

Abstract
Toll-like receptors (TLRs) play a key role in the innate immune system. The TLR7, 8, and 9 compose a family of intracellularly localized TLRs that signal in response to pathogen-derived nucleic acids. So far, there are no crystallographic structures for TLR7, 8, and 9. For this reason, their ligand-binding mechanisms are poorly understood. To enable first predictions of the receptor–ligand interaction sites, we developed three-dimensional structures for the leucine-rich repeat ectodomains of human TLR7, 8, and 9 based on homology modeling. To achieve a high sequence similarity between targets and templates, structural segments from all known TLR ectodomain structures (human TLR1/2/3/4 and mouse TLR3/4) were used as candidate templates for the modeling. The resulting models support previously reported essential ligand-binding residues. They also provide a basis to identify three potential receptor dimerization mechanisms. Additionally, potential ligand-binding residues are identified using combined procedures. We suggest further investigations of these residues through mutation experiments. Our modeling approach can be extended to other members of the TLR family or other repetitive proteins.
Keywords: Toll-like receptor, leucine-rich repeats, protein-nucleic acid interaction, homology modeling
Introduction
Toll-like receptors (TLRs) play an essential role in the innate immunity, recognizing invasion of microbial pathogens and initiating intracellular signal transduction pathways to trigger expression of genes, the products of which can control innate immune responses.1 To understand how these receptors work, it is crucial to investigate them from a structural perspective. To date, only the crystal structures of the ectodomains of human TLR1/2/3/4 and mouse TLR3/4 have been determined.2–6 The progress of genome projects, however, already led to the identification of 13 TLRs in mammalian and more than 20 TLRs in nonmammalian. A total of more than 2000 TLR proteins has been sequenced.7 Thus, the structures of most TLRs are still unknown because structure determination by X-ray diffraction or nuclear magnetic resonance spectroscopy experiments remains time-consuming. Here, computational methods can help to bridge the gap between sequence determination and structure determination. To this end, homology modeling is a powerful tool to predict the three-dimensional structure of proteins.
Homology modeling is based on the assumption that similar sequences among evolutionarily related proteins share an overall structural similarity. The modeling procedure can be divided into a number of steps.8,9 First, selection of suitable template(s) related to the target sequence. A template segment assembly can usually improve the model quality.10 Second, alignment of the target sequence to the template(s). Third, building coordinates of the three-dimensional model based on the alignment. Fourth, evaluation of the model and its refinement. The resulting model can then be used to infer biological functionalities or to generate hypotheses for new experiments. A recent study on TLR411 highlighted the reliability and the significance of homology modeling applied to TLRs.
The structure of a TLR consists of a leucine-rich repeat (LRR) ectodomain, a helical transmembrane domain, and an intracellular Toll/IL-1 receptor homology (TIR) signaling domain.12 The ectodomain contains varying numbers of LRRs and resembles a solenoid bent into a horseshoe shape. At both ends there is a terminal LRR that shields the hydrophobic core of the horseshoe. These ectodomains are highly variable. They are directly involved in the recognition of a variety of pathogen-associated motifs including lipopolysaccharide, lipopeptide, cytosine–phosphate–guanine (CpG) DNA, flagellin, imidazoquinoline, and ds/ssRNA.13 Upon receptor activation, a TIR signaling complex is formed between the receptor and adaptor TIR domains.14
The receptors TLR7, 8, and 9 compose a family15 with a longer amino acid sequence than other TLRs. They are localized intracellularly and signal in response to nonself nucleic acids. They also contain an irregular segment between their LRR14 and 15. A recent study showed that the ectodomains of TLR9 and 7 are cleaved in the endolysosome to recognize ligands.16 Only the cleaved forms can recruit MyD88 on activation. In the absence of the crystallographic structures, we developed structural models of cleaved ligand-binding domains of TLR7/8/9 by homology modeling. From the structural model we predict potential ligand-binding sites and infer possible configurations of the receptor–ligand complex.
Results
Template identification
Our target structures are the cleaved functional ectodomains of the human TLR7/8/9 comprising LRR15–25 and N/C-terminal LRRs. All the six structure-known TLR homologues were employed as template sources: human TLR1/2/3/4 and mouse TLR3/4. The TLR ectodomain is composed of strictly organized LRRs. Nevertheless, the LRR number of cleaved ligand-binding domain of human TLR7/8/9 is 13 (LRR15–25 and N/C-terminal LRR),16 whereas the LRR number of the structure-known TLRs varies from 20 to 25. Therefore, none of the structure-known TLRs is suitable to serve as a full length template. To overcome this limitation, LRR segments with higher sequence similarity to the individual LRRs in the target were selected from the six complete homologous structures. The segments were then combined into the multiple templates. Figure 1 shows the multiple alignment models for the three proteins TLR 7/8/9, presenting the relationship between target and template segments. The sequence similarity between each LRR pair (target/template LRR) is listed in Table I. The average target–template similarities of TLR7/8/9 are 47.70, 47.20, and 46.78%, respectively.
Figure 1.

Models of multiple alignments between targets and templates. The numbers 01–25 denote the canonical LRRs; NT and CT denote N-/C-terminal LRRs. (A) Five segments selected from four structures (2Z80 chain A: human TLR2; 3CIG chain A: mouse TLR3; 2Z64 chain A: mouse TLR4; 2A0Z chain A: human TLR3) were used as templates for the human TLR7 ectodomain. (B) Six segments selected from three structures (2Z66 chain A: human TLR4; 2A0Z chain A: human TLR3; 2Z7X chain A: human TLR2) were used as templates for the human TLR8 ectodomain. (C) Four segments selected from two structures (2A0Z chain A: human TLR3; 2Z63 chain A: human TLR4) were used as templates for the human TLR9 ectodomain.
Table I.
Sequence Similarities (%) of Target–Template LRR Pairs
| NT | 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 | 23 | 24 | 25 | CT | Avg | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TRL7 | 28.60 | 58.30 | 60.00 | 41.70 | 47.10 | 58.30 | 46.20 | 52.00 | 50.00 | 33.30 | 46.20 | 48.30 | 50.00 | 47.70 |
| TRL8 | 29.40 | 41.70 | 52.00 | 50.00 | 57.70 | 60.00 | 40.60 | 44.00 | 52.00 | 36.00 | 60.00 | 42.30 | 53.30 | 47.20 |
| TRL9 | 32.30 | 58.30 | 48.00 | 50.00 | 44.40 | 41.70 | 46.90 | 51.90 | 36.00 | 54.20 | 50.00 | 52.00 | 42.40 | 46.78 |
In the header line, 15–25 denote canonical LRRs. NT and CT denote N-/C-terminal LRRs. Avg denotes the average values.
Remarkably, the group of TLR7/8/9 has a unique structural character that is absent in other TLRs. A specific segment (26–32 residue long) is located before LRR15, which was described as an undefined region.17–19 The sequence similarity search against Protein Data Bank (PDB) provided no significant results. Thus, we carried out secondary structure predictions for this region with four different methods. As example, the results for TLR9 are shown in Figure 2. All methods indicated a short β-sheet at position 3–5 of the segment, which is a prominent characteristic of LRRs. In addition, we compared its amino acid sequence with the consensus sequence of LRRs. The most significant positions of the LRR consensus sequence, LxxLxLxxNxL, are the four L residues which form the hydrophobic core of a LRR structure. Here, the letter L not only stands for leucine but also for other highly hydrophobic residues. As illustrated in Figure 2, the specific segment of TLR9 contains three of the four highly hydrophobic residues. Also, the corresponding segment of TLR7/8 has the same features. Thus we regard this segment as an irregular LRR. Because the N-terminal LRR together with LRR1–14 of the receptor ectodomain are deleted upon arriving in endolysosome, this irregular LRR may become a new N-terminal LRR of the truncated structure. Moreover, multiple alignments of all known mammalian sequences showed that this region is very variable within each of the TLR7/8/9 groups. The structure of this LRR may be relatively relaxed, because it lacks the first L residue that participates in forming the hydrophobic core of a LRR structure and the N residue that forms hydrogen bonds between neighboring LRRs. These features also support the hypothesis that this irregular LRR is an N-terminal LRR. For this reason, a N-terminal LRR with known structure was selected as corresponding template (Fig. 1).
Figure 2.

Irregular region analysis of TLR9. Four methods (PredictProtein,20 NNPREDICT,21 SSPro,22 and GOR IV23) were used to predict the secondary structures of the irregular region of TLR9. The results (italic letters) indicate a short β-sheet at position 3–5 of this region. Besides, this region matches the LRR pattern at three important positions (bold letters). These features support the presumption that this irregular region is a beginning N-terminal LRR after the ectodomain cleavage.
Structure modeling and evaluation
The three-dimensional coordinates of the models were created by MODELLER24 and modified by ModLoop.25 The final structures of the ectodomains of TLR7/8/9 reveal a large, arc-shaped assembly consisting of 11 canonical LRRs and two terminal LRRs, which adopted a right-handed solenoid structure (Fig. 3). The TLRs are distinct from other LRR proteins in that their LRR consensus motifs are often interrupted by extended insertions.26 Two 4–7-residue-long insertions protuberate from the structure surface at LRR18 and LRR20, respectively. These insertions are well conserved in length and position on the sequence level in the three TLRs. The models show that the insertions are all located on one face of the arc, whereas the other face is insertion-free (Fig. 3). The convex site β-sheets are directed toward the insertion face. This feature is consistent with the known structures of TLR1/2/3/4. Because all the known ligand-binding sites of TLR1/2/3/4 are on the insertion face of the structure, the insertions suggest some functional significance. In addition, the human TLR7/8/9 are glycosylated as it is the case for other TLRs. The glycans were shown to be nonfunctional for ligand binding.2–6 The NCBI protein database provides seven predicted N-linked glycosylation sites for TLR7/8 cleaved form and six for TLR9. All sites are located on the insertion-free faces. The PDB format files of the three final models are provided as Supporting Information Files 1–3. Evaluation of the models involved analysis of geometry, stereochemistry, and energy distributions in the models. The evaluation results (Table II) are indicative of a good quality of all three models.
Figure 3.
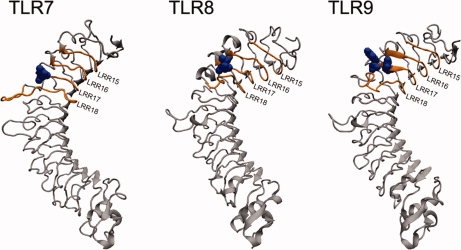
Structural models and ligand-binding regions of TLR7/8/9. Insertions are located on one face of the horseshoe, whereas the other face is insertion-free. The reported essential residues are located on the insertion face (labeled in blue). The orange regions are potential ligand-binding regions on the insertion face. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Table II.
Model Evaluation
| TLR7 | TLR8 | TLR9 | TLR3 | |
|---|---|---|---|---|
| ProQ_LG/MS | 5.340/0.461 | 4.613/0.402 | 4.355/0.339 | 7.923/0.526 |
| PROCHECK | 97.4% | 96.2% | 97.5% | 99.6% |
| ModFOLD_Q/P | 0.7588/0.01 | 0.7100/0.0126 | 0.7166/0.0121 | 0.7116/0.0124 |
| MetaMQAP_GDT/RMSD | 57.534/3.049 Å | 53.908/3.121 Å | 54.645/3.244 Å | 79.322/1.566 Å |
All these displayed scores indicate the models to be reliable in terms of overall packing. For comparison purpose, the values of TLR3 crystal structure (PDB code: 2A0Z) were also listed. ProQ_LG: >1.5 fairly good; >2.5 very good; >4 extremely good. ProQ_MS: >0.l, fairly good; >0.5, very good; >0.8, extremely good. PROCHECK: percentage of residues in most favored regions and additional allowed regions. ModFOLD_Q: >0.5, medium confidence; >0.75, high confidence. ModFOLD_P: <0.05, medium confidence; <0.01, high confidence. MetaMQAP_GDT/RMSD: an ideal model has a GDT score over 59 and a RMSD around 2.0 Å.
Potential ligand-binding residues
Several residues are essential for the ligand recognition: Asp543 in TLR8; Asp535 and Tyr537 in TLR9.18,27 Our models can help to understand the biological function of these residues (Fig. 3). According to these reported residues and the sequence comparison of TLR7/8/9, we inferred a ligand-binding region for TLR7/8/9, respectively (detailed in the Discussion section). It is located at the insertion face of the ectodomain around LRR17 (Fig. 3). Because of the considerable size of the nucleic acids, the ligand-binding region should contain more interacting residues. We identified potential ligand-binding residues in the ligand-binding region aside from the experimentally determined ones. To accomplish this goal we integrated results from manual analyses and automatic docking programs.
TLR3 is closely related to the TLR7/8/9 family because of its intracellular localization and nucleic acid ligand. Therefore, we used the recently published crystal structure of the mTLR3-dsRNA 2:1 complex6 as a guide to predict the essential interacting residues in TLR7/8/9. From all interacting residues of mTLR3, we identified three principles for the essential residues:
The essential residues are located on the protein surface and spatially close to each other.
They are highly conserved among species.
They create a nonnegatively charged environment.
On basis of these principles, we searched for additional residues that might be essential for ligand recognition. At first, surface residues that were spatially close (within two LRRs) to the experimentally determined essential residues were marked on the predicted models (orange regions in Fig. 3). These residues can be far from each other on the sequence level. Then, multiple alignments of all known mammalian TLR7/8/9 sequences were generated to select the highly conserved residues (columns with an asterisk in Fig. 4) from the marked ones. Notably, the L (or I, V) and N residues of the LRR consensus sequence LxxLxLxxNxL are conserved, but they cannot interact with ligands, because they are buried to form the hydrophobic core of an LRR. These residues are not labeled with asterisks in Figure 4.
Figure 4.
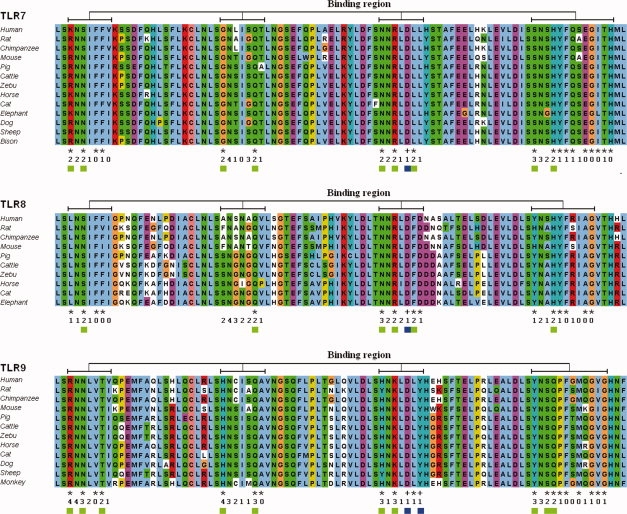
Partial multiple sequence alignments of different mammalian TLR7/8/9. The multiple sequence alignments represent the conservation of each residue in the potential ligand-binding regions (corresponding to the orange regions in Fig. 3). In the first line below the alignments, plus signs designate important residues as reported in the literature and the asterisks designate highly conserved positions. In the second line, the number of positive docking predictions of each position is indicated. In the third line, blue squares designate important residues as reported in the literature and green squares indicate the suggested ligand-binding residues. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Four protein-RNA docking programs and five protein-DNA docking programs (listed in the Materials and Methods section) were used to predict ligand-binding residues in TLR7/8 and TLR9. A residue from the prefiltered regions was marked as a ligand-binding residue, if it was positively predicted by at least two programs. In Figure 4, the number of positive predictions is listed for each target residue. The surface charge distributions of the regions of interest were calculated to verify the charge pattern in the predicted ligand-binding regions [Fig. 5(A)]. The resulting residues correspond to positively charged or neutral environments.
Figure 5.
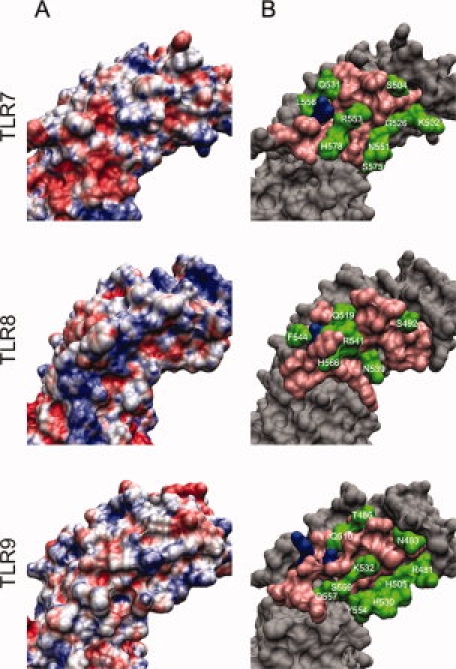
Surface analysis of ligand-binding regions of TLR7/8/9. (A) Surface charge distribution (APBS electrostatics) of ligand-binding regions of TLR7/8/9. Blue: positive charge; white: neutral; red: negative charge. (B) Important residues in ligand-binding regions of TLR7/8/9. Blue: important residues as reported in the literature; pink: residues close the blue ones but excluded from the potential ligand-binding residues through investigating processes; green: suggested potential ligand-binding residues (residue name and number are labeled). [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Figure 5(B) illustrates the protein surface residues from the different steps of our investigation for TLR7/8/9, respectively. All final predicted ligand-binding residues are summarized in Table III. These residues are indicated in green in both Figures 4 and 5(B).
Table III.
Potential Ligand-Binding Residues of TLR7/8/9
| TLR7 | K502 S504 G526 Q531 N551 R553 L556 S575 H578 |
| TLR8 | S492 Q519 N539 R541 F544 H566 |
| TLR9 | R481 N483 T486 H505 Q510 H530 K532 Y554 S556 Q557 |
Discussion
All three resulting models revealed similar conformations. This supports the assumption that TLR7/8/9 share a common ligand-binding and signaling mechanism.18 We compared and analyzed the predicted structures to suggest the receptor–ligand 2:1 complex models.
Ligand-binding region
The mouse TLR9 contains a short fragment in its LRR17 that is homologous to the methyl CpG DNA binding domain protein.27 The mutant of Asp535 and Tyr537 in this fragment abolished the TLR9 function.27 In the human TLR8, the Asp543 that corresponds to TLR9's Asp535 was determined to be required for the TLR8 function.18 Through sequence comparison, the Asp residue was found to be highly conserved in the TLR7/8/9 family but not in other TLRs. We considered this Asp to be significant for TLR7, because the TLR7/8/9 are highly homologous and their ligands are all pathogen-derived nucleic acids. In particular, the TLR7 and 8 are present as tandem duplication in many studied genomes discussed by Roach et al.15 In this regard, TLR7/8/9 have a ligand-binding region located spatially around the Asp residue.
We can further exclude the necessity of other ligand-binding regions on the ectodomains, because the minimum size of stimulatory oligonucleotides is six bases.28 These oligonucleotides are not large enough to reach another ligand-binding region on the receptor.
Receptor dimerization
The signaling mechanism of all TLRs is likely to involve dimerization of the ectodomains.18 However, this can be achieved in various ways by using different receptors and stimuli. TLR9 is a preformed dimer. The distance between both monomers is reduced upon contact with CpG DNA.29 TLR1/2 are activated and connected into a heterodimer by triacylated lipopeptide.4 TLR4 recognizes lipopolysaccharide indirectly through the coreceptor protein MD-2 and is induced to form a TLR4-MD-2 homodimer.5 In the TLR3 homodimer the dsRNA interacts with two regions of each receptor ectodomain. Direct protein–protein interactions between both receptors occur at their C-terminal LRRs, whereas the other regions are separated by the dsRNA.6
The structures obtained by the homology modeling together with the identification of possible ligand-binding sites can be used to derive a working hypothesis for the structure of the receptor–ligand complex. We propose three possible receptor–ligand 2:1 complex models for the TLR7/8/9 family (Fig. 6). In all three models, the ssRNA or CpG DNA ligand interacts with the binding region on the insertion surface of both receptor ectodomains. The ectodomains are on opposite sides of the ligand. Simultaneously, the intracellular TIR domains are also in a dimer configuration. Thus the C-terminal LRRs of each monomer, which are connected to the TIR through a 20-amino-acid-long transmembrane stretch, are spatially close to each other. The main difference between the three models is the relative position of the ectodomains. In the first model [Fig. 6(A)], both C-terminal LRRs are brought into proximity, forming a protein–protein contact. Both binding regions sandwich the ligand. In the second model [Fig. 6(B)], both receptors are shifted apart along the ligand extending directions back to back. In the third model [Fig. 6(C)], both receptors are shifted in opposite directions face to face. Obviously, the minimum ligand size required by the first model is the smallest. Therefore, a CpG DNA of six bases is already long enough to stimulate TLR9.28 The minimum size required by the second and third models is larger. These two models, however, cannot be excluded, because there is so far no evidence that TLRs have only one dimer form. Without the crystal structure of their ligands, it is difficult to determine a more precise model for the receptor dimerization. Hence, it remains interesting to study the atomic structure of the stimulatory ssRNA/CpG DNA and to further determine the detailed interactions between ligands and receptors.
Figure 6.
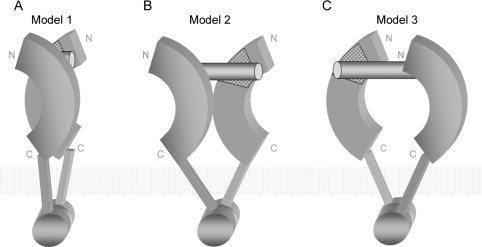
Proposed models of receptor–ligand 2:1 complex.
Materials and Methods
Template identification and sequence alignments
Amino acid sequences with LRR motif partitions of human TLR7/8/9 ectodomain were extracted from TollML.7 TollML is a specialized database of TLR sequence motifs, derived from the NCBI protein database.30 Multiple sequence alignments of all individual LRRs of TLR7/8/9 to the LRR consensus sequence are provided as Supporting Information File 4. Because the TLR ectodomain is a repetitive protein (LRRs), we selected and combined segments from all the six known TLR ectodomain structures into multiple templates to optimize the sequence similarity between targets and templates. The six candidate templates were human TLR1/2/3/4 and mouse TLR3/4 and were obtained from the PDB.31 The PDB codes are 2Z7X , 2Z80, 2A0Z, 2Z63, 2Z66, 3CIG and 2Z64, respectively. Three steps led to the identification of structural templates. First, we partitioned the known structures into a total of 136 individual LRRs. Because of the irregularity of the LRR sequences, the partition according to the LRR consensus sequences was performed manually. Second, the LRRs were collected into the LRRML database,32 which can return the most similar LRR for an input LRR sequence through similartiy search. Third, optimal template pieces for each target were found and combined to generate multiple alignments. Because the TLR LRRs follow common characteristic consensus sequences, target–template alignments were generated more accurately by hand than through software.
Structure construction and analysis
The initial three-dimensional coordinates of the models were generated by the fully automated program MODELLER 9v3.24 The input files were the multiple alignment file and the coordinate files of the templates. The ligand-binding domains of TLR7/8/9 contain two 4–7-residue-long insertion regions, which correspond to gaps in the multiple alignment. During the modeling these regions became loop structures, which limited the model accuracy. ModLoop25 was used to modify these loop regions. The resulting models were evaluated by PROCHECK,33 ProQ,34 ModFOLD,35 and MetaMQAP.36
The detection of potential ligand-binding sites was achieved through residue conservation analysis, surface charge analysis, and several automatic docking programs. BindN,37 DP-Bind,38 DBS-PRED,39 DBS-PSSM,40 and PreDs41 were used for protein-DNA docking of TLR9. BindN, Pprint,42 RNAbindR,43 and RISP44 were used for protein-RNA docking of TLR7/8.
Conclusions
We predicted three-dimensional structures of the closely related TLR7/8/9 ligand-binding domains by homology modeling. LRR segments were selected from known TLR structures, which are locally optimal for the target sequences. These segments were then combined into multiple templates.
To predict essential residues in the ligand-binding region, sequence conservation and charge distributions were examined. Only highly conserved nonnegative residues that are positively predicted by at least two docking programs can be considered as potential ligand-binding residues. Based on these models we also suggest three possible receptor dimerization schemes which require different minimum ligand sizes.
In summary, our models provide a structural framework that can act as a guide to develop a functional hypothesis to interpret experimental data of TLR7/8/9. They may also facilitate efforts to design further site-directed mutagenesis to learn the ligand recognition and the downstream signaling mechanisms. The presented modeling approach can be extended to other repetitive protein domains.
References
- 1.Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol. 2005;17:1–14. doi: 10.1093/intimm/dxh186. [DOI] [PubMed] [Google Scholar]
- 2.Bell JK, Botos I, Hall PR, Askins J, Shiloach J, Segal DM, Davies DR. The molecular structure of the Toll-like receptor 3 ligand-binding domain. Proc Natl Acad Sci USA. 2005;102:10976–10980. doi: 10.1073/pnas.0505077102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Choe J, Kelker MS, Wilson IA. Crystal structure of human toll-like receptor 3 (TLR3) ectodomain. Science. 2005;309:581–585. doi: 10.1126/science.1115253. [DOI] [PubMed] [Google Scholar]
- 4.Jin MS, Kim SE, Heo JY, Lee ME, Kim HM, Paik SG, Lee H, Lee JO. Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell. 2007;130:1071–1082. doi: 10.1016/j.cell.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 5.Kim HM, Park BS, Kim JI, Kim SE, Lee J, Oh SC, Enkhbayar P, Matsushima N, Lee H, Yoo OJ, Lee JO. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell. 2007;130:906–917. doi: 10.1016/j.cell.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 6.Liu L, Botos I, Wang Y, Leonard JN, Shiloach J, Segal DM, Davies DR. Structural basis of toll-like receptor 3 signaling with double-stranded RNA. Science. 2008;320:379–381. doi: 10.1126/science.1155406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gong J, Wei T, Jamitzky F, Heckl WM, Rössle SC. TollML-a user editable database for Toll-like receptors and ligands. 2007. Proceedings of the Second IAPR International Workshop on Pattern Recognition in Bioinformatics, International Association of Pattern Recognition, Singapore, Paper ID 231.
- 8.Ginalski K. Comparative modeling for protein structure prediction. Curr Opin Struct Biol. 2006;16:172–177. doi: 10.1016/j.sbi.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 9.Kopp J, Schwede T. Automated protein structure homology modeling: a progress report. Pharmacogenomics. 2004;5:405–416. doi: 10.1517/14622416.5.4.405. [DOI] [PubMed] [Google Scholar]
- 10.Bujnicki JM. Protein-structure prediction by recombination of fragments. Chembiochem. 2006;7:19–27. doi: 10.1002/cbic.200500235. [DOI] [PubMed] [Google Scholar]
- 11.Kubarenko A, Frank M, Weber AN. Structure-function relationships of Toll-like receptor domains through homology modelling and molecular dynamics. Biochem Soc Trans. 2007;35:1515–1518. doi: 10.1042/BST0351515. [DOI] [PubMed] [Google Scholar]
- 12.Brodsky I, Medzhitov R. Two modes of ligand recognition by TLRs. Cell. 2007;130:979–981. doi: 10.1016/j.cell.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 13.Gay NJ, Gangloff M. Structure and function of Toll receptors and their ligands. Annu Rev Biochem. 2007;76:141–165. doi: 10.1146/annurev.biochem.76.060305.151318. [DOI] [PubMed] [Google Scholar]
- 14.O'Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007;7:353–364. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- 15.Roach JC, Glusman G, Rowen L, Kaur A, Purcell MK, Smith KD, Hood LE, Aderem A. The evolution of vertebrate Toll-like receptors. Proc Natl Acad Sci USA. 2005;102:9577–9582. doi: 10.1073/pnas.0502272102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ewald SE, Lee BL, Lau L, Wickliffe KE, Shi GP, Chapman HA, Barton GM. The ectodomain of Toll-like receptor 9 is cleaved to generate a functional receptor. Nature. 2008;456:658–662. doi: 10.1038/nature07405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bell JK, Mullen GE, Leifer CA, Mazzoni A, Davies DR, Segal DM. Leucine-rich repeats and pathogen recognition in Toll-like receptors. Trends Immunol. 2003;24:528–533. doi: 10.1016/s1471-4906(03)00242-4. [DOI] [PubMed] [Google Scholar]
- 18.Gibbard RJ, Morley PJ, Gay NJ. Conserved features in the extracellular domain of human toll-like receptor 8 are essential for pH-dependent signaling. J Biol Chem. 2006;281:27503–27511. doi: 10.1074/jbc.M605003200. [DOI] [PubMed] [Google Scholar]
- 19.Matsushima N, Tanaka T, Enkhbayar P, Mikami T, Taga M, Yamada K, Kuroki Y. Comparative sequence analysis of leucine-rich repeats (LRRs) within vertebrate toll-like receptors. BMC Genomics. 2007;8:124. doi: 10.1186/1471-2164-8-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rost B, Yachdav G, Liu J. The PredictProtein server. Nucleic Acids Res. 2004;32:W321–W326. doi: 10.1093/nar/gkh377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kneller DG, Cohen FE, Langridge R. Improvements in protein secondary structure prediction by an enhanced neural network. J Mol Biol. 1990;214:171–182. doi: 10.1016/0022-2836(90)90154-E. [DOI] [PubMed] [Google Scholar]
- 22.Cheng J, Sweredoski M, Baldi P. Accurate prediction of protein disordered regions by mining protein structure data. Data Min Knowl Discov. 2005;11:213–222. [Google Scholar]
- 23.Garnier J, Gibrat JF, Robson B. GOR method for predicting protein secondary structure from amino acid sequence. Methods Enzymol. 1996;266:540–553. doi: 10.1016/s0076-6879(96)66034-0. [DOI] [PubMed] [Google Scholar]
- 24.Fiser A, Do RK, Sali A. Modeling of loops in protein structures. Protein Sci. 2000;9:1753–1773. doi: 10.1110/ps.9.9.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fiser A, Sali A. ModLoop: automated modeling of loops in protein structures. Bioinformatics. 2003;19:2500–2501. doi: 10.1093/bioinformatics/btg362. [DOI] [PubMed] [Google Scholar]
- 26.Bell JK, Askins J, Hall PR, Davies DR, Segal DM. The dsRNA binding site of human Toll-like receptor 3. Proc Natl Acad Sci USA. 2006;103:8792–8797. doi: 10.1073/pnas.0603245103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rutz M, Metzger J, Gellert T, Luppa P, Lipford GB, Wagner H, Bauer S. Toll-like receptor 9 binds single-stranded CpG-DNA in a sequence- and pH-dependent manner. Eur J Immunol. 2004;34:2541–2550. doi: 10.1002/eji.200425218. [DOI] [PubMed] [Google Scholar]
- 28.He G, Patra A, Siegmund K, Peter M, Heeg K, Dalpke A, Richert C. Immunostimulatory CpG oligonucleotides form defined three-dimensional structures: results from an NMR study. ChemMedChem. 2007;2:549–560. doi: 10.1002/cmdc.200600262. [DOI] [PubMed] [Google Scholar]
- 29.Latz E, Verma A, Visintin A, Gong M, Sirois CM, Klein DC, Monks BG, McKnight CJ, Lamphier MS, Duprex WP, Espevik T, Golenbock DT. Ligand-induced conformational changes allosterically activate Toll-like receptor 9. Nat Immunol. 2007;8:772–779. doi: 10.1038/ni1479. [DOI] [PubMed] [Google Scholar]
- 30.Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K, Chetvernin V, Church DM, Dicuccio M, Edgar R, Federhen S, Feolo M, Geer LY, Helmberg W, Kapustin Y, Khovayko O, Landsman D, Lipman DJ, Madden TL, Maglott DR, Miller V, Ostell J, Pruitt KD, Schuler GD, Shumway M, Sequeira E, Sherry ST, Sirotkin K, Souvorov A, Starchenko G, Tatusov RL, Tatusova TA, Wagner L, Yaschenko E. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2008;36:D13–D21. doi: 10.1093/nar/gkm1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wei T, Gong J, Jamitzky F, Heckl WM, Stark RW, Roessle SC. LRRML: a conformational database and an XML description of leucine-rich repeats (LRRs) BMC Struct Biol. 2008;8:47. doi: 10.1186/1472-6807-8-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 34.Wallner B, Elofsson A. Identification of correct regions in protein models using structural, alignment, and consensus information. Protein Sci. 2006;15:900–913. doi: 10.1110/ps.051799606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McGuffin LJ. The ModFOLD server for the quality assessment of protein structural models. Bioinformatics. 2008;24:586–587. doi: 10.1093/bioinformatics/btn014. [DOI] [PubMed] [Google Scholar]
- 36.Pawlowski M, Gajda MJ, Matlak R, Bujnicki JM. MetaMQAP: a meta-server for the quality assessment of protein models. BMC Bioinformat. 2008;9:403. doi: 10.1186/1471-2105-9-403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang L, Brown SJ. BindN: a web-based tool for efficient prediction of DNA and RNA binding sites in amino acid sequences. Nucleic Acids Res. 2006;34:W243–W248. doi: 10.1093/nar/gkl298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hwang S, Gou Z, Kuznetsov IB. DP-Bind: a web server for sequence-based prediction of DNA-binding residues in DNA-binding proteins. Bioinformatics. 2007;23:634–636. doi: 10.1093/bioinformatics/btl672. [DOI] [PubMed] [Google Scholar]
- 39.Ahmad S, Gromiha MM, Sarai A. Analysis and prediction of DNA-binding proteins and their binding residues based on composition, sequence and structural information. Bioinformatics. 2004;20:477–486. doi: 10.1093/bioinformatics/btg432. [DOI] [PubMed] [Google Scholar]
- 40.Ahmad S, Sarai A. PSSM-based prediction of DNA binding sites in proteins. BMC Bioinformat. 2005;6:33. doi: 10.1186/1471-2105-6-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsuchiya Y, Kinoshita K, Nakamura H. PreDs: a server for predicting dsDNA-binding site on protein molecular surfaces. Bioinformatics. 2005;21:1721–1723. doi: 10.1093/bioinformatics/bti232. [DOI] [PubMed] [Google Scholar]
- 42.Kumar M, Gromiha MM, Raghava GP. Prediction of RNA binding sites in a protein using SVM and PSSM profile. Proteins. 2008;71:189–194. doi: 10.1002/prot.21677. [DOI] [PubMed] [Google Scholar]
- 43.Terribilini M, Sander JD, Lee JH, Zaback P, Jernigan RL, Honavar V, Dobbs D. RNABindR: a server for analyzing and predicting RNA-binding sites in proteins. Nucleic Acids Res. 2007;35:W578–W584. doi: 10.1093/nar/gkm294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tong J, Jiang P, Lu ZH. RISP: a web-based server for prediction of RNA-binding sites in proteins. Comput Methods Programs Biomed. 2008;90:148–153. doi: 10.1016/j.cmpb.2007.12.003. [DOI] [PubMed] [Google Scholar]