fig. 1, panels B–E). These water sites will be referred to hence forth at w-1a, w-1b, and w-1c. The central water (w-1b, shown in red) has the highest energy (5.8 kcal/mol), while the two flanking waters (w-1a and w-1c, shown in orange) have moderately unfavorable energies (1.5 and 2.3 kcal/mol). Amongst the experimentally tested variants, only Trp [Fig. 1(B)] has the ability to displace these waters effectively, while Phe [Fig. 1(C)] can only partially displace them. Pro [Fig. 1(D)] and Ala [Fig. 1(E)] do not overlap with these water sites.
The presence of high-energy water molecules in the P-0 pocket is consistent with the strongly hydrophobic character of this pocket; the P-0 Val residue is in direct contact with the side chains of Leu23, Phe25, Ile27 in the βB strand, and Val83, Leu86 in the αB helix. However, the P-1 high-energy water site is more surprising, given its partial exposure to solvent, and the proximity of three polar residues Ser26, Arg49, and Gln51. Closer inspection of the environment reveals that aside from the Ser, no protein hydrogen bonding partners are available, since the β-sheet already has satisfied all the backbone hydrogen bonds, and the Arg and Gln polar groups point away from the water site toward solvent. This also explains why dramatic affinity can be gained with Trp while at the same time alanine scans of Erbin do not have a significant effect on the affinity of the Trp P-1 variant (see next section for details).
Prediction of peptide affinity
To determine whether WaterMap could be used to retrospectively predict the peptide affinities in Table I, the NMR structure was used as a template to create models of all the PDZ-peptide complexes from Table I (see Materials and Methods for details). The total energies were then estimated by summing over all water sites overlapping with the different peptides, according to the formula described in Abel et al.,26 with no adjustable parameters (see Materials and Methods). The WaterMap energies of the WETAV and WETFV peptides calculated using this formula are −27.8 kcal/mol and −31.3 kcal/mol, relative to −37.2 kcal/mol for WETWV. Thus, the trend of the calculated energy differences is in good agreement with the experimental results. Not surprisingly, there is a significant difference between the absolute values of the predicted and measured affinities, which is likely due to the neglect of entropy loss and induced strain in both the ligand and receptor. The WaterMap energies for the entire set of peptides in Table I were calculated and compared with experimental data. As can be seen in Figure 2, there is a good correlation (R2 = 0.67) between WaterMap energies and experimental peptide affinities, suggesting that displacement of water from the surface of the PDZ domain is an important contributor to the affinity of PDZ-peptides complexes.
Figure 2.
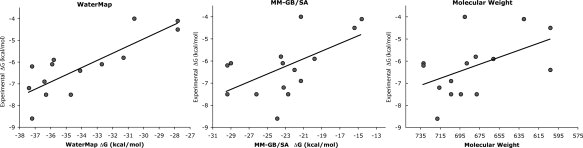
Correlations between computationally estimated energies and experimental peptide affinities [RTln (IC50)]. (a) WaterMap (R2 = 0.67), (b) MM–GB/SA (R2 = 0.33), (c) molecular weight (R2 = 0.25).
We also computed peptide affinities using the molecular mechanics–Generalized Born/surface area (MM–GB/SA) method, as implemented in Prime.32–35 This method has been used to successfully predict binding affinities in diverse systems such as thrombin, factor Xa, HIV-RT, and several kinases.34,36 In the case of Erbin studied here, there is only modest correlation between peptide affinities and MM–GB/SA energies [R2 = 0.33, see Fig. 2(B)]. Indeed, the MM–GB/SA correlation only slightly exceeds the correlation of molecular weight to affinity of the peptides [R2 = 0.24, see Fig. 2(C)]. Thus, while implicit solvation and non-bonded interaction energy terms provide some explanation of the affinity of this series of peptides, more reliable predictions are obtained when the role of explicit water is considered.
Virtual amino acid screening
To further assess the ability of WaterMap to explain and predict relative binding affinities, we performed a single residue modification to each of the 20 amino acid types at the five different positions P-0 to P-4. As shown in Figure 3, the WaterMap energies predict that the optimal residues at the P-0, P-1, and P-4 sites are V, W, and W, respectively, in agreement with the optimal motif identified through phage display. For P-0, the β-branched side-chains Ile and Thr have similar energies to the optimal Val. For P-2 and P-3, there appears to be no specific preference for Thr and Glu respectively in terms of water displacement. Although there is significant water overlap of these side chains with water sites, many other amino acids can give comparable or even improved WaterMap energies. This suggests that other structural criteria are involved in determining selectivity at these positions in Erbin (e.g., hydrogen bond formation at P-2 or charge–charge interactions at P-3).
Figure 3.
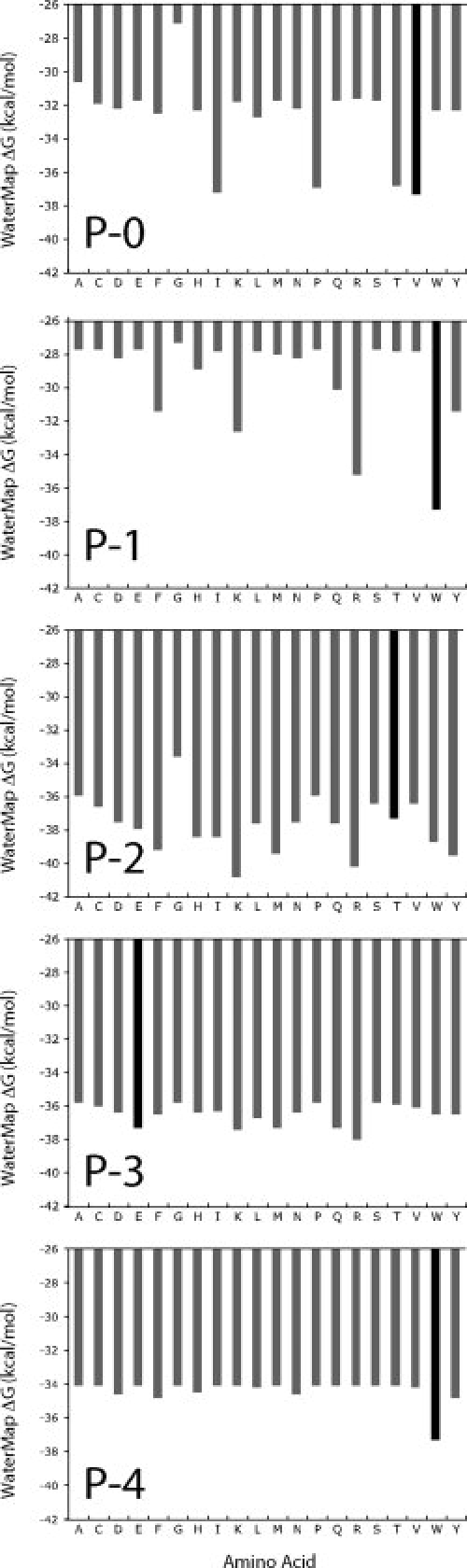
Virtual amino acid screening of the WETWV peptide. All 20 amino-acid types were introduced in the WETWV reference peptide at all five positions. For positions P-0, P-1, and P-4, the phage-display derived optimal residues (black) were identified among the most favorable based on the WaterMap energies. For P-2 and P-3, many different amino-acid types were comparable to or better than native residues, suggesting that energetic factors other than water displacement are important for affinity at these positions.
Erbin mutants S26A, R49A, and Q51A
Skelton et al. (ref.16) probed the dependence of the peptide affinity on specific side-chain interactions with the PDZ domain by mutating all residues in the peptide binding site to Ala.16 They discovered that none of the mutations in the proximity of the Trp (i.e., S26A, R49A, and Q51A) strongly affected peptide affinity. In particular, the S26A mutation resulted in only a 1.5-fold loss of affinity and R49A affinity was 3.4-fold less, while the Q51A mutant affinity was 9-fold higher than wild-type. To investigate whether these variants contain conserved high-energy water sites, we generated models for all three mutants (see Materials and Methods). Locations and energies of water sites for the mutants were calculated using WaterMap and the difference in affinity of the WETWV and WETAV peptides was predicted. The results from WaterMap for these three mutants reveal that the w-1b high-energy water site predicted in the wild type is also present in the structures of the Ala mutants (see Fig. 4). The energies of the w-1b water site are 6.1 kcal/mol in S26A, 5.2 kcal/mol in Q51A, and 4.6 kcal/mol in R49A, compared to 5.8 kcal/mol in wild-type Erbin. In all three systems, there is a large difference between the energies of the WETWV and WETAV peptides, due to the overlap of the Trp side chain with multiple high-energy water sites. The values of these differences are 9.9 kcal/mol for S26A, 8.2 kcal/mol for Q51A, and 7.1 kcal/mol for R49A, compared to 9.4 kcal/mol for wild-type. Thus, these results are in good agreement with the observation that specific side-chain interactions are not important for Trp binding in Erbin.
Figure 4.
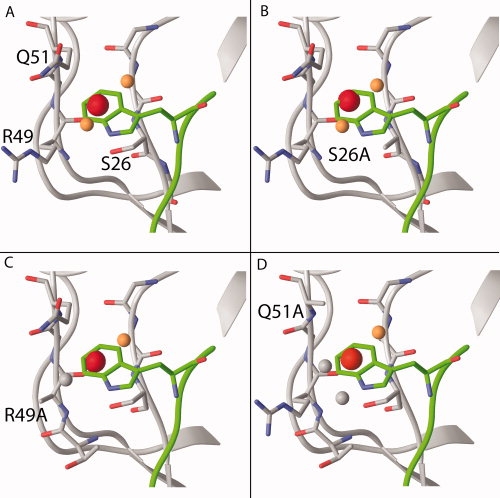
Conservation of the P-1 water sites in Erbin wild-type (A), S26A (B), R49A (C), and Q51A (D) mutants. The color scheme of the water sites is described in Figure 1. The w-1b high-energy water site (red sphere) is present in all three mutants. The entropic and enthalpic contributions to ΔG are similar in wild-type (−TΔS 2.6 kcal/mol, ΔH 3.2 kcal/mol), R49A (−TΔS 2.3 kcal/mol, ΔH 2.3 kcal/mol), and Q51A (−TΔS 2.4 kcal/mol, ΔH 2.8 kcal/mol), while the site in the S26A has a strong unfavorable enthalpic term (−TΔS 1.5 kcal/mol, ΔH 4.6 kcal/mol). Removal of the Trp side chain in the peptide leads to a large reduction in predicted affinity in all three mutants (from 7.1 to 9.9 kcal/mol).
Although the absolute free energy of the w-1b high-energy water site is similar in wild type (5.8 kcal/mol) and S26A (6.1 kcal/mol), there is an interesting difference in the thermodynamic characteristics of the sites. The wild type free energy has both large entropic and enthalpic contributions (−TΔS = 2.6 kcal/mol, ΔH = 3.2 kcal/mol). In the S26A mutant, the removal of the hydroxyl group increases the enthalpic penalty (ΔH = 4.6 kcal/mol) but this is compensated by a decrease in the entropy (−TΔS = 1.5 kcal/mol). Thus, the water site has approximately equal free energy in the wild type and S26A, but with a different balance of enthalpy and entropy (a case of entropy/enthalpy compensation).37
Finally, there are significant differences between the WaterMap free energies of the entire peptide between wild-type and the Ala mutants. Thus, while the wild-type and the S26A mutant calculated free energies are similar (−35.8 kcal/mol vs. −35.7 kcal/mol), the values of Q51A (−32.9 kcal/mol), and R49A (−29.7 kcal/mol) are significantly reduced. These differences can be traced to the energies of the water sites in the P-1 pocket and to the proximity of the P-3 side chain (especially in R49A). The differences in energies increase with the more dramatic mutations (R49A>Q51A>S26A). It is possible that the R49 and Q51 could induce local changes in the peptide side-chain orientations, which might attenuate the differences.
Conservation of the P-1 water sites in other PDZ domains
Application of WaterMap to a series of PDZ domains other than Erbin suggests that an unfavorable water site at the P-1 pocket may be present in all PDZ domains. The PDZ domains that were investigated here include ZO-1, disheveled (dvl), AF-6, and the HTRA family. The results for the HTRA family are described in detail below. In the case of ZO-1, dvl, and AF-6, the simulations indicate that the P-1 pocket accommodates at least one moderately or highly unfavorable water within 2 Å of the Erbin w-1b site (data not shown). Thus, the displacement of high-energy water from the P-1 site might be a general mechanism for affinity in PDZ domains. There are significant differences in the magnitudes of the free energies of the water sites among the PDZ domains that were tested, and it is possible that these differences might correlate with differences in specificity.
To explore this further, WaterMap was run on the HTRA family of PDZ domains, including HTRA1, 2, and 3. Phage display peptide profiles are available for the members of this family,19,20 and scanning Ala mutations of the peptide have revealed differences in sensitivity to Trp at P-1. As can be seen in Table II, the mutation of Trp (P-1) to Ala from the HTRA2 and HTRA3 super binding peptides (SWTMFWV and FGRWV, respectively), leads to a dramatic reduction in affinity (>300 fold and ∼450 fold, or 3.2 kcal/mol and 3.4 kcal/mol, respectively). In contrast, mutation of Trp (P-1) to Ala only attenuates the affinity of the DSRIWWV peptide for HTRA1 by 6-fold (1.1 kcal/mol). These differences correlate well with qualitative and quantitative differences among the water site locations and energies of the three HTRA PDZ domains.
Table II.
Trp-Sensitivity of HTRA PDZ Domains
| PDZ domain | Peptide | IC50 (μM) | RTln (IC50) | WaterMap ΔG (kcal/mol) |
|---|---|---|---|---|
| HTRA1 | DSRIWWV | 0.9 | 7.8 | −28.3 |
| DSRIWAV | 6 | 6.7 | −24.6 | |
| HTRA2 | SWTMFWV | 3.3 | 7.1 | −34.1 |
| SWTMFAV | >1000 | <3.9 | −22.6 | |
| HTRA3 | FGRWV | 0.6 | 8.0 | −28.5 |
| FGRAV | 270 | 4.6 | −17.6 |
Peptide affinities for super-binding peptides against the three HTRA domains show a small difference between Trp and Ala at P-1 for HTRA1 (∼6-fold, 0.9 kcal/mol), but a large difference for HTRA2 (>300-fold, >3.2 kcal/mol) and HTRA3 (∼450-fold, 3.4 kcal/mol). Correspondingly, WaterMap ΔG values are much smaller for HTRA1 (3.7 kcal/mol) than for HTRA2 (11.5 kcal/mol) and HTRA3 (10.9 kcal/mol).
Similar to Erbin, calculations for HTRA2 and HTRA3 suggest several unfavorable water molecules in the P-1 pocket (see Fig. 5), with a central w-1b-like water that is highly unfavorable (5.9 kcal/mol and 4.8 kcal, respectively). In HTRA2, mutation of Trp to Ala leads to a computed loss of affinity of 11.5 kcal/mol, while in HTRA3, the computed loss is 10.9 kcal/mol. In HTRA1, there is only one moderately unfavorable water site, with a free energy of 3.3 kcal/mol, and the difference between Trp and Ala peptides is only 3.5 kcal/mol. Indeed, the rotameric states of the Arg386 and Glu416 residues in HTRA1 change the shape of the P-1 pocket relative to HTRA2/3, which limits the number of water sites in the pocket. Thus, differences in the energetics of bound water molecules among members of the HTRA family are consistent with the observed differences in specificity.
Figure 5.
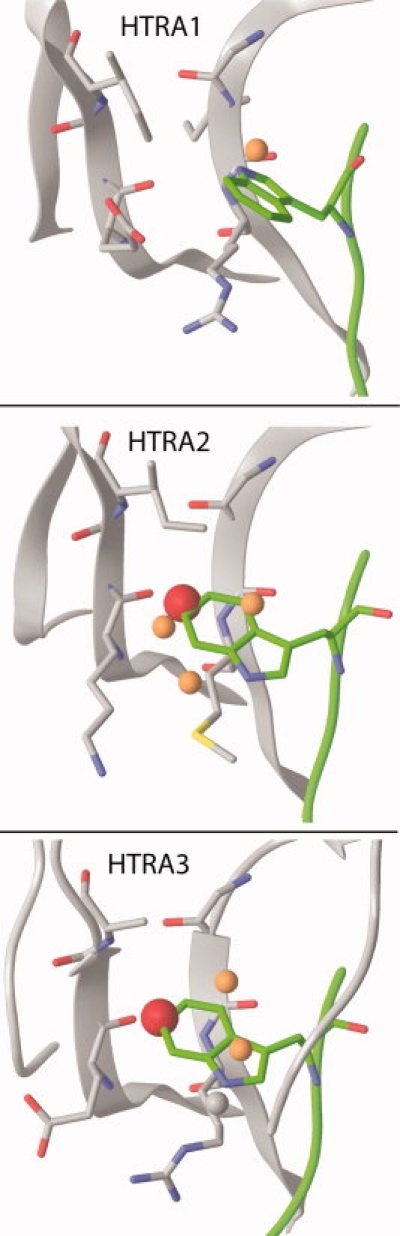
Predicted P-1 water-sites in the HTRA family. The color scheme of the water sites is described in Figure 1. HTRA2 (middle) and HTRA3 (bottom) both have several unfavorable water sites overlapping with the P-1 Trp side chain, with the high-energy w-1b site present in both pockets. HTRA1 (top) has only a single, moderately unfavorable water site overlapping with Trp. This is in good agreement with the observed sensitivity of HTRA2 and HTRA3, but not HTRA1, to the mutation of Trp to Ala.
Discussion
The molecular and cellular basis of specificity of PDZ-peptide interactions has been a field of intense research in recent years. One aspect of elucidating the mechanism of specificity is an accurate description of the molecular determinants of affinity of PDZ domains to peptides. A key technology to address this issue experimentally has been the high-throughput generation of peptide profiles using phage display.21 An important insight that has resulted from these experiments is that in addition to the P-0 and P-2 positions, the P-1 position is responsible for high-affinity binding of peptides. Specifically, several PDZ domains, including Erbin, ZO-1, and members of the HTRA family have been shown to bind optimally to peptides with Trp residues at P-1. However, the mechanism of high-affinity Trp binding has remained unknown, as no specific side-chain interactions have been identified that confer Trp specificity to the PDZ domain.16 The prediction of a high-energy water site in the Trp binding pocket described here now suggests a molecular basis for this effect.
The WaterMap technology used here has been used previously to identify specific 3D-protein motifs that generate high-energy water sites in proteins, for example the hinge region of kinases and the binding site of streptavidin.25 Here, we show that the exposed surface of a β-sheet can create a particularly unfavorable environment for a water molecule. Thus, in the case of Erbin, the β-sheet and the polar groups of Ser26, Arg49, and Gln51 contribute to a polar environment that might traditionally be classified as hydrophilic, but this region is nonetheless unfavorable for water because of the absence of specific hydrogen bonding partners. Indeed, removal of these polar side chains by mutation to Ala maintains the unfavorable water site, in agreement with experimental observations.
Strong correlations between WaterMap energies and experimental binding affinities of small molecules have been observed for factor Xa.26 It is encouraging that in this work the methodology has proven to be amenable to biologically endogenous protein-peptide interactions. In fact, the PDZ domains appears to be well-suited to this approach, since induced-fit effects are minimal in the binding event of peptides,9 and the interactions are predominantly non-polar in nature. While free energy trends in this case are captured by WaterMap—leading to strong correlations between computed free energies and experimental binding affinities—the computed WaterMap energies are considerably more favorable than the experimental peptide affinities. Neglect of the ligand entropy terms (translational, rotational, and configurational) associated with the binding event is likely to be the primary source of these differences. Indeed, the magnitude of this entropy term was estimated using quasi-harmonic analysis to be between 5 and 24 kcal/mol for a series of 12 PDZ/peptide complexes.38 Other neglected components of the free energy that disfavor binding include induced strain of the peptide and protein reorganization energy of the PDZ domain. We also note a general overestimation of the ΔΔG between pairs of peptides [i.e., the slope of the line in Fig. 2(A) is less than one]. For example, the difference in experimental binding between the WETWV and WETAV peptides for Erbin is 4.1 kcal/mol while the calculated difference is 9.5 kcal/mol. It is likely that this overestimation is caused by the neglect of second order terms in the calculation of the entropy. In cases where multiple water sites are found in close proximity (e.g., the P-1 pocket), this neglect of water–water correlation could lead to overestimation of the free energies of these sites. Regardless of the absolute values of the calculated binding affinity, the predicted trends in affinity presented in this work suggest that the WaterMap methodology may be useful for predicting relative ligand binding affinities in other systems.
To estimate the free energy differences of the peptides in Table I, substitutions were introduced in the structure of the WETWV peptide by maintaining the χ1 and χ2 angles. This appears to be a reasonable assumption, since most mutations were either conservative or to Ala. Thus, the models of the peptide-PDZ domain complex can be expected to be reasonable, and indeed there is a good correlation between the peptide affinities and the computed energies. The introduction of mutations in the PDZ domains is more complicated, especially in the case of R49A and Q51A. While the measured affinities of WETWV against wild-type and the three mutants are approximately constant, there are differences in the total WaterMap energies for the peptide. These differences can be attributed not only to the energies of water sites in the P-1 pocket, but also more distal effects. For example, the R49A mutation changes the water environment around Glu (P-3). However, the removal of a positive charge from the protein surface undoubtedly has an effect on the orientation of Glu (P-3), and therefore, the current model may be inappropriate to assess the total water displacement free energy of the entire peptide. In the absence of an X-ray structure for the mutants, the induced changes in the binding mode of peptides needs to be more carefully modeled, which is beyond the scope of this study.
The differences in Trp sensitivity among members of the HTRA family were found to be in good agreement with the differences in energies of water sites in the P-1 pocket. The structures of the HTRA family reveal qualitative differences in the side-chain orientation of the P-1 pocket. In HTRA1, the β-branched side chains of Ile415 and Ile418 pack in a manner that appears to favor the formation of a salt bridge between Arg386 and Glu416, and this creates a different shape of the P-1 pocket. On the basis of our results, we suggest that these differences have an effect on the peptide affinity by modulating the water energies in the unbound state of the PDZ domain.
Although the biological and structural properties of PDZ domains are increasingly well described, the development of small molecule inhibitors is still in its infancy. PDZ domains are potentially relevant pharmaceutical targets because of their important role in signaling pathways.39 Given their abundant interaction with GPCRs, modulation of PDZ-receptor interactions could be an indirect approach to target this important class of targets. Several PDZ domains have been implicated in disease, including the PDZ domain containing protein PICK1 in schizophrenia40 and the disheveled (dvl) protein in cancer. Indeed, a dvl inhibitor named FJ9 was shown to have anti-tumor properties.41 Several studies have focused on developing improved compounds for inhibiting dvl and the structure-activity relationships for some of these compounds suggest that water displacement is an important mechanism for small molecule binding as well. For example, compounds with large aromatic substituents at a position that is expected to overlap with the Trp-1 site were shown to have improved affinity over the lead compound.42,43 In general, the methodology described in this work to quantify the energetics of water molecules in the binding process and the application thereof should aid in the understanding of molecular recognition in PDZ domains and potentially other pharmaceutically relevant targets.
Materials and Methods
The WaterMap methodology has been described in detail in Abel et al.26 Molecular dynamics simulations were performed with the Desmond molecular dynamics engine44 using the OPLS2005 force field.45,46 The starting structures were obtained from the Protein Data Bank47 and prepared with the Protein Preparation Wizard in Maestro 8.5.48 The simulation system was generated using the System Builder module of Desmond. Briefly, atoms of the protein were truncated beyond 15 Å of the peptide binding site and the resulting protein construct was solvated in a TIP4P water box extending at least 5 Å beyond the protein in all directions. The peptide was not included in the system. For all simulations, a 9 ns production MD simulation with positional restraints on the protein non-hydrogen atoms was performed following the default relaxation protocol, which involved successive stages of minimization and heating. Water molecules in the proximity of the peptide binding site from approximately 2000 equally spaced snapshots from the simulation were clustered to form hydration sites. The enthalpy was computed from the average non-bonded energy of each hydration site. The excess entropy was computed by numerically integrating a local expansion of spatial and orientational correlation functions49,50 as implemented in Abel et al.26 As an approximation, only contributions from the first order term of the expansion were included in the entropy calculation. Ligand binding energies were then estimated as the sum of hydration site (hs) free energies that are displaced by ligand atoms (lig) upon binding. The function for the ΔG of binding is
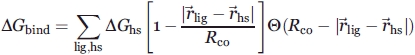 |
(1) |
where, Rco is the distance cutoff for a ligand atom to begin displacing a hydration site, ΔGhs is the computed free energy of transferring the water molecule in a given hydration site from the active site to the bulk fluid, and θ is the Heaviside step function. The value of Rco was chosen to be 2.24.26
The best representative conformer of the NMR ensemble (the structure with the least violations of the experimental restraints) of the Erbin/peptide complex (PDB ID: 1N7T ,16) was used to calculate the Erbin WaterMap. To assess consistency and reproducibility of the results, calculations were repeated for additional structures from the ensemble, as well as with a crystal structure of the Erbin PDZ domain (PDB ID: 1MFG).51 The WaterMap results (hydration site locations and corresponding free energies) from these structures were quantitatively similar and only the representative structure was used to estimate the peptide energies. For the HTRA1/DSRIWWV complex, the best representative NMR conformer of PDB entry 2JOA19 was used, while for HTRA2/WTMFWV and HTRA3/FGRWV complexes, X-ray structures were available (PDB ID: 2PZD20 and 2P3W19).
Models of the S26A, R49A, and Q51A mutants were generated by deleting all side-chain atoms except for the Cβ carbon. The structural effects of these mutations were assumed to be minimal; hence no optimization of the backbone and proximal side chains was performed. Models of PDZ/peptide complexes were based on the Erbin/WETWV structure and substitutions were introduced by keeping the χ1 and χ2 dihedral angles constant. For non-conservative mutations, the side chains were manually reoriented to maximize the overlap with the WETWV residues, while minimizing steric overlap with the PDZ domain.
MM–GB/SA energies were calculated using Prime32,33,35 by taking the difference between the bound state and unbound state energies of the protein–peptide system after minimization.34 Reported energies include the molecular mechanics energy (intramolecular and non-bonded terms) as well as the solvation energy (polar and non-polar terms). Peptide and protein configurational entropies were not included in these calculations; however, an approximation to the solvent entropy is included as an implicit term in the Generalized Born methodology.
Glossary
Abbreviations:
- dvl
dishevelled
- GPCR
G-protein coupled receptor
- HTRA
high temperature requirement A
- MD
molecular dynamics
- MM–GB/SA
molecular mechanics–Generalized Born surface area
- NMR
nuclear magnetic resonance
- PDZ
PSD-95/Discs-large/ZO-1.
REFERENCES
- 1.Nourry C, Grant SG, Borg JP. PDZ domain proteins: plug and play! Sci STKE RE7. 2003 doi: 10.1126/stke.2003.179.re7. [DOI] [PubMed] [Google Scholar]
- 2.Bilder D, Schober M, Perrimon N. Integrated activity of PDZ protein complexes regulates epithelial polarity. Nat Cell Biol. 2003;5:53–58. doi: 10.1038/ncb897. [DOI] [PubMed] [Google Scholar]
- 3.Hurd TW, Gao L, Roh MH, Macara IG, Margolis B. Direct interaction of two polarity complexes implicated in epithelial tight junction assembly. Nat Cell Biol. 2003;5:137–142. doi: 10.1038/ncb923. [DOI] [PubMed] [Google Scholar]
- 4.Kim E, Sheng M. PDZ domain proteins of synapses. Nat Rev Neurosci. 2004;5:771–781. doi: 10.1038/nrn1517. [DOI] [PubMed] [Google Scholar]
- 5.Madsen KL, Eriksen J, Milan-Lobo L, Han DS, Niv MY, Ammendrup-Johnsen I, Henriksen U, Bhatia VK, Stamou D, Sitte HH, McMahon HT, Weinstein H, Gether U. Membrane localization is critical for activation of the PICK1 BAR domain. Traffic. 2008;9:1327–1343. doi: 10.1111/j.1600-0854.2008.00761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harris BZ, Lim WA. Mechanism and role of PDZ domains in signaling complex assembly. J Cell Sci. 2001;114:3219–3231. doi: 10.1242/jcs.114.18.3219. [DOI] [PubMed] [Google Scholar]
- 7.Schultz J, Copley RR, Doerks T, Ponting CP, Bork P. SMART: a web-based tool for the study of genetically mobile domains. Nucleic Acids Res. 2000;28:231–234. doi: 10.1093/nar/28.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beuming T, Skrabanek L, Niv MY, Mukherjee P, Weinstein H. PDZBase: a protein-protein interaction database for PDZ-domains. Bioinformatics. 2005;21:827–828. doi: 10.1093/bioinformatics/bti098. [DOI] [PubMed] [Google Scholar]
- 9.Doyle DA, Lee A, Lewis J, Kim E, Sheng M, MacKinnon R. Crystal structures of a complexed and peptide-free membrane protein-binding domamolecular basis of peptide recognition by PDZ. Cell. 1996;85:1067–1076. doi: 10.1016/s0092-8674(00)81307-0. [DOI] [PubMed] [Google Scholar]
- 10.Oschkinat H. A new type of PDZ domain recognition. Nat Struct Biol. 1999;6:408–410. doi: 10.1038/8203. [DOI] [PubMed] [Google Scholar]
- 11.Lockless SW, Ranganathan R. Evolutionarily conserved pathways of energetic connectivity in protein families. Science. 1999;286:295–299. doi: 10.1126/science.286.5438.295. [DOI] [PubMed] [Google Scholar]
- 12.Niv MY, Weinstein H. A flexible docking procedure for the exploration of peptide binding selectivity to known structures and homology models of PDZ domains. J Am Chem Soc. 2005;127:14072–14079. doi: 10.1021/ja054195s. [DOI] [PubMed] [Google Scholar]
- 13.Songyang Z, Fanning AS, Fu C, Xu J, Marfatia SM, Chishti AH, Crompton A, Chan AC, Anderson JM, Cantley LC. Recognition of unique carboxyl-terminal motifs by distinct PDZ domains. Science. 1997;275:73–77. doi: 10.1126/science.275.5296.73. [DOI] [PubMed] [Google Scholar]
- 14.Wiedemann U, Boisguerin P, Leben R, Leitner D, Krause G, Moelling K, Volkmer-Engert R, Oschkinat H. Quantification of PDZ domain specificity, prediction of ligand affinity and rational design of super-binding peptides. J Mol Biol. 2004;343:703–718. doi: 10.1016/j.jmb.2004.08.064. [DOI] [PubMed] [Google Scholar]
- 15.Kurakin A, Swistowski A, Wu SC, Bredesen DE. The PDZ domain as a complex adaptive system. PLoS ONE. 2007;2:e953. doi: 10.1371/journal.pone.0000953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Skelton NJ, Koehler MF, Zobel K, Wong WL, Yeh S, Pisabarro MT, Yin JP, Lasky LA, Sidhu SS. Origins of PDZ domain ligand specificity. Structure determination and mutagenesis of the Erbin PDZ domain. J Biol Chem. 2003;278:7645–7654. doi: 10.1074/jbc.M209751200. [DOI] [PubMed] [Google Scholar]
- 17.Appleton BA, Zhang Y, Wu P, Yin JP, Hunziker W, Skelton NJ, Sidhu SS, Wiesmann C. Comparative structural analysis of the Erbin PDZ domain and the first PDZ domain of ZO-1. Insights into determinants of PDZ domain specificity. J Biol Chem. 2006;281:22312–22320. doi: 10.1074/jbc.M602901200. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Y, Yeh S, Appleton BA, Held HA, Kausalya PJ, Phua DC, Wong WL, Lasky LA, Wiesmann C, Hunziker W, Sidhu SS. Convergent and divergent ligand specificity among PDZ domains of the LAP and zonula occludens (ZO) families. J Biol Chem. 2006;281:22299–22311. doi: 10.1074/jbc.M602902200. [DOI] [PubMed] [Google Scholar]
- 19.Runyon ST, Zhang Y, Appleton BA, Sazinsky SL, Wu P, Pan B, Wiesmann C, Skelton NJ, Sidhu SS. Structural and functional analysis of the PDZ domains of human HtrA1 and HtrA3. Protein Sci. 2007;16:2454–2471. doi: 10.1110/ps.073049407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Y, Appleton BA, Wu P, Wiesmann C, Sidhu SS. Structural and functional analysis of the ligand specificity of the HtrA2/Omi PDZ domain. Protein Sci. 2007;16:1738–1750. doi: 10.1110/ps.072833207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tonikian R, Zhang Y, Sazinsky SL, Currell B, Yeh JH, Reva B, Held HA, Appleton BA, Evangelista M, Wu Y, Xin X, Chan AC, Seshagin S, Lasky LA, Sander C, Boone C, Bader GD, Sidhu SS. A specificity map for the PDZ domain family. PLoS Biol. 2008;6:e239. doi: 10.1371/journal.pbio.0060239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bezprozvanny I, Maximov A. Classification of PDZ domains. FEBS Lett. 2001;509:457–462. doi: 10.1016/s0014-5793(01)03214-8. [DOI] [PubMed] [Google Scholar]
- 23.Karthikeyan S, Leung T, Birrane G, Webster G, Ladias JA. Crystal structure of the PDZ1 domain of human Na(+)/H(+) exchanger regulatory factor provides insights into the mechanism of carboxyl-terminal leucine recognition by class I PDZ domains. J Mol Biol. 2001;308:963–973. doi: 10.1006/jmbi.2001.4634. [DOI] [PubMed] [Google Scholar]
- 24.Wang L, Piserchio A, Mierke DF. Structural characterization of the intermolecular interactions of synapse-associated protein-97 with the NR2B subunit of N-methyl-D-aspartate receptors. J Biol Chem. 2005;280:26992–26996. doi: 10.1074/jbc.M503555200. [DOI] [PubMed] [Google Scholar]
- 25.Young T, Abel R, Kim B, Berne BJ, Friesner RA. Motifs for molecular recognition exploiting hydrophobic enclosure in protein-ligand binding. Proc Natl Acad Sci USA. 2007;104:808–813. doi: 10.1073/pnas.0610202104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abel R, Young T, Farid R, Berne BJ, Friesner RA. Role of the active-site solvent in the thermodynamics of factor Xa ligand binding. J Am Chem Soc. 2008;130:2817–2831. doi: 10.1021/ja0771033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bryant PJ, Huwe A. LAP proteins: what's up with epithelia? Nat Cell Biol. 2000;2:E141–E143. doi: 10.1038/35019616. [DOI] [PubMed] [Google Scholar]
- 28.Borg JP, Marchetto S, Le Bivic A, Ollendorff V, Jaulin-Bastard F, Saito H, Fournier E, Adelaide J, Margolis B, Birnbaum D. ERBa basolateral PDZ protein that interacts with the mammalian ERBB2/HER2 receptor. Nat Cell Biol. 2000;2:407–414. doi: 10.1038/35017038. [DOI] [PubMed] [Google Scholar]
- 29.Laura RP, Witt AS, Held HA, Gerstner R, Deshayes K, Koehler MF, Kosik KS, Sidhu SS, Lasky LA. The Erbin PDZ domain binds with high affinity and specificity to the carboxyl termini of delta-catenin and ARVCF. J Biol Chem. 2002;277:12906–12914. doi: 10.1074/jbc.M200818200. [DOI] [PubMed] [Google Scholar]
- 30.Kim DY, Kim KK. Structure and function of HtrA family proteins, the key players in protein quality control. J Biochem Mol Biol. 2005;38:266–274. doi: 10.5483/bmbrep.2005.38.3.266. [DOI] [PubMed] [Google Scholar]
- 31.Ekert PG, Vaux DL. The mitochondrial death squad: hardened killers or innocent bystanders? Curr Opin Cell Biol. 2005;17:626–630. doi: 10.1016/j.ceb.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 32.Jacobson MP, Friesner RA, Xiang Z, Honig B. On the role of the crystal environment in determining protein side-chain conformations. J Mol Biol. 2002;320:597–608. doi: 10.1016/s0022-2836(02)00470-9. [DOI] [PubMed] [Google Scholar]
- 33.Jacobson MP, Kaminski GA, Friesner RA, Rapp CS. Force field validation using protein side chain prediction. J Phys Chem B. 2002;106:11673–11680. [Google Scholar]
- 34.Lyne PD, Lamb ML, Saeh JC. Accurate prediction of the relative potencies of members of a series of kinase inhibitors using molecular docking and MM-GBSA scoring. J Med Chem. 2006;49:4805–4808. doi: 10.1021/jm060522a. [DOI] [PubMed] [Google Scholar]
- 35.Yu Z, Jacobson MP, Friesner RA. What role do surfaces play in GB models? A new-generation of surface-generalized born model based on a novel gaussian surface for biomolecules. J Comput Chem. 2006;27:72–89. doi: 10.1002/jcc.20307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guimaraes CRW, Cardozo M. MM-GB/SA rescoring of docking poses in structure-based lead optimization. J Chem Inf Model. 2008;48:958–970. doi: 10.1021/ci800004w. [DOI] [PubMed] [Google Scholar]
- 37.Gallicchio E, Kubo MM, Levy RM. Entropy-enthalpy compensation in solvation and ligand binding revisited. J Am Chem Soc. 1998;120:4526–4527. [Google Scholar]
- 38.Basdevant N, Weinstein H, Ceruso M. Thermodynamic basis for promiscuity and selectivity in protein-protein interactions: PDZ domains, a case study. J Am Chem Soc. 2006;128:12766–12777. doi: 10.1021/ja060830y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang NX, Lee HJ, Zheng JJ. Therapeutic use of PDZ protein-protein interaction antagonism. Drug News Perspect. 2008;21:137–141. [PMC free article] [PubMed] [Google Scholar]
- 40.Dev KK, Henley JM. The schizophrenic faces of PICK1. Trends Pharmacol Sci. 2006;27:574–579. doi: 10.1016/j.tips.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fujii N, You L, Xu Z, Uematsu K, Shan J, He B, Mikami I, Edmondson LR, Neale G, Zheng J, Guy RK, Jablons DM. An antagonist of dishevelled protein-protein interaction suppresses beta-catenin-dependent tumor cell growth. Cancer Res. 2007;67:573–579. doi: 10.1158/0008-5472.CAN-06-2726. [DOI] [PubMed] [Google Scholar]
- 42.Shan J, Shi DL, Wang J, Zheng J. Identification of a specific inhibitor of the dishevelled PDZ domain. Biochemistry. 2005;44:15495–15503. doi: 10.1021/bi0512602. [DOI] [PubMed] [Google Scholar]
- 43.Shan J, Zheng JJ. Optimizing Dvl PDZ domain inhibitor by exploring chemical space. J Comput Aided Mol Des. 2009;23:37–47. doi: 10.1007/s10822-008-9236-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bowers KJ, Chow E, Xu H, Dror RO, Eastwood MP, Gregersen BA, Klepeis JL, Kolossvary I, Moraes MA, Sacerdoti FD, Salmon JK, Shan Y, Shaw DE. 2006. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the ACM/IEEE Conference on Supercomputing (SC06). New York: ACM Press.
- 45.Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc. 1996;118:11225–11236. [Google Scholar]
- 46.Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J Phys Chem B. 2001;105:6474–6487. [Google Scholar]
- 47.Deshpande N, Addess KJ, Bluhm WF, Merino-Ott JC, Townsend-Merino W, Zhang Q, Knezevich C, Xie L, Chen L, Feng Z, Green RK, Flippen-Anderson JL, Westbrook J, Berman HM, Bourne PE. The RCSB protein data bank: a redesigned query system and relational database based on the mmCIF schema. Nucleic Acids Res. 2005;33:D233–D237. doi: 10.1093/nar/gki057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maestro. 8.5 ed. LLC: Schrodinger; 2008. [Google Scholar]
- 49.Lazaridis T. Inhomogeneous fluid approach to solvation thermodynamics. 1 theory. J Phys Chem B. 1998;102:3531–3541. [Google Scholar]
- 50.Lazaridis T. Inhomogeneous fluid approach to solvation thermodynamics. 2. Applications to simple fluids. J Phys Chem B. 1998;102:3542–3550. [Google Scholar]
- 51.Birrane G, Chung J, Ladias JA. Novel mode of ligand recognition by the Erbin PDZ domain. J Biol Chem. 2003;278:1399–1402. doi: 10.1074/jbc.C200571200. [DOI] [PubMed] [Google Scholar]