

Abstract
Loop 181–197 of human thymidylate synthase (hTS) populates two major conformations, essentially corresponding to the loop flipped by 180°. In one of the conformations, the catalytic Cys195 residue lies distant from the active site making the enzyme inactive. Ligands stabilizing this inactive conformation may function as allosteric inhibitors. To facilitate the search for such inhibitors, we have expressed and characterized several mutants designed to shift the equilibrium toward the inactive conformer. In most cases, the catalytic efficiency of the mutants was only somewhat impaired with values of kcat/Km reduced by factors in a 2–12 range. One of the mutants, M190K, is however unique in having the value of kcat/Km smaller by a factor of ∼7500 than the wild type. The crystal structure of this mutant is similar to that of the wt hTS with loop 181–197 in the inactive conformation. However, the direct vicinity of the mutation, residues 188–194 of this loop, assumes a different conformation with the positions of Cα shifted up to 7.2 Å. This affects region 116–128, which became ordered in M190K while it is disordered in wt. The conformation of 116–128 is however different than that observed in hTS in the active conformation. The side chain of Lys190 does not form contacts and is in solvent region. The very low activity of M190K as compared to another mutant with a charged residue in this position, M190E, suggests that the protein is trapped in an inactive state that does not equilibrate easily with the active conformer.
Keywords: thymidylate synthase, chemotherapy, colon cancer, conformational switching
Introduction
Thymidylate synthase (TS) catalyses the reaction that forms 2′-deoxythymidine 5′-monophosphate (dTMP) from 2′-deoxyuridine 5′-monophosphate (dUMP), using 5,10-methylenetetrahydrofolate (mTHF) as a cosubstrate.1 Inhibitory analogs of nucleotide- and folate-substrates are used in cancer chemotherapy because of the cytotoxic effects of thymidylate depletion.2,3 Human TS (hTS) differs from Escherichia coli TS (ecTS) by an N-terminal extension of 29 residues and two insertions at positions 117 and 146 of 12- and 8-residues, respectively.4 Upon comparison of native (unliganded) three-dimensional structures of hTS with its bacterial homologues, it was observed that loop 181–197 containing the cysteine nucleophile crucial in catalysis (Cys195 in hTS) is rotated ∼180° in hTS.5,6 Consequently, the catalytic thiol is ∼10 Å away from the active site, indicating that the human enzyme is in an inactive conformation. In contrast, upon formation of inhibitory ternary complexes with substrate analogues, the catalytic cysteines of hTS and ecTS are in the same orientation.7,8 Also, Cys195 is oriented within the active site upon deletion (Δ7-29) of 23 residues from the N-terminus of hTS.9 Collectively, the data show that loop 181–197 of hTS populates two major conformations: active and inactive.
Loop 181–197 contains a conserved tryptophan at position 182; its position differs between the active and inactive conformations by about 5 Å, whereas the positions of other tryptophan residues are essentially unchanged in the two conformers. Tryptophan fluorescence has been used as a tool to monitor conformational changes in proteins. In contrast to the inhibitory complex, four phosphate/sulfate-binding sites were observed in unliganded hTS, suggesting that binding of these ions stabilizes the inactive conformation of hTS. Titration with phosphate resulted in a concentration-dependent increase in the intrinsic fluorescence of native hTS in solution6 while addition of the substrate, dUMP, to phosphate-bound protein reversed this intrinsic fluorescence signal enhancement. In contrast, the intrinsic fluorescence of ecTS was refractory to phosphate concentration.6 The fluorescence data suggest that hTS, unlike ecTS, populates two states, active and inactive, which are in equilibrium modulated by phosphate or sulfate ions.
The physiological advantages of the existence and population of the inactive conformer in solution are not obvious. One possibility is the protection of the catalytic thiol from oxidation by removing it from the environment that promotes the thiolate state.10 It is also possible that this conformer plays a role in nonenzymatic functions of hTS. The inactive conformer binds 3-4 phosphate/sulfate ions, with the geometry that may reflect the enzyme binding to its own mRNA. This was proposed to be an autoregulatory translational mechanism that contributes to the controlling of intracellular levels of hTS.11 TS inhibitors utilized clinically interfere with translational repression and elevate steady-state TS levels, which is postulated to contribute to drug resistance12 although only small fractions of both hTS protein and TS mRNA are capable of binding each other.13,14 Stabilization of the inactive conformer as a way to inhibit hTS activity and alter the resistance mechanisms observed with current inhibitors was proposed,5 and it was argued that allosteric inhibition may lead in some cases to lesser resistance than that typically observed for currently used active site inhibitors.6,15 Such inhibitors were identified and showed positive cooperativity with some antifolate-based TS inhibitors.15 However, their polarity leads to poor membrane transport properties, and search for alternative inhibitory compounds would benefit from mutants with stabilized inactive or active conformations.
Previously, a R163K variant of hTS that was stabilized in the active conformation has been obtained.10 The major objective of this investigation was the generation of hTS variants that are stabilized in the inactive conformation of loop 181–197, with a long-term goal of using these proteins in drug discovery. On the basis of our analysis of the roles of Met190, Ala191, and Leu198 in the inactive and active conformations (see mutant design), we targeted these sites and characterized the resulting mutant proteins.
Results
Mutant design
Superpositioning of the structures of native hTS (inactive conformation) and the hTS/dUMP/ZD1694 complex (active conformation) generated hypotheses regarding the role of some residues in enzyme conformation. A striking difference in the active and inactive conformers is the environment of Met190 and Ala191. These amino acids are hydrophobic and invariant at their corresponding positions in all TSs. In the active conformer, Met190 and Ala191 are located in a hydrophobic pocket and sequestered away from the solvent [Fig. 1(a)]. However, in the inactive conformer, residues Met190 and Ala191 are exposed to solvent and lie in a very hydrophilic environment [Fig. 1(b)] with the Met190 side chain extended into the area occupied by loop 111–128 in the active conformation. Collectively, the data suggested that these residues may play a critical role in altering the conformational equilibrium of hTS and that substitution of Met190 and Ala191 with polar residues would destabilize the active conformation and shift the conformational equilibrium toward the inactive state.
Figure 1.
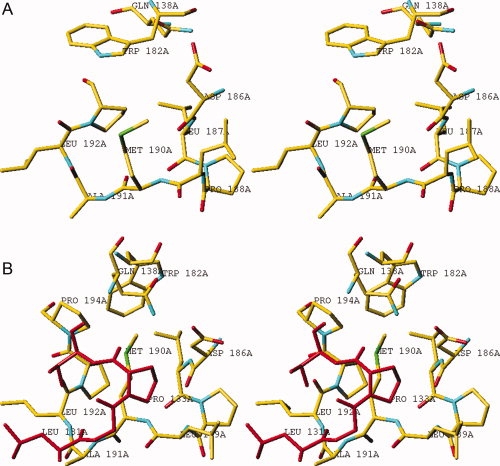
A stereoview of Met190 and Ala191 environment in the inactive (a) and active (b) conformation. (a) The side chains of 190 and 191 residues are directed toward solvent. (b) In contrast, in the active conformation residues 131–134, shown in red, screen the side chains of Met190 and Ala191 from the solvent. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
A different principle was used to design L198P. Ala197 changes its conformation from extended to helical upon the transition of loop 181–197 from the inactive to the active conformation. Since residues preceding a proline show strong preference for the extended conformation, the introduction of a proline in position 198 was thought to destabilize the active conformer.
Creation of mutants and complementation of thymidine auxotrophy
Mutant proteins with substitutions at positions 190 and 191 were created to destabilize a hydrophobic pocket in the active conformation, shifting the equilibrium toward the inactive conformation, in which residues of this region reside in an aqueous environment [Fig. 1(b)]. Mutant L198P was created to employ conformational restrictions present for residues preceding a proline residue to achieve the same goal. All mutant proteins were expressed as recombinant proteins in a TS-deficient bacterial strain. Catalytic competency was assessed by growth studies in standard medium or one supplemented with thymidine. In the absence of thymidine, all transformants were able to support bacterial growth, except the M190K transformant (Supplemental Fig. 1). Of transformants complemented for growth in medium lacking thymidine, A191K transformants exhibited a growth delay. The level of expression of the A191K mutant was not significantly lower than that of the other mutants (Supplemental Fig. 2). All transformants exhibited similar growth rates in thymidine-supplemented medium.
Figure 2.
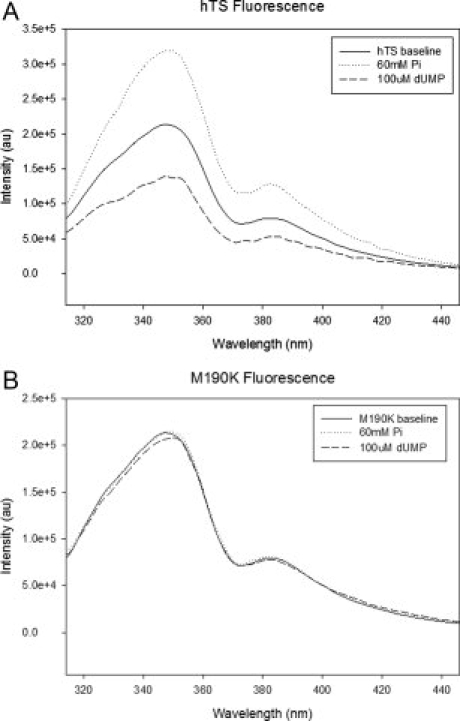
Intrinsic fluorescence of the wild-type hTS (a) and M190K (b) in the presence of 60 mM phosphate or 100 μM dUMP.
Steady-state kinetics characterization of TS enzymes
Steady-state kinetic parameters were determined for wild-type and mutant proteins. The kcat and Km values for dUMP are displayed in Table I. Mutants that were predicted to destabilize the active conformation, M190K, M190E, A191K, and L198P, displayed kcat values of 103-, 5.9-, 6, and 2-fold lower than wt-hTS, respectively. The kcat/Km values for M190E, and A191K were similar, approximately twofold lower than wt-hTS while L198P has tenfold lower catalytic efficiency. The mutant with the lowest turnover rate, M190K, exhibited a 7500-fold lower kcat/Km value. For this mutant, at least four preparations were obtained in two laboratories and the results were consistent.
Table I.
Steady-State Kinetic Constants for Wild-Type and Mutant hTS
| Enzyme | kcat (s-1) | Km (μM) | kcat/Km (s−1 μM−1) |
|---|---|---|---|
| wt-hTS | 3.11 ± 0.04 | 2.59 ± 0.21 | 1.20 |
| M190K | 0.030 ± 0.01 | 179 ± 9 | 1.6 × 10−4 |
| M190E | 0.53 ± 0.25 | 0.96 ± 0.15 | 0.55 |
| A191K | 0.52 ± 0.06 | 0.85 ± 0.12 | 0.61 |
| L198P | 1.64 ± 0.09 | 15.63 ± 1.13 | 0.10 |
Kinetic constants were determined as described in “Methods.” Data are the means of at least three independent determinations.
Ligand binding studies
Loop 181–197 contains a tryptophan residue at position 182. Intrinsic fluorescence has been used to monitor the environment of Trp182, which is altered in the active and inactive conformations.6 It was reported previously that dUMP induced a decrease in the intrinsic fluorescence of wt hTS while phosphate induced an increase. For M190K phosphate concentrations up to 60 mM or dUMP concentrations up to 100 μM had no effect on its intrinsic fluorescence (Fig. 2). Also, numerous crystal soaking experiments showed that hTS in the inactive conformation does not bind ligands other than sulfate or phosphate, while crystals with hTS in the active conformation readily bind dUMP or FdUMP.
Crystallography of A191K and M190K variants
A191K crystallized at essentially the same high-salt conditions, 2.0M ammonium sulfate, at which wt hTS yields crystals in the inactive conformation; the crystals were isomorphic. They scatter to 2.0-Å resolution, as is observed for wt hTS crystals.6 Crystallographic refinement statistics are included in Table II. The structure of A191K is very similar to that of the wt hTS (PDB entry 1hw3); least-squares superposition based on the positions of carbon-α yielded a 0.19 Å r.m.s. displacement with a maximal displacement of 0.75 Å. In particular, the environment of residue 191 is very similar to that in wt hTS, with a displacement of 0.38 Å. The ammonium group of Lys191 side chain interacts with a sulfate ion that is hydrogen bonded to Arg274 and His304 from another dimer (Supplemental Fig. 3). It is unlikely that the Lys191-sulfate coordination is present in solution, as this interaction is stabilized by crystal contacts; a sulfate ion is also observed in a similar position, bound to Arg274 and His304, in wt hTS, where an alanine is in position 191.
Table II.
Crystallographic Data and Refinement Statistics for A191K and M190K Variants of hTS
| hTS mutant | A191K | M190K |
| X-ray source | APS 19BM (SBC-CAT) | APS 22BM (SER-CAT) |
| Detector | 3 × 3 mosaic | 3 × 3 mosaic |
| Wavelength (Å) | 0.97929 | 1.00000 |
| Temperature | 100K | 100K |
| No. of frames | 120 | 180 (90) |
| Oscillation range (degree) | 0.5 | 0.5 (1.0) |
| Detector to crystal distance (mm) | 220 | 300 |
| Space group | P3121 | P3121 |
| Unit cell dimensions | ||
| a (Å) | 95.87 | 96.05 |
| c (Å) | 82.81 | 80.06 |
| Mosaicity, from HKL2000 (degree) | 0.40 | 1.06 |
| Resolution range (Å) (outer shell)a | 50.0–2.18 (2.26–2.18) | 50.0–2.0 (2.07–2.0) |
| Average I/σ(I) | 16.5 | 36.2 |
| Number of unique reflections | 16,116 (1,269) | 26,514 (1,107) |
| Redundancy | 3.1 (3.0) | 11.7 (2.4) |
| Completeness (%) | 92.5 (95.0) | 88.4 (40.8) |
| Low-resolution linear R factor in shell | 0.025 (50.0–4.7) | 0.034 (50.0–4.3) |
| Total linear and square R-mergeb | 0.056 (0.444) 0.040 (0.397) | 0.047 (0.459) 0.042 (0.381) |
| Number of reflections in refinement (with |F|/σ(|F|)>0) | 20,359 | 24,651 |
| R-valuec | 0.180 | 0.190 |
| Rfree (percent of reflections used)d | 0.208 (5.2%) | 0.226 (5.1%) |
| msd, bond lengths (Å) | 0.02 | 0.02 |
| msd, bond angles (deg.) | 1.9 | 1.8 |
| Residues present in the model | 27–96, 134–143, 157–310 | 26–103, 115–310 |
| Number of water molecules | 137 | 211 |
| Matthews coefficient (% solvent) | 3.09 (60.2) | 3.00 (59.1) |
| Ramachandran statistics | ||
| Residues in most favoured regions (%) | 183 | 195 |
| Residues in additionally allowed regions (%) | 12 | 30 |
| Residues in generously allowed regions (%) | 2 | 6 |
| Residues in disallowed regions (%) | 0 | 0 |
| Average B-factors main chain (all atoms) | 40.3 (42.1) | 54.4 (55.4) |
Values in parentheses are for the outermost resolution shell.
Rmerge = (∑h|Ih − 〈I〉|)/(∑hIh).
R = (∑h||Fobs| − |Fcal||)/(∑h|Fobs|).
Rfree = crystallographic R-factor for test set.
Figure 3.
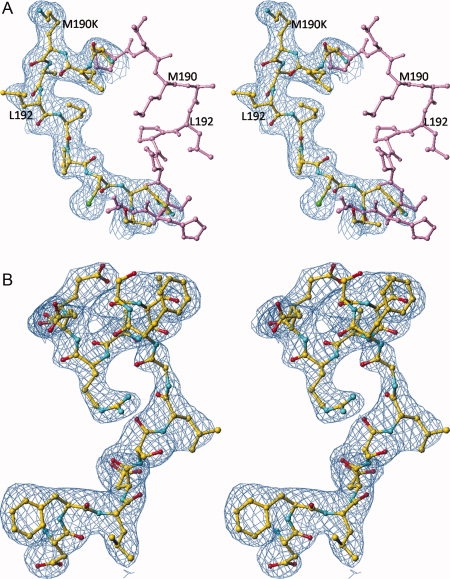
Comparison of the structures of loop 187–194 in wt hTS (PDB entry 1hw3) shown in lavender and in M190K (in yellow). (a) The electron density, contoured at 1.0 σ level, is from the final map calculated for M190K with 2Fo-Fc coefficients. The side chain of Lys190 in entirely in solvent and does not form any contacts. (b) Stereoview of the electron density for a part of eukaryotic insert 1 in M190K. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
The N-terminal extension in eukaryotic TS enzymes, residues 2–25, is disordered in all structures and the C-terminus, residues 311–313, is disordered in all unliganded structures. In addition, residues 107–128 were disordered in the structure of wt hTS, determined at the same resolution 2.0 Å.6 In A191K, disorder was observed at both termini; however, disorder is observed for a much larger region, residues 99–133 and 145–157, which encompasses insert 2. At a low contouring level, 0.6 σ, electron density in a 2FoFc omit map (data not shown), although discontinuous, suggests that the conformation observed in the wt hTS structure for residues 99–106 is populated to some extent; alternative conformations, however, could not be identified. The vicinity of the mutation site, K191, displays good density, and the conformation of the main chain remains unchanged.
M190K crystallized at the same high salt conditions as A191K and wt hTS and the crystals were isomorphic, with a similar scattering power—2.0 Å resolution. The statistics from its structure refinements are in Table II. The side chain of Lys190 points toward solvent and is disordered, as only partial electron density is observed. Superpositioning of structures of wt hTS and M190K yielded a rms difference between the positions of Cα atoms of 0.90 Å and revealed a large conformational change for residues 189–192 in M190K, reaching a 7.2 Å displacement for Cα of Leu189 [Fig. 3(a)]. Associated with this change is a unique structure of the eukaryotic insert 1. Previously, two conformational states of insert 1 were identified and they were correlated with the conformation of loop 181–197. In the structures of wt hTS determined at either low salt16 or high salt conditions6 and in the A191K structure reported here, loop 181–197 is in the inactive conformation and region 107–128, which contains eukaryotic insert 1 (residues 117–128), is disordered. In the structures of the hTS•dUMP•raltitrexed complex8 and in R163K,10 loop 181–197 is in the active conformation and region 107–128 is ordered. In M190K, loop 181–197 is in the inactive conformation and region 116–128 is ordered while region 103–115 is disordered [Fig. 3(b)]. A superposition of wt hTS in the active and inactive conformations and M190K is shown in Figure 4. The conformation of residues 116–135 in M190K is different from that observed in the hTS•dUMP•raltitrexed complex or R163K. This novel conformation is induced by the structure of 188–194 region; these residues create a steric hindrance, which prevents residues 115–135 from adopting the conformation observed before in the inhibitory complex and in R163K.
Figure 4.
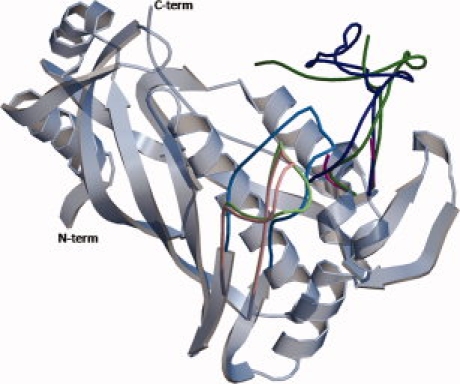
Positions of the loops in hTS. In gray is shown the invariant part which is based on hTS-R163K (PDB entry 2rda). The loops that change their position upon conformational transitions are shown in colors. Loop 181–197 is shown in light blue in the active conformation (R163K), in light pink in the inactive conformation (wt-hTS), and in light green in the alternative inactive conformation (M190K). Loop 94–135 is in dark blue in the active conformation observed in R163K, in purple in wt-hTS (residues 108–128 are disordered), and in dark green in M190K (residues 104–114 are disordered). [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Discussion
The major aim of this study was to create proteins that are stabilized in the inactive conformation of loop 181–197 for use as targets in drug discovery and for investigations of the physiological relevance of conformational switching. Based on analyses of three-dimensional structures of liganded and unliganded hTS, some substitutions at positions 190, 191, and 198 were predicted to shift the equilibrium toward the inactive conformer. We anticipated that these proteins will be defective in catalysis, particularly, as these residues are invariant in all TSs examined to date. Unexpectedly, only one of the mutants, M190K was highly defective, with kcat decreased by 100-fold relative to wt hTS and Km increased by a factor of 70. The other mutants, M190E and A191K, showed quite unexpected behavior, with lower kcat values, but also lower Km values for dUMP. The better dUMP binding properties cannot be attributed to simple electrostatics of binding the charged substrate since the net charges in the two mutants are changed in the opposite directions. One explanation of relatively low effect seen for M190E and A191K is the flexibility of the loop containing residues 131–134 which form the hydrophobic pocket in the active conformation (see Fig. 1). A likely explanation is that the loop containing residues 131–134 in the two mutants is unable to close the hydrophobic pocket because of water molecules hydrating the charged side chains at position 190 or 191. Such interpretation is consistent with the observed flexibility of residues 131–134, which have high temperature factors and are close to disordered region 107–108. Thus, the mutations may not lead to a completely buried electrostatic charge, and the destabilization of the active conformation apparently is not as strong as anticipated.
While 190 and 191 mutant design exploited charge burying, mutant L198P has been designed to destabilize the active conformation via disallowed main chain conformation. This approach actually worked moderately well yielding an increase in Km by a factor of 6 and a decrease in kcat by factor of 2. It appears that the large dUMP binding energy overpowers the steric hindrance generated by the presence of a proline in position 198.
In the background of modest activity reduction in other mutants studied here, the observed large loss of activity in M190K is unexpected. A priori, one would expect that the replacement of a buried neutral methionine with a positively charged lysine in M190K will have a similar effect as replacement with a negatively charged glutamate in M190E. This is especially so as the side chain of Lys190 in M190K is directed toward solvent and does not form any stabilizing contacts. The catalytic properties of M190K and M190E are however very different. The kinetic data are strongly supported by the fluorescence data. Even 60 mM phosphate did not significantly affect the intrinsic fluorescence of M190K. This strongly suggests that the fraction of M190K that populates the active conformer is low. For wt-hTS, dUMP binding was able to reverse the effect of 64 mM phosphate on the protein fluorescence and induce strong quenching.6 In contrast, in M190K, there is no quenching at even higher concentrations of dUMP and in the absence of phosphate. This strongly suggests that the dUMP binding energy cannot drive M190K into the active conformation, unlike what was observed for wt-hTS. Finally, M190K was expressed and purified in fairly good yields and readily crystallized, features consistent with a well-folded protein. One explanation of the observed kinetic and fluorescence data is that, in M190K, equilibration between the conformations of loop 181–197 does not take place due to a large energy barrier between the two states. Thus, M190K folds preferentially into the inactive conformation and remains trapped in the inactive state. Alternatively, the inactive conformation in M190K has a lower energy than the inactive conformation in wt-hTS and the folding process of the latter does not allow the protein to populate this global energy minimum. This is however not supported by preliminary differential scanning calorimetric data. They show that although protein stability is increased in the M190K mutant when compared to wild-type hTS by 2.6°C (with melting temperature for wt-hTS 57.1°C) a comparable Tm increase of 2.3°C is observed for dUMP binding to wt hTS. Formation of a ternary inhibitory complex of hTS-dUMP-antifolates typically increases Tm by more than 15°C.17
The low catalytic activity of M190K shows that the main design objective, a strong stabilization of the inactive conformer, has been achieved. A sobering fact is that serendipity played a large role, as for other mutants designed using the same underlying principle, namely, that charge burying is unfavorable, the effect was modest. It appears that for hTS inhibition, the version of the inactive conformation of loop 181–197 that is observed in M190K and associated with the unique conformation of region 107–128 is a better target for stabilization than the one that is observed in wt-hTS and the other mutants, since it does not equilibrate with the active conformer. It is not unlikely that ligands binding to this inactive conformer will also bind to a folding intermediate and “guide” it to the stable inactive conformation. Thus, the challenge that remains to achieve such allosteric inhibition of hTS via stabilization of the inactive conformer is to come up with a ligand that will drive hTS into the conformation observed for M190K. It appears that return from this conformation to the active one may not be easy.
Methods
Modeling
The structures of hTS in its active conformations were those of ternary inhibitory complex (PDB entry 1hvy),8 and R163K mutant (PDB entries 2rda and 2rd8).10 The structures of hTS in the inactive conformations were those obtained at high salt (PDB entry 1hw3)6 and low salt (PDB entry 1ypv) conditions.16 They were superposed using the LSQKAB program18 from the CCP4 software system.19 Interactive molecular graphics studies were carried out using the TURBO software.20
Bacterial expression system for hTS
The Escherichia coli strain TX61 (thyA−) containing kanamycin resistance and the pTS080 plasmid containing tetracycline and ampicillin resistance, expressing wild-type hTS, were generously provided by W. S. Dallas (Glaxo Wellcome, Research Triangle Park, NC), and have been described previously.21,22 TX61 was created by transposon-mediated mutagenesis and lacks detectable TS activity.21
Creation, expression, and purification of mutant hTSs
Derivatives of pTS080 with mutations creating substitutions at amino acid positions, 190, 191 and 198 of hTS, were produced by site-directed mutagenesis according to the QuickChange method (Stratagene, LaJolla, CA). Mutagenic primers were synthesized and purified by Integrated DNA Technology (IDT, Coralville, IA) (Supplemental Table 1). Supercompetent JM109 cells (New England Biolabs, Bedford, MA) were transformed with the resulting PCR products, screened on LB plates containing 50 μg tetracycline, and mutations were confirmed by LiCor DNA sequence analysis (MUSC Biotechnology Resource Laboratory, Charleston, SC) of the entire thyA gene. Plasmids containing the correct mutations were transformed into TX61. Transformants were grown and TSs purified by AKTA® FPLC system (Pharmacia, Piscataway, NJ) using Blue-Sepharose and Q-Sepharose columns as described previously.23 For studies of complementation of thymidine auxotrophy, TX61 transformants were grown in minimal medium as described previously.23
Enzymatic assays
Enzyme activity was measured spectrophotometrically at 37°C by monitoring the absorbance change accompanying the conversion of mTHF to DHF (ɛ340 = 6.4 mM−1 cm−1) using a Shimadzu UV 1601 spectrophotometer (Shimadzu Corporation, Japan) as described previously.22 For determination of kinetic constants for dUMP, initial velocities were measured by utilizing 40–400 nM of purified protein, 0.15 mM of mTHF, and dUMP ranging for 0.1–100 μM. Kinetic constants were determined as described previously, using the program KaleidaGraph WIN (v.3.6x Synergy Software, Reading, PA).
Fluorescence studies
Intrinsic fluorescence data were obtained as described previously by Phan et al.6 Samples were excited at 295 nm and emission scanned from 310 to 446 nm at a rate of 3 nm sec−1 at room temperature (SLM-Aminco 8100 Spectrofluorometer, Urbana, IL). A 2 μL sample containing a 1 μM protein in buffer A was titrated with 60 mM potassium phosphate pH 7.0, followed by subsequent quenching with 100 μM dUMP. All measurements were performed in triplicate, and total volume added during titration was less than 5% of the final volume. Graphs were prepared using SigmaPlot version 9.0.1(www.systat.com/products/SigmaPlot/).
Crystallization of mutant TSs
Crystals of hTS mutants M190K and A191K were grown as described previously6 from 2.0M ammonium sulfate, 0.1M Tris, pH 8.5, 20 mM β-mercaptoethanol by vapor diffusion in a hanging-drop protocol. Crystals were transferred to a cryoprotectant solution made from mother liquor and containing 5% ethylene glycol and then flash frozen in a cryostream.
X-ray diffraction data measurement and processing
Crystallographic diffraction experiments were carried out at the SER-CAT BM beamline at APS, Argonne National Laboratory. Data were collected on a single crystal of hTS with approximate dimensions of 0.30 × 0.30 × 0.25 mm3 for M190K and 0.45 × 0.50 × 0.45 mm3 for A191K. The data were indexed and processed with HKL2000.24 Data collection and processing statistics are shown in Table I.
Structure determination and refinement
The structure of hTS (PDB code 1hw3), obtained by similar crystallization conditions, was used as the search model in the molecular replacement method carried out with CNS software25 to compensate for 1–2% differences in unit cell parameters. Crystallographic refinements were conducted iteratively using TURBO graphics software20 and CNS.25
Acknowledgments
We thank Dr. Catherine J. Murphy for use of her spectrofluorimeter. Data were collected at the Southeast Regional Collaborative Access Team (SER-CAT) 22-ID and 22-BM beamlines at the Advanced Photon Source, Argonne National Laboratory. Supporting institutions may be found at www.ser-cat.org/members.html.
Glossary
Abbreviations:
- dTMP
2′-deoxythymidine 5′-monophosphate
- dUMP
2′-deoxyuridine 5′-monophosphate
- ecTS
Escherichia coli TS
- hTS
human TS
- mTHF
5,10-methylenetetrahydrofolate
- PDB
Protein Data Bank
- TS
thymidylate synthase.
References
- 1.Carreras CW, Santi DV. The catalytic mechanism and structure of thymidylate synthase. Annu Rev Biochem. 1995;64:721–762. doi: 10.1146/annurev.bi.64.070195.003445. [DOI] [PubMed] [Google Scholar]
- 2.Rustum YM, Harstrick A, Cao S, Vanhoefer U, Yin MB, Wilke H, Seber S. Thymidylate synthase inhibitors in cancer therapy: direct and indirect inhibitors. J Clin Oncol. 1997;26:621–631. doi: 10.1200/JCO.1997.15.1.389. [DOI] [PubMed] [Google Scholar]
- 3.Zhang N, Yin Y, Xu S-J, Chen W-S. 5-fluorouracil: mechanisms of resistance and reversal strategies. Molecules. 2008;13:1551–1569. doi: 10.3390/molecules13081551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perry KM, Fauman EB, Finer-Moore JS, Montfort WR, Maley GF, Maley F, Stroud RM. Plastic adaptation towards mutations in proteins: structural comparison of thymidylate synthases. Proteins. 1990;8:315–333. doi: 10.1002/prot.340080406. [DOI] [PubMed] [Google Scholar]
- 5.Schiffer CA, Clifton IJ, Davisson VJ, Santi DV, Stroud RM. Crystal structure of thymidylate synthase: a structural mechanism for guiding substrates into the active site. Biochemistry. 1995;34:16279–16287. doi: 10.1021/bi00050a007. [DOI] [PubMed] [Google Scholar]
- 6.Phan J, Steadman DJ, Koli S, Ding WC, Minor W, Dunlap RB, Berger SH, Lebioda L. Structure of human thymidylate synthase suggests advantages of chemotherapy with noncompetitive inhibitors. J Biol Chem. 2001;276:14170–14177. doi: 10.1074/jbc.M009493200. [DOI] [PubMed] [Google Scholar]
- 7.Monfort WR, Weichsel A. Thymidylate synthase: structure, inhibition, and strained conformations during catalysis. Pharmacol Ther. 1997;76:29–43. doi: 10.1016/s0163-7258(97)00099-5. [DOI] [PubMed] [Google Scholar]
- 8.Phan J, Koli S, Minor W, Dunlap RB, Berger SH, Lebioda L. Human thymidylate synthase is in the closed conformation when complexed with dUMP and raltitrexed, an antifolate drug. Biochemistry. 2001;40:1897–1902. doi: 10.1021/bi002413i. [DOI] [PubMed] [Google Scholar]
- 9.Almog R, Waddling CA, Maley F, Maley GF, Van Roey P. Crystal structure of a deletion mutant of human thymidylate synthase Δ(7-29.) and its ternary complex with Tomudex and dUMP. Protein Sci. 2001;10:988–996. doi: 10.1110/ps.47601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gibson LM, Lovelace LL, Lebioda L. The R163K mutant of human thymidylate synthase is stabilized in an active conformation: asymmetry and reactivity of Cys195. Biochemistry. 2008;47:4636–3643. doi: 10.1021/bi7019386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chu E, Koeller DM, Casey JL, Drake JC, Zinn S, Allegra CJ. Autoregulation of human thymidylate synthase messenger RNA translation by thymidylate synthase. Proc Natl Acad Sci USA. 1993;88:8977–8981. doi: 10.1073/pnas.88.20.8977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chu E, Allegra CJ. The role of thymidylate synthase as an RNA binding protein. Bioessays. 1996;18:191–198. doi: 10.1002/bies.950180306. Review. [DOI] [PubMed] [Google Scholar]
- 13.Chu E, Koeller DM, Johnston PG, Zinn S, Allegra CJ. Regulation of thymidylate synthase in human colon cancer cells treated with 5-fluorouracil and interferon-gamma. Mol Pharmacol. 1993;43:527–533. [PubMed] [Google Scholar]
- 14.Ciesla J, Jagielska E, Skopinski T, Dabrowska M, Maley F, Rode W. Binding and repression of translation of the cognate mRNA by Trichinella spiralis thymidylate synthase differ from the corresponding interactions of the human enzyme. Biochem J. 2005;390:681–688. doi: 10.1042/BJ20050548. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15.Lovelace LL, Gibson LM, Lebioda L. Cooperative inhibition of human thymidylate synthase by mixtures of active site binding and allosteric inhibitors. Biochemistry. 2007;46:2823–2830. doi: 10.1021/bi061309j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lovelace LL, Minor W, Lebioda L. Structure of human thymidylate synthase under low-salt conditions. Acta Crystallogr D. 2005;61:622–627. doi: 10.1107/S0907444905005895. [DOI] [PubMed] [Google Scholar]
- 17.Johnson SR. 2007. Stabilization of an Active and Inactive Conformation and Investigation of a Potential Proton Relay System of Human Thymidylate Synthase. Ph.D. dissertation: Department of Pharmaceutical & Biomedical Sciences, University of South Carolina, Columbia, South Carolina. [Google Scholar]
- 18.Kabsch W. Solution for best rotation to relate 2 sets of vectors. Acta Crystallogr A. 1976;32:922–923. [Google Scholar]
- 19.Collaborative computational project, number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 20.Roussel A, Cambillau C. “Turbo Frodo” silicon graphics geometry partners directory. Mountain View, CA: Silicon Graphics; 1991. p. 86. [Google Scholar]
- 21.Dev IK, Yates BB, Leong J, Dallas WS. Functional role of cysteine-146 in Escherichia coli thymidylate synthase. Proc Natl Acad Sci USA. 1988;85:1472–1476. doi: 10.1073/pnas.85.5.1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dev IK, Dallas WS, Ferone R, Hanlon M, McKee DD, Yates BB. Mode of binding of folate analogs to thymidylate synthase—evidence for a 2 asymmetric but interactive substrate-binding sites. J Biol Chem. 1994;269:1873–1882. [PubMed] [Google Scholar]
- 23.Williams AW, Dunlap RB, Berger SH. A hydroxyl group at residue 216 is essential for catalysis by human thymidylate synthase. Biochemistry. 1998;37:7096–7102. doi: 10.1021/bi972562+. [DOI] [PubMed] [Google Scholar]
- 24.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 25.Brunger AT, Adams PD, Clore GM, Delano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]