

Abstract
Cellular processes are highly interconnected and many proteins are shared in different pathways. Some of these shared proteins or protein families may interact with diverse partners using the same interface regions; such multibinding proteins are the subject of our study. The main goal of our study is to attempt to decipher the mechanisms of specific molecular recognition of multiple diverse partners by promiscuous protein regions. To address this, we attempt to analyze the physicochemical properties of multibinding interfaces and highlight the major mechanisms of functional switches realized through multibinding. We find that only 5% of protein families in the structure database have multibinding interfaces, and multibinding interfaces do not show any higher sequence conservation compared with the background interface sites. We highlight several important functional mechanisms utilized by multibinding families. (a) Overlap between different functional pathways can be prevented by the switches involving nearby residues of the same interfacial region. (b) Interfaces can be reused in pathways where the substrate should be passed from one protein to another sequentially. (c) The same protein family can develop different specificities toward different binding partners reusing the same interface; and finally, (d) inhibitors can attach to substrate binding sites as substrate mimicry and thereby prevent substrate binding.
Keywords: protein–protein interaction, multibinding interfaces, promiscuous sites, domain–domain interaction, conserved binding mode
Introduction
Proteins, nucleic acids, and other biomolecular compounds function while interacting with each other. Such biochemical interactions are astonishing in their magnitude and diversity and are involved in signal transduction, transcriptional, and translational regulation and other important cellular processes. Protein interactions form intricate interaction networks where some proteins create connections to many other proteins (so-called hub proteins) with about one-third of all proteins participating in multiple complexes or pathways.1–3 Indeed, cell processes are highly interconnected and many proteins can perform diverse tasks bridging different pathways, classical examples of such complexes being RNA polymerase II and various transcription factors.4,5 The analysis of yeast complexes showed that many consist of proteins not found to be stable in isolation, so-called “obligate” complexes.6 A higher level of organization of cellular processes can also be achieved through transient protein interactions when the same protein is shared between different complexes (crosstalk between complexes).7 Such shared components might have regulatory and auxiliary functions as was reported earlier.3 To draw conclusions about the functions of shared components, it is important to know the structures of the complexes, the structural details of protein interaction interfaces, and the locations of binding sites.
Similarity between ligand-binding sites in structures of nonhomologous proteins was assessed previously.8 Most recent papers report large-scale analyses of equivalent protein binding sites in unrelated proteins and their connections with protein interaction network properties.9–12 These studies describe examples of similar binding patterns in the absence of sequence or structure similarity between proteins, pointing to the possible convergent evolution between binding interfaces.9 Recently, similarity between interaction interfaces was assessed by clustering them using interface sequence profiles or tags13 and structure superpositions in the interface regions.14–17 Structural classification of interaction interfaces of protein chains allowed for the examination of common interface motifs, which bind to proteins with different functions and structures.14,15 These multibinding interface regions were not homogeneously packed but rather had highly packed conserved regions. The authors suggested that these conserved regions are consistent with the concept of hot spots, which are optimized for binding and contribute the most to the free energy of binding.18,19 In addition, these multibinding interfaces were found to be smaller, more planar, and contain more α-helices.
Promiscuous protein binding is not well understood, and gaining this knowledge would have a practical impact on the treatment of many diseases associated with promiscuous binding. Various experimental and computational endeavors have been undertaken to address this issue.20 Protein design protocols, for example, proposed two major mechanisms to achieve multispecificity on promiscuous interfaces.21,22 According to one mechanism the same set of highly optimized hot spots is used, and the other mechanism employs the multifaceted larger interfaces where each partner uses a slightly different subset of interface residues. One recent study showed that hot spots in promiscuous sites are more distributed among different modules compared with the specific sites.23 At the same time, molecular dynamics simulations point to the fact that a promiscuous protein should provide alternative contact points at its interface to increase binding entropy and consequently binding affinity.24
The main goal of our study is to attempt to decipher the mechanisms of specific molecular recognition of multiple diverse partners by promiscuous protein regions. To address this, we first ask how common such mechanisms are and how frequently the same interface is utilized in evolution by apparently nonhomologous and functionally different interacting partners. Second, we attempt to highlight the major mechanisms of functional switches realized through multibinding. One advantage of our approach is that we use alignments which are based on explicit structural superpositions of shared components (domains) of the protein complexes. Such alignments allow us to map binding interfaces of different binding partners on one reference frame. To ensure that the interactions between chains/domains are biologically relevant and evolutionary conserved, we perform conserved binding mode (CBM) analysis introduced previously.25 With our approach we assess amino acid usage on multibinding interfaces and the mechanisms of multibinding specificity. A great deal of research is dedicated to studying the properties of so-called hub or social proteins, usually defined as proteins with many different protein-binding partners. In this regard, we explore a slightly different definition of hub proteins as those having a large number of different binding modes (not necessarily a large number of different interaction partners) and analyze the properties of networks upon purging the interacting partners with the same binding mode.
Results
Interface overlap ratio among different interaction types
Among 1098 conserved domain database (CDD) superfamilies considered in our analysis, 64% had at least one homodimer interaction (both interacting partners belonged to the same superfamily) and 65% of the superfamilies had at least one heterodimer interaction (interacting partners belonged to different superfamilies). Overall, our data set was equally balanced with both types of interactions. We performed comparisons of all the interactions for a given domain superfamily as described in the Methods and calculated the interface overlap ratio. Figure 2 shows the interface overlap ratio distribution for cases where homodimer interaction overlaps with heterodimer [Fig. 2(a)] and where two heterodimer interactions overlap [Fig. 2(b)]. As can be seen from this figure, in both cases only a few interactions have high overlap with only 2% of cases where homo- and heterodimers share the same interface and 4% of cases where different heterodimers share the same interface (overlap is larger than 50%).
Figure 2.
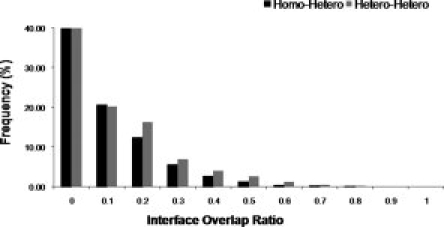
Interface overlap ratio between homo- and heterodimers (black bar) and between different heterodimers (gray bar).
Degree distributions for interaction networks with multibinding interfaces
We calculate the number of distinct interacting superfamilies with nonoverlapping CBMs as a node degree for a given CDD superfamily in the interaction network. For example, if family A interacts with families B and C using the same interface (more than 50% interface overlap), the node degree of family A in the collapsed network will be equal to one. Supporting Information Figure S1 shows the degree distribution before and after the collapse of multibinding interfaces. We compared our results with the previous study on multibinding interfaces of yeast interactions.26 Despite the differences in the test sets and definitions of multibinding interfaces, there is a consistent observation in both studies that the degree distribution has a shorter tail when multibinding interactions are collapsed into one. At the same time, the node degree does not change much for the nodes with a few interactions implying that multibinding sites occur mostly in the hub proteins forming a dense set of interactions. The difference between “collapsed” and original degree distributions in our case is statistically significant (P-value is less than 0.001). We found that only 49 CDD superfamilies contribute to these differences (Supporting Information Tables S1 and S3; the shorter list of such superfamilies from the human interaction set is given in Table I); these superfamilies have at least two different interacting partners with the largely overlapping interfaces. The average node degree of these superfamilies drops from 5.4 to 4.4 in the collapsed network, while the maximum number of interactions decreases from 28 to 22. The superfamilies with the maximum drop off in the node degree include ubiquitin, immunoglobulin, trypsin-like serine protease, and small GTPases. Overall, the degree distributions of original and collapsed networks follow a power law although parameters of the distribution change upon taking into account multibinding patterns (Supporting Information Fig. S2).
Table I.
List of Domain Superfamilies (Containing the Related CDD Domains) with Multibinding Interfaces and Interaction Partners
| Interacting domain partners |
Interacting chains |
||||
|---|---|---|---|---|---|
| Domain superfamily name | Partner1 | Partner2 | PDB1 | PDB2 | Interface overlap ratio |
| 17, Small GTPases | Arfaptin domain, cd00011 | GEF Rho/Rac/ Cdc42-like GTPases, cd00160 | 1I4T:A-D | 1X86:E-F | 58 |
| GEF Ras-like small GTPases, cd00155 | GEF Rho/Rac/ Cdc42-like GTPases, cd00160 | 1NVV:S-R | 1X86:E-F | 50 | |
| Sec7 domain, smart00222 | GEF Rho/Rac/ Cdc42-like GTPases, cd00160 | 1REO:B-A | 1X86:E-F | 53 | |
| RasGEF-N, smart00229 | GRIP domain, pfam01465 | 1XD2:C-A | 1UPT:B-A | 52 | |
| Rab family, cd00154, 17 | Raf-like Ras-binding domain, smart00455 | 1OIV:B-A | 1CIY:B-A | 65 | |
| Rab family, cd00154, 17 | GEF Ras-like small GTPases, cd00155 | 1T91:C-D | 1NVX:S-Q | 60 | |
| 1990, Trypsin-like Serine protease | BPTI/Kunitz serine protease inhibitors, cd00109 | Protease inhibitor ecotin, cd00242 | 1YCO:I-A | 1XX9:D-B | 68 |
| AT_III like, cd02045 | Protease inhibitor ecotin, cd00242 | 1TB6:I-H | 1XX9:D-B | 63 | |
| BPTI/Kunitz serine protease inhibitors, cd00109 | AT_III like, cd02045 | 1YCO:I-A | 1TB6:I-H | 69 | |
| 558, Ubiquitin | RAS small GTPases. smart00173 | Uracil DNA glycosylase, pfam03167 | 1C1Y:A-B | 1WYW:A-B | 56 |
| Peptidase_C12, pfam01088 | Peptidase_C48, pfam02902 | 1XD3:A-B | 1XT9:A-B | 54 | |
| 2129, MHC_I | IG, smart00409 | MHC_I, pfam00129 | 2BNQ:E-A | 1A6Z:C-A | 52 |
| 2263, Transthyretin | Transthyretin, smart00095 | Lipocalin/cytosolic fatty acid-binding protein, pfam00061 | 1ICT:A-F | 1QAB:A-E | 75 |
| 3592, Malic_M | Malic enzyme, N-terminal domain, pfam00390 | Malic enzyme, NAD-binding domain, pfam03949 | 1PJ2:C-B | 1PJ3:C-B | 50 |
| 1936, Immunoglobulin | MHC_I, pfam00129 | MHC_II_alpha, pfam00993 | 1T8O:A-A | 1UVQ:A-B | 64 |
| 2100, EFhand | S-100/ICaBP, calcium-binding domain, pfam01023 | EF hand, pfam00036 | 1M31:B-B | 1XFY:O-O | 88 |
| 1979, Serpin | Heparin cofactor II (HCII) inhibits thrombin, cd02047 | Trypsin-like serine protease, cd00190 | 1LK6:L-I | 1TB6:H-I | 70 |
This is a subset from overall dataset for entries corresponding to human proteins. The first column gives domain family name and its corresponding cluster-id number (See Supporting Information).
Analysis of amino acid composition and conservation at multibinding sites
Previous analysis of amino acid frequencies on protein binding interfaces showed that Arg, Tyr, and Trp may be energetically important for binding27,28 and occur frequently in hot spot regions.18,21 Our analysis of amino acid frequencies on multibinding interfaces showed that the multibinding interfaces are statistically enriched with Gly, Tyr, Trp, Cys, Thr, and Ser compared with the background of protein interfaces using two binomial models (Fig. 3, Supporting Information Table S2). The chi-square contingency test with Bonferroni correction confirms that multibinding interfaces are enriched with Gly and Tyr and under-represented with Ile, Asp, and Glu compared with protein interfaces. Glycine might be important for conformational flexibility, which in turn can promote multibinding while aromatic amino acids (Tyr, Trp) may provide stacking interactions on the interfaces.29
Figure 3.
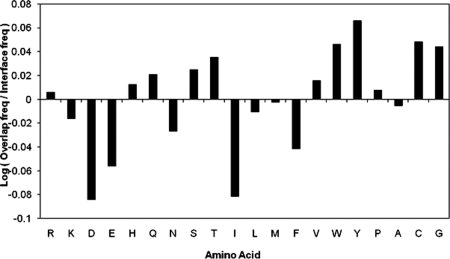
Amino acid composition at multibinding sites.
We also checked whether promiscuous binding sites are more conserved in terms of the sequence compared with other interface sites (see Methods). We did not find any significant difference in sequence conservation between multibinding and all interface sites (Supporting Information Fig. S3). The absence of sequence conservation compared with the background interface might point to the multifaceted organization of multibinding interfaces, where each binding partner uses a slightly different subset of interface residues.
Several functional mechanisms of multibinding
Analysis of families with multibinding interfaces allowed us to distinguish several major functional mechanisms of protein binding promiscuity.
Switching between different functional pathways
The first mechanism of how overlap between different functional pathways can be prevented by the post-translational modifications of closely located residues is illustrated with the example of the histidine-containing phosphocarrier protein domain (HPr, cd00367). It participates in two different pathways and separately interacts with transcription regulators (pfam00532, 1RZRD) and PEP-utilizing enzymes (pfam05524, 3EZAA), using the same interface region with the interface fraction overlap of 64% (Fig. 4, Supporting Information Table S1). In glucose replete conditions, Hpr-Ser46 is phosphorylated, which activates binding of carbon catabolite protein A (CcpA) to the DNA sites to regulate genes encoding carbon metabolism proteins. This carbon catabolite repression pathway (CCR) represents a regulatory mechanism allowing bacteria to utilize the most efficiently catabolized carbon sources. HPr also has a catalytic function in sugar uptake in the sugar phosphotransferase system (PTS) pathway through the phosphorylation of His15. Recently, it has been elegantly demonstrated that phosphorylation of Ser46 disrupts interactions between HPr and enzymes of the PTS pathway30; and at the same time, phosphorylation of His15 prevents binding of HPr to CcpA.31 Clearly, phosphorylation events occurring on nearby residues of the same interfacial region of HPr proteins prevent overlap between CCR and PTS pathways.32
Figure 4.
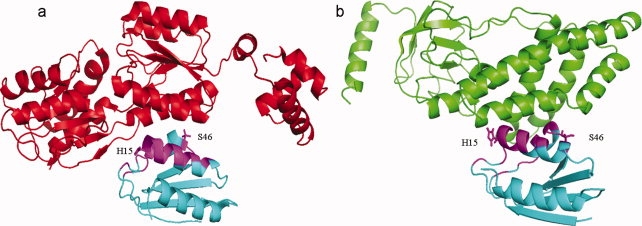
Functional switch between different functional pathways. (a) Interaction between histidine-containing phosphocarrier protein domain (HPr, cd00367, 1RZRY, cyan) and transcription regulators (pfam00532, 1RZRD, red). (b) Interaction between Hpr domain (cd00367, 3EZAB, cyan) and PEP-utilizing enzymes (pfam05524, 3EZAA, green). An overlap of 64% is observed on the Hpr domain interface between the above two interactions, as shown in magenta color. Side chains of key interface residues like H15 and S46 are also shown. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Preventing simultaneous binding to different partners in sequential reactions
The multibinding mechanism is important in preventing the simultaneous interaction with different binding partners when a protein should be passed through sequential reactions and interacts at each stage with a specific binding partner. An example is ubiquitin (cd01803), which interacts with ubiquitin-binding domains, ubiquitin ligase, ubiquitin-activating enzymes, and other molecules using multibinding interfaces, which all include an important residue Ile44.33 This multibinding site around residue Ile44 is illustrated in Figure 5, where ubiquitin-conjugating enzyme E2 (smart00212, 1ZGUA) and ubiquitin-associated domain, UBA (pfam00627, 2G3QA) share 55% of the total interface region. (Supporting Information Table S1).
Figure 5.
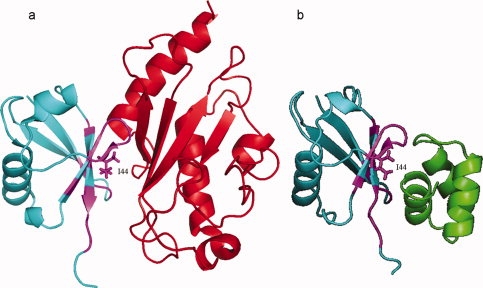
Multibinding interface on ubiquitin domain. (a) Interaction between ubiquitin domain, UBQ (cd01803, 1ZGUB, cyan) and ubiquitin-conjugating enzyme E2 (smart00212, 1ZGUA, red). (b) Interactions between ubiquitin domain (cd01803, 2G3QB, cyan) and ubiquitin-associated domain, UBA (pfam00627, 2G3QA, green). An overlap of 55% is observed on UBQ domain interface and is shown in magenta. Key interface residue I44 on promiscuous interface is shown with the side chain. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Developing binding specificity to new interaction partners
Interactions of small guanosine triphosphatases (GTPases) with their effectors represent an example of how different families of the same superfamily can develop binding specificities to new interaction partners reusing the same interface region. Small GTPases work as GDP/GTP-regulated molecular switches (switch I residues 32–38 and switch II residues 59–67)34 and movement of these switch regions is important to facilitate nucleotide release, which enables interaction with effector molecules. Guanine nucleotide exchange factor (GEFs) are regulatory proteins that promote formation of the active GTP bound form of small GTPases and are very specific to GTPase families.35 We looked at two such families; the Ras GTPase domain interacting with Ras guanine nucleotide exchange factor (RasGEF cd00155, 1NVVS) and the Rho GTPase domain interacting with Rho GEF (RhoGEF, cd00160, 1X86E) sharing the same interface region on GTPases (Fig. 6, Table I). RasGEF and RhoGEF belong to different nonhomologous families of proteins with different folds, which probably emerged through convergent evolution.36 As shown in Figure 6, although Ras and Rho domains share similar structural features and similar interface regions, they differ on the key interfacial residues that are located in and around the switch I and switch II regions, namely, Ras residues Ile36, Glu37, and Arg68 and Rho residues Try58, Arg68, and His105. These results are in line with previous observations.37,38
Figure 6.
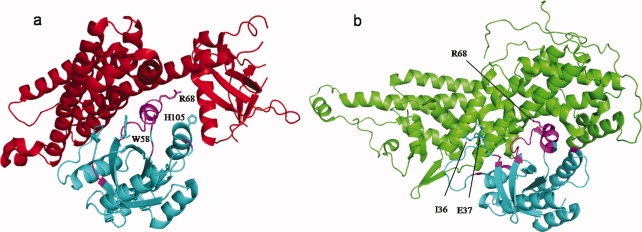
Specific binding on promiscuous site. (a) Interaction between Rho GTPase domain (cd00157, 1X86F, cyan) and GEF for Rho-like GTPases (RhoGEF, cd00160, 1X86E, red). (b) Interaction between Ras GTPase domain (smart00173, 1NVVR, cyan) and GEF for Ras-like GTPases (RasGEF, cd00155, !NVVS, green). GTPase domain in both interactions shares similar interface region and overlap on 50% of the interface residues, shown in magenta color. Specific interface residues in each of the interactions are shown with the side chains, for example, I36 and E37 on Ras GTPases and W58 and H105 on Rho GTPases. Residues like R68 are shared by both interactions and are part of the critical region of the interface site. Families cd00157 and smart00173 belong to one superfamily cluster (see Methods). [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Mimicking substrates with inhibitors
The trypsin-like serine protease family (cd00190) has a major functional role in blood clotting, immune system, and inflammation. Proteases are found to be in complex with a wide variety of inhibitors from different families, which bind to the active site of proteases in a substrate-like manner, mimicking substrates/peptides. An example of two different inhibitors, antithrombin (cd02045, 1TB6:I) and ecotin (cd00242, 1XX9:D) binding to the same active site is shown in Figure 7. We mapped all multibinding residues on the protease domain and showed that the multibinding region is located on a region with many cavities (Supporting Information Fig. S5) and enriched with charged and aromatic residues.
Figure 7.
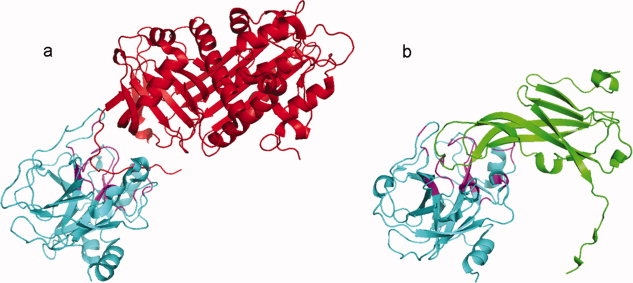
Inhibitors mimic substrate binding in serine proteases. (a) Interaction between trypsin-like serine protease (cd00190, 1TB6H, cyan) and antithrombin from serpin superfamily (cd02045, 1TB6I, red). (b) Interaction between serine protease (cd00190, 1XX9B, cyan) and protease inhibitor ecotin (cd00242, 1XX9D, green). The active site of proteases is targeted by different inhibitors; both of them mimic the substrate resulting in inactive proteases. In this case, 67% of the interface residues are shared between two inhibitors as shown in magenta color. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Discussion
Here, we addressed the question of how a protein or protein family can utilize multibinding interfaces to bind to diverse partners and perform different functions. Protein functional moonlighting can be, in general, achieved through differential expression in different cell types, different cellular localization or oligomeric states. We report other mechanisms of functional switches through the use of promiscuous interfaces. (a) Overlap between functional pathways can be prevented by post-translational modifications occurring in nearby residues on the same interfacial region. (b) Interfaces can be reused in the pathways where the substrate should be passed from one protein to another sequentially and simultaneous binding to different proteins in a pathway is not permitted. (c) The same protein family can differ in key interacting residues on a multibinding interface to develop different specificities to structurally and evolutionary diverse protein partners, which might share similar function. (d) Finally, inhibitors can mimic substrates by attaching to their binding sites to prevent substrate binding.
Surprisingly, we showed a rather limited number of protein families with multibinding interfaces in the structure database (49 of 1098, 4.5%). It is consistent with previous studies on classification of domain interaction interfaces using sequence tags, which showed that only 4–6% of interface tags would interact with multiple families or superfamilies.39,40 One possible explanation is that promiscuous binding can be achieved through intrinsically disordered regions.41–43,50 According to another, so-called “conformational selection hypothesis,” there can exist an ensemble of conformations in dynamic equilibrium and evolution can select new binding interactions out of this ensemble rather than developing new ones.44 Moreover, promiscuous binding involves mostly transient complexes, which are under-represented in PDB as well as disordered and flexible regions. These other mechanisms are the subject of our future studies.
Methods
Construction of interaction data set
The interacting chains/domains have been selected from the CBM ftp site ftp://ftp.ncbi.nlm.nih.gov/pub/cbm,25 which maps protein domains defined by the CDD45 onto a set of structurally interacting domains from MMDB46 and verifies the interactions via CBMs. The CBM analysis uses conserved geometric interfaces to reduce the possibility of including nonbiological interactions. Two chains/domains qualify as interacting if they have at least five residue–residue contacts. A contact takes place between a residue from one domain and a residue from the other when the distance between any nonhydrogen atom of one residue is within 6 Å of any nonhydrogen atom of the other residue. The set of residues which make contacts between the chains form the interface. The CBM interaction data provide a list of interacting chains/domains categorized by CDD domain family type and by mode of interaction. Each domain family can interact with multiple domains and each domain pair can interact through multiple modes (distinct spatial orientations). To deal with redundancy of similarly defined protein domains, we record interactions between superfamilies, which represent clusters of CDD families based on the overlap in sequence space.47 It should be mentioned that different superfamilies usually encompass unrelated proteins with different folds and functions; families within superfamilies can be also quite diverse with the average percent identity being about 40% (for the set of 49 families with promiscuous interfaces, see next section). The list of families and superfamilies together with their average sequence identity can be accessed through the ftp site ftp://ftp.ncbi.nih.gov/pub/cbm/overlap_interface. Altogether we analyzed 1098 domain superfamilies and 3623 CBMs.
Defining multibinding interfaces
To define multibinding interfaces, we mapped all CBMs from all different interacting partners on one structure representative of a given domain superfamily (a common reference frame). To calculate the number of different binding partners and the node degree of interaction networks, it is important to correctly account for the redundancy not only in terms of the sequence similarity of shared domains but also redundancy of binding modes. To do this, we first collect all interactions involving a given CDD superfamily and group them by their binding partners (different binding partners are defined as those belonging to different CDD superfamilies and by their CBM (Fig. 1, row A). Within each of these CBMs, we choose a single representative structure as the one with the largest interaction interface (Fig. 1, row B). Then, we choose a template structure among the CBM representatives and thereafter map all of their interaction interfaces onto this template (Fig. 1, row C). The structural similarity and mapping is calculated using the VAST48 structural alignment algorithm. “Promiscuous” or “multibinding” interfaces are defined as those interfaces shared by at least two different binding partners (different CDD superfamilies) and having more than 50% interface overlap ratio. “Interface overlap ratio” is defined as the number of interfacial sites shared by both interacting partners divided by the union of interfacial sites from both interactions. The list of CDD superfamilies with multibinding interfaces together with their characteristics is given in Supporting Information Table S1; the shorter list for human interactions is given in Table I. Multibinding interfaces can be accessed through the ftp site ftp://ftp.ncbi.nih.gov/pub/cbm/overlap_interface/.
Figure 1.
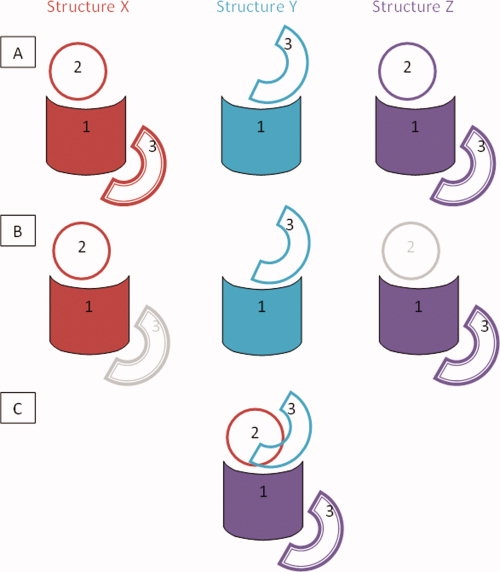
Procedure of mapping of multibinding interaction interface on the domain family. At the first stage (row A), all interactions involving a given domain family “1” are collected, which in this case involve interactions with domains “2” and “3” from structures “X”, “Y,” and “Z”. The interactions are categorized into CBMs and interaction 1:3 is found to occur in two unique CBMs (the second of which is distinguished with the compound border). At the second stage, the representatives for each CBM of each interaction are chosen (row B). At the third stage, all structures X, Y, and Z are structurally superimposed and interfaces from all CBMs are mapped onto a template of domain “1” (row C). [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Calculation of amino acid composition and sequence conservation
Amino acid composition is computed for the overall interface and multibinding interface positions for all CDD superfamilies in our data set. We calculate amino acid frequencies using all structurally similar proteins belonging to a given CDD superfamily. We distinguish three different groups of sites: noninterface, interface, and multibinding interface sites. “Noninterface sites” are defined as those sites which are outside the interface in all structural members of a given superfamily. Knowing the number of different amino acid types on the interface and multibinding region, we can estimate from the binomial distribution the probability of observing a given number (or higher) of a particular type of amino acid on the multibinding interfaces purely by chance. Two different probabilistic models were used, and we report the results on amino acid compositions which are consistent with both models (see Supporting Information Table S2 footnote for model descriptions). To calculate the association between amino acid type and interface type, we also used a chi-square contingency test with the Bonferroni correction for multiple testing.
Sequence conservation of interface and multibinding interface sites was calculated based on the alignment of structures from a given superfamily (see previous section) using the AL2CO program49 with the weighted frequency count and sum of pairs measure options. These two options were utilized to compensate for redundancy in the sequences and presence of similar amino acids in the aligned column. Conservation was reported as a Z-score and columns having more than 50% gaps were not considered for the calculation.
References
- 1.Gavin AC, Bosche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick JM, Michon AM, Cruciat CM, Remor M, Hofert C, Schelder M, Brajenovic M, Ruffner H, Merino A, Klein K, Hudak M, Dickson D, Rudi T, Gnau V, Bauch A, Bastuck S, Huhse B, Leutwein C, Heurtier MA, Copley RR, Edelmann A, Querfurth E, Rybin V, Drewes G, Raida M, Bouwmeester T, Bork P, Seraphin B, Kuster B, Neubauer G, Superti-Furga G. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature. 2002;415:141–147. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- 2.Krogan NJ, Peng WT, Cagney G, Robinson MD, Haw R, Zhong G, Guo X, Zhang X, Canadien V, Richards DP, Beattie BK, Lalev A, Zhang W, Davierwala AP, Mnaimneh S, Starostine A, Tikuisis AP, Grigull J, Datta N, Bray JE, Hughes TR, Emili A, Greenblatt JF. High-definition macromolecular composition of yeast RNA-processing complexes. Mol Cell. 2004;13:225–239. doi: 10.1016/s1097-2765(04)00003-6. [DOI] [PubMed] [Google Scholar]
- 3.Krause R, von Mering C, Bork P, Dandekar T. Shared components of protein complexes—versatile building blocks or biochemical artefacts= Bioessays. 2004;26:1333–1343. doi: 10.1002/bies.20141. [DOI] [PubMed] [Google Scholar]
- 4.Maniatis T, Reed R. An extensive network of coupling among gene expression machines. Nature. 2002;416:499–506. doi: 10.1038/416499a. [DOI] [PubMed] [Google Scholar]
- 5.Beckett D. Functional switches in transcription regulation; molecular mimicry and plasticity in protein-protein interactions. Biochemistry. 2004;43:7983–7991. doi: 10.1021/bi049890b. [DOI] [PubMed] [Google Scholar]
- 6.Nooren IM, Thornton JM. Diversity of protein-protein interactions. EMBO J. 2003;22:3486–3492. doi: 10.1093/emboj/cdg359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aloy P, Bottcher B, Ceulemans H, Leutwein C, Mellwig C, Fischer S, Gavin AC, Bork P, Superti-Furga G, Serrano L, Russell RB. Structure-based assembly of protein complexes in yeast. Science. 2004;303:2026–2029. doi: 10.1126/science.1092645. [DOI] [PubMed] [Google Scholar]
- 8.Russell RB, Sasieni PD, Sternberg MJ. Supersites within superfolds. Binding site similarity in the absence of homology. J Mol Biol. 1998;282:903–918. doi: 10.1006/jmbi.1998.2043. [DOI] [PubMed] [Google Scholar]
- 9.Henschel A, Kim WK, Schroeder M. Equivalent binding sites reveal convergently evolved interaction motifs. Bioinformatics. 2006;22:550–555. doi: 10.1093/bioinformatics/bti782. [DOI] [PubMed] [Google Scholar]
- 10.Kim PM, Lu LJ, Xia Y, Gerstein MB. Relating three-dimensional structures to protein networks provides evolutionary insights. Science. 2006;314:1938–1941. doi: 10.1126/science.1136174. [DOI] [PubMed] [Google Scholar]
- 11.Higurashi M, Ishida T, Kinoshita K. Identification of transient hub proteins and the possible structural basis for their multiple interactions. Protein Sci. 2008;17:72–78. doi: 10.1110/ps.073196308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nobeli I, Favia AD, Thornton JM. Protein promiscuity and its implications for biotechnology. Nat Biotechnol. 2009;27:157–167. doi: 10.1038/nbt1519. [DOI] [PubMed] [Google Scholar]
- 13.Kim WK, Ison JC. Survey of the geometric association of domain-domain interfaces. Proteins. 2005;61:1075–1088. doi: 10.1002/prot.20693. [DOI] [PubMed] [Google Scholar]
- 14.Tsai CJ, Lin SL, Wolfson HJ, Nussinov R. A dataset of protein-protein interfaces generated with a sequence-order-independent comparison technique. J Mol Biol. 1996;260:604–620. doi: 10.1006/jmbi.1996.0424. [DOI] [PubMed] [Google Scholar]
- 15.Keskin O, Nussinov R. Similar binding sites and different partners: implications to shared proteins in cellular pathways. Structure. 2007;15:341–354. doi: 10.1016/j.str.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 16.Tuncbag N, Gursoy A, Guney E, Nussinov R, Keskin O. Architectures and functional coverage of protein-protein interfaces. J Mol Biol. 2008;381:785–802. doi: 10.1016/j.jmb.2008.04.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keskin O, Tsai CJ, Wolfson H, Nussinov R. A new, structurally nonredundant, diverse data set of protein-protein interfaces and its implications. Protein Sci. 2004;13:1043–1055. doi: 10.1110/ps.03484604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bogan AA, Thorn KS. Anatomy of hot spots in protein interfaces. J Mol Biol. 1998;280:1–9. doi: 10.1006/jmbi.1998.1843. [DOI] [PubMed] [Google Scholar]
- 19.Moreira IS, Fernandes PA, Ramos MJ. Hot spots—a review of the protein-protein interface determinant amino-acid residues. Proteins. 2007;68:803–812. doi: 10.1002/prot.21396. [DOI] [PubMed] [Google Scholar]
- 20.Khersonsky O, Roodveldt C, Tawfik DS. Enzyme promiscuity: evolutionary and mechanistic aspects. Curr Opin Chem Biol. 2006;10:498–508. doi: 10.1016/j.cbpa.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 21.Humphris EL, Kortemme T. Design of multi-specificity in protein interfaces. PLoS Comput Biol. 2007;3:e164. doi: 10.1371/journal.pcbi.0030164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shifman JM, Mayo SL. Modulating calmodulin binding specificity through computational protein design. J Mol Biol. 2002;323:417–423. doi: 10.1016/s0022-2836(02)00881-1. [DOI] [PubMed] [Google Scholar]
- 23.Carbonell P, Nussinov R, del Sol A. Energetic determinants of protein binding specificity: insights into protein interaction networks. Proteomics. 2009;9:1744–1753. doi: 10.1002/pmic.200800425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chang CE, McLaughlin WA, Baron R, Wang W, McCammon JA. Entropic contributions and the influence of the hydrophobic environment in promiscuous protein-protein association. Proc Natl Acad Sci USA. 2008;105:7456–7461. doi: 10.1073/pnas.0800452105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shoemaker BA, Panchenko AR, Bryant SH. Finding biologically relevant protein domain interactions: conserved binding mode analysis. Protein Sci. 2006;15:352–361. doi: 10.1110/ps.051760806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim PM, Lu LJ, Xia Y, Gerstein MB. Relating three-dimensional structures to protein networks provides evolutionary insights. Science. 2006;314:1938–1941. doi: 10.1126/science.1136174. [DOI] [PubMed] [Google Scholar]
- 27.Ofran Y, Rost B. Analysing six types of protein-protein interfaces. J Mol Biol. 2003;325:377–387. doi: 10.1016/s0022-2836(02)01223-8. [DOI] [PubMed] [Google Scholar]
- 28.Villar HO, Kauvar LM. Amino acid preferences at protein binding sites. FEBS Lett. 1994;349:125–130. doi: 10.1016/0014-5793(94)00648-2. [DOI] [PubMed] [Google Scholar]
- 29.James LC, Tawfik DS. The specificity of cross-reactivity: promiscuous antibody binding involves specific hydrogen bonds rather than nonspecific hydrophobic stickiness. Protein Sci. 2003;12:2183–2193. doi: 10.1110/ps.03172703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Audette GF, Engelmann R, Hengstenberg W, Deutscher J, Hayakawa K, Quail JW, Delbaere LT. The 1.9 A resolution structure of phospho-serine 46 HPr from Enterococcus faecalis. J Mol Biol. 2000;303:545–553. doi: 10.1006/jmbi.2000.4166. [DOI] [PubMed] [Google Scholar]
- 31.Reizer J, Bergstedt U, Galinier A, Kuster E, Saier MH, Jr, Hillen W, Steinmetz M, Deutscher J. Catabolite repression resistance of gnt operon expression in Bacillus subtilis conferred by mutation of His-15, the site of phosphoenolpyruvate-dependent phosphorylation of the phosphocarrier protein HPr. J Bacteriol. 1996;178:5480–5486. doi: 10.1128/jb.178.18.5480-5486.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schumacher MA, Allen GS, Diel M, Seidel G, Hillen W, Brennan RG. Structural basis for allosteric control of the transcription regulator CcpA by the phosphoprotein HPr-Ser46-P. Cell. 2004;118:731–741. doi: 10.1016/j.cell.2004.08.027. [DOI] [PubMed] [Google Scholar]
- 33.Hicke L, Schubert HL, Hill CP. Ubiquitin-binding domains. Nat Rev Mol Cell Biol. 2005;6:610–621. doi: 10.1038/nrm1701. [DOI] [PubMed] [Google Scholar]
- 34.Bourne HR, Sanders DA, McCormick F. The GTPase superfamily: conserved structure and molecular mechanism. Nature. 1991;349:117–127. doi: 10.1038/349117a0. [DOI] [PubMed] [Google Scholar]
- 35.Wennerberg K, Rossman KL, Der CJ. The Ras superfamily at a glance. J Cell Sci. 2005;118:843–846. doi: 10.1242/jcs.01660. [DOI] [PubMed] [Google Scholar]
- 36.Schlumberger MC, Friebel A, Buchwald G, Scheffzek K, Wittinghofer A, Hardt WD. Amino acids of the bacterial toxin SopE involved in G nucleotide exchange on Cdc42. J Biol Chem. 2003;278:27149–27159. doi: 10.1074/jbc.M302475200. [DOI] [PubMed] [Google Scholar]
- 37.Margarit SM, Sondermann H, Hall BE, Nagar B, Hoelz A, Pirruccello M, Bar-Sagi D, Kuriyan J. Structural evidence for feedback activation by Ras.GTP of the Ras-specific nucleotide exchange factor SOS. Cell. 2003;112:685–695. doi: 10.1016/s0092-8674(03)00149-1. [DOI] [PubMed] [Google Scholar]
- 38.Kristelly R, Gao G, Tesmer JJ. Structural determinants of RhoA binding and nucleotide exchange in leukemia-associated Rho guanine-nucleotide exchange factor. J Biol Chem. 2004;279:47352–47362. doi: 10.1074/jbc.M406056200. [DOI] [PubMed] [Google Scholar]
- 39.Kim WK, Ison JC. Survey of the geometric association of domain-domain interfaces. Proteins. 2005;61:1075–1088. doi: 10.1002/prot.20693. [DOI] [PubMed] [Google Scholar]
- 40.Henschel A, Kim WK, Schroeder M. Equivalent binding sites reveal convergently evolved interaction motifs. Bioinformatics. 2006;22:550–555. doi: 10.1093/bioinformatics/bti782. [DOI] [PubMed] [Google Scholar]
- 41.Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 42.Tompa P, Szasz C, Buday L. Structural disorder throws new light on moonlighting. Trends Biochem Sci. 2005;30:484–489. doi: 10.1016/j.tibs.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 43.Fong J, Shoemaker BA, Garbuzynskiy SO, Lobanov MY, Galzitskaya OV, Panchenko AR. Intrinsic disorder in protein interactions: insights from a comprehensive structural analysis. PLoS Comput Biol. 2009;5:e1000316. doi: 10.1371/journal.pcbi.1000316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boehr DD, Wright PE. Biochemistry. How do proteins interact? Science. 2008;320:1429–1430. doi: 10.1126/science.1158818. [DOI] [PubMed] [Google Scholar]
- 45.Marchler-Bauer A, Anderson JB, Cherukuri PF, DeWeese-Scott C, Geer LY, Gwadz M, He S, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Liebert CA, Liu C, Lu F, Marchler GH, Mullokandov M, Shoemaker BA, Simonyan V, Song JS, Thiessen PA, Yamashita RA, Yin JJ, Zhang D, Bryant SH. CDD: a conserved domain database for protein classification. Nucleic Acids Res. 2005;33:D192–D196. doi: 10.1093/nar/gki069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen J, Anderson JB, DeWeese-Scott C, Fedorova ND, Geer LY, He S, Hurwitz DI, Jackson JD, Jacobs AR, Lanczycki CJ, Liebert CA, Liu C, Madej T, Marchler-Bauer A, Marchler GH, Mazumder R, Nikolskaya AN, Rao BS, Panchenko AR, Shoemaker BA, Simonyan V, Song JS, Thiessen PA, Vasudevan S, Wang Y, Yamashita RA, Yin JJ, Bryant SH. MMDB: Entrez's 3D-structure database. Nucleic Acids Res. 2003;31:474–477. doi: 10.1093/nar/gkg086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Geer LY, Domrachev M, Lipman DJ, Bryant SH. CDART: protein homology by domain architecture. Genome Res. 2002;12:1619–1623. doi: 10.1101/gr.278202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gibrat JF, Madej T, Bryant SH. Surprising similarities in structure comparison. Curr Opin Struct Biol. 1996;6:377–385. doi: 10.1016/s0959-440x(96)80058-3. [DOI] [PubMed] [Google Scholar]
- 49.Pei J, Grishin NV. AL2CO: calculation of positional conservation in a protein sequence alignment. Bioinformatics. 2001;17:700–712. doi: 10.1093/bioinformatics/17.8.700. [DOI] [PubMed] [Google Scholar]
- 50.Oldfield CJ, Meng J, Yang JY, Yang MQ, Uversky VN, Dunker AK. Flexible nets: disorder and induced fit in the associations of p53 and 14-3-3 with their partners. BMC Genomics. 2008;9:S1. doi: 10.1186/1471-2164-9-S1-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]